

Abstract
Paternal deletion of the imprinting control region (ICR) KvDMR1 results in loss of expression of the Kcnq1ot1 noncoding RNA and derepression of flanking paternally silenced genes. Truncation of Kcnq1ot1 also results in the loss of imprinted expression of these genes in most cases, demonstrating a role for the RNA or its transcription in gene silencing. However, enhancer-blocking studies indicate that KvDMR1 also contains chromatin insulator or silencer activity. In this report we demonstrate by electrophoretic mobility shift assays and chromatin immunoprecipitation the existence of two CTCF binding sites within KvDMR1 that are occupied in vivo only on the unmethylated paternally derived allele. Methylation interference and mutagenesis allowed the precise mapping of protein-DNA contact sites for CTCF within KvDMR1. Using a luciferase reporter assay, we mapped the putative transcriptional promoter for Kcnq1ot1 upstream and to a site functionally separable from enhancer-blocking activity and CTCF binding sites. Luciferase reporter assays also suggest the presence of an additional cis-acting element in KvDMR1 upstream of the putative promoter that can function as an enhancer. These results suggest that the KvDMR1 ICR consists of multiple, independent cis-acting modules. Dissection of KvDMR1 into its functional components should help elucidate the mechanism of its function in vivo.
The KvDMR1 imprinting control region (ICR; also known as IC2) is differentially methylated on the maternal chromosome and is associated with a noncoding RNA (ncRNA) of unknown function termed Kcnq1ot1 (8, 21, 41) (see Fig. 1a). Paternal inheritance of a deletion of KvDMR1 results in loss of expression of Kcnq1ot1 and derepression in cis of imprinted genes both telomeric (Osbpl5, Phlda2, Slc22a18, and Cdkn1c) and centromeric (Kcnq1, Tssc4, Cd81, and Ascl2) to the mutation, indicating that the unmethylated paternal allele of KvDMR1 and/or the expression of the Kcnq1ot1 RNA regulates imprinted expression by silencing genes on the paternal chromosome (11, 23, 28). Although the molecular mechanisms by which KvDMR1 and most other ICRs function are not completely defined, it has been proposed that these regulatory elements employ two distinct mechanisms (24, 35), one which utilizes enhancer-blocking or “insulation” that relies on the protein CTCF and another which operates through ncRNAs.
FIG. 1.

CTCF binding sites map within KvDMR1. (a) Imprinted domain in mouse distal chromosome 7. Imprinted genes are shown as white or gray (imprinted expression only in placenta) boxes, while black boxes represent nonimprinted genes. The arrows indicate the direction of transcription. (b) Restriction map of the mouse KvDMR1 locus with the nucleotide positions indicated as in AF119385; the thick bars above the map are the two CpG islands within KvDMR1. The thick bars below the map indicate the genomic region deleted in mice described by Fitzpatrick et al. (11) (black) and the fragment tested for enhancer blocking activity by Kanduri et al. (17) (gray). The lines with double arrowheads above the restriction map show the location of the regions analyzed in the CTCF ChIP assay (see Fig. 2a). The positions of 11 overlapping DNA fragments used as probes in an EMSA are shown as black lines below the map of the locus; the numbers refer to the names (e.g., mKD1) of forward and reverse PCR primers used to generate each EMSA probe. The asterisks indicate the positions of two CTCF binding sites (CTS) detected by EMSA and fine-mapped by methylation interference (see below). (c) EMSA analysis of CTCF binding to mouse KvDMR1 locus. DNA fragments shown in panel b were screened for binding to in vitro translated full-length CTCF (ivtFLCTCF) and 11-ZF domain of this protein. A DNA fragment (H19 DMD4) from the H19 ICR which is known to bind CTCF was used as a positive control for CTCF-binding. Only probes 1-2 and 7-8 were able to form DNA-protein complexes with full-length CTCF in these experiments. (d) Probes 1-2 (CTS1) and 7-8 (CTS2) form a complex with CTCF from HeLa cell nuclear extract. In the last lane, the complex between the extract and probes for CTS1 and CTS2 is supershifted with mouse monoclonal-CTCF antibody (36). The panels labeled “mutant” show gel shift experiments with the in vitro translated protein and nuclear extract demonstrating the effect of mutations introduced in CTS1 and CTS2 (see panel g) on the ability of these sites to bind CTCF. (e) SssI methyltransferase was used to in vitro methylate cytosine residues in CpG dinucleotides in the CTCF-positive DNA fragments (CTS1 [1-2] and CTS2 [7-8]). Prior methylation completely abrogated binding of CTCF at CTS2 and almost completely at CTS1. Digestion of unmethylated or SssI-methylated probe with methylation-sensitive enzyme BstUI demonstrates that probes were completely methylated following SssI treatment (+BstUI). (f) Methylation interference analysis of CTS1 and CTS2 sequences. Lanes F, free DNA probes separated from the CTCF-bound probes (lanes B). The nucleotides in contact with CTCF are shown as black dots. (g) CTS1 and CTS2 were mutagenized in the context of the 1-10 fragment. Most guanine nucleotides that are in direct contact with CTCF were replaced by adenosines or thymidines (shown in italics above and below the original sequence). (h) Sequence alignment of mouse CTS1 and CTS2 with sequences within fragments (KvDMR-HBi and KvDMR-FBi) previously shown to bind in vitro transcribed/translated CTCF (7). Identical nucleotides are shaded. Larger, bold nucleotides represent G (C when on opposite strand) residues in contact with CTCF in vitro as determined by methylation interference (above).
The most extensively studied ICR, the H19/Igf2 differentially methylated region (DMR) is of the insulator type. The capacity of this regulatory element to block enhancer-promoter interactions has been demonstrated in transgenes (14), by positioning it downstream of the H19 gene between the endodermal and mesodermal enhancers (16), and in several cell culture-based systems (3, 14, 16, 18). Importantly, it has been shown that the multifunctional transcriptional regulator CTCF binds to the H19/Igf2 DMR in a methylation-sensitive (14, 18) and parent-of-origin-dependent manner (19, 42). Mutation of CTCF binding sites results in a loss of insulator activity in cell culture-based assays (14, 16, 19) and in the mouse (33, 34, 39).
Due to the precedent of the H19/Igf2 DMR, we initially tested KvDMR1 for insulator (position dependent) and silencer (position independent) activity in two independent cell culture-based enhancer-blocking assays. One system was an episomal enhancer-blocking assay using the hepatoma cell line Hep3B (18); the second assay involved integration of an expression construct into the genome of Jurkat cells (50). In both assays, a 3.6-kb fragment containing KvDMR1 showed significant activity by repressing reporter gene expression by more than 95% in a position-dependent fashion (i.e., only when the test fragment was inserted between the reporter gene's enhancer and promoter), suggesting that this locus can function as an insulator. Interestingly, the insulator activity depended on the orientation of the 3.6-kb fragment with respect to the reporter gene's promoter (17). In other enhancer-blocking systems, smaller fragments of KvDMR1 function as silencers (i.e., in a position-independent manner) (27, 43); these contrasting results most likely reflect differences in the size and location of DNA fragments used in the assays and/or the cell type used (see Discussion).
The case for the involvement of an ncRNA in imprinted gene expression was first described for the mouse Igf2r locus where the noncoding transcript Air was shown to be required for silencing of three genes on the paternal chromosome (40). KvDMR1 also contains the promoter for the 60-kb-long Kcnq1ot1 ncRNA (21, 29, 41). Truncation of Kcnq1ot1 in both episomal expression vectors in cell culture (44) and in the mouse (28; J.-Y. Shin, G. V. Fitzpatrick, and M. J. Higgins, unpublished data) suggests that this ncRNA or its transcription does, in fact, function in paternal gene silencing in this domain. However, these more recent finding do not exclude the possibility that KvDMR1 might also silence genes by mechanisms other than those involving transcription of Kcnq1ot1. In this report, we demonstrate methylation-sensitive binding of the insulator-associated protein CTCF to KvDMR1 at two sites within a 1,050-bp sequence exhibiting the maximum repressive activity in an enhancer-blocking assay. Furthermore, we show that in vivo CTCF binds to KvDMR1 in a parent-of-origin manner only to the unmethylated paternal allele. Moreover, we provide unequivocal evidence that the repressive activity of KvDMR1 in an enhancer-blocking assay is completely separable from the Kcnq1ot1 promoter. Finally, we provide evidence that a transcriptional enhancer is located just upstream of the Kcnq1ot1 promoter. Our results demonstrate that KvDMR1 contains distinct repressive, promoter, and enhancer modules, defining it as a multipartite, multifunctional regulatory element.
MATERIALS AND METHODS
EMSA.
Eleven overlapping fragments covering the mouse KvDMR1 locus were amplified by PCR (primer sequences available on request); one primer of each pair was labeled with [γ-32P]ATP at the 5′ end by T4 polynucleotide kinase. To test for methylation sensitivity of protein binding, some probes were methylated using SssI methyltransferase (New England BioLabs [NEB]) by the following protocol: 20 ml of PCR product was combined with 2.7 ml of NEB buffer 2, 3 μl (12 U) of SssI methylase, and 1 ml of S-adenosylmethionine (32 mM) and incubated at 37°C. After 3 h, we added an additional 0.5 μl of NEB buffer 2, 3 ml (12 U) of SssI methylase, and 1 μl of S-adenosylmethionine (32 mM) and incubated the mixture at 37°C for three more hours. Complete methylation of the DNA fragments was assessed by digesting them with the methylation-sensitive enzyme BstUI. DNA fragments were gel purified, and equal amounts of each fragment were used for electrophoretic mobility shift assays (EMSAs) as described previously (2). Briefly, binding reactions for EMSA were carried out for 30 min at room temperature with 10.0 ml of in vitro synthesized CTCF or 1 ml of nuclear extract in polyethylene glycol buffer [the 2× buffer contains 100 mM HEPES, 500 mM NaCl, 5 mM MgCl2, 10 mM dithiothreitol, 1 mM ZnSO4, 100 μg/ml poly(dI-dC), 0.02% NP-40, 20% polyethylene glycol 6000]. DNA-protein complexes were resolved on 5% nondenaturing polyacrylamide gels run in 0.5× Tris borate-EDTA buffer. Full-length (FL) human CTCF and the 11-zinc finger (ZF) domain of CTCF were synthesized from pCITE-FL and pCITE-11ZF expression vectors, respectively (2), using the TNT reticulocyte lysate-coupled in vitro transcription-translation system (Promega). HeLa nuclear extracts were prepared as described previously (9). In supershift EMSA experiments, binding reactions included 1 ml of a mixture of nine mouse monoclonal antibodies raised against human recombinant CTCF expressed in Pichia pastoris (36). EMSA with nuclear extract was performed in the presence of cold, double-stranded competitor DNAs: poly(dI-dC), poly(dG) · poly(dC), and oligonucleotides containing strong binding sites for both Sp1 and Egr1 proteins (9).
Derivation of mouse primary lung and embryonic fibroblasts.
Primary lung fibroblasts were derived from 4-week-old C57BL/6J mice carrying maternally or paternally derived deletions of KvDMR1 (11) and wild-type littermates. Intact lungs were minced in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum, and the drained pieces were distributed in a plastic culture dish. Glass slides were positioned on top of the tissue pieces to immobilize them while 10 ml of medium was added to the plate. Following 2 weeks of incubation at 37°C, cells that had migrated out of the tissue pieces were harvested by mild trypsinization. Following inactivation and removal of the trypsin, the cells were plated in fresh DMEM plus 10% fetal calf serum. Mouse embryonic fibroblasts (MEFs) were derived from embryonic day 13.5 embryos from reciprocal crosses between C57BL/6J and SD7 mice. SD7 mice are congenic for distal chromosome 7 from Mus spretus in a C57BL/6J background and were kindly provided by W. Reik (Babraham Institute). Eviscerated embryos were placed in separate wells of a six-well tissue culture dish containing a small volume of trypsin-EDTA. Embryos were minced with a sterile razor blade, and the mixture was incubated at 37°C for 30 min; during the incubation, embryonic tissue was dissociated by vigorous pipetting. MEF culture medium (DMEM-high glucose supplemented with 10% fetal bovine serum, 1× penicillin-streptomycin-l-glutamine, and 0.5 mg/ml Fungizone) was added, and the cell suspensions were spun down, resuspended in fresh medium, and transferred to T75 flasks. MEFs were frozen down after the passage 1 cells reached confluence.
ChIP.
Chromatin immunoprecipitation (ChIP) assays were carried out according to the protocol supplied by Upstate Biotechnology. Briefly, cross-linked chromatin from 5 × 106 primary lung fibroblasts or MEFs was sonicated, diluted 10-fold in ChIP dilution buffer (10 ml), and precleared with 80 ml of salmon sperm DNA-protein A/G agarose for 1 h at 4°C with rotation. A portion of the protein A-purified chromatin (200 ml) was used to prepare DNA as the “input” sample. Antibodies (2 to 10 ml) were added to 4.5 ml of clarified chromatin and incubated overnight with rotation. For CTCF a mixture of nine monoclonal antibodies (36) was used; anti-dimethyl-histone H3 (Lys 9) (catalogue no. 07-441) was obtained from Upstate Cell Signaling Solutions. Sixty milliliters of protein A/G agarose beads was then added to the antibody-chromatin mix and incubated at 4°C for 1 to 2 h with rotation. The complex was collected by gentle centrifugation and washed three times, and the bound chromatin was eluted twice in 500 ml of elution buffer. After 20 μl of 5 M NaCl was added, protein-DNA cross-links were reversed by heating at 65°C for 4 h. Samples were treated with proteinase K, and the DNA was purified by phenol-chloroform extraction and ethanol precipitation. DNA pellets were dissolved in 50 ml of Tris-EDTA buffer, further purified using a MiniElute Reaction Cleanup kit (QIAGEN, Valencia, CA), and eluted in 20 ml of 2 mM Tris, pH 8.0. The purified DNA was used as a template in quantitative real-time PCR assays or for endpoint PCR followed by direct sequencing for allele-specific analysis.
Primers used in the real-time PCR analysis for KvDMR1 CTCF target site 1 (CTS1) were CTS1F (5′-GGCTGCCACGTCACCAA-3′) and CTS1R (5′-CCTGACTGGACCAAAATGCA-3′), and for KvDMR1 CTS2 they were CTS2F (5′-TTTTTCACGGTGAGGTCATATCAGC-3′) and CTS2R (5′-GAGGTGTAGTGCTCAAGTGATCCGA-3′). As a positive control for the ChIP experiments, a known insulator site of the mouse c-myc oncogene was employed using the following primers: mycN_IP2f, 5′-AAGGAAGCATCTTCCCAGAACCTG-3′; and mycN_IP2r, 5′-AAAGTAAGTGTGCCCTCTACTGGCC-3′ (12). To control for amplification due to nonspecific binding of long chromatin fragments to agarose beads, fluorescence levels for CTS1 and CTS2 were normalized using fluorescence levels generated by primer pairs amplifying loci 1.0 kb upstream (NC2F, 5′-AGTCATCTTTGGCAAGAGAGCTTCC-3′; NC2R, 5′-CAAACCACCCCTACCCAGAATTGA-3′) and 1.3 kb downstream (NC1F, 5′-TCCCTATGAAAGGGTTGTTCAAG-3′; NC1R, 5′-TCTGCTATTCCCCCAATTCAGA-3′) of the KvDMR1 CTCF binding sites; generally, only very low amplification was detected at these flanking loci. Real-time PCR analysis of samples obtained by immunoprecipitation of the chromatin fraction with CTCF and H3 dimethyl K9 (H3K9) antibodies and a no-antibody control were performed using the Applied Biosystems 7900HT Fast Real-Time PCR System and SYBR Green PCR Master Mix. PCR was carried out in triplicate on equal amounts of ChIP, control (no antibody), and input DNA samples at the following thermal cycling parameters: 95°C for 10 min and 40 cycles of 95°C for 15 s and 60°C for 1 min. Data were analyzed by the comparative cycle threshold (CT) method as described in the ABI User's Bulletin (1a) and by Litt et al. (25). The relative enrichment for a particular target sequence was determined by calculating the ratio of the amount of the target sequence in the immunoprecipitation to the amount of the target sequence in the input DNA. Briefly, we used the following equation for relative enrichment: 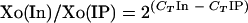 , where Xo is the initial DNA concentration of a target sequence in immunoprecipitation (IP) and input (In) and CT is the number of cycles required to reach the threshold. Each value was normalized with respect to the no-antibody control.
, where Xo is the initial DNA concentration of a target sequence in immunoprecipitation (IP) and input (In) and CT is the number of cycles required to reach the threshold. Each value was normalized with respect to the no-antibody control.
Primers used for direct sequencing of KvDMR1 in ChIP analysis of MEFs were YY236 (5′-CACCATCTGTCCAATCAACAGTGTC-3′) and YY238 (5′-ATCCAAAATGAGGCCGACCACACCG-3′). The 193-bp PCR product generated with these primers was directly sequenced to detect a single-nucleotide polymorphism between C57BL/6J (allele A) and SD7 mice (allele G) (position 1975 in AF119385).
Plasmid constructs.
For enhancer-blocking experiments, different fragments of KvDMR1 were amplified by PCR from mouse genomic DNA using primer pairs (primer sequences available on request) with either a ClaI or SalI restriction enzyme recognition site at their 5′ ends. The fragments were named based on the two primers used to amplify them (e.g., fragment 1-22 was amplified using primers mKD1 and mKD22). The PCR products were subcloned into the pCRII-TOPO vector (Invitrogen), and the sequence was verified. The PCR-amplified fragments were released from the pCRII-TOPO vector by digestion with SalI and ClaI, gel purified, and cloned into the SalI and ClaI site of E-p-neo (50) (i.e., in the insulator position, between the Eδ enhancer and the Vδ promoter) for use in an enhancer-blocking assay. Plasmids used in the enhancer-blocking assay were linearized with NotI or AhdI and purified using a Wizard DNA Clean-Up kit (Promega). For luciferase reporter assays, different fragments of KvDMR1 were PCR amplified from mouse genomic DNA (primer sequences available on request) and cloned into the pCRII-TOPO vector (Invitrogen), and sequences were verified. Restriction fragments were released from the pCRII-TOPO vector by digestion with KpnI and XhoI, gel purified, and inserted into KpnI/XhoI-digested pGL3-Basic vector (Promega). Orientation of the inserts was confirmed by restriction digest and sequencing. Before transfection, all plasmids were purified using an S.N.A.P MidiPrep kit (Invitrogen).
Soft-agar colony-forming assay.
The assay was performed as described by Zhong and Krangel (50). Briefly, equimolar amounts (5.0 to 10.0 mg) of each linearized plasmid were transfected into the human T-cell leukemia cell line Jurkat grown in RPMI 1640 medium supplemented with 10% fetal calf serum. Plasmids were introduced into 0.8 ml of cell suspension (5 × 106 cells/ml) in a 4-mm gap cuvette (Bio-Rad Laboratories) by electroporation using a Gene Pulser II (Bio-Rad Laboratories) at 250 V and 960 μF. Following electroporation, cells were cultured in 10 ml of RPMI 1640 medium for 48 h at 37°C, harvested, and plated on 10-cm cell culture dishes in 30 ml of soft-agar plating medium containing 950 μg/ml active G418 (Invitrogen). Antibiotic-resistant colonies were counted after 3 to 4 weeks. In a given experiment, each construct was tested in triplicate, and experiments were repeated two or three times.
Methylation interference and mutagenesis.
Methylation interference was carried out as previously described by Lobanenkov et al. (26). Mutagenesis of CTCF binding sites was carried out using a QuikChange Site-Directed Mutagenesis Kit (Stratagene).
Mapping of Kcnq1ot1 transcriptional start site.
The transcription initiation sites of the mouse Kcnq1ot1 gene were mapped by 5′ rapid amplification of cDNA ends (5′-RACE). A FirstChoice RNA ligase-mediated RACE Kit (Ambion) was used with mouse placental or brain total RNA and the gene-specific primers RACEI and RACEII (RACEI, 5′-GGAAGGACCATGCAGAGAAA-3′; RACEII, 5′-ACTGGACCAAAATGCACCAT-3′). The PCR conditions were 94°C for 3 min, then 35 cycles of 94°C for 30 s, 60°C for 30 s, 72°C for 3 min, and finally 72°C for 7 min. Each of the amplicons from the PCR was purified using a QIAquick PCR purification kit (QIAGEN), cloned into pCR 2.1 TOPO (Invitrogen), and sequenced.
Luciferase reporter assay.
Equimolar amounts (1 to 2 mg) of pGL3-Basic vector-based constructs along with 100 ng of pRL-TK, the internal control plasmid, were transiently cotransfected into Jurkat cells using TransIT Jurkat reagent (Mirus Bio). After culturing in 2 ml of RPMI 1640 medium in six-well plates for 24 h at 37°C, cells were harvested, washed twice in PBS, resuspended in lysis buffer (Promega), and lysed by three cycles of freezing in liquid nitrogen and thawing. Luciferase and Renilla activities were measured in 20 μl of each lysate using a Dual-Luciferase Reporter Assay System kit (Promega). Luciferase activity was normalized to the Renilla activity. All transfections were performed in triplicate. HeLa cells used in enhancer mapping experiments were transiently transfected using Lipofectamine 2000 (Invitrogen). The day before transfection, HeLa cells were plated into 12-well tissue culture plates at a density of 1 × 105 cells per well. Cells were cotransfected with 1 mg of pGL-promoter-based experimental constructs and 10 ng of the Renilla reporter construct pRL-TK. Twenty-four hours after transfection, the reporter activities were assayed as described above.
RESULTS
Methylation-sensitive binding of the chromatin insulator protein CTCF to KvDMR1.
We have previously shown that a 3.6-kb fragment encompassing the mouse KvDMR1 locus (Fig. 1b, fragment 1A4) functioned in a position-dependent manner in two independent cell culture-based enhancer-blocking assays (17). Since the function of all known vertebrate enhancer-blocking elements is mediated by the DNA-binding protein CTCF (4, 13, 32), we asked if CTCF also binds to KvDMR1. Because chromatin insulators tend to be C+G rich (6, 46) and the 1A4 fragment includes two CpG islands, we speculated that the identified insulator activity, and therefore CTCF binding sites, might be confined to this CpG-rich region. However, the nature of CTCF binding precludes simply scanning KvDMR1 sequences for a consensus sequence. The term “multivalent 11 ZF” for the DNA-binding domain of CTCF describes the multiplicity of dissimilar target sequences to which CTCF interacts (1, 9, 10, 12, 19, 32, 34) and underscores the notion that no single consensus sequence exists that would match all CTCF target sequences. The unusual capability of CTCF to specifically form complexes with different target sequences is attributed to the differential contribution of distinct subsets of its 11 zinc fingers (9, 32). Since any attempt to predict CTCF-binding sequences based solely on homology with the known targets is likely to fail (31) or lead to false positives (20), we carried out a systematic search for CTCF-binding sites by using recombinant CTCF protein in EMSAs with a set of 11 overlapping DNA probes.
CTCF binding sites can remain undetected by conventional gel shift analysis with 20- to 60-bp double-stranded oligonucleotides because these probes are often too short to fully accommodate the typical length of a CTCF DNase I footprint (50 to 60 bp) and do not provide the additional footprint-flanking DNA sequence necessary for the DNA-bending required for efficient CTCF binding in vitro (26). With these considerations in mind, an overlapping set of 32P-end-labeled DNA probes was designed such that each probe was longer than 100 bp and began approximately in the middle of adjacent fragments. The fragments were named based on the two primers used to amplify them (e.g., fragment 1-2 was amplified using primers mKD1 and mKD2).Therefore, although the set of overlapping probes spanning KvDMR1 was designed without prior knowledge of a CTCF target location, this strategy ensured that CTCF ZF-contacting bases would not be too close to the end of any DNA probe to result in decreased CTCF-binding efficiency, while at the same time providing additional flanking DNA around any putative CTCF target site in one of the two overlapping fragments. Each of the 11 DNA fragments (Fig. 1b) was generated by PCR using 32P-labeled primers and utilized in an EMSA with two in vitro translated CTCF polypeptides, representing either full-length CTCF (FL CTCF) or the 11 ZF domains of the protein. Probes 1-2, 3-4, and 7-8 gave specific binding shifts with the CTCF 11-ZF protein, although only fragments 1-2 and 7-8 formed DNA-protein complexes with the FL CTCF (Fig. 1c). The band generated by probe 5-6 is likely nonspecific since it is also present in the minus (−) protein lane. Why probe 3-4 bound the ZF polypeptide and not the FL CTCF is likely explained by the results of methylation interference (see below) which showed that the core binding site in this case is located in the overlap region between EMSA probes 1-2 and 3-4. This core sequence appears sufficient to bind the 11-ZF CTCF protein, but presumably sequences unique to probe 1-2 are required to bind the full-length protein. The gel shifts obtained with in vitro translated CTCF protein were also seen using nuclear extracts from HeLa cells as the protein source; supershift with a mixture of nine monoclonal antibodies to CTCF (36) confirmed that the bound protein factor was CTCF (Fig. 1d). These putative CTCF binding sites were designated CTS1 and CTS2, respectively. Most CTCF binding sites are methylation sensitive, including CTCF insulator sites (3, 10, 14, 19, 38, 49). To determine whether CpG methylation affects KvDMR1 CTCF binding sites, we performed the EMSA with SssI-methylated PCR products corresponding to CTS1 and CTS2. As seen in Fig. 1e, CpG methylation of the probes abolishes CTCF binding.
To map CTS1 and CTS2 precisely, we used the methylation interference approach (26) to identify guanine residues critical for CTCF binding (Fig. 1f). Multiple G residues (marked by dots) come into contact with in vitro translated CTCF at each of the two detected CTS. Since these guanines could be important for the binding of CTCF in vivo, the majority of them were changed to A or T residues, and the mutated sequences were tested for their ability to form complexes with CTCF in gel shift assays. The mutations in CTS2 completely abrogated binding by both in vitro translated full-length CTCF and the 11-ZF protein as well as to proteins in HeLa cell extract, indicating that these sequences were indeed necessary for CTCF binding (Fig. 1d). While preventing binding to CTCF in nuclear extracts and in in vitro translated full-length CTCF protein, mutations in CTS1 were not 100% effective in preventing binding of the 11-ZF truncated CTCF protein. Nevertheless, these results allowed the exact mapping of CTS1 and CTS2 at the nucleotide level. As mentioned above, a consequence of using multiple combinations of zinc fingers is that no strong consensus sequence exists for CTCF binding sites (9, 20, 31, 32). The CTCF sites mapped here are unique to the KvDMR locus; no similarity was found between KvDMR1 CTS1 and CTS2 and other CTCF sites known to have enhancer-blocking activity. In contrast, weak homology does exist between the mouse and human sequences. Previous gel shift analysis of human KvDMR identified three fragments that bound in vitro transcribed/translated CTCF (KvDMR F, G, and H) (7). Approximately 50% homology exists between mouse CTS1 and CTS2 and two of the human CTCF binding sites; moreover, the majority of the contact residues identified by methylation interference are conserved between mouse CTS1 and CTS2 and human KvDMR-HBi and KvDMR-FBi, respectively (Fig. 1 h). No homology was found between either CTS1 or CTS2 and the sequence of the KvDMR-GBi fragment.
CTCF binds to KvDMR1 in an allele-specific manner.
To determine whether CTCF binds to the endogenous KvDMR1 locus, we carried out a ChIP assay on mouse embryonic fibroblasts and analyzed precipitated chromatin by real-time PCR. As a control, quantitative PCR was also performed on the same immunoprecipitated material using primers designed to anonymous DNA sequences approximately 1 kb upstream or 1.3 kb downstream of KvDMR1 CTS1 and CTS2 (Fig. 1b). Enrichment for CTCF binding sequences was found at CTS1 but not at the flanking control sequences (Fig. 2a). Similar enrichment was observed at CTS2 (data not shown). Another set of primers that amplify across CTS1 and a known polymorphism between C57BL/6J (A) and SD7 (G) mice was used, and the amplified material was directly sequenced. This analysis demonstrated that enrichment of CTCF at KvDMR1 was specific to the unmethylated paternal allele (Fig. 2b, SD7 allele in MEFs from C57 × SD7 embryos and C57 allele in MEFs from SD7 × C57 offspring). Allele-specific binding of CTCF to KvDMR1 was confirmed by ChIP analysis of primary lung fibroblasts (PLFs) derived from mice carrying a 2.8-kb deletion of KvDMR1 (11). Based on the findings described above, CTCF binding would be predicted to be absent at KvDMR1 in cells from mice with a paternal deletion and present in cells with a maternal deletion (Fig. 2c). Indeed, real-time PCR showed no enrichment of CTCF at KvDMR1 in PLFs from mice with a paternal deletion of the locus but significant enrichment in cells with a maternal KvDMR1 deletion (Fig. 2c, middle and lower histograms, respectively). Allele-specific histone modifications have previously been detected at KvDMR1 in both placenta and embryonic tissues such that the maternal allele is enriched for methylation at H3K9 and H3K27 (thought to be repressive marks), while the paternal allele is enriched for methylation at H3K4, a modification associated with active chromatin (45). This raises the possibility that the preferential binding of CTCF to the paternal allele simply reflects its more accessible chromatin structure compared to the maternal allele which could, for example, lead to preferential retention on agarose beads during the ChIP procedure. To exclude this possibility, we carried out a similar experiment on the same cells using an antibody to dimethyl-H3K9. This time, quantitative PCR demonstrated enrichment only in PLFs from wild-type mice and mice with a paternal deletion of KvDMR1 (Fig. 2c). These results argue against any artifactual precipitation of the paternal KvDMR1 locus due to differential chromatin structure. Thus, consistent with EMSA results using methylated PCR products and the methylation sensitivity of most characterized CTCF binding sites (3, 10, 14, 19, 38, 49), our results demonstrate that CTCF binds KvDMR1 only at the unmethylated paternal allele.
FIG. 2.

CTCF binds to the unmethylated paternal allele of KvDMR1 in vivo. (a) Chromatin immunoprecipitated from C57BL/6J × SD7 F1 MEFs with monoclonal CTCF antibody was analyzed by real-time PCR using PCR primers flanking CTS1 or primers amplifying two control regions (1.2 kb upstream or 1.6 kb downstream of CTS1, as shown in Fig. 1b). (b) The same CTCF ChIP material and that prepared from SD7 × C57BL/6J F1 MEFs was amplified using primers that flank a single-nucleotide polymorphism (SNP) between C57BL/6J and SD7 mice and directly sequenced. While both A and G alleles are present in input chromatin, only the paternal allele (G in the C57 × SD7 cross and A in the SD7 × C57 cross) was detected in the immunoprecipitated chromatin. (c) Real-time PCR analysis of chromatin immunoprecipitated from PLFs derived from mice heterozygous for a deletion of KvDMR1 (11). ChIP was carried out using a monoclonal CTCF antibody or polyclonal antibody against dimethylated lysine 9 of histone 3. A region from Myc promoter (12) served as a positive control for CTCF binding. Enrichment of CTCF-bound chromatin at KvDMR1 was much less in PLFs from mice with a maternal deletion than in PLFs from wild-type mice (lower and upper histograms, respectively). Since the enrichment of CTCF was also drastically reduced at the internal control Myc locus in the same cells, it is likely that this reduction is due to loss of chromatin during the washing of protein A/G agarose beads or the recovery of the immunoprecipitated DNA. Ab, antibody.
CTS1 and CTS2 are located within the minimal repressive element in KvDMR1.
To determine whether the two CTCF binding sites described above are responsible for the insulator activity previously reported in KvDMR1, we analyzed subfragments of the original test fragment (Fig. 3a, 1A4) in the soft-agar colony-forming enhancer-blocking assay (Fig. 3b) (50) used previously (17). As for the EMSA experiments, in most cases fragments were named based on the two primers used to amplify them (e.g., fragment 1-22 was amplified using primers mKD1 and mKD22). Each subfragment was tested only in the “insulator” position (i.e., the “in” position in Fig. 3b) since the goal of this study was to determine the relationship between the CTCF binding sites and the repressive activity in KvDMR1; it is possible that smaller fragments of KvDMR1 may also function in the “out” or “silencer” position (see Discussion). It should also be noted that the repressive activity of fragment 1A4 depended on the orientation of the 3.6-kb fragment with respect to the reporter gene's promoter (Fig. 3c, compare the two 1A4 constructs) (17). We first tested the 1-22 fragment which encompasses the two CpG islands as well as CTS1 and CTS2 (Fig. 3a). Inclusion of this fragment in E-p-neo resulted in a reduction of 75 to 90% in the number of neo-resistant colonies compared to the E-p-neo control (Fig. 3c). Most of the remainder of the large 1A4 fragment is contained in fragments up16 and dn12 (Fig. 3a), neither of which exhibited enhancer-blocking activity (Fig. 3c). Thus, the majority of the repressive activity observed in fragment 1A4 is present in fragment 1-22. To narrow the region with enhancer-blocking activity further, the 1-22 sequence was divided into four overlapping fragments. The 11-22 and 7-16 fragments each displayed roughly half the activity of larger 1-22 fragment, suggesting that sequences near the 5′ end of 1-22 are necessary for full repressive function. Indeed, the 1-16 fragment, which extends upstream of fragment 7-16 repressed reporter gene expression at least as well as fragment 1-22. Although some of this activity was lost in the shorter 1-10 fragment, the latter sequence behaved as a very strong enhancer blocker. We conclude that the core repressor activity resides within the 1-10 sequence, while sequences in the 3′ half of fragment 1-16 enhances the activity of the adjacent 1-10 fragment without having strong repressive activity itself. Importantly, this region is within or overlaps with DNA segments shown to function as silencers in other enhancer-blocking assays (27, 43) (Fig. 3a). Interestingly, none of the smaller fragments tested demonstrated orientation dependence in the enhancer-blocking assay (Fig. 3c), suggesting that the sequence responsible for the polarity of the larger 1A4 fragment must be located within its 5′ portion, upstream of the KvDMR1 CpG islands. Significantly, the 670-bp 1-10 fragments encompass CTCF binding sites CTS1 and CTS2, suggesting that a large proportion of the repressive activity of this locus is attributable to the binding of CTCF. To examine their functional significance, the mutated CTCF binding sites (Fig. 1f) were introduced into fragment 1-10 and tested in the enhancer-blocking assay individually and together. Although the mutant constructs did exhibit significant reductions in repressive activity in some experiments, the effect was variable (data not shown). This somewhat surprising result may be due to residual binding activity at CTS1 as indicated in Fig. 1d or CTCF-independent repressive activity in fragment 1-10 (see Discussion).
FIG. 3.

Mapping of KvDMR1 repressive activity in an enhancer-blocking assay. (a) The extent of DNA fragments tested for different functional activities are shown as black lines with the name of each fragment indicated. For comparison, the genomic region deleted in mice described in Fitzpatrick et al. (11) is shown as a black bar. The light gray bars are regions of the locus tested for insulator/silencer activity by others (27, 43). (b) Schematic representation of the E-p-neo basic vector used for generation of the experimental constructs (see text for details; SCS is a Drosophila insulator element [SO]). (c) The test-fragments indicated in panel a were inserted in the SalI/ClaI (“in” position) site in the indicated orientations with respect to the endogenous locus, stably transfected into Jurkat cells, and plated on soft agar; neo-resistant colonies were counted after 3 to 4 weeks. Enhancer-blocking activity was assessed as the number of the neo-resistant colonies for a given construct relative to the number of colonies formed with the E-p-neo construct (taken as 100%). Each transfection was done in triplicate. Individual constructs were transfected in two to three independent experiments.
The KvDMR1 promoter and transcriptional start sites are distinct from its repressive activity.
To facilitate further genetic investigation of the KvDMR1 locus, it is important to determine whether the promoter for Kcnq1ot1 and the CTCF binding sites operate as a single unit or are independent functional modules. The promoter for Kcnq1ot1 was mapped previously to a 600-bp fragment (27) that includes part of CTS1 at its 3′ end and overlaps with the 1-10 repressive fragment functionally characterized in Fig. 3. We wanted to map the promoter more precisely to see specifically if we could separate the promoter and repressive functions of KvDMR1. To confirm previous RNase protection assays (27), the Kcnq1ot transcriptional start site was mapped using 5′ RACE on total RNA from embryonic day 15.5 mouse brain and placenta. More than half (11/20) of the sequenced clones had 5′ ends terminating 36 to 37 bp upstream of CTS1 (as defined by the methylation interference) (Fig. 1f), with another six clones terminating further upstream (Fig. 4a). We cloned a 433-bp fragment that contained the major and upstream transcriptional start sites but not CTS1 (Fig. 4a) in both orientations upstream of the luciferase reporter vector, pGL3-Basic pGL/400 and pGL/400-R, respectively), and transfected the constructs into Jurkat cells since these were the same cells used in the enhancer-blocking experiments. Over 100-fold induction of promoter activity was detected when the fragment was inserted in the same orientation (relative to the reporter) as the endogenous locus (pGL/400) but not in the reverse orientation (pGL/400-R) (Fig. 4b). No larger fragment from the KvDMR1 locus, including the full-length 1A4 fragment, exhibited as much promoter activity. Serial deletion of pGL/400 from the 5′ end resulted in decreased activity, indicating that in this assay pGL/400 is the minimal Kcnq1ot1 promoter. Similar results were obtained when the same constructs were tested in a second human cell line (HEK293) and in two mouse cell lines (NIH 3T3 and MEFs) (data not shown). It should also be noted that the repressive fragment 1-16 (which contains both CTCF sites) had little or no promoter activity in the same assay despite containing approximately 90 bp of the minimal promoter including the major and upstream transcription start sites at its 5′ end (Fig. 4a and b, construct pGL/1-16). Consistent with this observation is the finding that fragment pGL/100, roughly corresponding to the overlap between fragment 1-16 and the minimal promoter fragment pGL/400, displayed virtually no promoter activity. When the strong minimal promoter fragment pGL/400 was tested in the enhancer-blocking assay, no repressive activity was observed even though this fragment carries the major and upstream transcriptional start sites as well as the promoter-associated DNase I-hypersensitive sites identified by others (27) (Fig. 4c). In fact, the number of G418-resistant colonies was threefold higher than the E-p-neo control construct, presumably due to the influence of the strong promoter. These results clearly show that, in these assays, Kcnq1ot1 promoter sequences do not contribute to the repressive activity in KvDMR1, making these two elements functionally separable. Furthermore, these results exclude the possibility that the repressive activity of KvDMR1 observed in reporter constructs is due to promoter competition.
FIG. 4.

A transcriptional promoter within KvDMR1 can be uncoupled from enhancer-blocking activity. (a) Restriction map of KvDMR1 locus. The arrow above the map points toward direction of Kcnq1ot1 transcription. The sequence of the locus harboring promoter activity is shown below the restriction map with arrows facing down indicating the 5′ ends of individual fragments tested for promoter activity; all these fragment have the same 3′ end specified by an arrow pointing up. The broken arrows show transcription start sites as determined by RACE analysis. The heavy broken arrow shows the position of the major transcription start site (see text). The bolded and italicized sequence represents the overlap between the fragments tested in the promoter assay and those representing the minimal repressive element. (b) Promoter activity of different fragments from KvDMR1 locus (positions of the fragments are shown in Fig. 3a) was evaluated by a luciferase reporter assay. All test fragments were cloned into pGL3-Basic vector upstream of a luciferase gene. The luciferase activity is shown as the increase in activation relative to the activity from the vector alone. All constructs were transiently transfected into Jurkat cells in triplicate. (c) Enhancer-blocking activity of the fragment containing the full promoter sequence (pGL/400) was measured in enhancer-blocking assay as described in the legend of Fig. 3c.
A sequence with properties of an enhancer is located upstream of the KvDMR1 CpG island.
During the mapping of the minimal repressive element in KvDMR1, fragments upstream and downstream of the CpG-islands were tested in the enhancer-blocking assay. As shown in Fig. 3c, neither the up16 fragment, which corresponds to a 5′ part of the 1A4 fragment, nor the dn12 fragment possesses enhancer-blocking activity; indeed the number of the neo-resistant colonies consistently increased 15 to 30% compared to the E-p-neo control (Fig. 3c). This increase of the reporter activity could be explained by the presence of positive regulatory elements (i.e., a promoter or an enhancer) within these sequences. To assess this possibility, we cloned these two fragments 5′ of a luciferase reporter gene driven by the simian virus 40 promoter (pGL3-promoter vector). Because we found high reporter expression in Jurkat cells with the parent pGL3-promoter vector alone, these experiments were done using HeLa cells. The results presented in Fig. 5b indicate that, whereas the dn12 does not exhibit enhancer activity in HeLa cells, the up16 fragment does since it induces transcription of the reporter more than fivefold over that of the promoter-only vector (construct up16 SmaI/pGL3-promoter). Similar to a classical transcriptional enhancer, the up16 fragment also stimulated reporter expression when placed downstream of the promoter (construct up16 SalI/pGL3-promoter). This finding also excludes the possibility that the induction of reporter gene expression by up16 sequences is simply due to the presence of a strong promoter. Four subfragments of up16 were tested in an attempt to narrow the region containing enhancer activity (Fig. 5a). Three smaller fragments (up12, up34, and up56) exhibited 30 to 40% of the enhancer activity observed with up16 (Fig. 5b) while the larger up76 fragment gave activity similar to the up16 fragment.
FIG. 5.

An enhancer-like element located 5′ of the KvDMR1 CpG island. (a) Restriction map of KvDMR1 region as shown in Fig. 1b. The gray bars below the sequence represent the fragments tested for enhancer activity; for comparison, fragments with repressive activity (1-10 and 1-16) are shown in black. A summary of the different functional elements defined in this study is presented in the lower panel. (b) Fragments shown in panel were inserted in pGL3-promoter vector upstream (SmaI site) or downstream (SalI site) of the luciferase reporter. Luciferase activity of each test construct is shown as the increase in activation relative to the activity of the vector alone. Because of the high background level obtained with the vector alone in Jurkat cells, the experiment was carried out in HeLa cells. Each transfection was done in triplicate.
DISCUSSION
Deletion of KvDMR1 in the mouse leads to the activation in cis of the normally silent paternal alleles of eight genes on mouse distal chromosome 7 (11, 23, 28). These results demonstrate that KvDMR1 mediates imprinted expression in this domain by silencing genes on the paternal chromosome. Initial studies aimed at determining the mechanism by which KvDMR1 silences these genes consisted of testing this locus in several enhancer-blocking assays. Our first analysis indicated that KvDMR1 can act as a position-dependent enhancer-blocker or insulator (17). Because known vertebrate insulators function through CTCF, we scanned the KvDMR1 locus for the presence of CTCF binding sites. EMSAs using recombinant protein suggested that the insulator-associated protein CTCF binds to KvDMR1; this notion was supported by the finding that DNA/nuclear extract complexes could be supershifted using antibodies to CTCF. Gel shift experiments also demonstrated that the interaction of CTCF with KvDMR1 is methylation sensitive. The presence of CTCF at KvDMR1 in vivo was shown by ChIP analysis using F1 MEFs from reciprocal crosses between C57BL/6J and SD7 mice, which showed that CTCF binds only to the unmethylated allele of KvDMR1, similar to the situation at the H19 DMR (19, 42). Allele-specific binding of CTCF to KvDMR1 was confirmed using PLFs from adult mice carrying either a maternal or paternal deletion of KvDMR1. Significantly, the two CTCF binding sites (CTS1 and CTS2) are located within the minimal repressive region of the KvDMR1 ICR. Unexpectedly, mutation of the two CTCF sites did not reproducibly result in a reduction of activity in the enhancer-blocking assay. This result is in contrast to the observation that the majority of repressive activity is lost from the 460-bp silencer region defined by Mancini-DiNardo et al. when the sequence encompassing CTS1 was deleted (27). The absence of a reproducible effect of CTS mutations in the present assay may reflect residual binding of CTCF to CTS1 (partial binding of the 11-ZF protein in vitro, as shown in Fig. 1d, may indicate that native CTCF in Jurkat cells might still be able bind). Alternatively, the 1-10 fragment may carry an undetected binding site for CTCF or binding sites for other factors that contribute to its suppressive activity (30).
The minimal repressive activity of KvDMR1, as well as CTS1 and CTS2, are localized to fragment 1-10 that is within the 1,500-bp silencer region defined by Thakur et al. (43) and overlaps (at CTS1) with the 460-bp silencer region determined by Mancini-DiNardo et al. (27). This observation suggests that binding of CTCF may also be responsible for the silencing activity of KvDMR1 demonstrated in these earlier studies. Thus, it is surprising that fragments from KvDMR1 behave as an insulator (position dependent) in some enhancer-blocking assays but as a silencer (position independent) in others. These discrepancies probably arise from differences in the cell types used in the enhancer-blocking assays and/or in the length and positions of the fragments tested. In the first instance, it is noteworthy that the same episomal construct containing the 1A4 fragment behaves as an insulator in Hep3B cells (17) but as a silencer in Jeg3 cells (43). Thus, in Thakur et al. (43), the silencer designation was assigned to KvDMR1 using a cell line not capable of revealing the insulator activity previously observed in this fragment (17). Secondly, none of the fragments tested in an enhancer-blocking assay by Mancin-DiNardo et al. (27) included the 5′ half of 1A4 (Fig. 3a), the fragment which exhibits position-dependent enhancer-blocking activity (17). We have not tested fragments 1-10 or 1-16 (which also lack this region) in the “out” or silencer position in the enhancer-blocking assay used in this study. It is possible that, if tested, these smaller fragments would also exhibit repressive activity in a position-independent fashion. Thus, it is conceivable that the insulator activity of KvDMR1 is only manifested when the upstream half of the 1A4 fragment is included, as it would be at the endogenous locus. This view is consistent with the notion that the insulator function of CTCF may be context dependent (13). Finally, it is far from certain whether insulator or silencer activity defined in cell culture-based enhancer-blocking assays accurately reflects the situation at the endogenous locus. The question of whether KvDMR1 in its natural context functions as an insulator or silencer, or both, can only be addressed once the enhancers for the genes under its control are identified and their positions (with respect to KvDMR1) changed.
Using a luciferase reporter we refined the location of the putative promoter for the Kcnq1ot1 transcript to the pGL/400 fragment which is within the promoter region identified previously (27). Targeted deletion in the mouse has recently provided confirmation that this region is the bona fide promoter for Kcnq1ot1 (28). Importantly, the promoter fragment pGL/400 has no repressive activity in our enhancer-blocking assay, and the maximal repressive fragment 1-16 has little or no promoter activity. Furthermore, compared to fragment 1-10, the level of enhancer blocking was not changed when a larger fragment including both the core repressive element and the adjacent promoter sequences was tested (data not shown). Analysis of KvDMR1 in an episomal enhancer-blocking assay led Thakur et al. to propose that transcription of Kcnq1ot1 is prerequisite to its silencing activity (44). Although we have not determined whether low-level transcription is detectable from fragment 1-16, it is unlikely to be significant since we observed little or no promoter activity in this sequence. Thus, in the system used here, transcription from the putative promoter for Kcnq1ot1 does not appear necessary for repressive activity. Since fragment pGL/400 exhibits no repressive activity, these results also exclude the possibility that the polarity of the KvDMR1 insulator activity observed in the 3.6-kb fragment was due to the presence of an active promoter (17). Finally, the uncoupling of the two cis-regulatory elements (promoter and repressive element) within KvDMR1 will enable us to address the role of each of these sequences in regulating imprinted expression in vivo by introducing mutations inactivating only one but not both activities. In this regard it should be noted that, unbeknownst to the authors, the promoter deletion described by Mancini-DiNardo et al. (28) includes part of CTS1.
The region upstream of the presumptive Kcnq1ot1 promoter and repressive region significantly increases the transcription levels in a luciferase reporter assay, suggesting that it may contain an enhancer. If this region functions as a transcriptional enhancer in vivo, the gene(s) on which it acts is unknown. It could drive the expression of one or more of the maternally expressed genes in the domain or could be an enhancer for Kcnq1ot1 itself. Our earlier deletion of KvDMR1 does not include this region, so this possibility could not be tested (11). However, a larger deletion that does include this putative enhancer had no effect on the expression of any of these maternally expressed genes in midgestation placenta and embryos when inherited maternally (28). It is also possible that this sequence does not function as a typical enhancer of gene expression in vivo but may nevertheless work in a cell-type-specific fashion, possibly regulating the mechanism by which KvDMR1 functions in different cell lineages. The presence of an enhancer upstream of the repressive element in KvDMR1 could explain the orientation effect observed when the 1A4 fragment was used in the enhancer-blocking assay (Fig. 3) (17). The 1A4 fragment blocked enhancer-promoter interactions in the forward orientation but not in the reverse. This could be explained if the 1A4 fragment contained enhancer activity at the 5′ end and insulator activity at the 3′ end. In the reverse orientation the putative enhancer would be juxtaposed to the reporter gene's promoter, thereby bypassing any effect of the insulator. It has been noted previously that the function of some enhancer blocking sequences was orientation dependent (3, 5, 14). The basis for this polarity has not been clarified, but it was suggested that orientation dependence could be explained by the presence of an enhancer-like element next to an insulator (48). Indeed, a combination of an enhancer and an insulator has been described upstream of the human apoB gene (1). Our work shows that the KvDMR1 locus could be yet another example of a compound enhancer-blocking element (Fig. 5a).
A number of mechanisms of action of KvDMR1 have been discussed (reviewed in reference 47). To date, a role for the noncoding Kcnq1ot1 RNA or its transcription in gene silencing has been demonstrated. Previously, this mechanism has been proposed for the AIR ncRNA which is transcribed from region 2 of the Igf2r (40) gene. Evidence for a role of Kcnq1ot1 in gene silencing was initially obtained using a cell culture model (44). More recently, it has been shown that truncation of the Kcnq1ot1 RNA at the mouse endogenous locus results in loss of imprinted expression of the genes known to be under the control of KvDMR1 (28; Shin et al., unpublished data), although the mechanism by which the Kcnq1ot1 ncRNA or its transcription elicit gene silencing remains unclear (35). Despite these findings, it is still possible that more than one mechanism is operational in the KvDMR1 domain, perhaps regulating different subsets of genes or functioning in different cell lineages or at different times during development. In support of this notion are recent findings that, in the imprinted subdomain controlled by KvDMR1, allele-specific differences in chromatin structure at most genes are found only in the placenta and not in the embryo proper (23, 45). Furthermore, genes such as Cdkn1c and Kcnq1 already exhibit imprinted expression in preimplantation embryos while Tssc4 and Cd81, which only exhibit imprinted expression in the placenta, are biallelic until differentiation of the trophoblast lineage (22). Moreover, we have found that, unlike deletion of KvDMR1 (11), truncation of the Kcnq1ot1 RNA affects Cdkn1c imprinted expression in only a subset of embryonic tissues, indicating the existence of a second mechanism elicited by KvDMR1 that is capable of regulating imprinted expression of this gene (Shin et al., unpublished data). Considering the enhancer-blocking studies discussed above (17, 27, 43), additional mechanisms may include KvDMR1 functioning as a bidirectional silencer independent of the Kcnq1ot1 noncoding RNA. In this model, KvDMR1 nucleates repressive chromatin which then spreads bidirectionally to neighboring genes (27, 37, 43). Another possibility is that KvDMR1 functions as a chromatin insulator in a manner similar to the H19 DMR. However, a prerequisite to this model is that the enhancer and promoter for a particular gene under the control of this locus must be located on opposite sides of KvDMR1. At present, the locations of the enhancers for the eight genes regulated by KvDMR1 are unknown; however, analysis of Cdkn1c transgenes suggests that at least some tissue-specific enhancers lie between the gene and KvDMR1 (15). Thus, if KvDMR1 does function as an insulator at the endogenous locus in vivo, it is unlikely it would employ this mechanism for all genes in all cell types.
It remains to be seen if the sequences initially defined as an insulator by us (17) or as a silencer by others (27, 43) possess similar activity in vivo at their natural genomic locations. Based on enhancer-blocking and luciferase reporter assays, however, it appears that the repressive activity of KvDMR1 may function independently of its promoter activity. Regardless of whether this repressive activity represents a silencer or an insulator, it is most likely regulated at least in part by methylation-sensitive binding of CTCF. The results of our Kcnq1ot1 truncation mutant mice suggest that in some tissues the imprinted expression of Cdkn1c is not regulated by the Kcnq1ot1 ncRNA (Shin et al., unpublished data). Thus, the repressive element identified in KvDMR1 is a prime candidate for this function.
Acknowledgments
This research utilized core facilities supported in part by Roswell Park Cancer Institute's NCI-funded Cancer Center Support Grant (CA16056). This work was supported by the Roswell Park Alliance Foundation, by Public Service Grant CA89426 from the National Cancer Institute awarded to M.J.H., and in part by the intramural NIAID funding to V.V.L.
Footnotes
Published ahead of print on 22 January 2007.
REFERENCES
- 1.Antes, T. J., S. J. Namciu, R. E. Fournier, and B. Levy-Wilson. 2001. The 5′ boundary of the human apolipoprotein B chromatin domain in intestinal cells. Biochemistry 40:6731-6742. [DOI] [PubMed] [Google Scholar]
- 1a.Applied Biosystems. 2001. ABI user's bulletin no. 933. Applied Biosystems, Foster City, CA.
- 2.Awad, T. A., J. Bigler, J. E. Ulmer, Y. J. Hu, J. M. Moore, M. Lutz, P. E. Neiman, S. J. Collins, R. Renkawitz, V. V. Lobanenkov, and G. N. Filippova. 1999. Negative transcriptional regulation mediated by thyroid hormone response element 144 requires binding of the multivalent factor CTCF to a novel target DNA sequence. J. Biol. Chem. 274:27092-27098. [DOI] [PubMed] [Google Scholar]
- 3.Bell, A. C., and G. Felsenfeld. 2000. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 405:482-485. [DOI] [PubMed] [Google Scholar]
- 4.Bell, A. C., A. G. West, and G. Felsenfeld. 2001. Insulators and boundaries: versatile regulatory elements in the eukaryotic genome. Science 291:447-450. [DOI] [PubMed] [Google Scholar]
- 5.Chao, W., K. D. Huynh, R. J. Spencer, L. S. Davidow, and J. T. Lee. 2002. CTCF, a candidate trans-acting factor for X-inactivation choice. Science 295:345-347. [DOI] [PubMed] [Google Scholar]
- 6.Chung, J. H., A. C. Bell, and G. Felsenfeld. 1997. Characterization of the chicken beta-globin insulator. Proc. Natl. Acad. Sci. USA 94:575-580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Du, M., L. G. Beatty, W. Zhou, J. Lew, C. Schoenherr, R. Weksberg, and P. D. Sadowski. 2003. Insulator and silencer sequences in the imprinted region of human chromosome 11p15.5. Hum. Mol. Genet. 12:1927-1939. [DOI] [PubMed] [Google Scholar]
- 8.Engemann, S., M. Strodicke, M. Paulsen, O. Franck, R. Reinhardt, N. Lane, W. Reik, and J. Walter. 2000. Sequence and functional comparison in the Beckwith-Wiedemann region: implications for a novel imprinting centre and extended imprinting. Hum. Mol. Genet. 9:2691-2706. [DOI] [PubMed] [Google Scholar]
- 9.Filippova, G. N., S. Fagerlie, E. M. Klenova, C. Myers, Y. Dehner, G. Goodwin, P. E. Neiman, S. J. Collins, and V. V. Lobanenkov. 1996. An exceptionally conserved transcriptional repressor, CTCF, employs different combinations of zinc fingers to bind diverged promoter sequences of avian and mammalian c-myc oncogenes. Mol. Cell. Biol. 16:2802-2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Filippova, G. N., C. P. Thienes, B. H. Penn, D. H. Cho, Y. J. Hu, J. M. Moore, T. R. Klesert, V. V. Lobanenkov, and S. J. Tapscott. 2001. CTCF-binding sites flank CTG/CAG repeats and form a methylation-sensitive insulator at the DM1 locus. Nat. Genet. 28:335-343. [DOI] [PubMed] [Google Scholar]
- 11.Fitzpatrick, G. V., P. D. Soloway, and M. J. Higgins. 2002. Regional loss of imprinting and growth deficiency in mice with a targeted deletion of KvDMR1. Nat. Genet. 32:426-431. [DOI] [PubMed] [Google Scholar]
- 12.Garrett, F. E., A. V. Emelyanov, M. A. Sepulveda, P. Flanagan, S. Volpi, F. Li, D. Loukinov, L. A. Eckhardt, V. V. Lobanenkov, and B. K. Birshtein. 2005. Chromatin architecture near a potential 3′ end of the Igh locus involves modular regulation of histone modifications during B-cell development and in vivo occupancy at CTCF sites. Mol. Cell. Biol. 25:1511-1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gaszner, M., and G. Felsenfeld. 2006. Insulators: exploiting transcriptional and epigenetic mechanisms. Nat. Rev. Genet. 7:703-713. [DOI] [PubMed] [Google Scholar]
- 14.Hark, A. T., C. J. Schoenherr, D. J. Katz, R. S. Ingram, J. M. Levorse, and S. M. Tilghman. 2000. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 405:486-489. [DOI] [PubMed] [Google Scholar]
- 15.John, R. M., J. F. Ainscough, S. C. Barton, and M. A. Surani. 2001. Distant cis-elements regulate imprinted expression of the mouse p57(Kip2) (Cdkn1c) gene: implications for the human disorder, Beckwith-Wiedemann syndrome. Hum. Mol. Genet. 10:1601-1609. [DOI] [PubMed] [Google Scholar]
- 16.Kaffer, C. R., M. Srivastava, K. Y. Park, E. Ives, S. Hsieh, J. Batlle, A. Grinberg, S. P. Huang, and K. Pfeifer. 2000. A transcriptional insulator at the imprinted H19/Igf2 locus. Genes Dev. 14:1908-1919. [PMC free article] [PubMed] [Google Scholar]
- 17.Kanduri, C., G. Fitzpatrick, R. Mukhopadhyay, M. Kanduri, V. Lobanenkov, M. Higgins, and R. Ohlsson. 2002. A differentially methylated imprinting control region within the Kcnq1 locus harbors a methylation-sensitive chromatin insulator. J. Biol. Chem. 277:18106-18110. [DOI] [PubMed] [Google Scholar]
- 18.Kanduri, C., C. Holmgren, M. Pilartz, G. Franklin, M. Kanduri, L. Liu, V. Ginjala, E. Ulleras, R. Mattsson, and R. Ohlsson. 2000. The 5′ flank of mouse H19 in an unusual chromatin conformation unidirectionally blocks enhancer-promoter communication. Curr. Biol. 10:449-457. [DOI] [PubMed] [Google Scholar]
- 19.Kanduri, C., V. Pant, D. Loukinov, E. Pugacheva, C. F. Qi, A. Wolffe, R. Ohlsson, and V. V. Lobanenkov. 2000. Functional association of CTCF with the insulator upstream of the H19 gene is parent of origin-specific and methylation-sensitive. Curr. Biol. 10:853-856. [DOI] [PubMed] [Google Scholar]
- 20.Klenova, E., I. Chernukhin, T. Inoue, S. Shamsuddin, and J. Norton. 2002. Immunoprecipitation techniques for the analysis of transcription factor complexes. Methods 26:254-259. [DOI] [PubMed] [Google Scholar]
- 21.Lee, M. P., M. R. DeBaun, K. Mitsuya, H. L. Galonek, S. Brandenburg, M. Oshimura, and A. P. Feinberg. 1999. Loss of imprinting of a paternally expressed transcript, with antisense orientation to KVLQT1, occurs frequently in Beckwith-Wiedemann syndrome and is independent of insulin-like growth factor II imprinting. Proc. Natl. Acad. Sci. USA 96:5203-5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lewis, A., K. Green, C. Dawson, L. Redrup, K. D. Huynh, J. T. Lee, M. Hemberger, and W. Reik. 2006. Epigenetic dynamics of the Kcnq1 imprinted domain in the early embryo. Development 133:4203-4210. [DOI] [PubMed] [Google Scholar]
- 23.Lewis, A., K. Mitsuya, D. Umlauf, P. Smith, W. Dean, J. Walter, M. Higgins, R. Feil, and W. Reik. 2004. Imprinting on distal chromosome 7 in the placenta involves repressive histone methylation independent of DNA methylation. Nat. Genet. 36:1291-1295. [DOI] [PubMed] [Google Scholar]
- 24.Lewis, A., and W. Reik. 2006. How imprinting centres work. Cytogenet. Genome Res. 113:81-89. [DOI] [PubMed] [Google Scholar]
- 25.Litt, M. D., M. Simpson, F. Recillas-Targa, M. N. Prioleau, and G. Felsenfeld. 2001. Transitions in histone acetylation reveal boundaries of three separately regulated neighboring loci. EMBO J. 20:2224-2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lobanenkov, V. V., R. H. Nicolas, V. V. Adler, H. Paterson, E. M. Klenova, A. V. Polotskaja, and G. H. Goodwin. 1990. A novel sequence-specific DNA binding protein which interacts with three regularly spaced direct repeats of the CCCTC-motif in the 5′-flanking sequence of the chicken c-myc gene. Oncogene 5:1743-1753. [PubMed] [Google Scholar]
- 27.Mancini-DiNardo, D., S. J. Steele, R. S. Ingram, and S. M. Tilghman. 2003. A differentially methylated region within the gene Kcnq1 functions as an imprinted promoter and silencer. Hum. Mol. Genet. 12:283-294. [DOI] [PubMed] [Google Scholar]
- 28.Mancini-DiNardo, D., S. J. Steele, J. M. Levorse, R. S. Ingram, and S. M. Tilghman. 2006. Elongation of the Kcnq1ot1 transcript is required for genomic imprinting of neighboring genes. Genes Dev. 20:1268-1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mitsuya, K., M. Meguro, M. P. Lee, M. Katoh, T. C. Schulz, H. Kugoh, M. A. Yoshida, N. Niikawa, A. P. Feinberg, and M. Oshimura. 1999. LIT1, an imprinted antisense RNA in the human KvLQT1 locus identified by screening for differentially expressed transcripts using monochromosomal hybrids. Hum. Mol. Genet. 8:1209-1217. [DOI] [PubMed] [Google Scholar]
- 30.Moon, H., G. Filippova, D. Loukinov, E. Pugacheva, Q. Chen, S. T. Smith, A. Munhall, B. Grewe, M. Bartkuhn, R. Arnold, L. J. Burke, R. Renkawitz-Pohl, R. Ohlsson, J. Zhou, R. Renkawitz, and V. Lobanenkov. 2005. CTCF is conserved from Drosophila to humans and confers enhancer blocking of the Fab-8 insulator. EMBO Rep. 6:165-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mukhopadhyay, R., W. Yu, J. Whitehead, J. Xu, M. Lezcano, S. Pack, C. Kanduri, M. Kanduri, V. Ginjala, A. Vostrov, W. Quitschke, I. Chernukhin, E. Klenova, V. Lobanenkov, and R. Ohlsson. 2004. The binding sites for the chromatin insulator protein CTCF map to DNA methylation-free domains genome-wide. Genome Res. 14:1594-1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ohlsson, R., R. Renkawitz, and V. Lobanenkov. 2001. CTCF is a uniquely versatile transcription regulator linked to epigenetics and disease. Trends Genet. 17:520-527. [DOI] [PubMed] [Google Scholar]
- 33.Pant, V., S. Kurukuti, E. Pugacheva, S. Shamsuddin, P. Mariano, R. Renkawitz, E. Klenova, V. Lobanenkov, and R. Ohlsson. 2004. Mutation of a single CTCF target site within the H19 imprinting control region leads to loss of Igf2 imprinting and complex patterns of de novo methylation upon maternal inheritance. Mol. Cell. Biol. 24:3497-3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pant, V., P. Mariano, C. Kanduri, A. Mattsson, V. Lobanenkov, R. Heuchel, and R. Ohlsson. 2003. The nucleotides responsible for the direct physical contact between the chromatin insulator protein CTCF and the H19 imprinting control region manifest parent of origin-specific long-distance insulation and methylation-free domains. Genes Dev. 17:586-590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pauler, F. M., and D. P. Barlow. 2006. Imprinting mechanisms—it only takes two. Genes Dev. 20:1203-1206. [DOI] [PubMed] [Google Scholar]
- 36.Pugacheva, E. M., V. K. Tiwari, Z. Abdullaev, A. A. Vostrov, P. T. Flanagan, W. W. Quitschke, D. I. Loukinov, R. Ohlsson, and V. V. Lobanenkov. 2005. Familial cases of point mutations in the XIST promoter reveal a correlation between CTCF binding and pre-emptive choices of X chromosome inactivation. Hum. Mol. Genet. 14:953-965. [DOI] [PubMed] [Google Scholar]
- 37.Richards, E. J., and S. C. Elgin. 2002. Epigenetic codes for heterochromatin formation and silencing: rounding up the usual suspects. Cell 108:489-500. [DOI] [PubMed] [Google Scholar]
- 38.Rosa, A. L., Y. Q. Wu, B. Kwabi-Addo, K. J. Coveler, V. Reid Sutton, and L. G. Shaffer. 2005. Allele-specific methylation of a functional CTCF binding site upstream of MEG3 in the human imprinted domain of 14q32. Chromosome Res. 13:809-818. [DOI] [PubMed] [Google Scholar]
- 39.Schoenherr, C. J., J. M. Levorse, and S. M. Tilghman. 2003. CTCF maintains differential methylation at the Igf2/H19 locus. Nat. Genet. 33:66-69. [DOI] [PubMed] [Google Scholar]
- 40.Sleutels, F., R. Zwart, and D. P. Barlow. 2002. The non-coding Air RNA is required for silencing autosomal imprinted genes. Nature 415:810-813. [DOI] [PubMed] [Google Scholar]
- 41.Smilinich, N. J., C. D. Day, G. V. Fitzpatrick, G. M. Caldwell, A. C. Lossie, P. R. Cooper, A. C. Smallwood, J. A. Joyce, P. N. Schofield, W. Reik, R. D. Nicholls, R. Weksberg, D. J. Driscoll, E. R. Maher, T. B. Shows, and M. J. Higgins. 1999. A maternally methylated CpG island in KvLQT1 is associated with an antisense paternal transcript and loss of imprinting in Beckwith-Wiedemann syndrome. Proc. Natl. Acad. Sci. USA 96:8064-8069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Szabo, P. E., S.-H. E. Tang, A. Rentsendorj, G. P. Pfeifer, and J. R. Mann. 2000. Maternal-specific footprints of putative CTCF sites in the H19 imprinting control region give evidence for insulator function. Curr. Biol. 10:607-610. [DOI] [PubMed] [Google Scholar]
- 43.Thakur, N., M. Kanduri, C. Holmgren, R. Mukhopadhyay, and C. Kanduri. 2003. Bidirectional silencing and DNA methylation-sensitive methylation-spreading properties of the Kcnq1 imprinting control region map to the same regions. J. Biol. Chem. 278:9514-9519. [DOI] [PubMed] [Google Scholar]
- 44.Thakur, N., V. K. Tiwari, H. Thomassin, R. R. Pandey, M. Kanduri, A. Gondor, T. Grange, R. Ohlsson, and C. Kanduri. 2004. An antisense RNA regulates the bidirectional silencing property of the Kcnq1 imprinting control region. Mol. Cell. Biol. 24:7855-7862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Umlauf, D., Y. Goto, R. Cao, F. Cerqueira, A. Wagschal, Y. Zhang, and R. Feil. 2004. Imprinting along the Kcnq1 domain on mouse chromosome 7 involves repressive histone methylation and recruitment of Polycomb group complexes. Nat Genet. 36:1296-1300. [DOI] [PubMed] [Google Scholar]
- 46.Vazquez, J., and P. Schedl. 1994. Sequences required for enhancer blocking activity of scs are located within two nuclease-hypersensitive regions. EMBO J. 13:5984-5993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Verona, R. I., M. R. Mann, and M. S. Bartolomei. 2003. Genomic imprinting: intricacies of epigenetic regulation in clusters. Annu. Rev. Cell Dev. Biol. 19:237-259. [DOI] [PubMed] [Google Scholar]
- 48.West, A. G., M. Gaszner, and G. Felsenfeld. 2002. Insulators: many functions, many mechanisms. Genes Dev. 16:271-288. [DOI] [PubMed] [Google Scholar]
- 49.Yoon, B., H. Herman, B. Hu, Y. J. Park, A. Lindroth, A. Bell, A. G. West, Y. Chang, A. Stablewski, J. C. Piel, D. I. Loukinov, V. V. Lobanenkov, and P. D. Soloway. 2005. Rasgrf1 imprinting is regulated by a CTCF-dependent methylation-sensitive enhancer blocker. Mol. Cell. Biol. 25:11184-11190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhong, X. P., and M. S. Krangel. 1997. An enhancer-blocking element between alpha and delta gene segments within the human T cell receptor alpha/delta locus. Proc. Natl. Acad. Sci. USA 94:5219-5224. [DOI] [PMC free article] [PubMed] [Google Scholar]