

Abstract
Artemis is a phospho-protein that has been shown to have roles in V(D)J recombination, nonhomologous end-joining of double-strand breaks, and regulation of the DNA damage-induced G2/M cell cycle checkpoint. Here, we have identified four sites in Artemis that are phosphorylated in response to ionizing radiation (IR) and show that ATM is the major kinase responsible for these modifications. Two of the sites, S534 and S538, show rapid phosphorylation and dephosphorylation, and the other two sites, S516 and S645, exhibit rapid and prolonged phosphorylation. Mutation of both of these latter two residues results in defective recovery from the G2/M cell cycle checkpoint. This defective recovery is due to promotion by mutant Artemis of an enhanced interaction between unphosphorylated cyclin B and Cdk1, which in turn promotes inhibitory phosphorylation of Cdk1 by the Wee1 kinase. In addition, we show that mutant Artemis prevents Cdk1-cyclin B activation by causing its retention in the centrosome and inhibition of its nuclear import during prophase. These findings show that ATM regulates G2/M checkpoint recovery through inhibitory phosphorylations of Artemis that occur soon after DNA damage, thus setting a molecular switch that, hours later upon completion of DNA repair, allows activation of the Cdk1-cyclin B complex. These findings thus establish a novel function of Artemis as a regulator of the cell cycle in response to DNA damage.
The cellular response to DNA damage results in a pleiotropic activation of numerous pathways including cell cycle checkpoints, DNA repair, transcription, and apoptosis. In eukaryotic cells these responses are mediated to a large degree by a family of phosphatidylinositol-3-OH kinase-like kinases (PIKKs) that include DNA-dependent protein kinase catalytic subunit (DNA-PKcs), ATM, and ATR (reviewed in references 1, 6, 34, and 44). In particular, ATM and ATR are well established as upstream initiators of the checkpoint response emanating from various types of DNA lesions. Although these two kinases may have some overlap in function, ATM primarily responds to DNA double-strand breaks (DSBs) while ATR is activated in response to agents such as UV and blocks to DNA replication forks as well as ionizing radiation (IR). In addition, it has also been shown that ATM can be activated by alterations to chromatin structure such as occur under hypotonic conditions or after exposure to inhibitors of histone deacetylation (2). Numerous downstream targets of ATM and/or ATR have been identified including p53, Brca1, Nbs1, Smc1, Chk1, Chk2, 53BP1, Mdc1, Rad17, and Artemis. These proteins are involved in transducing or mediating checkpoint signals and may in some cases also play a direct role in the repair of DNA damage. On the other hand, DNA-PKcs has no identified role in mediating cell cycle checkpoints but, rather, has a well-described function in the repair of DSBs mediated by the nonhomologous end-joining (NHEJ) pathway.
Artemis is a member of the SNM1 gene family, all members of which contain conserved metallo-β-lactamase and β-CASP domains but are otherwise distinct (3, 5, 9, 21, 32). Artemis is a known substrate of PIKKs and has been shown to be a phosphorylation target of DNA-PKcs, ATM, and ATR in vitro and in vivo (4, 18, 19, 28, 29, 41, 43). This modification is dependent upon the nature of the DNA damaging agent as IR induces phosphorylation of Artemis by both DNA-PKcs and ATM, and UV induces phosphorylation by ATR (43). Artemis appears to play multiple roles in cells. Its original characterization was based on the finding that mutations in Artemis lead to a human severe combined immunodeficiency syndrome due to a defect in V(D)J recombination (21, 22). In addition, Artemis-deficient cells were shown to be radiosensitive, leading to the designation of the complete phenotype as radiosensitive severe combined immunodeficiency syndrome (23, 24). The inability of Artemis-deficient cells to complete V(D)J recombination is caused by a failure to open hairpins that occur at coding joints in a step that is required for subsequent end-joining by NHEJ (19). The hairpin opening activity of Artemis is mediated by an endonuclease that is activated in the presence of DNA-PKcs (19, 26). Artemis has also been reported to be required in NHEJ for the repair of DSBs that possess damaged termini, and this function has been shown to require activation by either ATM (29) and/or DNA-PK (19). In addition, we have previously reported that Artemis plays a role in the cell cycle since a defect was observed in the maintenance or recovery of the G2/M checkpoint upon DNA damage in human cancer cell lines depleted of Artemis (43).
The resumption of the cell cycle or recovery from DNA damage-induced arrest is an area of investigation not well understood. In mammalian cells recovery from the G2/M checkpoint has been shown to be mediated by Polo-like kinase-1 (Plk1) in that, upon completion of DNA repair, Plk1 causes destruction of the Wee1 kinase, a negative regulator of Cdk1 (39). This same study showed that Cdc25B, but not Cdc25A or Cdc25C, was required for resumption of the cell cycle from the G2/M checkpoint. In addition, a number of genes have been implicated in checkpoint recovery in the budding yeast Saccharomyces cerevisiae including Srs2, Ptc2, Ptc3, and Pph3 (12, 14, 40). Ptc2 and Ptc3 were shown to act by dephosphorylating the checkpoint kinase Rad53, and Pph3 was shown to dephosphorylate γ-H2AX in a step required for efficient recovery from the G2/M checkpoint.
Artemis is well established as a phospho-protein that is modified in response to genotoxic agents; however, the functional significance of these phosphorylations has not been elucidated. Here, we report on the identification of four serine residues in Artemis that are subject to phosphorylation after exposure of cells to IR in vivo. We show that after a 3-Gy dose of IR, phosphorylation at these sites is almost solely dependent on ATM, while at a 10-Gy dose of IR it is dependent upon both ATM and DNA-PK. Phosphorylation and dephosphorylation at subsets of these sites have distinct kinetic profiles. We therefore examined one of these subsets and showed that phosphorylation at these sites is required for normal recovery from G2/M cell cycle arrest via regulation of the cyclin B-Cdk1 activation and nuclear import. This finding shows that ATM regulates checkpoint recovery through modifications of Artemis that occur soon after DNA damage, thus setting a molecular switch that acts upon completion of DNA repair. These studies define a novel function for Artemis as a cell cycle regulator after exposure of cells to DNA damage.
MATERIALS AND METHODS
Construction of plasmids.
The coding region of human Artemis was introduced into the Gateway pENTR11 vector (Invitrogen), and mammalian expression plasmids pDEST12.2-Artemis and pDEST27-Artemis were subsequently constructed by interplasmid recombination. To prepare an Artemis construct resistant to small interfering RNA (siRNA; termed pDEST12.2-Artemis-R), three silent mutations were introduced at the sites 1129 (A to C), 1131 (A to C), and 1134 (T to A) using a QuickChange site-directed mutagenesis kit (Stratagene). Other Artemis plasmids expressing mutant versions of the protein were prepared by the same methods.
Cell culture and DNA transfection.
HCT116 and HEK293 cells were cultured in Dulbecco's modified Eagle's medium plus 10% fetal calf serum. Stable cell lines were established by transfection of HEK293 cells with plasmid DNA using FuGENE 6 (Roche), selected in medium containing 400 μg/ml neomycin, and subsequently maintained in 200 μg/ml neomycin. siRNAs were transfected into cells using Oligofectamine reagent (Invitrogen).
GST-Artemis expression and purification.
HEK293 cells stably expressing glutathione S-transferase (GST)-Artemis were washed twice with ice-cold phosphate-buffered saline (PBS) and lysed in 1× EBC buffer, 10 mM sodium fluoride, and 5 mM dithiothreitol. The lysate was spun for 15 min at 10,000 rpm, and the supernatant was transferred and mixed with 100 μl of a 50% slurry of GST beads previously well equilibrated with PBS. After incubation for 2 h with rotation at 4°C, the beads were spun down and washed three times with 10 volumes of PBS. GST-Artemis was eluted by adding sodium dodecyl sulfate sample buffer, and subsequently separated from contaminating proteins by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Gels were stained with Coomassie blue, and the band containing GST-Artemis was excised for subsequent mass spectrometry analysis.
Antibodies and siRNA.
Artemis polyclonal antibodies were developed as described previously (43). Antibodies for other proteins were obtained from commercial sources: ATM (2c-1; GeneTex), ATR (Ab-2; Oncogene), DNA-PKcs (Ab-4; NeoMarkers), phospho-histone H3 (Ser10; Cell Signaling), Cdc25C (Ser216 and 63F9; Cell Signaling), and γ-H2AX (Ser139; Upstate). Antibodies for Cdk1 (C-19), cyclin B (sc-245), Wee1 (B11), Cdc25A (F6), Cdc25C (H-6), and actin (I19) were obtained from Santa Cruz. Antibodies for lamin B (101-B7) and glyceraldehyde-3-phosphate dehydrogenase (6c-5) were from Calbiochem. A monoclonal antibody for bromodeoxyuridine (BrdU; B44) was obtained from Becton Dickinson. Affinity-purified antibody against cyclin B1-pS126 was purchased from Rockland.
Phospho-specific peptide antibodies for Artemis were prepared by Bethyl Laboratories. The peptide sequences synthesized for S516, S534, S538, and S645 were TVAGGS(p)QSPKLFS, CGESTHISS(p)QNSS, CHISSQNSS(p)QSTHI, and CLSTNADS(p)QSSSD, respectively (p indicates phosphorylation site).
siRNA oligonucleotides were synthesized by Dharmacon. The sequence of the coding strand of ATR siRNA was CCUCCGUGAUGUUGCUUGA and as described previously for Artemis, ATM, and DNA-PKcs (43).
Immunoprecipitation, BrdU labeling, cell cycle analysis and phospho-histone H3 staining.
Immunoprecipitation, cell cycle analysis, and phospho-histone H3 staining were performed as described previously (43).
For BrdU labeling, cells were pulse-labeled with BrdU at a concentration of 10 μM for 15 min, and then cells were irradiated with 3 Gy of IR or mock treated. Immediately after treatment, cells were washed twice with PBS and harvested at various time points for cell cycle analysis by flow cytometry.
Small-scale biochemical fractionation.
Approximately 1.5 × 107 cells were scraped into an Eppendorf tube, and 600 μl of cold PBS was added to resuspend the cells. The cells were pelleted by centrifugation at 1,000 rpm at 4°C for 2 min and were subsequently washed twice with 500 μl of PBS. The pellet was resuspended in 200 μl of fractionation buffer containing 10 mM HEPES (pH 7.9), 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 1 mM dithiothreitol, 10 mM NaF, 100 μM Na3VO4, 1× protease inhibitor cocktail, and 0.1% Triton X-100. The suspensions were left on ice for 8 min and subsequently centrifuged for 5 min at 1,300 × g at 4°C. The supernatants were then clarified by centrifugation at 20,000 × g at 4°C for 15 min, and an equal volume of 2× sample buffer was added to the supernatants to form the cytosolic fraction. The pellet was further washed twice with PBS, and a volume of 1× sample buffer equal to the cytosolic fraction was added to form the nuclear fraction.
Immunofluorescence.
HEK293 cells stably transfected with wild-type Artemis or its mutants were harvested and fixed with ethanol. The fixed cells were permeabilized with blocking solution consisting of 4% bovine serum albumin and 0.15% Triton X-100 in PBS for 1 h. Cells were subsequently incubated with primary antibody in blocking solution for 1 h and then washed with blocking buffer. Cells were incubated with either fluorescein isothiocyanate or Alexa Fluor 555 conjugated immunoglobulin G for 30 min and then washed with PBS. DNA was stained with 0.5 μg/ml of 4′,6′-diamidino-2-phenylindole and then observed by fluorescence microscopy.
RESULTS
Identification of sites of phosphorylation in Artemis and the responsible kinases.
ATM, ATR, and DNA-PKcs have been shown to have a preference for phosphorylation of proteins at (S/T)Q motifs (13). As shown in Fig. 1A, Artemis contains nine of these motifs that are conserved between mice and humans. One each of these motifs is located in the metallo-β-lactamase and β-CASP domains, and the other seven are clustered together in the carboxy-terminal half of the protein. Such a cluster of these (S/T)Q motifs is referred to as an (S/T)Q cluster domain (SCD) and has been shown to be preferred sites of phosphorylation by ATM, ATR, and DNA-PK (13, 38). We along with others have shown previously that Artemis undergoes a gel mobility shift after exposure of cells to IR and UV and that this shift is due to phosphorylation (4, 28, 29, 41, 43). In addition to IR and UV, mitomycin C and hydroxyurea also induced this shift (data not shown). These results indicate that Artemis modification is responsive to a broad range of genotoxic agents that induce different types of DNA damage.
FIG. 1.

The role of ATM and DNA-PK in the phosphorylation of Artemis at specific SQ sites after IR treatment is dose dependent. (A) Schematic of Artemis showing (S/T)Q motifs conserved between human and mouse proteins. (B) Specificity of Artemis phospho-specific peptide antibodies to pS516, pS534, pS538, and pS645 as shown by immunoblot analysis. GST fusions of wild-type and the indicated Artemis S-to-A mutants (GST-Artemis mt) were stably expressed in HEK293 cells and exposed to either IR or UV. Rows A, D, G, and J show the signal detected from wild-type and mutant Artemis using the phospho-specific antibodies; rows B, E, H, and K show the lack of signal when the phospho-specific antibodies were preincubated with the appropriate phospho-peptide; and rows C, F, I, and L were probed with an Artemis polyclonal antibody. (C) Phospho-specific antibodies were used to probe the modification of Artemis at either low-dose (2 Gy) or high-dose (10 Gy) IR in the presence or absence of caffeine or wortmannin (wort). (D) Cells were depleted of ATM or ATM and ATR by siRNA treatment, and lysates were probed with the indicated antibodies. (E) Cells were depleted of DNA-PKcs by siRNA treatment and exposed to either low-dose or high-dose IR. Cell lysates were subsequently probed with the indicated antibodies. UT, untreated cells.
To identify specific phosphorylated residues in Artemis, we initiated an analysis by mass spectroscopy. GST-Artemis was overexpressed in HEK293 cells, and GST fusion proteins were purified with or without exposure to IR. This analysis indicated with a high probability that the S516 residue was phosphorylated upon exposure of cells to IR (results not shown). This residue has been previously reported to be a site of phosphorylation in Artemis (18, 36). No other phosphorylated (S/T)Q sites were identified by this analysis. Since it is common for more than one site to be phosphorylated in an SCD, we used gel mobility shift analysis to identify additional sites.
This analysis identified three additional sites: S534, S538, and S645 (data not shown). It should be noted that other investigators have also reported a change in the gel mobility of Artemis after mutation of S645 (4, 28, 36). The S534 and S538 sites have also previously been shown to be phosphorylated in vitro by DNA-PK (36). Taken together, these results suggested that S516, S534, S538, and S645 were candidate sites for phosphorylation in Artemis in vivo after DNA damage.
In order to further analyze these four candidate sites, we prepared affinity purified phospho-specific peptide antibodies for each site. These antibodies were then examined for specificity by immunoblot analysis against either GST-Artemis wild-type protein or the appropriate S-to-A mutant. As shown (Fig. 1B), all four antibodies were able to detect wild-type Artemis but not the mutant protein. As a further control, the antibodies were preincubated with the appropriate phospho-peptide prior to incubation on the membrane, and in each case the signal against wild-type Artemis was ablated (Fig. 1B). These experiments indicated that all four antibodies were specific for phosphorylation at the targeted site and that the reactivity of the antibodies increased after exposure of cells to IR or UV. We have noted in previous experiments (43) that overexpression of GST-Artemis usually results in a noticeable constitutive level of phosphorylation that is not observed for endogenous Artemis. This presumably accounts for the signal detected against GST-Artemis derived from untreated cells.
To determine the kinase(s) responsible for IR-induced phosphorylation at each of the four identified sites, we used the PIKK inhibitors caffeine and wortmannin. Caffeine is an inhibitor of ATM and ATR but not DNA-PK, while wortmannin inhibits all three enzymes (10, 33). For these experiments we examined the phosphorylation of endogenous Artemis. Interestingly, at a relatively low dose of IR (2 Gy), caffeine and wortmannin inhibited phosphorylation at each site to approximately the same degree, while at a high dose of IR (10 Gy) wortmannin was the more potent inhibitor (Fig. 1C). These results suggest that at the lower dose of IR, the phosphorylation is primarily carried out by ATM/ATR, while at the higher dose of IR, DNA-PK may play a significant role. To further examine this issue, we used siRNA transfection to deplete ATM, ATR, or DNA-PKcs and then examined phosphorylation at each site. At the low-dose IR, siRNA-mediated knock-down of ATM significantly reduced the level of phosphorylation at each site (Fig. 1D). A knock-down of ATM and ATR together showed that there was no additional reduction in phosphorylation at any of the four sites, indicating that ATR does not measurably contribute to the phosphorylation at these four sites after low-dose IR (Fig. 1D). The residual phosphorylation at each of the sites may have been due to incomplete knock-down of ATM or to phosphorylation by another kinase such as DNA-PK. To examine the role of DNA-PKcs, we depleted the protein by siRNA and analyzed phosphorylation at each of the four sites at both low- and high-dose IR (Fig. 1E). Knock-down of DNA-PKcs at low-dose IR had little or no effect on the phosphorylation at any of the four sites, while at high-dose IR a significant decrease in signal was observed at all four sites. Taken together, these findings indicate that ATM is the principal kinase that modifies Artemis at these sites after the lower and more physiologically relevant induction of DSBs, while at high levels of DNA damage, Artemis also becomes a substrate for DNA-PK. This conclusion is also consistent with the recent report that an Artemis mutant defective in interaction with DNA-PKcs is, nevertheless, proficient in the joining of DSBs created during V(D)J recombination (36). To eliminate any confounding effects of phosphorylation of Artemis by multiple kinases, all of the IR experiments to be described below were carried out using a 3-Gy dose.
Mutation of S516 and S645 results in prolonged G2/M arrest.
We examined the kinetics of phosphorylation at each of the four SQ sites after IR treatment. As shown (Fig. 2A), phosphorylation at S534 and S538 showed rapid phosphorylation that was readily apparent by 30 min; however, this phosphorylation was highly transient and had disappeared by 2 h. In other experiments (not shown) we have observed dephosphorylation at S534 and S538 as early as 1 h after irradiation. On the other hand, phosphorylation at S516 and S645 was observable at 30 min, remained strong until 6 h, and then slowly declined up to 24 h. It should also be noted that in our prior work we showed that phosphorylation of Artemis, as observed by altered gel migration, could be observed as early as 5 min after IR treatment (43). These findings indicate that residues S516 and S645 have a different kinetic signature of IR-induced phosphorylation and dephosphorylation compared to the S534 and S538 residues.
FIG. 2.

Mutation of the S516 and S645 phosphorylation sites in Artemis causes a prolonged G2/M delay after IR. (A) Immunoblot showing kinetics of phosphorylation and dephosphorylation of GST-Artemis at the indicated sites after exposure to IR. (B) Immunoblot showing expression of the indicated Artemis wild-type and mutant proteins in HEK293 cells. (C) Cell cycle analysis after IR of HEK293 cells stably expressing wild-type Artemis or the S516/645A or S516/645D mutant. (D) Graphical analysis of results shown in panel C. (E) Phospho-histone H3 (P-H3) staining analysis of wild-type and mutant Artemis cell lines after exposure to IR. (F) Cell cycle analysis of Artemis-expressing cells pulse-labeled with BrdU and immediately exposed to IR. WT, wild type; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Since the S516 and S645 residues displayed similar kinetics of phosphorylation and dephosphorylation after DNA damage, we prepared a double mutant in which both residues were changed to alanine (S516A and S645A). We will refer to this double mutant as S516/645A. Since we have previously shown that siRNA-mediated knock-down of Artemis affected the regulation of the G2/M checkpoint (43), we examined the effects of the S516/645A mutant on this checkpoint. For these experiments we prepared HEK293 cells representing a population that stably expressed this mutant protein and control cells that overexpressed wild-type Artemis (Fig. 2B). The sequence of each of the constructs used to express these proteins was modified to render them resistant to a siRNA-mediated knock-down of Artemis. Interestingly, cells expressing the S516/645A mutant showed an accumulation of cells in G2/M that was readily detected between 12 and 24 h after IR treatment compared to cells expressing wild-type Artemis (Fig. 2C and D). In addition, expression of a mutant Artemis in which the serines at 516 and 645 were mutated to aspartic acid residues (S516/645D) in order to mimic phosphorylation resulted in a wild-type phenotype (Fig. 2C and D). This latter result confirms that the observed effect on the G2/M arrest is due to the inability to phosphorylate the S516 and S645 residues after IR. Also, a vector-only control showed that overexpression of Artemis per se did not perturb the cell cycle (Fig. 2D). To determine if the accumulation observed with the S516/645A mutant was due to an effect on the G2/M checkpoint, we examined phospho-H3 staining in each of the three cell lines. As shown (Fig. 2E), the transition from G2 to M phase is delayed in the S516/645A mutant as opposed to cells expressing wild-type Artemis or the S516/645D mutant. The experiments shown in Fig. 2C to E were conducted with knock-down of endogenous Artemis with siRNA (43); however, the prolonged G2 delay was observed whether or not endogenous Artemis was depleted, suggesting that the S516/645A mutant is dominant to wild-type Artemis. Also, as we have shown previously with knock-down of Artemis (43), the initial enforcement of the checkpoint is unaffected by the S516/645A mutant.
To ensure that the accumulation in G2 observed after IR treatment was not the result of effects in earlier stages of the cell cycle, we labeled cells with BrdU, removed the label, and then immediately treated cells with IR. As shown (Fig. 2F), labeled cells expressing the S516/645D mutant were found to transit into G1 as early as 9 h after IR treatment, while G1 phase cells were not observed in the S516/645A-expressing cells even at 12 h after IR, thus confirming the prolonged G2 delay induced by this mutant of Artemis.
We also examined whether the observed G2 accumulation in cells expressing the S516/645A mutant required alteration of both serine residues. Interestingly, cells expressing either the S516A or S645A mutant and exposed to IR did not exhibit the accumulation phenotype (results not shown). Thus, mutation of both serine residues is required for the observed delay in the transition from G2 to M phase after IR treatment.
Finally, we also prepared a double mutant at the S534 and S538 sites, which, as shown above, exhibited transient phosphorylation after IR exposure; however, this mutant did not exhibit any detectable defects in recovery from the G2/M checkpoint after DNA damage.
The prolonged arrest in the S516/645A mutant is not due to a failure to repair.
There are at least two plausible explanations for the observed prolonged delay in the G2 phase in cells expressing the S516/645A Artemis mutant. Such cells may have a reduced ability to repair DSBs and, thus, a prolonged checkpoint or may be defective in recovery or release from the checkpoint. As discussed above, several previous studies have indicated a role for Artemis in NHEJ (17, 19, 29), although this role is apparently limited to the repair of a small fraction of DSBs that have incompatible ends. If the observed G2 accumulation in cells expressing the S516/645A mutant is due to a defect in a pathway of repair of DSBs that requires Artemis, then absence of Artemis should give rise to the same phenotype. However, depletion of Artemis by siRNA (Fig. 3B) actually gave rise to an accelerated transition from G2 to M phase after IR treatment as determined by both cell cycle (Fig. 3A, compare the 12-h time points) and phospho-H3 staining (Fig. 3D), which is contrary to the result obtained with cells expressing the S516/645A mutant (Fig. 2). We also pulse-labeled cells with BrdU and observed that depletion of Artemis by siRNA led to a more rapid entry into the G1 phase than in control cells (Fig. 3C). To further confirm that the observed prolonged G2 arrest with the S516/645A mutant is not due to a DNA repair defect, we examined γ-H2AX levels after IR treatment. The levels of γ-H2AX are an indicator of the levels of DSBs (30, 31). As shown (Fig. 3E), levels of γ-H2AX higher than in control cells were not sustained during the 12- to 24-h time period after IR in cells depleted of Artemis. Similarly, cells expressing the S516/645A mutant did not show an extended duration of H2AX phosphorylation compared to cells expressing Artemis wild type or the S516/645D mutant (Fig. 3F). These results indicate that although the S516/645A mutant induces a prolonged G2 arrest, the cause is not due to a failure to repair DSBs.
FIG. 3.

The effects of Artemis dysfunction on the G2/M checkpoint are not due to a failure to repair DSBs. (A) Cell cycle analysis of HEK293 cells after exposure to IR upon depletion of Artemis by siRNA. (B) Immunoblot showing knockdown of Artemis in HEK293 cells by siRNA. (C) Cell cycle analysis of cells depleted of Artemis by siRNA, pulse-labeled with BrdU, and exposed to IR. (D) Phospho-histone H3 (P-H3) staining analysis of cells depleted of Artemis by siRNA and exposed to IR. (E and F) Levels of γ-H2AX after IR in HEK293 cells were determined in cells depleted of Artemis by siRNA or stably expressing the S516/645A or S516/645D mutants. WT, wild type; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Artemis is a regulator of Cdk1-cyclin B during IR-induced G2/M arrest.
The ultimate target of the G2/M checkpoint is the cyclin-dependent kinase Cdk1 (7, 16). Cdk1 activity is subject to inhibition by the Wee1 and Myt1 kinases that phosphorylate Cdk1 at residues T14 and Y15 (20, 25). These inhibitory phosphorylations are readily detected by shifts in gel migration. To substantiate our cell cycle data, we examined the phosphorylation status of Cdk1 after IR in cells expressing the S516/645A mutant and observed an extended duration of phosphorylation of this kinase compared to cells expressing wild-type Artemis or the S516/645D mutant, particularly at the 12- to 24-h time period (Fig. 4A). We also examined Cdk1 phosphorylation status after knock-down of Artemis by siRNA and found that the phosphorylated forms were greatly reduced compared to control cells (Fig. 4B), consistent with the accelerated transition from G2 to M described above (Fig. 3). Thus, both of these results validate the cell cycle data shown above and suggest that Artemis is a regulator of Cdk1 after exposure of cells to DNA damage.
FIG. 4.

Artemis affects Cdk1 inhibitory phosphorylation through regulation of cyclin B after IR. (A) Immunoblot analysis showing Cdk1 isoforms in HEK293 cells stably expressing the S516/645A mutant compared to cells expressing wild-type Artemis or the S516/645D mutant. (B) Immunoblot showing Cdk1 and cyclin B after depletion of Artemis by siRNA. (C) Immunoprecipitations (IP) of cyclin B from the indicated cell lysates were blotted with antibodies to Cdk1 and Wee1. (D) Lysates from the indicated cell lines were fractionated into cytoplasmic (C) or nuclear (N) fractions and probed with antibodies to the indicated proteins. Cyclin B-P indicates the phosphorylated protein. (E) Immunoprecipitations of Artemis from the indicated cell lysates were blotted with cyclin B antibody. WT, wild type; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
There are a number of direct regulators of Cdk1 including the aforementioned kinases Wee1 and Myt1 and the opposing phosphatases Cdc25A, Cdc25B, and Cdc25C. We examined a number of these proteins in cells expressing the S516/645A mutant and found that either no changes in the levels occurred or the changes that did occur were not in the direction predicted by an extended G2 delay (data not shown). A similar result was observed with Plk1 (data not shown) which is an effector of G2/M checkpoint recovery by mediating degradation of Wee1 (39).
Cyclin B is the principal cyclin that regulates Cdk1 activity during the transition from G2 to M, and we noted in the experiments shown in Fig. 4A and B that Artemis status had a significant impact on the levels of this protein. This observation is particularly apparent in the siRNA knock-down experiment but is also noticeable at the 24-h time points shown in Fig. 4A, where cyclin B was stabilized in cells expressing the S516/645A mutant. Unphosphorylated cyclin B has previously been shown to bind to Cdk1 and to promote the inhibitory phosphorylation at residues T14 and Y15, resulting in an inactive cyclin B-Cdk1 complex (20, 35). Phosphorylation of cyclin B at its cytoplasmic retention sequence allows nuclear accumulation of cyclin B-Cdk1 (8), subsequent dephosphorylation of Cdk1 by Cdc25, and activation of the complex. To determine if Artemis status has an effect on the cyclin B-Cdk1 interaction, we performed reciprocal coimmunoprecipitation experiments. As shown (Fig. 4C, upper right and left blots), expression of the S516/645A mutant had a dramatic impact on the interaction between these two mitotic regulators compared to the expression of wild-type Artemis or the S516/645D mutant. This increased interaction was not entirely due to increased levels of cyclin B since these were modest (Fig. 4C, lower blots) compared to the highly enhanced coimmunoprecipitation. Also note that cyclin B appeared to interact with the phosphorylated form of Cdk1 in cells expressing the S516/645A mutant compared to cells expressing the wild-type protein (Fig. 4C, long exposure). In addition, the coimmunoprecipitation of Wee1 was enhanced by expression of the S516/645A mutant (Fig. 4C), consistent with previous results indicating that unphosphorylated cyclin B promotes the inhibitory phosphorylation of Cdk1 by Wee1 (20, 35). Next, we examined the localization and phosphorylation status of cyclin B after IR. Compared to cells expressing wild-type Artemis, expression of the S516/645A mutant resulted in a significant increase in cytoplasmic levels of cyclin B and a decreased level of phosphorylation (Fig. 4D). Finally, immunoprecipitation of Artemis after exposure of cells to IR shows that cyclin B weakly interacts with wild type or the S516/645D mutant of Artemis but that this interaction is significantly stronger in the presence of the S516/645A mutant (Fig. 4E). Taken together, these results suggest that Artemis regulates cyclin B by affecting its phosphorylation and cytoplasmic retention and that the S516/645A mutant enhances the accumulation of inactive cyclin B-Cdk1 complexes.
Cyclin B is located in the cytoplasm in an unphosphorylated and inactive state during G2. In prophase cyclin B accumulates in the centrosome, where it is activated by phosphorylation at four different sites (11). The kinases responsible for this phosphorylation have not been completely identified although Plk1 appears to phosphorylate at least one of the sites (11, 37). Activation of cyclin B then promotes dephosphorylation and activation of Cdk1 and nuclear import. To determine if this process is affected by mutation of Artemis, we used immunofluorescence to examine the localization of cyclin B after exposure to IR. For these experiments we used an antibody to cyclin B and a phospho-specific antibody that recognizes phospho-S126 of cyclin B, which is one of the sites required for activation of cyclin B. As shown (Fig. 5A), activation and nuclear import of cyclin B occurred normally in cells expressing wild-type Artemis and the S516/645D mutant, whereas the phospho-S126 form of cyclin B appeared to be preferentially localized to the centrosome in cells expressing the S516/645A mutant. Gamma-tubulin staining was used to confirm the centrosomal localization of phospho-S126-cyclin B (Fig. 5B). We also stained for the presence of Artemis in the centrosome. In wild-type-expressing cells the occurrence of Artemis in the centrosome was rare but detectable (approximately 1%), while significantly more cells exhibited this localization in cells expressing the S516/645A mutant (approximately 10%) (Fig. 5C). In addition, the colocalization of Artemis and phospho-S126-cyclin B was significantly enhanced in the S516/645A mutant-expressing cells compared to cells expressing wild-type Artemis (Fig. 5D). This latter result may explain the significantly enhanced coimmunoprecipitation between Artemis and cyclin B observed in cells expressing the S516/645A mutant. The low incidence of wild-type Artemis in the centrosome suggests that, once phosphorylated at S516 and S645, it is excluded from this organelle. Taken together, these results indicate that the S516/645A mutant impedes the nuclear import of cyclin B from the centrosome into the nucleus. This finding thus explains the delayed activation of Cdk1-cyclin B and the resulting extended G2 arrest observed in these cells after IR.
FIG. 5.


Artemis regulates the nuclear import of Cdk1-cyclin B from the centrosome. Shown are immunohistochemical analyses of cells expressing wild-type Artemis or the S516/645A and S516/645D mutants using the indicated antibodies. All cells were fixed 18 h after IR exposure. Arrowheads indicate centrosomes in panels C and D.
DISCUSSION
ATM is the major regulator of Artemis after IR.
We have shown here that four SQ sites, located in the Artemis SCD at positions SQ516, SQ534, SQ538, and SQ645, are modified in vivo in response to IR. These results were confirmed by the use of phospho-specific peptide antibodies in immunoblotting experiments. These experiments showed that at a relatively low dose of IR, ATM is the primary kinase involved in the phosphorylation of Artemis at these four sites, while at a high dose Artemis is also a substrate of DNA-PK. These findings suggest that ATM rather than DNA-PK is the major or most important regulator of Artemis function upon physiologically relevant doses of IR, consistent with our previous findings and reports from other investigators (28, 29, 41, 43).
Phosphorylation at S516 and S645 regulates recovery from the G2/M cell cycle checkpoint.
In our previous studies of Artemis, we showed that knock-down of Artemis did not affect the initial enforcement of the G2/M checkpoint but that there was a failure to sustain the checkpoint (43). These results were supported by our findings that Artemis does not function upstream or downstream of the Chk1/Chk2 kinases. We theorized in that report that Artemis was therefore required for the maintenance of the G2/M checkpoint. Our findings reported here, however, suggest a different conclusion: that Artemis is a negative regulator of recovery from the G2/M checkpoint. This conclusion is based on the following argument. Knock-down of Artemis results in a phenotype in which we observed accelerated release from the IR-induced G2/M checkpoint. This phenotype is consistent with Artemis as either a positive regulator of checkpoint maintenance or a negative regulator of checkpoint recovery. However, if Artemis is a positive regulator of maintenance, then phosphorylation at S516 and S645 would presumably activate Artemis for this function, and the S516/645A mutant would be predicted to have the same phenotype as observed with a lack of Artemis. In fact, the S516/645A mutant has the exact opposite phenotype in that a prolonged G2/M delay was observed. In addition, the dominant nature of this mutant is not consistent with the maintenance model. These observations are, however, consistent with a model in which Artemis is a negative regulator of G2/M checkpoint recovery and in which the phosphorylations at S516 and S645 inhibit Artemis function, allowing recovery from the G2/M checkpoint to occur upon completion of DNA repair. Finally, our findings have also shown that these observed phenotypes are not likely due to a failure of DNA repair since knock-down of Artemis showed accelerated recovery from G2 after IR, and γ-H2AX levels were not affected by either Artemis knock-down or expression of the S516/645A mutant.
A model of G2/M checkpoint recovery mediated by ATM and Artemis.
Previous findings have shown that Plk1 regulates recovery from the G2/M checkpoint by phosphorylation of Wee1, which induces polyubiquitination and degradation of this kinase by the 26S proteosome (39). We were unable to find evidence that Artemis was involved in this pathway in a manner that would negatively regulate recovery. In fact, the S516/645A mutant which exhibits a prolonged G2/M arrest paradoxically showed upregulation of Plk1. This result could be due to a feedback or adaptation pathway that induces Plk1 in response to the inhibition of recovery caused by the S516/645A mutant. Rather, we found that expression of the S516/645A mutant caused disregulation of the activation of the Cdk1-cyclin B complex. Inhibitory phosphorylation of Cdk1 was maintained for a longer time, accounting for the extended G2 arrest in cells expressing the S516/645A mutant. The extended inhibitory phosphorylation of Cdk1 appears to be due to defective activation of cyclin B since in the inactive or unphosphorylated configuration, cyclin B promotes phosphorylation of Cdk1 by Wee1 (20, 35). This conclusion is supported by our finding that the normal activation of cyclin B that occurs in the centrosome and the subsequent export to the nucleus were defective upon expression of the S516/645A mutant. Two possible mechanisms come to mind for the observed abnormal centrosomal retention of cyclin B. Artemis may bind directly to Cdk1-cyclin B and affect it migration from the centrosome into the nucleus, or it may regulate the activating phosphorylations of cyclin B. Cyclin B is known to be phosphorylated on four sites, all of which are required for full activation and nuclear import (8, 11, 15, 27, 37, 42). Clearly, the phosphorylation of the S126 site is not affected by expression of the S516/645A mutant; however, it remains possible that phosphorylation at one or more of the other sites is regulated by Artemis.
A model for the pathway by which ATM and Artemis mediate recovery from the G2/M checkpoint is shown schematically in Fig. 6. Our model envisions that in response to DNA damage ATM immediately phosphorylates Artemis on residues S516 and S645, thereby inhibiting its negative regulation of Cdk1-cyclin B activation. Whether or not this regulation of Cdk1-cyclin B is coupled to completion of DNA repair is unclear; however, it does suggest that Artemis likely has a role in the regulation of Cdk1-cyclin B in the unperturbed cell cycle. The inability to phosphorylate the S516 and S645 sites in the Artemis mutant maintains Artemis in an active state, thereby preventing timely activation of Cdk1-cyclin B in the centrosome. This model thus proposes that ATM regulates recovery from the G2/M checkpoint by an early or immediate inhibitory phosphorylation of Artemis. This phosphorylation essentially constitutes a molecular switch that manifests itself hours later upon completion of DNA repair to allow normal checkpoint recovery.
FIG. 6.
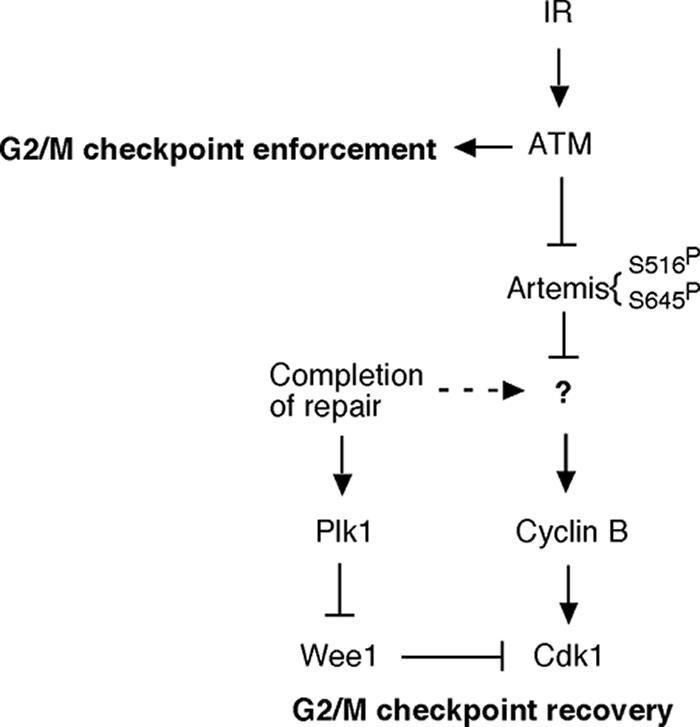
A schematic model depicting the role of Artemis in the cellular response to IR. Dashed line indicates postulated pathway. See the text for additional details.
Functions of Artemis in response to IR.
Previous reports have described a role for Artemis in NHEJ-mediated repair of DSBs and suggested that Artemis is regulated in this function by either DNA-PKcs (19) or ATM (29). Artemis is proposed to facilitate the repair of DSBs that possess incompatible termini that constitute approximately 10% of all DSBs induced by IR (29). Our findings do not directly address the issue of a role for Artemis in NHEJ but do clearly indicate that Artemis has a definitive role in regulating recovery from the G2/M checkpoint that is independent of a role in DSB repair and that ATM is the major regulatory kinase. Finally, Artemis phosphorylation is induced by agents other than IR including UV, mitomycin C, and hydroxyurea and by other checkpoint kinases such as ATR (41, 43). These findings indicate that Artemis is a general stress-responsive protein whose modification is induced by many forms of DNA damage.
Acknowledgments
We thank Lei Li for comments on the manuscript.
This work was supported by grants from the NIH (CA096574 and CA052461).
Footnotes
Published ahead of print on 22 January 2007.
REFERENCES
- 1.Abraham, R. T. 2001. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 15:2177-2196. [DOI] [PubMed] [Google Scholar]
- 2.Bakkenist, C. J., and M. B. Kastan. 2003. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421:499-506. [DOI] [PubMed] [Google Scholar]
- 3.Callebaut, I., D. Moshous, J. P. Mornon, and J. P. de Villartay. 2002. Metallo-beta-lactamase fold within nucleic acids processing enzymes: the beta-CASP family. Nucleic Acids Res. 30:3592-3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen, L., T. Morio, Y. Minegishi, S. Nakada, M. Nagasawa, K. Komatsu, L. Chessa, A. Villa, D. Lecis, D. Delia, and S. Mizutani. 2005. Ataxia-telangiectasia-mutated dependent phosphorylation of Artemis in response to DNA damage. Cancer Sci. 96:134-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dronkert, M. L., J. de Wit, M. Boeve, M. L. Vasconcelos, H. van Steeg, T. L. Tan, J. H. Hoeijmakers, and R. Kanaar. 2000. Disruption of mouse SNM1 causes increased sensitivity to the DNA interstrand cross-linking agent mitomycin C. Mol. Cell. Biol. 20:4553-4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Durocher, D., and S. P. Jackson. 2001. DNA-PK, ATM and ATR as sensors of DNA damage: variations on a theme? Curr. Opin. Cell Biol. 13:225-231. [DOI] [PubMed] [Google Scholar]
- 7.Ferrari, S. 2006. Protein kinases controlling the onset of mitosis. Cell Mol. Life Sci. 63:781-795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hagting, A., M. Jackman, K. Simpson, and J. Pines. 1999. Translocation of cyclin B1 to the nucleus at prophase requires a phosphorylation-dependent nuclear import signal. Curr. Biol. 9:680-689. [DOI] [PubMed] [Google Scholar]
- 9.Henriques, J. A., and E. Moustacchi. 1980. Isolation and characterization of pso mutants sensitive to photo-addition of psoralen derivatives in Saccharomyces cerevisiae. Genetics 95:273-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Izzard, R. A., S. P. Jackson, and G. C. Smith. 1999. Competitive and noncompetitive inhibition of the DNA-dependent protein kinase. Cancer Res. 59:2581-2586. [PubMed] [Google Scholar]
- 11.Jackman, M., C. Lindon, E. A. Nigg, and J. Pines. 2003. Active cyclin B1-Cdk1 first appears on centrosomes in prophase. Nat. Cell Biol. 5:143-148. [DOI] [PubMed] [Google Scholar]
- 12.Keogh, M. C., J. A. Kim, M. Downey, J. Fillingham, D. Chowdhury, J. C. Harrison, M. Onishi, N. Datta, S. Galicia, A. Emili, J. Lieberman, X. Shen, S. Buratowski, J. E. Haber, D. Durocher, J. F. Greenblatt, and N. J. Krogan. 2006. A phosphatase complex that dephosphorylates γH2AX regulates DNA damage checkpoint recovery. Nature 439:497-501. [DOI] [PubMed] [Google Scholar]
- 13.Kim, S. T., D. S. Lim, C. E. Canman, and M. B. Kastan. 1999. Substrate specificities and identification of putative substrates of ATM kinase family members. J. Biol. Chem. 274:37538-37543. [DOI] [PubMed] [Google Scholar]
- 14.Leroy, C., S. E. Lee, M. B. Vaze, F. Ochsenbien, R. Guerois, J. E. Haber, and M. C. Marsolier-Kergoat. 2003. PP2C phosphatases Ptc2 and Ptc3 are required for DNA checkpoint inactivation after a double-strand break. Mol. Cell 11:827-835. [DOI] [PubMed] [Google Scholar]
- 15.Li, J., A. N. Meyer, and D. J. Donoghue. 1997. Nuclear localization of cyclin B1 mediates its biological activity and is regulated by phosphorylation. Proc. Natl. Acad. Sci. USA 94:502-507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lukas, J., C. Lukas, and J. Bartek. 2004. Mammalian cell cycle checkpoints: signalling pathways and their organization in space and time. DNA Repair 3:997-1007. [DOI] [PubMed] [Google Scholar]
- 17.Ma, Y., H. Lu, B. Tippin, M. F. Goodman, N. Shimazaki, O. Koiwai, C. L. Hsieh, K. Schwarz, and M. R. Lieber. 2004. A biochemically defined system for mammalian nonhomologous DNA end joining. Mol. Cell 16:701-713. [DOI] [PubMed] [Google Scholar]
- 18.Ma, Y., U. Pannicke, H. Lu, D. Niewolik, K. Schwarz, and M. R. Lieber. 2005. The DNA-dependent protein kinase catalytic subunit phosphorylation sites in human Artemis. J. Biol. Chem. 280:33839-33846. [DOI] [PubMed] [Google Scholar]
- 19.Ma, Y., U. Pannicke, K. Schwarz, and M. R. Lieber. 2002. Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell 108:781-794. [DOI] [PubMed] [Google Scholar]
- 20.Meijer, L., L. Azzi, and J. Y. Wang. 1991. Cyclin B targets p34cdc2 for tyrosine phosphorylation. EMBO J. 10:1545-1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moshous, D., I. Callebaut, R. de Chasseval, B. Corneo, M. Cavazzana-Calvo, F. Le Deist, I. Tezcan, O. Sanal, Y. Bertrand, N. Philippe, A. Fischer, and J. P. de Villartay. 2001. Artemis, a novel DNA double-strand break repair/V(D)J recombination protein, is mutated in human severe combined immune deficiency. Cell 105:177-186. [DOI] [PubMed] [Google Scholar]
- 22.Moshous, D., L. Li, R. Chasseval, N. Philippe, N. Jabado, M. J. Cowan, A. Fischer, and J. P. de Villartay. 2000. A new gene involved in DNA double-strand break repair and V(D)J recombination is located on human chromosome 10p. Hum. Mol. Genet. 9:583-588. [DOI] [PubMed] [Google Scholar]
- 23.Nicolas, N., N. J. Finnie, M. Cavazzana-Calvo, D. Papadopoulo, F. Le Deist, A. Fischer, S. P. Jackson, and J. P. de Villartay. 1996. Lack of detectable defect in DNA double-strand break repair and DNA-dependent protein kinase activity in radiosensitive human severe combined immunodeficiency fibroblasts. Eur. J. Immunol. 26:1118-1122. [DOI] [PubMed] [Google Scholar]
- 24.Nicolas, N., D. Moshous, M. Cavazzana-Calvo, D. Papadopoulo, R. de Chasseval, F. Le Deist, A. Fischer, and J. P. de Villartay. 1998. A human severe combined immunodeficiency (SCID) condition with increased sensitivity to ionizing radiations and impaired V(D)J rearrangements defines a new DNA recombination/repair deficiency. J. Exp. Med. 188:627-634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Norbury, C., J. Blow, and P. Nurse. 1991. Regulatory phosphorylation of the p34cdc2 protein kinase in vertebrates. EMBO J. 10:3321-3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pannicke, U., Y. Ma, K. P. Hopfner, D. Niewolik, M. R. Lieber, and K. Schwarz. 2004. Functional and biochemical dissection of the structure-specific nuclease Artemis. EMBO J. 23:1987-1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pines, J., and T. Hunter. 1994. The differential localization of human cyclins A and B is due to a cytoplasmic retention signal in cyclin B. EMBO J. 13:3772-3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poinsignon, C., R. de Chasseval, S. Soubeyrand, D. Moshous, A. Fischer, R. J. Hache, and J. P. de Villartay. 2004. Phosphorylation of Artemis following irradiation-induced DNA damage. Eur. J. Immunol. 34:3146-3155. [DOI] [PubMed] [Google Scholar]
- 29.Riballo, E., M. Kuhne, N. Rief, A. Doherty, G. C. Smith, M. J. Recio, C. Reis, K. Dahm, A. Fricke, A. Krempler, A. R. Parker, S. P. Jackson, A. Gennery, P. A. Jeggo, and M. Lobrich. 2004. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Mol. Cell 16:715-724. [DOI] [PubMed] [Google Scholar]
- 30.Rogakou, E. P., C. Boon, C. Redon, and W. M. Bonner. 1999. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 146:905-916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rogakou, E. P., D. R. Pilch, A. H. Orr, V. S. Ivanova, and W. M. Bonner. 1998. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 273:5858-5868. [DOI] [PubMed] [Google Scholar]
- 32.Ruhland, A., M. Kircher, F. Wilborn, and M. Brendel. 1981. A yeast mutant specifically sensitive to bifunctional alkylation. Mutat. Res. 91:457-462. [DOI] [PubMed] [Google Scholar]
- 33.Sarkaria, J. N., E. C. Busby, R. S. Tibbetts, P. Roos, Y. Taya, L. M. Karnitz, and R. T. Abraham. 1999. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 59:4375-4382. [PubMed] [Google Scholar]
- 34.Shiloh, Y. 2003. ATM and related protein kinases: safeguarding genome integrity. Nat. Rev. Cancer 3:155-168. [DOI] [PubMed] [Google Scholar]
- 35.Solomon, M. J., M. Glotzer, T. H. Lee, M. Philippe, and M. W. Kirschner. 1990. Cyclin activation of p34cdc2. Cell 63:1013-1024. [DOI] [PubMed] [Google Scholar]
- 36.Soubeyrand, S., L. Pope, R. De Chasseval, D. Gosselin, F. Dong, J. P. de Villartay, and R. J. Hache. 2006. Artemis phosphorylated by DNA-dependent protein kinase associates preferentially with discrete regions of chromatin. J. Mol. Biol. 358:1200-1211. [DOI] [PubMed] [Google Scholar]
- 37.Toyoshima-Morimoto, F., E. Taniguchi, N. Shinya, A. Iwamatsu, and E. Nishida. 2001. Polo-like kinase 1 phosphorylates cyclin B1 and targets it to the nucleus during prophase. Nature 410:215-220. [DOI] [PubMed] [Google Scholar]
- 38.Traven, A., and J. Heierhorst. 2005. SQ/TQ cluster domains: concentrated ATM/ATR kinase phosphorylation site regions in DNA-damage-response proteins. Bioessays 27:397-407. [DOI] [PubMed] [Google Scholar]
- 39.van Vugt, M. A., A. Bras, and R. H. Medema. 2004. Polo-like kinase-1 controls recovery from a G2 DNA damage-induced arrest in mammalian cells. Mol. Cell 15:799-811. [DOI] [PubMed] [Google Scholar]
- 40.Vaze, M. B., A. Pellicioli, S. E. Lee, G. Ira, G. Liberi, A. Arbel-Eden, M. Foiani, and J. E. Haber. 2002. Recovery from checkpoint-mediated arrest after repair of a double-strand break requires Srs2 helicase. Mol. Cell 10:373-385. [DOI] [PubMed] [Google Scholar]
- 41.Wang, J., J. M. Pluth, P. K. Cooper, M. J. Cowan, D. J. Chen, and S. M. Yannone. 2005. Artemis deficiency confers a DNA double-strand break repair defect and Artemis phosphorylation status is altered by DNA damage and cell cycle progression. DNA Repair 4:556-570. [DOI] [PubMed] [Google Scholar]
- 42.Yang, J., H. Song, S. Walsh, E. S. Bardes, and S. Kornbluth. 2001. Combinatorial control of cyclin B1 nuclear trafficking through phosphorylation at multiple sites. J. Biol. Chem. 276:3604-3609. [DOI] [PubMed] [Google Scholar]
- 43.Zhang, X., J. Succi, Z. Feng, S. Prithivirajsingh, M. D. Story, and R. J. Legerski. 2004. Artemis is a phosphorylation target of ATM and ATR and is involved in the G2/M DNA damage checkpoint response. Mol. Cell. Biol. 24:9207-9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou, B. B., and S. J. Elledge. 2000. The DNA damage response: putting checkpoints in perspective. Nature 408:433-439. [DOI] [PubMed] [Google Scholar]