

Abstract
We compared the transcriptomes of Saccharomyces cerevisiae cells growing under steady-state conditions on 21 unique sources of nitrogen. We found 506 genes differentially regulated by nitrogen and estimated the activation degrees of all identified nitrogen-responding transcriptional controls according to the nitrogen source. One main group of nitrogenous compounds supports fast growth and a highly active nitrogen catabolite repression (NCR) control. Catabolism of these compounds typically yields carbon derivatives directly assimilable by a cell's metabolism. Another group of nitrogen compounds supports slower growth, is associated with excretion by cells of nonmetabolizable carbon compounds such as fusel oils, and is characterized by activation of the general control of amino acid biosynthesis (GAAC). Furthermore, NCR and GAAC appear interlinked, since expression of the GCN4 gene encoding the transcription factor that mediates GAAC is subject to NCR. We also observed that several transcriptional-regulation systems are active under a wider range of nitrogen supply conditions than anticipated. Other transcriptional-regulation systems acting on genes not involved in nitrogen metabolism, e.g., the pleiotropic-drug resistance and the unfolded-protein response systems, also respond to nitrogen. We have completed the lists of target genes of several nitrogen-sensitive regulons and have used sequence comparison tools to propose functions for about 20 orphan genes. Similar studies conducted for other nutrients should provide a more complete view of alternative metabolic pathways in yeast and contribute to the attribution of functions to many other orphan genes.
Nitrogen is an essential nutrient for all life forms. Evolutionary selective pressure has thus likely favored the early emergence of cells able to transport and catabolize a wide variety of nitrogenous compounds as well as to synthesize endogenously all essential nitrogen-containing molecules. These properties, typical of free-living unicellular organisms, have been particularly well studied in the bacterium Escherichia coli (86) and the yeast Saccharomyces cerevisiae (24, 84, 136). Yeast can use almost 30 distinct nitrogen-containing compounds, including amino acids, urea, ammonium, nitrogen bases, and purine derivatives (Fig. 1) (24). These molecules enter cells via permeases (51) and are immediately used as building blocks in biosynthesis reactions or catabolized to release nitrogen in the form of ammonium (via deamination), glutamate (via transamination), or both (24, 84, 136). Glutamine is then synthesized by the condensation of glutamate and ammonium, a reaction catalyzed by glutamine synthetase (GNL1). Glutamate and glutamine are the two major nitrogen donors in biosynthesis reactions. As illustrated in Fig. 1, ammonium, glutamate, and glutamine are interlinked by specific enzyme systems forming the hub of nitrogen metabolism (24, 84, 136).
FIG. 1.

Main reactions involved in the utilization of nitrogen sources in the yeast S. cerevisiae. The nitrogenous compounds indicated in gray boxes are the 27 nitrogen sources utilizable by the yeast S. cerevisiae. Those tested in this study are indicated in bold. Thick gray arrows represent the catabolic pathways leading to either ammonium or glutamate, and narrower arrows point to carbonaceous products resulting from these catabolic pathways. Among these compounds, those which cannot be metabolized by the cell are circled. The enzymatic reactions involved in the central pathway of nitrogen metabolism and associating ammonium, glutamate, and glutamine are also indicated and correspond to the anabolic NADPH glutamate dehydrogenase Gdh1 (1), the catabolic NAD+ glutamate dehydrogenase Gdh2 (2), the glutamine synthetase Gln1 (3), and the glutamate synthase Glt1 (4). TCA cycle, tricarboxylic acid cycle; CoA, coenzyme A.
Nitrogen transport, anabolism, and catabolism are subject to tight control according to the nitrogen content of the medium. These controls act on gene transcription and on the synthesis, activity, and/or degradation rates of enzymes and permeases (85). Transcriptional controls of genes involved in nitrogen anabolism are of two types: specific ones affecting the genes of one anabolic pathway and the general control of amino acid biosynthesis (GAAC) affecting many amino acid biosynthetic pathways (57, 69). Transcriptional repression of genes involved in a specific biosynthetic pathway has been described in the cases of arginine (39), branched-chain amino acids (leucine, isoleucine, valine) (74), methionine (124), and lysine (10, 103). In the case of leucine, repression is mediated through the inhibition of the Leu3 transcription factor (74), and the corresponding target genes have recently been identified on the whole-genome scale (13). For genes of other anabolic pathways, there appears to be no specific transcriptional control. This is notably the case for the biosynthetic pathways for histidine and aromatic amino acids (phenylalanine, tryptophan, and tyrosine) (69).
GAAC is mediated by the Gcn4 transcription factor (57). This protein is most active in cells starved of amino acids, a situation in which the Gcn2 protein (eIF2α kinase) causes the translation of Gcn4 mRNA to be derepressed (56). Other conditions also promote increased Gcn4 synthesis (139). Gcn4 activates the expression of a large number of genes involved in amino acid biosynthesis. Furthermore, in the absence of any amino acid deficiency, a basal level of Gcn4 mRNA translation is required for the transcription of several genes (e.g., HIS3, ARG4, ARG3, and ARO3) (57). Two genomic studies have aimed to identify the whole set of GAAC target genes (68, 90). In one of them (90), 539 genes whose expression is activated in a Gcn4-dependent manner in amino acid-starved cells were listed.
Many different amino acids can be used as general sources of nitrogen (Fig. 1). Most amino acids present in the external medium are detected by yeast cells via a membrane-associated sensor complex (Ssy1-Ptr3-Ssy5 [SPS]) made of three proteins, including Ssy1, an amino acid permease homolog devoid of transport activity (15, 46). This SPS complex in turn activates the transcription of several amino acid permease genes via the Stp1, Stp2, and Uga35/Dal81 transcription factors (1, 5, 63). Putative additional target genes of this transcriptional control system have emerged from several genomic studies aiming to list all genes induced by amino acids in a Ssy1-, Stp1-, and/or Stp2-dependent manner (43, 45, 73).
Like the transcriptional controls acting on genes involved in nitrogen anabolism, those regulating genes involved in transport and catabolism are of two types: specific ones affecting only a limited number of genes and nitrogen catabolite repression acting on a wide variety of genes (26, 84, 136). Thus, several nitrogenous compounds, such as arginine (39), proline (18), serine, threonine (59), urea, allantoin (25), γ-aminobutyric acid (GABA) (121), and the aromatic amino acids (62), specifically induce the transcription of the genes involved in their utilization. On the other hand, nitrogen catabolite repression (NCR) is typically exerted on the many genes involved in the utilization of nonpreferential nitrogen sources when a good nitrogen source (e.g., ammonium, glutamine, and asparagine) is available in the medium (26, 84, 136). NCR in fact acts through the inhibition of two transcription factors of the GATA family (Gln3 and Gat1/Nil1) which typically bind to upstream 5′-GATA-3′ core sequences and activate gene transcription alone or in conjunction with inducer-specific transcription factors (26, 84). The Gln3 and Gat1 factors are thus most active under limiting nitrogen supply conditions (e.g., when cells grow on poor nitrogen sources like urea and proline) and are also transiently activated upon the addition of rapamycin to nitrogen-rich media (26, 84). Rapamycin inhibits the Tor proteins, which are proposed to govern the inhibition of Gln3 and Gat1 under good nitrogen supply conditions (9). The Tor-dependent inhibition of Gln3 involves the Ure2 protein (28), whereas the repression of Gat1-dependent expression under good nitrogen supply conditions is also dependent on Gzf3/Deh1/Nil2, another GATA family transcription factor (23, 109, 119). A fourth GATA factor encoded by the DAL80/UGA43 gene (27) also acts as an inhibitor of Gat1 in specific gene contexts but is specifically active under poor nitrogen supply conditions (4, 27, 31). Transcription of the GAT1, GZF3, and DAL80 genes is under the control of all four GATA factors. A network of auto- and cross-regulation systems thus links these four key transcriptional regulators of NCR target genes (12, 23, 109, 119). Several studies have focused on identifying in the complete yeast genome the genes subject to NCR or regulated by the GATA factors or those whose expression is activated under nitrogen starvation or by rapamycin (7, 14, 29, 112, 116, 138).
In this study we used a systematic approach to examine the influence of nitrogen on the yeast transcriptome. For this we compared the expression levels of the 5,690 yeast genes in cells growing on 21 distinct unique sources of nitrogen. This analysis has enabled us to identify more than 500 nitrogen-regulated genes and to derive a general scheme representing the status of each nitrogen-sensitive transcriptional control according to the nitrogen source. It has further enabled us to associate new genes with several nitrogen-sensitive regulons and to propose a function related to nitrogen metabolism for several nitrogen-regulated orphan genes. Our results offer a novel and complementary view of how yeast cells adapt their transcriptome and metabolism to the nitrogen supply.
MATERIALS AND METHODS
Strains and media.
The Saccharomyces cerevisiae strains used in this study are all isogenic with the wild-type Σ1278b strain (8) (Table 1). Cells were grown in a minimal buffered (pH 6.1) medium with 3% glucose as the carbon source and various nitrogen sources (Table 2). To nitrogen source-free medium, described in reference 66, each of the following was added as the sole nitrogen source: 10 mM urea (reference medium), (NH4)2SO4, alanine, arginine, asparagine, aspartate, citrulline, GABA, glutamate, glutamine, isoleucine, leucine, methionine, ornithine, phenylalanine, proline, serine, threonine, tyrosine, or valine or 5 mM tryptophan. Comparative analysis of the influence of nitrogen on the expression of the nitrogen-sensitive GAP1 gene in cells growing in the nonbuffered yeast nitrogen base medium versus the citrate-buffered medium (pH 6.1) used in this study revealed similar responses to the quality of the nitrogen source (see Fig. S1 in the supplemental material). The gcn2Δ strain was constructed by the PCR-based gene deletion method (134). The DNA segment used to introduce this mutation was generated with the kanMX2 gene from plasmid pFAa-kanMX2 (81) as a template and the D5-GCN2 and D3-GCN2 PCR oligonucleotide primers (Table 3). Yeast strain 23344c (ura3) was transformed with the PCR fragment by the lithium method described previously (49). Transformants were selected on rich medium containing 200 μg/ml G418 (Geneticin; GIBCO BRL, Gaithersburg, MD). The GAP1::lacZ fusion in plasmid pFL38 has been previously described (119).
TABLE 1.
Strains used in this study
TABLE 2.
Nitrogen sources used in this studya and corresponding generation times
| Nitrogen source (concn [mM]) | Generation time of Σ1278b (h, min) |
|---|---|
| Alanine (10) | 2, 30 |
| Ammonium [(NH4+)2SO4] (10) | 2, 00 |
| Arginine (10) | 2, 25 |
| Asparagine (10) | 2, 00 |
| Aspartate (10) | 2, 10 |
| Citrulline (10) | 4, 30 |
| γ-Aminobutyric acid (10) | 2, 35 |
| Glutamine (10) | 2, 05 |
| Glutamate (10) | 2, 15 |
| Isoleucine (10) | 3, 55 |
| Leucine (10) | 3, 25 |
| Methionine (10) | 4, 05 |
| Ornithine (10) | 4, 30 |
| Phenylalanine (10) | 3, 20 |
| Proline (10) | 3, 15 |
| Serine (10) | 2, 15 |
| Threonine (10) | 4, 20 |
| Tryptophan (5) | 4, 45 |
| Tyrosine (10) | 4, 10 |
| Urea (10) | 3, 35 |
| Valine (10) | 3, 00 |
The 21 nitrogen sources studied here were selected from a list of 27 possible nitrogen sources (the 21 shown plus allantoate, allantoin, cytosine, cytidine, adenine, and guanine) for their ability to support the growth of strain Σ1278b with a reproducible doubling time below 5 h and without any pronounced latency upon inoculation of cultures from a 24-hour preculture. The generation times of cells on cytidine, adenine, and guanine were above 5 h, and latency problems were observed on allantoin and allantoate.
TABLE 3.
Oligonucleotides used in this study
| Oligonucleotide | Sequence |
|---|---|
| D5-GCN2 | 5′-TCAATAATTTTCCGTTCCCCTTAACACATACTATGTATAAGCGGCCGCCAGCTGAAGCTT-3′ |
| D3-GCN2 | 5′-TTAACTGATGCGTTATAGCGCCGCACAGATCTTTAAAGGCGCGGCCGCATAGGCCACTAG-3′ |
| ACT1-left | 5′-CTGCCGGTATTGACCAAACT-3′ |
| ACT1-right | 5′-CGGTGATTTCCTTTTGCATTC-3′ |
| ZAP1-left | 5′-GTACGTTCCCCCAAAGACAA-3′ |
| ZAP1-right | 5′-GGTTTTTCGCCAGAGTGTGT-3′ |
| YGL258W-left | 5′-TCCACTCATCTTCACCACCA-3′ |
| YGL258W-right | 5′-AGCAGTGTCTGCCCAAGAGT-3′ |
| ARO9-left | 5′-GCCGCAACAGATGGATATTT-3′ |
| ARO9-right | 5′-GCATAGGCGATGGTGAGTCT-3′ |
| ESBP6-left | 5′-TACCGTTCTGATTGCCGATA-3′ |
| ESBP6-right | 5′-GCAATCACCTCACAAACCAA-3′ |
| ARO80-left | 5′-ATGCAATGTGGAGATGGTGA-3′ |
| ARO80-right | 5′-TTTACGCGTTATTGGCCTTG-3′ |
| SNQ2-left | 5′-CCTGGCTTCTGGACATTCAT-3′ |
| SNQ2-right | 5′-CCGTTTGGTGGGTTGAAGTA-3′ |
| UGA4-left | 5′-GTCCCGGTATCTGGATCCTT-3′ |
| UGA4-right | 5′-CGATAACATCAGCACCAACG-3′ |
| AMD2-left | 5′-ATTTAGGCCTGGTGACATGC-3′ |
| AMD2-right | 5′-CCATTAGCCCATCAATTTGG-3′ |
| MAE1-left | 5′-ACCAACTAGCCGAATTGTCG-3′ |
| MAE1-right | 5′-CTTGCAAGATCACAGCGGTA-3′ |
β-Galactosidase assays.
β-Galactosidase assays were performed as described previously (3). Results are expressed in nanomoles of o-nitrophenol formed per minute per milligram of protein. Protein concentrations were measured in assays using bovine serum albumin as the standard.
RNA preparation and qRT-PCRs.
Total RNA was purified as previously described (75). Quantitative reverse transcription-PCRs (qRT-PCRs) were used to measure the mRNA levels of the following genes: ACT1, YGL258W, ZAP1, ARO9, ESBP6, ARO80, SNQ2, UGA4, AMD2, and MAE1. For this we used the RT-RTCK05 and RT-SN10-05 kits (Eurogentec, Liège, Belgium) with the following primers: ACT1-left, ACT1-right, YGL258W-left, YGL258W-right, ZAP1-left, ZAP1-right, ARO9-left, ARO9-right, ESBP6-left, ESBP6-right, ARO80-left, ARO80-right, SNQ2-left, SNQ2-right, UGA3-left, UGA3-right, AMD2-left, AMD2-right, MAE1-left, and MAE1-right (Table 3).
Generation and analysis of microarray data.
To purify mRNAs, we used the poly(dT) Oligotex kit (QIAGEN, Westburg, The Netherlands). In each assay, 5 μg mRNA was converted to a labeled cDNA target with the Fairplay indirect labeling kit (Amersham-Pharmacia-Biotech, Gent, Belgium) as previously described (137). Microarrays corresponding to the genome of S. cerevisiae strain S288C were produced by Eurogentec (Liège, Belgium) (D290C and G250E series) (122). Cy3- and Cy5-labeled cDNA targets were combined in equal amounts (0.5 μg), vacuum dried (SpeedVac centrifugation), and resolubilized in 50 μl hybridization buffer composed of DIG Easy Hyb solution (Roche Diagnostics, Vilvoorde, Belgium) with 1 mg/ml salmon DNA. Hybridization with the solution of CyDye-labeled cDNA was performed at 42°C for approximately 24 h. Following hybridization, the slide was first washed in 2× SSC (1× SSC is 0.15 M NaCl plus 0.015 sodium citrate) for 30 seconds and then in 0.1× SSC-0.1% sodium dodecyl sulfate for 5 min and finally twice in 0.1× SSC for 5 min. It was then immediately dried by centrifugation (8 min at 800 × g).
The hybridized microarray was scanned with a GMS418 fluorescence reader (Genetic MicroSystems, Woburn, MA) with a resolution of 10 μm. The slide was scanned twice to get the Cy5 and Cy3 signals, once with a high-photo multiplier tube (PMT) gain and once with a low-PMT gain. Signal quantification for each probe on the microarray was performed with GenePix 4.01.17 image acquisition software (Axon Instruments, Union City, CA). Spots with a diameter greater than 210 μm or smaller than 80 μm were considered low-quality spots, as were spots having a median pixel intensity minus mean pixel intensity of more than 40% of the median pixel intensity for each channel and those having less than 95% of their spot pixels be more than 2 standard deviations above background in either the green or the red channel. Low-quality spots were excluded from further analysis. Intensity values from high-PMT-gain pictures were used, except in the case of saturated spots. In the latter case, intensity values from low-PMT-gain pictures were used after scale correction. Intensity-dependent within-tip group and scale normalization were applied as described in reference 140 using Bioconductor tools (48). Fluorescence ratios were computed on the basis of hybridization signals normalized with background corrections. Experiments were carried out independently twice, with dye swapping. For each experiment, we calculated for each gene the value M as log2(expressionM.NS/expressionM.urea) (where expressionM.NS is the level of expression of the gene on minimal medium with the considered nitrogen source at the concentration specified in Table 2 and expressionM.urea is that on minimal medium with urea). Genes that could not be measured (because of a low-quality spot) in at least one of the duplicate experiments were not considered in further analyses. Figure S2 in the supplemental material compares for each medium the results obtained in the two experiments. Calculated on the basis of a normal distribution of the SAM (significance analysis of microarray) test statistic S, S = Mg/c + SDg (where Mg and SDg are, respectively, the mean and the standard deviation of the M values for gene g and c is the 90th percentile of the SDg values) (30); the P value indicates the confidence level at which a gene can be considered differentially expressed on a given medium (M.NS) compared to the reference medium (M.urea). For each medium, we selected genes having a P value below 1/5,690 (5,690 is the total number of genes considered) to be differentially expressed. This value was chosen in the hope that there would be no more than one false positive per medium.
For genes not expressed on urea but highly expressed on other nitrogen media, M values are typically greater than 1 under all conditions for which a ratio could be calculated. Fifteen of the 506 differentially expressed genes are concerned: ARO9, BAP2, BAP3, SED1, ADH4, YGL258W, YPS5, DOG1, SPL2, YHR213W, YPS6, DAN1, YLL053c, ALD3, and YOR387C (see Table S1 in the supplemental material). The computed ratios calculated for these genes must be considered qualitative rather than quantitative. They were nevertheless considered in the data analysis, as they reflect a high level of expression on specific nitrogen media. Analysis of the distribution of gene expression levels on the 21 tested nitrogen media indicates that the number of unexpressed or poorly expressed genes is not particularly higher on urea than on the other media (see Fig. S3 in the supplemental material). Urea is thus an appropriate choice for the reference nitrogen source.
Clustering methods.
Hierarchical clustering was performed using TIGR MultiExperiment Viewer (111) on the 390 genes whose expression varies significantly under at least one nitrogen condition and for which an expression ratio could be computed for at least 13 media (N value = 13). The data presented here are those obtained using the complete linkage method and the average dot product as a measure of the distance between gene expression profiles. The tree was finally segmented into eight clusters, with the distance threshold between genes considered to be 0.137 in TIGR's MultiExperiment Viewer. Other settings (N value, clustering method, distance, distance threshold) were also tested and evaluated by comparing the obtained gene clusters with predefined lists of genes. Those which were finally chosen globally resulted in the best overlaps with several lists of nitrogen-regulated genes and thus optimal physiological coherence without too much loss of information. A second hierarchical clustering was performed on genes belonging to cluster 3 using the complete linkage method and Pearson's correlation. The resulting subtree was segmented into four subclusters, with a distance threshold between genes considered to be 0.548 in TIGR's MultiExperiment Viewer.
Gene list comparisons.
Comparison of groups or subgroups of coregulated genes to predefined gene lists (about 300 in total) were performed using the compare classes utility provided by the Regulatory Sequence Analysis Tools website (http://rsat.ulb.ac.be/rsat/) (130). To check the significance of the overlap between two lists, overlapping P values were computed on the basis of the following hypergeometric formula:
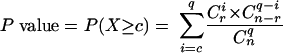 |
(1) |
The P value is the probability of observing at least c common elements between a given query class (the size is q) and a given reference class (the size is r), with consideration of the size of the population (n). Ckj is the number of possible combinations of j elements among k. An e value was also calculated according to the formula Ncomp × P value, where Ncomp is to the number of performed comparisons. The corresponding significance score (sig) is equal to −log10(e value). The predefined gene lists are those available in the MIPS functional catalogue (110) or Gene Ontology categories (6) and in many other lists, like those obtained by chromatin immunoprecipitation (ChIP)-chip (53) or transcriptomic analyses or gene lists available in the literature. The complete data set of these comparisons and the gene lists are accessible online (http://dbm.ulb.ac.be/PhysCell/data/Godard.htm).
Regulatory sequence analyses.
To analyze upstream noncoding sequences, we used the collection of software tools provided by the web resource Regulatory Sequence Analysis Tools (http://rsat.ulb.ac.be/rsat/) (130).
Sequence retrieval.
Upstream sequences of all the yeast genes were retrieved over 800 bp upstream from the start codon. When the upstream open reading frame (ORF) is closer than this distance, a shorter sequence is retrieved, which allows us to discard coding sequences.
NCR-related motifs.
We used the program oligo-analysis (131) to detect overrepresented oligonucleotides in the promoter sequences of the 41 genes annotated as NCR sensitive (A-NCR genes). This analysis was performed for all oligonucleotide sizes between 5 and 8, leading to a total of 56 significantly overrepresented oligonucleotides. Quite consistently, most of these motifs were variants of the GATA box, and the most significant among them was the canonical GATA box GATAAG. To these 56 discovered motifs, we added the auxiliary GATA box (GATTA), and six pairs of GATA boxes separated by a region from 0 to 60 base pairs. The program dna-pattern was used to count the occurrences of the 63 motifs in each yeast gene promoter.
Discriminant analysis.
We applied linear discriminant analysis to classify genes into two classes (NCR versus not NCR) on the basis of the pattern counts (the complete data set is available at http://dbm.ulb.ac.be/PhysCell/data/Godard.htm). As a positive training set (NCR), we used the 41 genes previously A-NCR (see Table S2 in the supplemental material). Since we did not dispose of a reliable negative set for the training (i.e., genes not regulated by NCR), we applied the same strategy as that described previously (117) by randomly selecting a set of 123 (3 × 41) genes in the yeast genome. Since the number of variables (63 in total) is greater than the number of genes in the positive training set, we applied forward stepwise selection to select the subset of variables giving the most accurate classification. The efficiency of a classification was estimated using leave-one-out cross-validation. After this phase of training and variable selection, the discriminant function was then applied to each yeast gene to estimate its posterior probability to be NCR sensitive and to assign it to a class (NCR or not NCR). The whole process was repeated 10 times with different negative groups in order to reduce the number of fluctuations due to random selection. A list of 100 genes predicted to be subject to NCR was finally obtained (see Table S3 in the supplemental material).
Nucleotide sequence accession number.
The microarray data set has been deposited at the Gene Expression Omnibus resource (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE4861.
RESULTS
General approach.
We have used whole-genome microarray hybridization to compare the transcriptomes of wild-type yeast strain Σ1278b during growth on a minimal medium containing 21 different single nitrogen sources, including urea used as a reference condition (Table 2). The 21 compounds were selected from a list of 27 nitrogen sources for their ability to support reproducible growth with a generation time of less than 5 h. The reasons for choosing urea as a reference nitrogen source to which the 20 other media were compared for genome expression were that urea catabolism and its regulation are well known (25), the generation time of strain Σ1278b on urea is near the middle of the studied generation time range (Table 2), and the major regulation systems like NCR and SPS-mediated control do not operate on this medium.
Strain Σ1278b was selected for this work because it is the reference strain of many previous studies of nitrogen regulation, including those having led to the concepts of NCR (136) and pseudohyphal growth induced by limiting ammonium supply to diploid cells (50). Furthermore, strain Σ1278b displays the negative regulatory effect exerted by ammonium on the expression of many genes involved in the use of poor nitrogen sources and on the activities of enzymes and permeases encoded by these genes (136). This negative control is less pronounced or even absent in strain S288C (whose genome has been sequenced) and its derivatives (106). This regulation by ammonium is also exerted in a diploid strain obtained by crossing Σ1278b with S288C, showing that the particular behavior of S288C with respect to ammonium is recessive (85).
The medium was buffered at pH 6.1, and the cells were harvested during exponential growth after at least 10 generations and at low cell density (∼106 cells per ml). We could thus consider that changes in medium composition were minimal during cell culture and that cells were harvested in a balanced state of growth. These precautions ensured highly reproducible growth conditions. The mRNA samples were hybridized to microarrays containing 5,690 known or predicted genes defined after reannotation of the yeast genome on the basis of comparative genome analyses of closely related Saccharomyces species (88, 122). Statistical analysis of collected data (see Materials and Methods) enabled us to identify 506 genes displaying significantly different levels of expression on at least one of the test media and on urea (see Table S1 in the supplemental material). This list of 506 genes was compared to multiple predefined gene lists, including those of the Gene Ontology and MIPS functional categories (see Materials and Methods). As expected, it was found to be highly enriched in genes involved in nitrogen and amino acid and/or sulfur metabolism or in genes whose expression is known to be under nitrogen regulation (the complete data set is available at http://dbm.ulb.ac.be/PhysCell/data/Godard.htm). Among the gene lists compared to the 506 nitrogen-regulated genes are those including the stress-responsive genes. The expression of these genes is typically up-regulated (between 200 and 300 genes) or down-regulated (between 400 and 600 genes) in response to a wide range of environmental changes, with this general control being variously named the environmental-stress response (ESR) or the common environment response (CER) (20, 47). For instance, the majority of genes repressed in response to environmental disturbances code for proteins involved in protein biosynthesis, notably, ribosomal proteins (20, 47). Although yeast cells grew at different rates according to the nitrogen source supplied, we observed no significant variation in levels of expression of the protein synthesis machinery genes from one medium to another (P value = 0.99 [see Fig. S4 in the supplemental material]). It thus seems that the ESR/CER is not differentially active on the 21 tested nitrogen media. This result most likely reflects the fact that, in our experiments, the cells were harvested during steady-state growth, without any significant perturbation in the medium.
We next used the transcriptome data set to classify both the nitrogen media and the genes and to identify the main transcriptional control circuits active on each tested medium.
Classification of nitrogen sources.
The gene expression data were used to establish a classification of the 20 tested nitrogen sources versus urea. For this, we started from the 506 genes and discarded those whose expression versus that on urea could not be computed on a significant number of nitrogen sources, e.g., because their expression level was too low or could not be measured. We thus selected 390 of the 506 genes displaying a significant expression level on at least 13 of the 20 media tested. We then applied hierarchical clustering (see Materials and Methods) to their expression values and derived a nitrogen source classification tree based on the average dot product and the complete linkage method (Fig. 2A). We compared this result with those obtained using other metrics and/or tree construction methods. It appeared that two main groups, together containing 14 nitrogen sources, were classified similarly by the majority of techniques applied. Group A (Fig. 2, left part of the tree) includes asparagine, glutamine, and ammonium, known to be good nitrogen sources supporting rapid growth (generation time, ∼2 h). Interestingly, serine also appears in this group and the corresponding generation time is one of the shortest (generation time, 2 h 13 min). Group A also includes aspartate, alanine, arginine, and glutamate, on which the generation time is below 3 h. It is noteworthy that transamination or deamination of the nitrogenous compounds of group A yields pyruvate or Krebs cycle intermediates (α-ketoglutarate, oxaloacetate), directly assimilable by cell metabolism (Fig. 1). On the media containing nitrogen sources not belonging to group A, the generation time exceeds 3 h. The only exception is the medium containing GABA, which supports fast growth despite significant differences at the level of the yeast transcriptome. Group B (Fig. 2, right part of the tree) includes leucine, isoleucine, methionine, threonine, tryptophan, and tyrosine. In contrast to the group A nitrogen sources and except for leucine, these nitrogen sources support slow growth (generation time, >4 h). Moreover, it is known that the catabolism of these compounds leads to nonmetabolizable products that the cell must excrete and from which fusel oils derive (115, 132, 135). Finally, the classification of the remaining nitrogen sources, namely, valine, phenylalanine, ornithine, proline, GABA, and citrulline, depends strongly on the clustering technique applied. This is why we were unable to associate these compounds with any group on the basis of transcription data. Surprisingly, this classification also indicates that arginine, ornithine, and citrulline are not similar as regards yeast growth or their effects on the transcriptome. Yet all three are urea cycle intermediates degraded largely via the same pathway (136) (Fig. 1). Likewise, among the aromatic amino acids, phenylalanine affects the growth and transcriptome of yeast differently from tryptophan and tyrosine. This is also true of valine, which behaves differently from the other two branched-chain amino acids, leucine and isoleucine.
FIG. 2.

Transcriptome-based classification of nitrogen supply conditions and status of wide-spectrum nitrogen transcriptional regulations. (A) Classification of nitrogen sources. The tree was obtained by applying a hierarchical clustering method (metric, average dot product; method, complete linkage) to the expression data obtained for 390 of the nitrogen-regulated genes (see the text). Group A and group B nitrogen sources are indicated in green and red, respectively. The lower part of the figure shows the expression profiles of the 390 nitrogen-regulated genes subdivided into 8 clusters (see the text). The M value of each gene (see details below and in Materials and Methods) on each tested nitrogen source is shown in a green-to-red color code system, in which M values above 0 are in red, values below 0 are in green, and values equal to 0 or not determined are in black. (B) Influence of the nitrogen source on the generation time (in hours) of yeast cells and on the average expression of genes subject to NCR or to general GAAC. The thick black line indicates the generation time of strain Σ1278b on the different nitrogen media. Bars indicate the means of M values obtained for two sets of genes. One (in green) corresponds to 18 genes known to be mainly regulated by the NCR circuit (see the text). The other corresponds to 32 genes defined as being under Gcn4 control (90). Group A nitrogen sources are indicated in green and group B nitrogen sources in red.
Nitrogen catabolite repression.
Two distinct approaches were used to identify the transcriptional regulations which are differentially active according to the nitrogen source and the associated target genes. One was to use hierarchical clustering to classify 390 genes according to their expression profiles and to compare the resulting groups of coexpressed genes with a large number of annotated gene lists available in databases or generated by previous genome-wide studies (see Materials and Methods). This first approach revealed mostly a limited number of transcriptional controls acting on large sets of genes. A second, complementary approach was to identify groups of genes which are up- or down-regulated on any medium compared to their expression on urea. This approach, applied to the 506 differentially expressed genes, revealed additional, smaller groups of coregulated genes which are the targets of transcriptional controls responding to more-specific signals, e.g., the presence of a single amino acid in the medium.
Below, we present the data obtained by hierarchical clustering based on the average dot product metric (see Materials and Methods). The resulting tree was segmented into eight clusters gathering 140, 14, 119, 37, 21, 23, 6, and 30 genes (Fig. 2A and see Table S1 in the supplemental material). These clusters of genes were systematically compared to predefined gene lists (see Materials and Methods; data available at http://dbm.ulb.ac.be/PhysCell/data/Godard.htm). The largest cluster (cluster 1, 140 genes) significantly overlapped the lists of NCR target genes. We thus describe below how the expression of this first group of genes varies according to the nitrogen conditions. The next sections are devoted to the analysis of the other large clusters. The four remaining smaller clusters significantly overlapped with gene lists of more-specific regulations (e.g., genes inducible by GABA or repressed by methionine), which are considered in the second part of Results.
Behavior of known NCR target genes.
We established a list of genes shown by classical molecular biology studies to be sensitive to NCR, considering a gene to be subject to this regulation if its expression level responds to at least one positive transcriptional regulator (GATA transcription factor Gln3 or Gat1) and to at least one negative one (Dal80, Gzf3, or Ure2) related to NCR. For example, the ZRT1 gene was not included in the list because, although its expression depends on the positive factor Gln3, it does not seem to be controlled by any NCR-related negative regulator (29). We thus propose a list of 41 A-NCR target genes (see Table S2 in the supplemental material). We obtained results for only 34 of the 41 A-NCR genes because ASP3-1, ASP3-2, ASP3-3, and ASP3-4 are not present in the genome of strain Σ1278b (136) and because we were unable to measure any significant expression of the genes ATG14, BAT2, and GDH3 under most conditions tested. Furthermore, two A-NCR genes (VID30 and PEP4) failed to show significant differential expression under our conditions. We thus focused our analysis on the 32 remaining A-NCR genes.
Figure 3A shows that the expression profiles of these 32 A-NCR genes are not all similar. Closer analysis led us to subdivide these genes into two categories. One comprises A-NCR genes which are subject to other transcriptional regulations in addition to NCR. For instance, the UGA1 and UGA4 genes are specifically inducible by GABA (121) and the CAR1 gene by arginine (40). Likewise, DAL4, DAL5, DAL7, DUR1,2, and DUR3 are inducible by allophanate (a product of urea degradation) (25) and PUT1 and PUT2 by proline (18). Other genes in this first category are subject to transcriptional regulations acting on wider sets of genes. This is the case for AGP1, whose expression is induced by extracellular amino acids via the SPS system (63). Likewise, GDH2 is subject to GAAC via transcription factor Gcn4 (57). Yet the expression of several of these inducible genes tends to be lower on nitrogen media that support optimal growth (Fig. 3A), which is consistent with the previous observation that the basal expression of these genes (i.e., monitored in inducer-free media) is subject to NCR.
FIG. 3.

Influence of nitrogen on the expression of known NCR target genes. (A) Expression profiles of 34 of the 41 A-NCR genes (see the text). The color code of the genes is the same as that used in the Venn diagram presented in Fig. 4A. The M value of each gene (see the details in the legend of Fig. 2 and under Materials and Methods) on each tested nitrogen source is shown in the green-to-red color code system used in Fig. 2. The nitrogen sources are sorted according to the classification presented in Fig. 2A. The left part of the figure indicates the number of 5′-GATAAG-3′ sequences found in the 800-bp upstream region of each gene. The brace corresponds to the 18 genes solely regulated by NCR. (B) Influence of the nitrogen source on the degree of NCR. The dotted line represents the Σ1278b strain's generation times on the different nitrogen sources. The black line marked with black squares represents the means of M values (see the details in the legend of Fig. 2 and under Materials and Methods) for the 18 genes known to be regulated mainly by NCR (A-NCR genes [see the text]). The dark-gray line marked with inverted triangles represents the M values obtained for the GAP1 gene alone in our expression data. The light-gray line marked with triangles represents the log2 ratio of the β-galactosidase activities measured on each nitrogen medium to those on urea in wild-type strain 23344c transformed with a centromere-based plasmid bearing a GAP1-lacZ fusion. The nitrogen sources are sorted by the increasing generation time of strain Σ1278b.
The second category includes 18 A-NCR genes all sharing the same expression profile (Fig. 3A) and for which NCR is the main (if not the sole) known transcriptional regulation. This category includes GAP1, which codes for the general amino acid permease (67) and has been the focus of many studies aimed at deciphering the molecular mechanisms of NCR (26, 85). It also includes three of the four genes (GAT1/NIL1, DAL80/UGA43, GZF3/DEH1/NIL2) coding for the GATA transcription factors involved in the transcriptional control of NCR target genes (26, 85), shown in previous studies to be under NCR control and interlinked in a network of trans- and auto-regulation systems (12, 23, 109, 119). Compared to the levels observed on urea, the expression of these 18 A-NCR genes is strongly repressed in yeast cells growing on group A nitrogen sources (Fig. 3). We thus used the average expression profile of these 18 genes as a measure of the degree of NCR on each medium tested. This measure turned out to be very similar to the expression profiles obtained for GAP1 both on microarrays and in experiments using a lacZ reporter gene (Fig. 3). We first noticed that the average expression of the 18 genes is lower on all 20 tested nitrogen media than on urea. This indicates that under the culture conditions used in this study, the reference nitrogen source (urea) is also the one on which NCR is least active. A clear NCR effect is observed with the group A nitrogen sources that support a generation time below 3 h (ammonium, asparagine, glutamine, aspartate, serine, glutamate, arginine, alanine, GABA). NCR is strongest with asparagine, glutamine, and serine. Although glutamine and asparagine are well known to exert a strong NCR effect, the finding that serine does too has been mentioned in only one previous study (17). Interestingly, despite the rapid growth supported by ammonium, glutamate, and aspartate (generation time, ≥2 h but ≤2 h 15 min), these nitrogen sources exert a lesser NCR effect than do asparagine, glutamine, and serine. Alanine, arginine, and GABA support slower growth (generation time, ≥2 h 20 min but ≤2 h 45 min) than glutamate or aspartate but exert equally strong NCR. When the nitrogen source supports growth at a generation time ranging from 3 to 4 h (this is the case for valine, proline, and phenylalanine), NCR is weaker than on the above-mentioned media, and when the generation time exceeds 4 h, there appears to be no NCR except on ornithine (generation time, 4 h 32 min). Surprisingly, as mentioned above, NCR is minimal on urea, despite the intermediate generation time (3 h 35 min). We can thus generally and qualitatively associate three generation time intervals, <3 h, ≥3 h but <4 h, and ≥4 h, with three levels of NCR: strong, weak, and essentially absent, but as shown by the exceptions highlighted above (ammonium, glutamate, aspartate, ornithine, urea), there is no perfect correlation between the growth rate observed with each nitrogen source and the degree of NCR. This point has been discussed in a previous review (24).
Towards an exhaustive list of NCR target genes.
As mentioned above, hierarchical clustering analysis of gene expression data defined a group of 140 coexpressed genes (cluster 1) (see Table S1 in the supplemental material). These 140 genes include 26 of the 32 A-NCR genes found to be differentially expressed in our study (overlap P value = 4.5 × 10−34; sig = 31.6). The six remaining genes were classified in other clusters of coexpressed genes as described below. The group of 140 genes likely includes other NCR target genes in addition to the 26 A-NCR genes. To identify these genes, we first compared the 140 genes with the lists of genes generated by two independent studies aimed at identifying in the whole genome genes controlled by the GATA transcription factors. One of these lists contains 83 yeast genes proposed on the basis of an analysis of ChIP-chip and genome expression data—including those reported in reference 116—to be associated with at least one of the four GATA transcription factors (7); these 83 genes include 17 of the 41 A-NCR genes (overlap P value = 2.3 × 10−22; sig = 19.8). The other list comprises 91 genes whose transcript levels are reported to be positively controlled by the Gln3 and Gat1 GATA factors (112); it includes 28 of the 41 A-NCR genes (overlap P value = 1 × 10−44; sig = 42.2). We thus compared our group of 140 genes with those two lists (Fig. 4A). Of the 16 genes found in all three studies, 4 do not appear on the list of A-NCR genes (GLT1, VBA1, DIP5, and YDR090C), nor do 26 of the 38 genes identified by two of the three studies (Fig. 4A). These 30 genes (4 plus 26) are thus highly probable novel NCR target genes (P-NCR) (Fig. 4B). Accordingly, the average expression profile of these P-NCR genes (Fig. 4C) is quite similar to that described for the A-NCR genes (Fig. 3B), although the amplitudes of differential expression according to the nitrogen source are smaller. We thus propose an updated list of 71 (41 plus 30) genes whose expression appears to be controlled by NCR (see Table S2 in the supplemental material).
FIG. 4.

Identification of additional P-NCR target genes. (A) Venn diagram showing the numbers of NCR target genes found in common in our study (140 genes [see the text]) and/or in two other genome-wide studies (7, 112). Thick black lines delimit the groups of genes belonging to at least two lists. The number of A-NCR genes included in each section of the diagram is also indicated. (B) Expression profiles of 30 highly P-NCR target genes (see the text). The P-NCR genes shown here are those belonging to at least two of the gene lists presented in panel A and which are not A-NCR genes. The color code of the genes is the same as that used in the Venn diagram presented in panel A. The M values and the numbers of GATAAG sequences are indicated as for Fig. 2A and 3A, respectively. (C) Influence of the nitrogen source on the expression levels (average of M values as described in the legend of Fig. 2B) of the 30 P-NCR genes shown in panel B. The nitrogen sources are sorted according to the classification presented in Fig. 2A.
Figure 4A shows that additional putative NCR target genes were found in only one of the three studies. This concerns 91 genes in our study, 52 genes in reference 7, and 47 genes in reference 112. As 11 of these 190 genes are A-NCR genes (Fig. 4A), we cannot simply dismiss them all as false positives. To identify among these 190 genes those which are most likely to be true NCR targets, we analyzed their upstream regions. The promoter regions of NCR target genes typically contain several 5′-GATA-3′ core sequences recognized by the GATA family transcription factors (26, 85). We thus counted, in the upstream regions of all yeast protein-encoding genes, the occurrences of oligonucleotides and spaced pairs corresponding to NCR-specific regulatory motifs and used these counts to classify genes into two classes (NCR or not NCR) using linear discriminant analysis with forward stepwise variable selection (see Materials and Methods). Leave-one-out cross-validation estimates the precision to 92%, with most errors corresponding to false negatives. The optimal discriminant function was then applied to all yeast gene upstream regions, resulting in a list of 100 putative NCR-regulated genes (see Table S3 in the supplemental material). This list included 30 of the 41 A-NCR genes and 4 of the 30 P-NCR genes. Among the remaining genes, 14 were included in the 190 genes found in one of the three above-described experimental studies. We thus considered these 14 genes as additional P-NCR targets potentially extending to 85, i.e., 41 (30 plus 14) A-NCR plus P-NCR genes, the number of yeast genes subject to NCR (see Table S2 in the supplemental material).
We used qRT-PCR to measure the transcript levels of 20 of the 44 P-NCR genes in the wild type and ura3 gzf3 dal80 mutant grown on glutamine. In this triple mutant strain, the nitrogen catabolite repression exerted on Gln3- and Gat1-dependent transcription is largely relieved (26, 119). We have obtained data for 18 genes, and in all cases, the gene was found reproducibly derepressed in the mutant, confirming their sensitivity to NCR (see Table S4 in the supplemental material). About two-thirds of the 44 P-NCR gene products are known to be involved in nitrogen metabolism, and some of them have previously been shown to be expressed to a lower level under good nitrogen supply conditions (see Table S2 in the supplemental material). Other P-NCR genes code for previously studied proteins which do not seem to be directly associated with nitrogen metabolism. The remaining P-NCR genes and three of the A-NCR genes have not been functionally characterized to date. Different methods of protein sequence comparison were applied to most of these proteins to try to infer a putative function. Among other data provided by this analysis (for details, see Table S2 in the supplemental material), we identified two probable amino acid racemases (YIR030C/DCG1 and YGL196W) and a putative vacuolar amino acid transporter (YDR090C). Furthermore, ORF YIL167W has been annotated as a pseudogene coding for part of a serine/threonine deaminase homolog (Sdl1) in strain S288C, with the adjacent gene YIL168W coding for the second part of this enzyme. We found that both YIL167W/SDL1 and YIL168W emerge as P-NCR genes similarly expressed in strain Σ1278b on all tested nitrogen sources. In closely related Saccharomyces species (S. paradoxus, S. mikatae, and S. bayanus), these two ORFs are fused and the corresponding gene probably encodes a functional protein (70). Similarly, the NIT1 gene, coding for a protein similar to nitrilases (98), appears to correspond to two adjacent ORFs, YIL164C and YIL165C, which are similarly expressed in our experiments. These two ORFs are also fused in S. paradoxus, S. mikatae, and S. bayanus (70), suggesting that they code for a functional enzyme in these species. We have sequenced the YIL167W/SDL1-YIL168W and NIT1/YIL164C-YIL165C ORFs isolated from strain Σ1278b used here. We found that, contrary to the situation in strain S288C, neither ORF is interrupted by any stop codon. This strongly suggests that both YIL167-168W/SDL1 and NIT1/YIL164-165C code for functional proteins in strain Σ1278b. The strain with the latter genotype thus appears suited for functional analysis of these genes. Finally, two other P-NCR genes (LEE1/YPL054W and YOR052C) code for proteins of unknown function that contain predicted zinc finger motifs. These genes are described in more detail in the last section of Results. More details on the primary sequences of all P-NCR gene products are available in Table S2 in the supplemental material.
GAAC.
Among the groups of genes found to be coexpressed on the various test media, cluster 4 and cluster 5 include 37 and 21 genes, respectively (see Table S1 in the supplemental material). Both of these clusters contain a significant number of genes subject to the general control of amino acid biosynthesis (GAAC): 7 (cluster 4) and 5 (cluster 5) are indeed among the 37 GAAC target genes originally defined on the basis of classical molecular studies (57) (respective overlap P values = 1.2 × 10−9 and 9.9 × 10−8; sig = 7.13 and 5.20), 16 (cluster 4) and 15 (cluster 5) are on the list of 539 Gcn4 target genes defined on the basis of whole-genome transcript analyses (90) (respective overlap P values = 6.1 × 10−9 and 1 × 10−12; sig = 6.42 and 7.13), and 15 (cluster 4) and 14 (cluster 5) are among the 187 genes found by ChIP-chip analyses (binding P value < 10−3) to be associated with Gcn4 (53) (respective overlap P values = 3.4 × 10−14 and 2.3 × 10−17; sig = 10.98 and 14.15). In addition, analysis of the sequences located upstream from the 37 and 21 genes reveals that the 5′-GAGTCA-3′ sequence is significantly overrepresented among them (sig = 2.77 and 1.98) (130). This consensus motif corresponds with UASGCRE, the Gcn4 binding site (95). Clusters 4 and 5 thus contain a subset of genes whose expression is controlled by this transcription factor. Interestingly, the genes of these groups are typically expressed to a higher level on leucine, isoleucine, methionine, threonine, tryptophan, and tyrosine than on urea, thus suggesting that GAAC is activated in cells growing on the group B nitrogen sources (Fig. 5). Accordingly, the average expression profiles of genes identified as GAAC targets in two previous studies (57, 90) reveal higher expression levels on the above-mentioned media (see Fig. S5 in the supplemental material). To ascertain the importance of GAAC on group B media, we examined the growth of a gcn2Δ mutant strain on all 21 nitrogen sources tested in our study. GCN2 is a key gene of GAAC, coding for the eIF2 kinase required to derepress the translation of Gcn4 mRNA (58). We found that the deletion of GCN2 specifically reduces growth when the nitrogen source is a group B compound (Fig. 6). Hence, the GAAC is not only more active but also important for optimal growth on group B media. Yeast on these nitrogen sources is thus characterized by a lack of NCR, slow growth, and a more active GAAC (Fig. 2B).
FIG. 5.

Influence of nitrogen on the expression of genes subject to GAAC. (A) Venn diagram showing the numbers of genes found in common in our study's clusters 4 and 5 of coregulated genes (58 genes [see the text]), in the list of genes found to be associated with the Gcn4 transcription factor (53), and/or in the list of Gcn4 target genes obtained by transcriptome analysis (90). For each section of the diagram, the number of Gcn4 target genes (H92) originally identified by classical molecular genetic studies (57) is indicated. (B) Expression profile of the 58 genes of clusters 4 and 5. The color code associated with the gene names comes from the Venn diagram presented in panel A. The genes indicated in bold are those classically known as GAAC targets, as reviewed in reference 57. The M value and its color code are as described for previous figures. A schematic of the number of 5′-GA[GC]TCA-3′ sequences found in the 800-bp upstream region of each of the 55 genes is indicated on the left. (C) Influence of nitrogen on the expression levels (averages of M values, as described for Fig. 2B) of the 58 genes shown in panel B. The nitrogen sources are sorted according to the classification presented in Fig. 2A.
FIG. 6.

The GCN2 gene is required for optimal growth on group B nitrogen sources. Strains 23344c (wild type [wt]) and JA766 (gcn2Δ) were streaked onto a solid minimal medium containing a single amino acid as the sole nitrogen source at the indicated concentration. The pictures were taken after 4 days of growth.
Other nitrogen-sensitive transcriptional controls.
Gene classification by hierarchical clustering revealed a third large group of 119 genes (cluster 3 [see Table S1 in the supplemental material]). These genes are expressed on most media better than on urea. Comparison of this cluster with predefined gene lists revealed significant overlaps with the target genes of the SPS system, those of the Zap1 transcription factor, and those of the unfolded-protein response (UPR) (data accessible at http://dbm.ulb.ac.be/PhysCell/data/Godard.htm). Moreover, different genes of this cluster display quite different expression profiles. We thus decided to subclassify them by means of a clustering strategy based on Pearson's correlation metric (see Materials and Methods). This led to subdividing the cluster 3 genes into four subclusters. The average expression profiles of these subclusters are presented in Fig. 7. As detailed below, three of these subclusters overlap previously identified regulons.
FIG. 7.

Many yeast genes are more highly expressed on most tested nitrogen sources than on urea. Influence of the nitrogen source on the expression levels (averages of M values, as described for Fig. 2B) of four subclusters of coregulated genes (see the text). The nitrogen sources are sorted according to the result of the classification presented in Fig. 2A.
SPS target genes.
A first subcluster (subcluster 3-1) comprises 15 genes which are expressed to a higher level on group B nitrogen sources, citrulline, phenylalanine, valine, alanine, and serine than on urea. They are poorly expressed on urea, GABA, or proline (Fig. 7A). The nitrogen sources promoting higher-level expression of these 15 genes are precisely the strongest amino acid inducers of the SPS sensor system (Ssy1-Ptr3-Ssy5 [see the introduction]) (1, 33, 63). Consistently, 3 of the 15 genes (AGP1, MUP1, and GNP1) of subcluster 3-1 encode amino acid permeases known to be targets of the SPS system. Five permease genes (BAP2, BAP3, PTR2, TAT1, and TAT2) in addition to AGP1, MUP1, and GNP1 have been described as SPS target genes in classical molecular studies (11, 38, 63, 72). Although these genes also show lower expression on urea, proline, and GABA than on most of the other media, they were not classified within subcluster 3-1, as their expression profiles significantly differ from those of AGP1, GNP1, and MUP1. The BAP2 and TAT1 genes were, respectively, classified in subcluster 3-2 and subcluster 3-3 (see below), and the three others are not among the 390 genes used in this hierarchical clustering analysis. That the responses to external amino acids differ significantly among SPS target genes has also been observed in experiments based on the use of lacZ reporter fusions for the AGP1, GNP1, BAP2, BAP3, TAT1, and TAT2 genes (our unpublished data). These differences are likely due to the fact that the SPS target genes are subject to other transcriptional regulations whose natures and potencies differ from one gene to another. For instance, PTR2 is also inducible by internal peptides via the Cup9 and Ubr1 factors (126), AGP1 is also subject to NCR (1), and BAP2 is also under the control of the Leu3 transcription factor responding to leucine (37).
We consider that other genes in subcluster 3-1 might be SPS targets, displaying an expression profile resembling that of AGP1, GNP1, and MUP1. Three previous studies based on whole-genome microarray hybridization have aimed to identify all yeast genes up-regulated by external amino acids in an SPS-dependent manner (43, 45, 73). In another study, the binding sites for the Stp1 and Stp2 transcription factors were mapped on the whole-genome scale in ChIP-chip experiments (53). Interestingly, although these reports propose lists comprising between 22 and 72 target genes, the overlap between them is mostly limited to the few amino acid permease genes mentioned above. The same applies to the overlap between these lists and the 15 genes of subcluster 3-1. This suggests that the number of genes up-regulated by amino acids via the SPS system might be more limited than generally thought, possibly mainly to amino acid permease genes.
Influence of nitrogen on the UPR regulon.
A second subcluster (subcluster 3-2) comprises 45 genes which are expressed to a higher level on media containing a good nitrogen source (asparagine, glutamine, serine, ammonium, aspartate, arginine, or glutamate) than on media containing a poor one (Fig. 7B). The expression profile of these genes is thus opposite to that of the NCR target genes. Among these 45 genes, 21 belong to the list of 381 targets of the UPR as defined by a previous whole-genome transcript analysis (125) (overlap P value = 1.4 × 10−14; sig = 12). KAR2, a well-known UPR target (93, 108) even though it does not appear in this list of 381 genes, also belongs to subcluster 3-2. The expression of UPR target genes typically increases when incorrectly folded proteins accumulate in the secretory pathway. This response can also be induced by exposing cells to dithiothreitol, a powerful reducing agent preventing the formation of disulfide bridges, or to tunicamycin, an inhibitor of N-glycosylation reactions. The transcription factor Hac1 and its regulator Ire2 are the two main protein mediators of the UPR (83, 99, 120). Using yMGV (the yeast Microarray Global Viewer) (77, 89), we compared the average expression profile of the 45 genes of subcluster 3-2 with that of the 85 above-defined NCR target genes in a wide set of whole-genome transcript analyses. We found these two gene groups to display opposite expression profiles, notably in two series of experiments aimed at inventorying stress-responsive genes (47). In this study, the NCR target genes were activated under nitrogen starvation and in the stationary phase of growth, while the 45 genes of subcluster 3-2 showed reduced expression under these conditions (see Fig. S6 in the supplemental material).
Influence of urea on the ZAP regulon.
Subcluster 3-3 comprises 42 genes showing lower expression on urea than on any other nitrogen source (Fig. 7C). Surprisingly we found among these genes 16 of the 46 established targets of the Zap1 transcription factor (overlap P value = 5.1 × 10−25; sig = 22.5). Expression of these 46 genes is activated by the Zap1 transcription factor in zinc-deprived cells (82). Interestingly, most of the 26 other genes of subcluster 3-2 also showed Zap1-dependent derepression (82), even though they lack the binding site for this factor in their respective promoter regions. We measured by qRT-PCR the expression of two known Zap1 target genes, YGL258W and ZAP1 itself, on urea and valine media. The results confirm that both genes are expressed to a lower level on urea than on valine (see Fig. S7 in the supplemental material). We also increased the zinc ion concentration from 0.05 μM (the Zn2+ concentration in the minimal medium used in our experiments) to 2.5 μM (the Zn2+ concentration in standard yeast nitrogen base medium); under these conditions, neither of the two genes is expressed to a significant level on either urea or valine medium (data not shown). The Zap1 regulon is thus active under our growth conditions but less so when urea is the sole nitrogen source. Yet we observed no growth defect on media containing as little as 0.05 μM Zn2+ (data not shown), indicating that the zinc ion concentration in the buffered minimal medium used in our experiments is not growth limiting. We do not understand the effect exerted by urea on the expression of Zap1-controlled genes. Other studies have likewise revealed links between nitrogen metabolism and cellular zinc ion homeostasis. On the one hand, expression of the Zap1 target gene ZRT1, coding for a plasma membrane zinc transporter, depends on the GATA transcription factor Gln3 (29). The expression of two other Zap1 target genes, ZRT2 and ZRC1, coding for two zinc transporters located, respectively, at the plasma membrane and the vacuolar membrane (44), is also subject to this control (112). Yet the relationship between zinc ion homeostasis and nitrogen metabolism remains unclear.
Genes most highly expressed on methionine and threonine.
Subcluster 3-4 comprises 17 genes showing higher expression on all media tested than on urea and their highest expression on methionine and threonine (Fig. 7D). Interestingly, eight of these genes encode proteins displaying a mitochondrial localization, according to annotations in the SGD (42).
Transcriptional regulations active on a single or a few nitrogen sources.
So far we have focused our analysis on 390 genes which are differentially expressed according to the nitrogen source, classifying them by clustering analysis. This has enabled us to identify several transcriptional regulations acting on large sets of genes and the nitrogen supply conditions under which these global controls operate (see Discussion). We next analyzed in detail the 506 genes over- or underexpressed on any medium compared to their expression on urea. For this, we again applied the hierarchical clustering technique, this time to the groups of genes which are up- or down-regulated on any of the 20 nitrogen sources versus urea. For several nitrogen sources such as asparagine, glutamine, ammonium, aspartate, alanine, and valine, the identified genes appear on lists of targets of the above-described transcriptional controls, e.g., NCR. Hence, none of these sources induced any detectable specific transcriptional regulation. For other nitrogen sources, there emerged groups of coregulated genes that were not identified in the above analysis and showed activated or repressed expression on only a limited number of nitrogen sources. These genes, described below, are targets of more-specific transcriptional regulations triggered by the nitrogen sources concerned.
Transcriptional responses triggered by aromatic amino acids.
Five genes show higher expression on phenylalanine, tryptophan, and tyrosine than on urea (see Fig. S8 in the supplemental material). As expected, these include ARO9 and ARO10, coding, respectively, for the transaminase and the decarboxylase involved in degrading these amino acids (62, 133) and known to be induced by aromatic amino acids via the Aro80 transcription factor (62). Among the three other genes displaying a similar expression profile, ESBP6 encodes a mitochondrial protein sharing sequence similarities with monocarboxylic acid transporters, but its function remains unknown (87). Another such gene is YDR379C-A, a gene adjacent to ARO10/YDR380W and separated from it by 701 bp. YDR379C-A and ARO10/YDR380W are divergent and thus share the same promoter region. The function of the YDR379C-A gene is unknown, and the sequence of its product is not indicative of any particular function. Lastly, ARO80 itself is expressed to a higher level on tryptophan than on urea, suggesting that Aro80 also controls the expression of its own gene. The promoter regions of these five genes have all been found to associate with the Aro80 transcription factor (53). In accordance with the view that Aro80 acts on ESBP6 and ARO80 in addition to ARO9 and ARO10, the upstream noncoding regions of these genes contain the cis-regulatory ARO upstream activation sequence through which Aro80 activates gene transcription (62). To verify that the ESBP6 and ARO80 genes are regulated by the Aro80 transcription factor, we used qRT-PCR to measure their transcript levels and that of ARO9 on urea medium with or without added tryptophan, tyrosine, or phenylalanine. The strains tested were a wild-type strain and an aro80 mutant (Fig. 8A). In these experiments, the Aro80-dependent induction of ARO9 and ESBP6 expression was observed in the presence of each aromatic amino acid. The same is true of ARO80, but the effect was much less pronounced. We also compared the growth of a strain lacking ESBP6 with that of the corresponding wild type in all media tested in this work but found no difference (data not shown).
FIG. 8.

Identification of novel target genes of the ARO and UGA regulons. (A) Verification by qRT-PCR that the ESBP6 and ARO80 genes are induced by aromatic amino acids in an Aro80-dependent manner. The relative amounts of the mRNA of each gene and of ARO9 used as a control were monitored by qRT-PCR analysis of wild-type (23344c; wt) and aro80-2 (IHI606) strains grown on urea with or without the addition for 1 hour of tryptophan, tyrosine, or phenylalanine (final concentration, 5 mM). The values were normalized with respect to the ACT1 reference gene. For each gene the mean M value is shown with its standard deviation. (B) Verification by qRT-PCR that the MAE1 and AMD2 genes are induced by GABA in a Uga3-dependent manner. The relative amounts of each gene transcript and of UGA4 mRNA used as a control were monitored and calculated as described for panel A in wild-type (23344c) and uga3 (26970a) strains grown on urea with or without the addition for 1 hour of GABA (final concentration, 5 mM).
Nine other genes (IMD2, FLR1, SNQ2, YOR1, ICT1, GRE2, YLR046C, YLR346C, and QDR3) showed higher expression on tryptophan than on other media (see Fig. S8 in the supplemental material). A similar though less pronounced effect was detected on tyrosine medium. All of these genes code for proteins involved in resistance to multiple drugs, and seven of them are known to be targets of Pdr1, Pdr3, Pdr8, Yrr1, and/or Yrm1, i.e., transcription factors involved in pleiotropic-drug resistance (PDR) (34, 55, 78, 96). We used qRT-PCR to measure the expression of the SNQ2 gene on urea with or without added tryptophan or tyrosine. We thus confirmed that SNQ2 is specifically expressed to a higher level in the presence of tryptophan. Furthermore, this induction was not dependent on the Aro80 transcription factor (see Fig. S8 in the supplemental material). These results suggest that the PDR transcriptional network is activated when cells use tryptophan as the sole nitrogen source.
Transcriptional responses triggered by methionine, leucine, and isoleucine.
Surprisingly, the five Aro80 target genes also show higher expression on methionine, leucine, isoleucine, and threonine than on urea (see Fig. S8 in the supplemental material). As the catabolism of these amino acids remains only partially characterized, our observation raises the interesting possibility that the genes of the ARO regulon might also be involved in the catabolism of these amino acids.
Eight other genes showed lower expression on methionine than on any other tested medium. Six of them are among the eight target genes of the Cbf1-Met4-Met28 transcription complex mediating the repression of transcription in response to methionine (124), and the two others are also known to be repressed by methionine (52, 123). The two remaining Cbf1-Met4-Met28 target genes (MET10 and MET2) are also expressed to a lower level on methionine medium in our study but are not among the 506 genes displaying significant variation of expression on one or more tested nitrogen sources from that on urea.
Transcriptional responses triggered by arginine.
The CAR1 and CAR2 genes, encoding arginase and ornithine transaminase, respectively, are up-regulated on arginine, whereas the ARG1, ARG3, ARG4, and ARG8 genes, encoding enzymes involved in arginine anabolism, are repressed on arginine medium. This is in accordance with the mechanisms of mutual exclusion of anabolism and catabolism of this amino acid (39). No other gene displaying arginine-dependent variation of expression was identified. The CAR1 and CAR2 genes are also highly expressed on citrulline and required for citrulline catabolism. Surprisingly, CAR1 is also more highly expressed on ornithine, isoleucine, leucine, methionine, threonine, tryptophan, and tyrosine even though arginase is not involved in the catabolism of any of these nitrogen sources and despite the fact that the arginine biosynthesis genes are GAAC activated only on group B nitrogen sources. A positive effect of these compounds on CAR1 expression has been observed previously, under conditions of nitrogen repression (41). The mechanism causing the higher expression of CAR1 under these conditions remains unknown.
Transcriptional responses triggered by proline, citrulline, and glutamate.
The PUT1 and PUT2 genes (encoding, respectively, proline oxidase and Δ-1-pyrroline-5-carboxylate dehydrogenase) are involved in proline catabolism and are targets of the Put3 transcription factor activated by intracellular proline (18, 35, 36, 60). As expected, both genes show higher expression on proline than on the other tested media. They also show higher expression on citrulline, and increased PUT2 expression is also observed on arginine and ornithine. A recent work has shown that the MCH5 gene encoding a riboflavin transporter (105) is also induced by proline in a Put3-dependent manner (Juergen Stolz, personal communication). Accordingly, MCH5 is more highly expressed on proline medium and also on citrulline, arginine, and ornithine. That citrulline, ornithine, and arginine have a positive effect on the expression of the PUT regulon is likely due to the fact that the catabolism of these amino acids leads to the formation of Δ-1-pyrroline-5-carboxylate, which is converted into proline (19). Apart from the PUT and MCH5 genes, no other gene displaying significant proline-dependent variation of expression was found.
CIT2 and DLD3 (encoding, respectively, a citrate synthase and a d-lactate dehydrogenase) show lower expression on glutamate, proline, and citrulline than on the other nitrogen sources. These two genes are subject to the retrograde (RTG) control (22, 79). This regulation is mediated by the Rtg1 and Rgt3 transcription factors, which are inhibited by glutamate and responsible for the expression of genes encoding enzymes involved in α-ketoglutarate synthesis. Other known RTG targets, i.e., CIT1 (citrate synthase), IDH1, IDH2 (isocitrate dehydrogenase), and ACO1 (aconitase), show the same profile of inhibition by glutamate, but their P value does not exceed the threshold set for selecting differentially expressed genes.
Transcriptional responses triggered by GABA.
Five genes show higher expression on GABA medium than on the other media (see Fig. S10 in the supplemental material). Among them, the genes UGA1 (GABA transaminase), UGA2 (succinate semialdehyde dehydrogenase), and UGA4 (GABA permease) are known to be induced specifically by GABA, via the Uga3 and Uga35/Dal81 transcription factors (3, 104, 121). The other two genes, AMD2 and MAE1, are thus new potential targets of this regulation. To test this hypothesis, we used qRT-PCR to measure their expression and that of UGA4 used as a control in a wild-type strain and a uga3Δ mutant growing on urea with or without added GABA (Fig. 8B). We observed Uga3-dependent induction by GABA of the expression of both genes. AMD2 codes for a putative amidase (21), and MAE1 encodes a mitochondrial malic enzyme catalyzing the oxidative decarboxylation of malate to pyruvate (16). It is known that the uga1 and uga2 mutants are unable to grow on GABA as the sole nitrogen source, and the same is true of a uga4 mutant if the strain lacks the two other GABA permeases (Gap1 and Put4) (3, 104). We deleted the AMD2 and MAE1 genes in the Σ1278b strain, but the resulting mutants displayed no growth defect on any of the tested media, including GABA medium (data not shown).
Transcriptional responses triggered by serine and threonine.
Two genes show much higher expression on serine and threonine than on the other nitrogen sources (see Fig. S11 in the supplemental material). The first, CHA1, encodes a serine/threonine deaminase and is required for growth when the nitrogen source is one of these amino acids (101). Expression of this gene is known to be induced by threonine and serine via the Cha4 transcription factor (59). The second gene, MMF1, encodes a mitochondrial protein of unknown function. This protein is required to maintain the mitochondrial genome and for isoleucine biosynthesis (71, 97). Orthologs of MMF1 are found in all domains of life. The ortholog in Escherichia coli, tdcF, belongs to the tdcABCDEFG operon. Interestingly, several genes of this operon are involved in serine and threonine catabolism in this organism (54). This suggests that MMF1 is involved in serine and/or threonine degradation in yeast. Yeast also possesses a paralog of this gene, HMF1, which is not differentially expressed under the conditions of this study.
Differential expression of genes encoding transcription factors and other regulatory proteins.
Among the 506 genes showing differential expression according to the nitrogen source, 22 code for transcription factors and 15 code for other proteins having a regulatory function. Generally speaking, most of these 37 genes show low-level expression and the variations that we observed are significant but slight. This is notably true of 14 genes that we were unable to associate with any of the above-described groups of coexpressed genes.
Eight of the 37 genes encode transcription factors directly involved in the regulation of nitrogen metabolism genes. As mentioned above, these include the genes coding for the positive GATA factor Gat1 and for the negative GATA factors Gzf3 and Dal80, which are involved in NCR and which are themselves targets of this regulation (23, 109, 119) (Fig. 3A). Another central regulator of nitrogen metabolism is Gcn4, the principal transcription factor of GAAC. Interestingly, our analysis classified GCN4 among the 30 probable new target genes of NCR. The expression profile of GCN4 is indeed typical of genes subject mainly to NCR (Fig. 4B). Furthermore, Gln3 has been associated with the GCN4 locus in ChIP-chip experiments (7), and our data of RT-qPCR experiments showed that GCN4 expression is significantly derepressed in a mutant strain defective in NCR (see Table S4 in the supplemental material). While these observations indicate that the transcription of GCN4 is subject to NCR, two other studies have highlighted a positive contribution of Gcn4 in NCR (118, 128), thus suggesting some cross-regulation between NCR and GAAC (see Discussion). Yet another transcription factor gene classified as an NCR target is UGA3, in keeping with a recent report (65). This factor is responsible for induction by GABA of the transcription of genes involved in GABA catabolism (2, 121). The expression of these genes is also subject to NCR (4, 32, 121). Finally, as already mentioned above, the Aro80 transcription factor seems to activate the expression of its own gene.
Among the 37 regulators differentially expressed in our experiments, 8 are involved in the cell cycle, filamentous growth, and/or pseudohyphal growth (Wtm1, Fus3, Kar4, Clb1, Tpk3, Sst2, Phd1, and Hms2), 7 are known to control glucose metabolism (Mig2, Pd7, Adr1, Nrg2, Hap4, Rgs2, and Mth1), 4 play a role in the cellular stress response (Haa1, Msn4, Hog1, and Ygk3), and 6 are associated with other regulations (Spl2, Spt4, Ino4, Tiss11, Zap1, and Hac1). It is possible that the nitrogen regulation of these regulatory genes allows other metabolic and signaling pathways of yeast to be modulated according to the nitrogen source. Only 4 of the 37 regulator-encoding genes differentially expressed in our experiments encode predicted regulatory proteins of unknown function. Two of them particularly draw our attention. One, encoded by the LEE1/YPL054W gene, codes for a protein containing two tandem repeats of the CCCH-type zinc finger domain. This domain is present in other proteins found in a wide range of organisms from yeast to the human species; it was shown in the case of Cth2 to promote binding to the 3′ end of mRNA and to accelerate its degradation under conditions of iron deficiency (102). The other gene (YOR052C) encodes a protein conserved in all eukaryotes and containing a zf-AN1-type zinc finger domain (80). This protein and its orthologs in many species (including other yeasts) contain an additional N-terminal ubiquitin-like domain whose presence in Yor052p, however, remains uncertain. Further experiments will be required to determine the functions of these potentially interesting putative regulatory proteins.
DISCUSSION
This study constitutes the first systematic analysis of the influence of nitrogen on the yeast transcriptome. We find that the levels of expression of almost 10% of all protein-encoding yeast genes (506 out of 5,690) vary significantly according to the nature of the unique nitrogen source available in the medium. This list of 506 genes is not significantly enriched in genes responding to stress. Even in cells grown on tryptophan, a nitrogen source supporting very slow growth, the expression levels of genes normally up- or down-regulated under stress conditions is not significantly different from that observed on nitrogen sources supporting fast growth. We believe that the lack of a stress response in the cells examined in this study is due to harvesting during steady-state growth. The variation in expression of the many genes subject to the ESR or CER (20, 47) is most visible in cells subjected to various perturbations of their environment, e.g., a shift to a different nutrient supply source, temperature, pH, or osmolarity. Also in support of this view, the transcriptome of yeast was recently examined in cells growing under steady-state conditions on proline or glutamine as the sole nitrogen source and in cells shifted for 2 hours from glutamine to proline (112). This analysis revealed a highly significant variation in the levels of expression of ESR/CER target genes in cells shifted from glutamine to proline but no significant difference in the expression levels of these genes between cells engaged in steady-state growth on one or the other medium.
We have used the expression data of nitrogen-regulated genes (i) to classify the nitrogen sources, (ii) to inventory the nitrogen-sensitive transcriptional controls and the corresponding target genes, and (iii) to evaluate the degree of activation of these transcriptional circuits on all tested nitrogen sources (Fig. 9). We have also tried to infer a function for some nitrogen-regulated orphan genes.
FIG. 9.

Influence of the nitrogen medium on the status of nitrogen-sensitive transcriptional controls. The average levels of expression of the target genes of each indicated regulon were calculated and used to evaluate the degree of activation of the corresponding transcriptional controls (see the text and previous figures). For simplicity's sake, we have defined four degrees of activation: highly active (black squares), moderately active (dark-gray squares), slightly active (light-gray squares), and inactive (white squares). Nitrogen sources are sorted according to the classification presented in Fig. 2A. The activation of transcriptional regulation generally leads to up-regulation of the corresponding target gene. NCR, however, is an exception; in the presence of a good nitrogen source, NCR is active, and this results in the lack of activation of the genes under the positive control of the Gln3 and Gat1/Nil1 transcription factors. ARO, specific induction of genes involved in aromatic amino acid catabolism; SPS, response to extracellular amino acids via the SPS system (Ssy1-Ptr3-Ssy5); PDR, response to drugs; DAL, specific induction of genes involved in allantoin, allantoate, and urea catabolism; UGA, specific induction of genes involved in GABA catabolism; CAR, specific induction of genes involved in arginine catabolism; PUT, specific induction of genes involved in proline catabolism; CHA, specific induction of genes involved in serine and threonine catabolism.
Most nitrogen sources fall into two main groups according to the supported growth rate and to the wide-spectrum transcriptional controls occurring in yeast cells growing on these sources (Fig. 2 and 9). Group A comprises asparagine, glutamine, serine, ammonium, aspartate, alanine, arginine, and glutamate. On these nitrogen sources, growth is rapid and NCR occurs. This result is essentially in agreement with the concept that NCR is active on good (preferential) and inactive on poor (nonpreferential) nitrogen sources (26, 85, 136). Yet we have also observed that the level of NCR does not correlate exactly with the generation time. For instance, NCR is partially relieved on glutamate and aspartate, despite rapid growth. Our study, furthermore, has revealed new genes potentially subject to NCR. Starting from a list of 41 established NCR target genes and using our expression data combined with data generated by other genome-wide studies or provided by the analysis of upstream gene sequences, we have extended this list with 44 additional genes, and experiments carried out on 18 of them confirmed that they are sensitive to NCR (see Table S2 in the supplemental material). Among the new NCR target genes are GDH1 and GLT1. The enzymes Gdh1 (anabolic glutamate dehydrogenase), Gdh2 (catabolic glutamate dehydrogenase), Glt1 (glutamate synthase), and Gln1 (glutamine synthetase) constitute the hub of yeast nitrogen metabolism (Fig. 1) (85, 136). Among them, GLN1 and GDH2 were previously established as being sensitive to NCR (84), but GDH1 and GLT1 were not, although observations consistent with this view have been reported (65, 129). It thus seems that the complete set of enzymes forming the heart of nitrogen metabolism in yeast is subject to NCR. Many of the new probable NCR target genes have known functions, and some code for proteins directly involved in nitrogen transport or metabolism. The functions of about 30 others, however, are still unknown. For 14 of these genes, clues to their roles in cell metabolism have been derived from in silico analyses of their protein sequences (see Table S2 in the supplemental material). Furthermore, we have found that two loci defined as pseudogenes in strain S288C are true protein-encoding genes in the Σ1278b strain (used in this study) and respond to NCR. One (YIL167-168W) encodes a homolog of serine/threonine deaminase and the other (YIL164-165C) a homolog of nitrilases. We also note that group A nitrogen sources are characterized by activation of the UPR pathway, a transcriptional control circuit typically activated when incorrectly folded proteins accumulate in the secretory pathway (83, 99, 120). Conversely, the expression of UPR target genes is reported to be reduced when cells are shifted to nitrogen starvation conditions (113). This suggests that the higher protein synthesis rate of cells growing on a nitrogen-rich medium is associated with a greater number of poorly folded proteins, resulting in UPR activation (113). Furthermore, other observations suggest that the UPR and signaling pathways involved in pseudohyphal growth or meiosis of diploid cells might be more connected than anticipated (113, 114).
The group B nitrogen sources comprise leucine, isoleucine, methionine, threonine, tryptophan, and tyrosine. On these nitrogen sources, growth is particularly slow and NCR does not occur. Furthermore, GAAC is activated on these nitrogen media (Fig. 2 and 9). The importance of GAAC in cells growing on group B amino acids is also illustrated by the specific growth defect of a gcn2Δ mutant on the corresponding media. In agreement with GAAC being particularly active under these conditions, the increased activities of two amino acid-biosynthetic enzymes, namely, indole-3-glycerol-phosphate synthase (TRP3) and arginosuccinate lyase (ARG4), have been observed on media containing isoleucine, leucine, methionine, threonine, or tyrosine as the sole nitrogen source (92). Furthermore, this activation depends on Ndr1/Gcn1 (92), a positive regulator of the function of Gcn2 (58). It has been proposed that the activation of GAAC during growth on these amino acids is due to amino acid imbalances caused, for instance, by feedback inhibition of enzymes shared by branched amino acid-biosynthetic pathways (92). In support of this view, the presence of an additional amino acid relieving the amino acid imbalance also leads to a reduction of GAAC (92).
Another difference between the group A and group B nitrogen sources lies in the fate of the carbon derivatives resulting from the catabolism of these compounds. Whereas the transamination or deamination of group A nitrogen sources yields derivatives directly assimilable by the cell metabolism (Fig. 1), the transamination of group B compounds leads to keto acids undergoing decarboxylation to aldehydes which are in turn converted by dehydrogenases into long-chain or complex alcohols. The amino acid derivatives generated by this so-called Ehrlich pathway (100, 115, 135) are toxic and thus excreted by the cell, and this contributes to the formation of fusel oils. The mechanisms involved in the excretion of aldehydes and higher alcohols remain unknown. In this respect it is noteworthy that growth on tryptophan (and to a lesser extent on tyrosine) leads to the up-regulation of several genes known to be sensitive to the PDR transcriptional control. These genes encode plasma membrane transporters known to promote the excretion of drugs: Snq2 and Yor1 are ATP binding cassette transporters (107), and Flr1 and Qdr3 are probable proton antiporters (91). Our observation that these genes are up-regulated on tryptophan medium suggests that their expression products may play a direct role in the excretion of certain derivatives of tryptophan catabolism, such as tryptophol.
The enzymes involved in the catabolism of the group B nitrogen sources are generally not nearly as well known as those contributing to the degradation of the other nitrogen sources. The main transaminase and the decarboxylase involved in degrading aromatic amino acids (phenylalanine, tryptophan, and tyrosine) have been identified (64, 133); the corresponding genes (ARO9 and ARO10) are controlled by the Aro80 transcription factor (62). Our results reveal that the ARO regulon is induced not only during growth on aromatic amino acids but also in cells grown on any other group B nitrogen source. This strongly suggests a possible involvement of the Aro9 and Aro10 enzymes in the catabolism of all these compounds. In agreement with this view, it was recently shown that Aro10 is a broad-spectrum decarboxylase (100, 132), and it is also known that Aro9 exhibits specificity for a wide range of substrates (76, 127). We have also observed that the ALT1/YLR089C gene, encoding an alanine transaminase homolog, shows higher expression on methionine, isoleucine, citrulline, valine, and alanine than on the other tested nitrogen sources. Hence, the protein encoded by this gene might be involved in the catabolism of these amino acids as well. The involvement of multiple, partially redundant enzymes displaying overlapping substrate specificities very likely explains why classical genetics-based approaches have not allowed effective dissection of the catabolic pathways of group B amino acids, in marked contrast to their successful use in dissecting amino acid anabolic pathways (69). We have also extended the ARO regulon involved in amino acid catabolism to two other genes, one of which (ESBP6/MCH3) encodes a putative transporter that localizes to the inner mitochondrial membrane (87). As Aro9 and Aro10 are cytoplasmic enzymes (61), Mch3 might be responsible for the transport of aldehydes into mitochondria, where several alcohol dehydrogenase activities have been detected (42).
A major difference between the two groups of nitrogen sources defined above is that NCR is active and GAAC is inactive on group A compounds, whereas the opposite is true on group B compounds. This raises the question of whether links might exist between the GAAC and NCR regulations. Our data reveal that the expression of GCN4, encoding the principal transcription factor of GAAC, depends on the nitrogen source supplied and is subject to NCR. On the other hand, previous work has highlighted a contribution of Gcn4 to NCR (118). Together these data suggest a possible general cross-regulation between the two major transcriptional controls of nitrogenous anabolism and catabolism in yeast, in which NCR (most active on good nitrogen sources) would down-regulate GAAC, which in turn would up-regulate NCR. Yet on several other nitrogen sources (valine, phenylalanine, ornithine, proline, citrulline, and urea), neither of these two major transcriptional control circuits appears to be very active (Fig. 9). Further experiments will be required to investigate possible molecular links between NCR and GAAC.
Some regulons are active on both group A and group B nitrogen sources (Fig. 9). This is the case, for instance, for the regulon formed by several amino acid permease genes induced by various external amino acids via the SPS sensor system and the Stp1, Stp2, and Uga35/Dal81 transcription factors (15, 46). We have observed that the target genes of the SPS regulon display quite divergent expression profiles when tested on the different nitrogen sources used here. This was not unexpected, since a similar result was obtained with a lacZ reporter fused to several of these permease genes (our unpublished data). It is likely that the different induction profiles of the genes of this regulon reflect their responsiveness to additional transcriptional controls, whose nature and importance vary from one gene to another. Many other nitrogen-responsive genes share this property of being under a combination of transcriptional controls rather than a single one. This likely explains why hierarchical clustering based on their expression profiles often show these genes to be scattered among distinct clusters of coexpressed genes (see below). Another regulon is less active on various nitrogen sources (especially glutamate and proline); it comprises the genes inhibited by glutamate via the RTG control (22, 79) (Fig. 9). These genes can be expected to be poorly expressed on proline medium, because the internal pool of glutamate is high on this medium. The average expression of genes subject to RTG control is also lower on group A nitrogen sources. This may again reflect a high glutamate pool, to be expected in the presence of a good nitrogen source.
Finally, several regulons are most active in cells grown on a more limited number of nitrogen sources (Fig. 9). Among them is the one inducible by GABA (121) and for which we identified two new target genes, MAE1 and AMD2. The MAE1 gene encodes a mitochondrial malic enzyme (16) that might contribute to optimizing the catabolism of succinate deriving from GABA catabolism (Fig. 1). We have also identified MMF1 as a new potential gene of the CHA regulon inducible by serine and threonine. A significant fraction of the 506 genes show weak though significant differential expression according to the nitrogen source code for transcription factors (22 genes) or for other proteins having a regulatory function (15 genes). Some of these regulatory genes encode proteins known to be directly involved in the regulation of nitrogen metabolism genes. For instance, we mention above a role of NCR in the transcriptional control of the GCN4 gene, while this gene appears to be required for optimal NCR (118). We have also identified two probable NCR target genes encoding potentially interesting regulatory factors, including one (LEE1/YPL054W) which may be involved in controlling mRNA stability. Experiments are in progress to test whether this protein plays such a role. Many other regulatory genes apparently controlled by nitrogen are involved in other metabolic pathways or cell processes. Further experiments will be needed to determine whether the apparent regulation by nitrogen of these regulatory genes contributes to the overall influence of nitrogen on other cellular pathways.
We have thus explored in a systematic manner the influence of different nitrogen sources on the yeast transcriptome. The same type of analysis could be used to acquire a general view of the transcriptional regulations involved in carbon, sulfur, and phosphorous metabolism. So far, genomic studies have focused on the mechanisms of adaptation to environmental change and especially to starvation (14, 47, 94, 138). Yet just as yeast is able to utilize many different nitrogen sources, it can also utilize different sources of carbon, sulfur, and/or phosphorous. A systematic study of the yeast transcriptome during growth on all these alternative nutrient sources should provide a more comprehensive view of the metabolic potentialities of yeast and of the associated transcriptional control mechanisms. It should also help to attribute potential functions to the still large number of orphan genes of yeast.
Supplementary Material
Acknowledgments
We are grateful to Sanna Venetvaara for her important contribution to the setting up of the microarray experiments in our laboratory and to Nasiha M'Rabet for her help in the carrying out of the first analyses. We are also very grateful to Catherine Jauniaux for her help in the sequencing the NIT1 and SDL1 loci and in the isolation of the gcn2Δ mutant. We also thank all members of the laboratory and the other participants of the ARC project (members of the laboratories of Jacques van Helden, Gianluca Bontempi, and Marcelline Kaufman) for fruitful discussions. Finally, we thank Juergen Stolz for communication of data before publication.
This work was supported by grant FRSM 3.4.605.05.F from the National Funds for Scientific Research, Belgium, grant Bioval 981/3861 (Région Wallonne), and the Communauté Française de Belgique (ARC grant number 04/09-307).
Footnotes
Published ahead of print on 16 February 2007.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Abdel-Sater, F., I. Iraqui, A. Urrestarazu, and B. Andre. 2004. The external amino acid signaling pathway promotes activation of Stp1 and Uga35/Dal81 transcription factors for induction of the AGP1 gene in Saccharomyces cerevisiae. Genetics 166:1727-1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andre, B. 1990. The UGA3 gene regulating the GABA catabolic pathway in Saccharomyces cerevisiae codes for a putative zinc-finger protein acting on RNA amount. Mol. Gen. Genet. 220:269-276. [DOI] [PubMed] [Google Scholar]
- 3.Andre, B., C. Hein, M. Grenson, and J. C. Jauniaux. 1993. Cloning and expression of the UGA4 gene coding for the inducible GABA-specific transport protein of Saccharomyces cerevisiae. Mol. Gen. Genet. 237:17-25. [DOI] [PubMed] [Google Scholar]
- 4.Andre, B., D. Talibi, S. Soussi Boudekou, C. Hein, S. Vissers, and D. Coornaert. 1995. Two mutually exclusive regulatory systems inhibit UASGATA, a cluster of 5′-GAT(A/T)A-3′ upstream from the UGA4 gene of Saccharomyces cerevisiae. Nucleic Acids Res. 23:558-564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andreasson, C., and P. O. Ljungdahl. 2002. Receptor-mediated endoproteolytic activation of two transcription factors in yeast. Genes Dev. 16:3158-3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ashburner, M., C. A. Ball, J. A. Blake, D. Botstein, H. Butler, J. M. Cherry, A. P. Davis, K. Dolinski, S. S. Dwight, J. T. Eppig, M. A. Harris, D. P. Hill, L. Issel-Tarver, A. Kasarskis, S. Lewis, J. C. Matese, J. E. Richardson, M. Ringwald, G. M. Rubin, and G. Sherlock. 2000. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25:25-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bar-Joseph, Z., G. K. Gerber, T. I. Lee, N. J. Rinaldi, J. Y. Yoo, F. Robert, D. B. Gordon, E. Fraenkel, T. S. Jaakkola, R. A. Young, and D. K. Gifford. 2003. Computational discovery of gene modules and regulatory networks. Nat. Biotechnol. 21:1337-1342. [DOI] [PubMed] [Google Scholar]
- 8.Béchet, J., M. Grenson, and J. M. Wiame. 1970. Mutations affecting the repressibility of arginine biosynthetic enzymes in Saccharomyces cerevisiae. Eur. J. Biochem. 12:31-39. [DOI] [PubMed] [Google Scholar]
- 9.Beck, T., and M. N. Hall. 1999. The TOR signalling pathway controls nuclear localization of nutrient-regulated transcription factors. Nature 402:689-692. [DOI] [PubMed] [Google Scholar]
- 10.Becker, B., A. Feller, M. el Alami, E. Dubois, and A. Pierard. 1998. A nonameric core sequence is required upstream of the LYS genes of Saccharomyces cerevisiae for Lys14p-mediated activation and apparent repression by lysine. Mol. Microbiol. 29:151-163. [DOI] [PubMed] [Google Scholar]
- 11.Bernard, F., and B. Andre. 2001. Genetic analysis of the signalling pathway activated by external amino acids in Saccharomyces cerevisiae. Mol. Microbiol. 41:489-502. [DOI] [PubMed] [Google Scholar]
- 12.Boczko, E. M., T. G. Cooper, T. Gedeon, K. Mischaikow, D. G. Murdock, S. Pratap, and K. S. Wells. 2005. Structure theorems and the dynamics of nitrogen catabolite repression in yeast. Proc. Natl. Acad. Sci. USA. 102:5647-5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boer, V. M., J. M. Daran, M. J. Almering, J. H. Winde, and J. T. Pronk. 2005. Contribution of the Saccharomyces cerevisiae transcriptional regulator Leu3p to physiology and gene expression in nitrogen- and carbon-limited chemostat cultures. FEMS Yeast Res. 5:885-897. [DOI] [PubMed] [Google Scholar]
- 14.Boer, V. M., J. H. Winde, J. T. Pronk, and M. D. Piper. 2003. The genome-wide transcriptional responses of Saccharomyces cerevisiae grown on glucose in aerobic chemostat cultures limited for carbon, nitrogen, phosphorus, or sulfur. J. Biol. Chem. 278:3265-3274. [DOI] [PubMed] [Google Scholar]
- 15.Boles, E., and B. Andre. 2004. Role of transporter-like sensors in glucose and amino acid signalling in yeast, p. 121-153. In E. Boles and K. Reinhard (ed.), Molecular mechanisms controlling transmembrane transport. Springer, Berlin, Germany.
- 16.Boles, E., P. D. Jong-Gubbels, and J. T. Pronk. 1998. Identification and characterization of MAE1, the Saccharomyces cerevisiae structural gene encoding mitochondrial malic enzyme. J. Bacteriol. 180:2875-2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bossinger, J., R. P. Lawther, and T. G. Cooper. 1974. Nitrogen repression of the allantoin degradative enzymes in Saccharomyces cerevisiae. J. Bacteriol. 118:821-829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brandriss, M. C. 1987. Evidence for positive regulation of the proline utilization pathway in Saccharomyces cerevisiae. Genetics 117:429-435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brandriss, M. C., and B. Magasanik. 1980. Proline: an essential intermediate in arginine degradation in Saccharomyces cerevisiae. J. Bacteriol. 143:1403-1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Causton, H. C., B. Ren, S. S. Koh, C. T. Harbison, E. Kanin, E. G. Jennings, T. I. Lee, H. L. True, E. S. Lander, and R. A. Young. 2001. Remodeling of yeast genome expression in response to environmental changes. Mol. Biol. Cell 12:323-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang, T. H., and J. Abelson. 1990. Identification of a putative amidase gene in yeast Saccharomyces cerevisiae. Nucleic Acids Res. 18:7180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chelstowska, A., Z. Liu, Y. Jia, D. Amberg, and R. A. Butow. 1999. Signalling between mitochondria and the nucleus regulates the expression of a new d-lactate dehydrogenase activity in yeast. Yeast 15:1377-1391. [DOI] [PubMed] [Google Scholar]
- 23.Coffman, J. A., R. Rai, D. M. Loprete, T. Cunningham, V. Svetlov, and T. G. Cooper. 1997. Cross regulation of four GATA factors that control nitrogen catabolic gene expression in Saccharomyces cerevisiae. J. Bacteriol. 179:3416-3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cooper, T. G. 1982. Nitrogen metabolism in Saccharomyces cerevisiae, p. 39-99. In J. N. Strathern, E. W. Jones, and J. R. Broach (ed.), The molecular biology of the yeast Saccharomyces cerevisiae: metabolism and gene expression, vol. 1. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 25.Cooper, T. G. 1996. Allantoin degradative system—an integrated transcriptional response to multiple signals, p. 139-169. In G. Marzluf and R. Bambrl (ed.), Mycota, vol. III. Springer-Verlag, Berlin, Germany. [Google Scholar]
- 26.Cooper, T. G. 2002. Transmitting the signal of excess nitrogen in Saccharomyces cerevisiae from the Tor proteins to the GATA factors: connecting the dots. FEMS Microbiol. Rev. 26:223-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coornaert, D., S. Vissers, B. Andre, and M. Grenson. 1992. The UGA43 negative regulatory gene of Saccharomyces cerevisiae contains both a GATA-1 type zinc finger and a putative leucine zipper. Curr. Genet. 21:301-307. [DOI] [PubMed] [Google Scholar]
- 28.Coschigano, P. W., and B. Magasanik. 1991. The URE2 gene product of Saccharomyces cerevisiae plays an important role in the cellular response to the nitrogen source and has homology to glutathione S-transferases. Mol. Cell. Biol. 11:822-832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cox, K. H., A. B. Pinchak, and T. G. Cooper. 1999. Genome-wide transcriptional analysis in S. cerevisiae by mini-array membrane hybridization. Yeast 15:703-713. [DOI] [PubMed] [Google Scholar]
- 30.Cui, X., and G. A. Churchill. 2003. Statistical tests for differential expression in cDNA microarray experiments. Genome Biol. 4:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cunningham, T. S., and T. G. Cooper. 1991. Expression of the DAL80 gene, whose product is homologous to the GATA factors and is a negative regulator of multiple nitrogen catabolic genes in Saccharomyces cerevisiae, is sensitive to nitrogen catabolite repression. Mol. Cell. Biol. 11:6205-6215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cunningham, T. S., V. V. Svetlov, R. Rai, W. Smart, and T. G. Cooper. 1996. G1n3p is capable of binding to UAS(NTR) elements and activating transcription in Saccharomyces cerevisiae. J. Bacteriol. 178:3470-3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De Boer, M., J. P. Bebelman, P. M. Goncalves, J. Maat, H. Van Heerikhuizen, and R. J. Planta. 1998. Regulation of expression of the amino acid transporter gene BAP3 in Saccharomyces cerevisiae. Mol. Microbiol. 30:603-613. [DOI] [PubMed] [Google Scholar]
- 34.DeRisi, J., B. van den Hazel, P. Marc, E. Balzi, P. Brown, C. Jacq, and A. Goffeau. 2000. Genome microarray analysis of transcriptional activation in multidrug resistance yeast mutants. FEBS Lett. 470:156-160. [DOI] [PubMed] [Google Scholar]
- 35.Des Etages, S. A., D. A. Falvey, R. J. Reece, and M. C. Brandriss. 1996. Functional analysis of the PUT3 transcriptional activator of the proline utilization pathway in Saccharomyces cerevisiae. Genetics 142:1069-1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Des Etages, S. A., D. Saxena, H. L. Huang, D. A. Falvey, D. Barber, and M. C. Brandriss. 2001. Conformational changes play a role in regulating the activity of the proline utilization pathway-specific regulator in Saccharomyces cerevisiae. Mol. Microbiol. 40:890-899. [DOI] [PubMed] [Google Scholar]
- 37.Didion, T., M. Grausland, C. Kielland-Brandt, and H. A. Andersen. 1996. Amino acids induce expression of BAP2, a branched-chain amino acid permease gene in Saccharomyces cerevisiae. J. Bacteriol. 178:2025-2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Didion, T., B. Regenberg, M. U. Jorgensen, M. C. Kielland-Brandt, and H. A. Andersen. 1998. The permease homologue Ssy1p controls the expression of amino acid and peptide transporter genes in Saccharomyces cerevisiae. Mol. Microbiol. 27:643-650. [DOI] [PubMed] [Google Scholar]
- 39.Dubois, E., D. Hiernaux, M. Grenson, and J. M. Wiame. 1978. Specific induction of catabolism and its relation to repression of biosynthesis in arginine metabolism of Saccharomyces cerevisiae. J. Mol. Biol. 122:383-406. [DOI] [PubMed] [Google Scholar]
- 40.Dubois, E., and F. Messenguy. 1997. Integration of the multiple controls regulating the expression of the arginase gene CAR1 of Saccharomyces cerevisiae in response to different nitrogen signals: role of Gln3p, ArgRp-Mcm1p, and Ume6p. Mol. Gen. Genet. 253:568-580. [DOI] [PubMed] [Google Scholar]
- 41.Dubois, E. L., and J. M. Wiame. 1976. Non specific induction of arginase in Saccharomyces cerevisiae. Biochimie 58:207-211. [DOI] [PubMed] [Google Scholar]
- 42.Dwight, S. S., M. A. Harris, K. Dolinski, C. A. Ball, G. Binkley, K. R. Christie, D. G. Fisk, L. Issel-Tarver, M. Schroeder, G. Sherlock, A. Sethuraman, S. Weng, D. Botstein, and J. M. Cherry. 2002. Saccharomyces Genome Database (SGD) provides secondary gene annotation using the Gene Ontology (GO). Nucleic Acids Res. 30:69-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eckert-Boulet, N., P. S. Nielsen, C. Friis, M. M. Dos Santos, J. Nielsen, M. C. Kielland-Brandt, and B. Regenberg. 2004. Transcriptional profiling of extracellular amino acid sensing in Saccharomyces cerevisiae and the role of Stp1p and Stp2p. Yeast 21:635-648. [DOI] [PubMed] [Google Scholar]
- 44.Eide, D. J. 2001. Functional genomics and metal metabolism. Genome Biol. 2:REVIEWS1028. [DOI] [PMC free article] [PubMed]
- 45.Forsberg, H., C. F. Gilstring, A. Zargari, P. Martinez, and P. O. Ljungdahl. 2001. The role of the yeast plasma membrane SPS nutrient sensor in the metabolic response to extracellular amino acids. Mol. Microbiol. 42:215-228. [DOI] [PubMed] [Google Scholar]
- 46.Forsberg, H., and P. O. Ljungdahl. 2001. Sensors of extracellular nutrients in Saccharomyces cerevisiae. Curr. Genet. 40:91-109. [DOI] [PubMed] [Google Scholar]
- 47.Gasch, A. P., P. T. Spellman, C. M. Kao, O. Carmel-Harel, M. B. Eisen, G. Storz, D. Botstein, and P. O. Brown. 2000. Genomic expression programs in the response of yeast cells to environmental changes. Mol. Biol. Cell 11:4241-4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gentleman, R. C., V. J. Carey, D. M. Bates, B. Bolstad, M. Dettling, S. Dudoit, B. Ellis, L. Gautier, Y. Ge, J. Gentry, K. Hornik, T. Hothorn, W. Huber, S. Iacus, R. Irizarry, F. Leisch, C. Li, M. Maechler, A. J. Rossini, G. Sawitzki, C. Smith, G. Smyth, L. Tierney, J. Y. Yang, and J. Zhang. 2004. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5:R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gietz, D., A. St. Jean, R. A. Woods, and R. H. Schiestl. 1992. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res. 20:1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gimeno, C. J., P. O. Ljungdahl, C. A. Styles, and G. R. Fink. 1992. Unipolar cell divisions in the yeast S. cerevisiae lead to filamentous growth: regulation by starvation and RAS. Cell 68:1077-1090. [DOI] [PubMed] [Google Scholar]
- 51.Grenson, M. 1992. Amino acid transporters in yeast: structure, function and regulation, p. 219-245. In J. J. L. L. M. De Pont (ed.), Molecular aspects of transport proteins. Elsevier Science, New York, NY.
- 52.Hansen, J., S. V. Bruun, L. M. Bech, and C. Gjermansen. 2002. The level of MXR1 gene expression in brewing yeast during beer fermentation is a major determinant for the concentration of dimethyl sulfide in beer. FEMS Yeast Res. 2:137-149. [DOI] [PubMed] [Google Scholar]
- 53.Harbison, C. T., D. B. Gordon, T. I. Lee, N. J. Rinaldi, K. D. Macisaac, T. W. Danford, N. M. Hannett, J. B. Tagne, D. B. Reynolds, J. Yoo, E. G. Jennings, J. Zeitlinger, D. K. Pokholok, M. Kellis, P. A. Rolfe, K. T. Takusagawa, E. S. Lander, D. K. Gifford, E. Fraenkel, and R. A. Young. 2004. Transcriptional regulatory code of a eukaryotic genome. Nature 431:99-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hesslinger, C., and G. Sawers. 1998. The tdcE gene in Escherichia coli strain W3110 is separated from the rest of the tdc operon by insertion of IS5 elements. DNA Seq. 9:183-188. [DOI] [PubMed] [Google Scholar]
- 55.Hikkel, I., A. Lucau-Danila, T. Delaveau, P. Marc, F. Devaux, and C. Jacq. 2003. A general strategy to uncover transcription factor properties identifies a new regulator of drug resistance in yeast. J. Biol. Chem. 278:11427-11432. [DOI] [PubMed] [Google Scholar]
- 56.Hinnebusch, A. 1996. Translational control of GCN4: gene-specific regulation by phosphorylation of eIF2, p. 199-244. In W. B. Hershey, M. B. Mathews, and N. Soneberg (ed.), Translational control. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 57.Hinnebusch, A. G. 1992. General and pathway-specific regulatory mechanisms controlling the synthesis of amino acid biosynthetic enzymes in Saccharomyces cerevisiae, p. 319-414. In E. W. Jones, J. R. Pringle, and J. R. Broach (ed.), The molecular biology of the yeast Saccharomyces—gene expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 58.Hinnebusch, A. G. 2005. Translational regulation of GCN4 and the general amino acid control of yeast. Annu. Rev. Microbiol. 59:407-450. [DOI] [PubMed] [Google Scholar]
- 59.Holmberg, S., and P. Schjerling. 1996. Cha4p of Saccharomyces cerevisiae activates transcription via serine/threonine response elements. Genetics 144:467-478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huang, H. L., and M. C. Brandriss. 2000. The regulator of the yeast proline utilization pathway is differentially phosphorylated in response to the quality of the nitrogen source. Mol. Cell. Biol. 20:892-899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huh, W. K., J. V. Falvo, L. C. Gerke, A. S. Carroll, R. W. Howson, J. S. Weissman, and E. K. O'Shea. 2003. Global analysis of protein localization in budding yeast. Nature 425:686-691. [DOI] [PubMed] [Google Scholar]
- 62.Iraqui, I., S. Vissers, B. Andre, and A. Urrestarazu. 1999. Transcriptional induction by aromatic amino acids in Saccharomyces cerevisiae. Mol. Cell. Biol. 19:3360-3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Iraqui, I., S. Vissers, F. Bernard, J. O. Craene, E. Boles, A. Urrestarazu, and B. Andre. 1999. Amino acid signaling in Saccharomyces cerevisiae: a permease-like sensor of external amino acids and F-box protein Grr1p are required for transcriptional induction of the AGP1 gene, which encodes a broad-specificity amino acid permease. Mol. Cell. Biol. 19:989-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Iraqui, I., S. Vissers, M. Cartiaux, and A. Urrestarazu. 1998. Characterisation of Saccharomyces cerevisiae ARO8 and ARO9 genes encoding aromatic aminotransferases I and II reveals a new aminotransferase subfamily. Mol. Gen. Genet. 257:238-248. [DOI] [PubMed] [Google Scholar]
- 65.Ishida, C., C. Aranda, L. Valenzuela, L. Riego, A. DeLuna, F. Recillas-Targa, P. Filetici, R. Lopez-Revilla, and A. Gonzalez. 2006. The UGA3-GLT1 intergenic region constitutes a promoter whose bidirectional nature is determined by chromatin organization in Saccharomyces cerevisiae. Mol. Microbiol. 59:1790-1806. [DOI] [PubMed] [Google Scholar]
- 66.Jacobs, P., J. C. Jauniaux, and M. Grenson. 1980. A cis-dominant regulatory mutation linked to the argB-argC gene cluster in Saccharomyces cerevisiae. J. Mol. Biol. 139:691-704. [DOI] [PubMed] [Google Scholar]
- 67.Jauniaux, J. C., and M. Grenson. 1990. GAP1, the general amino acid permease gene of Saccharomyces cerevisiae. Nucleotide sequence, protein similarity with the other bakers yeast amino acid permeases, and nitrogen catabolite repression. Eur. J. Biochem. 190:39-44. [DOI] [PubMed] [Google Scholar]
- 68.Jia, M. H., R. A. Larossa, J. M. Lee, A. Rafalski, E. Derose, G. Gonye, and Z. Xue. 2000. Global expression profiling of yeast treated with an inhibitor of amino acid biosynthesis, sulfometuron methyl. Physiol. Genomics 3:83-92.11015603 [Google Scholar]
- 69.Jones, E. W., and G. R. Fink. 1982. Regulation of amino acid and nucleotide biosynthesis in yeast, p. 181-299. In J. N. Strathern, E. W. Jones, and J. R. Broach (ed.), The molecular biology of the yeast Saccharomyces—metabolism and gene expression. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
- 70.Kellis, M., N. Patterson, M. Endrizzi, B. Birren, and E. S. Lander. 2003. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature 423:241-254. [DOI] [PubMed] [Google Scholar]
- 71.Kim, J. M., H. Yoshikawa, and K. Shirahige. 2001. A member of the YER057c/yjgf/Uk114 family links isoleucine biosynthesis and intact mitochondria maintenance in Saccharomyces cerevisiae. Genes Cells 6:507-517. [DOI] [PubMed] [Google Scholar]
- 72.Klasson, H., G. R. Fink, and P. O. Ljungdahl. 1999. Ssy1p and Ptr3p are plasma membrane components of a yeast system that senses extracellular amino acids. Mol. Cell. Biol. 19:5405-5416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kodama, Y., F. Omura, K. Takahashi, K. Shirahige, and T. Ashikari. 2002. Genome-wide expression analysis of genes affected by amino acid sensor Ssy1p in Saccharomyces cerevisiae. Curr. Genet. 41:63-72. [DOI] [PubMed] [Google Scholar]
- 74.Kohlhaw, G. B. 2003. Leucine biosynthesis in fungi: entering metabolism through the back door. Microbiol. Mol. Biol. Rev. 67:1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kohrer, K., and H. Domdey. 1991. Preparation of high molecular weight RNA. Methods Enzymol. 194:398-405. [DOI] [PubMed] [Google Scholar]
- 76.Kradolfer, P., P. Niederberger, and R. Hutter. 1982. Tryptophan degradation in Saccharomyces cerevisiae: characterization of two aromatic aminotransferases. Arch. Microbiol. 133:242-248. [DOI] [PubMed] [Google Scholar]
- 77.Le Crom, S., F. Devaux, C. Jacq, and P. Marc. 2002. yMGV: helping biologists with yeast microarray data mining. Nucleic Acids Res. 30:76-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Le Crom, S., F. Devaux, P. Marc, X. Zhang, W. S. Moye-Rowley, and C. Jacq. 2002. New insights into the pleiotropic drug resistance network from genome-wide characterization of the YRR1 transcription factor regulation system. Mol. Cell. Biol. 22:2642-2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liao, X., and R. A. Butow. 1993. RTG1 and RTG2: two yeast genes required for a novel path of communication from mitochondria to the nucleus. Cell 72:61-71. [DOI] [PubMed] [Google Scholar]
- 80.Linnen, J. M., C. P. Bailey, and D. L. Weeks. 1993. Two related localized mRNAs from Xenopus laevis encode ubiquitin-like fusion proteins. Gene 128:181-188. [DOI] [PubMed] [Google Scholar]
- 81.Longtine, M. S., A. Demarini, N. G. Shah, A. Wach, A. Brachat, P. Philippsen, and J. R. Pringle. 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14:953-961. [DOI] [PubMed] [Google Scholar]
- 82.Lyons, T. J., A. P. Gasch, L. A. Gaither, D. Botstein, P. O. Brown, and D. J. Eide. 2000. Genome-wide characterization of the Zap1p zinc-responsive regulon in yeast. Proc. Natl. Acad. Sci. USA 97:7957-7962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ma, Y., and L. M. Hendershot. 2001. The unfolding tale of the unfolded protein response. Cell 107:827-830. [DOI] [PubMed] [Google Scholar]
- 84.Magasanik, B. 1992. Regulation of nitrogen utilization, p. 238-317. In J. N. Strathern, E. W. Jones, and J. R. Broach (ed.), The molecular biology of the yeast Saccharomyces—metabolism and gene expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 85.Magasanik, B., and C. A. Kaiser. 2002. Nitrogen regulation in Saccharomyces cerevisiae. Gene 290:1-18. [DOI] [PubMed] [Google Scholar]
- 86.Magasanik, B., and F. C. Neidhardt. 1987. Regulation of carbon and nitrogen utilization, p. 1318-1325. In F. C. Neidhardt, J. L. Ingraham, K. B. Low, B. Magasanik, M. Schaechter, and H. E. Umbarger (ed.), Escherichia coli and Salmonella typhimurium, vol. 2. American Society for Microbiology, Washington, DC. [Google Scholar]
- 87.Makuc, J., S. Paiva, M. Schauen, R. Kramer, B. Andre, M. Casal, C. Leao, and E. Boles. 2001. The putative monocarboxylate permeases of the yeast Saccharomyces cerevisiae do not transport monocarboxylic acids across the plasma membrane. Yeast 18:1131-1143. [DOI] [PubMed] [Google Scholar]
- 88.Malpertuy, A., F. Tekaia, S. Casaregola, M. Aigle, F. Artiguenave, G. Blandin, M. Bolotin-Fukuhara, E. Bon, P. Brottier, J. de Montiqny, P. Durrens, C. Gaillardin, A. Lepingle, B. Llorente, C. Neuveglise, O. Ozier-Kalogeropoulos, S. Potier, W. Saurin, C. Toffano-Nioche, M. Wesolowski-Louvel, P. Wincker, J. Weissenbach, J. Souciet, and B. Dujon. 2000. Genomic exploration of the hemiascomycetous yeasts: 19. Ascomycetes-specific genes. FEBS Lett. 487:113-121. [DOI] [PubMed] [Google Scholar]
- 89.Marc, P., F. Devaux, and C. Jacq. 2001. yMGV: a database for visualization and data mining of published genome-wide yeast expression data. Nucleic Acids Res. 29:E63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Natarajan, K., M. R. Meyer, B. M. Jackson, D. Slade, C. Roberts, A. G. Hinnebusch, and M. J. Marton. 2001. Transcriptional profiling shows that Gcn4p is a master regulator of gene expression during amino acid starvation in yeast. Mol. Cell. Biol. 21:4347-4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nelissen, B., W. R. De, and A. Goffeau. 1997. Classification of all putative permeases and other membrane plurispanners of the major facilitator superfamily encoded by the complete genome of Saccharomyces cerevisiae. FEMS Microbiol. Rev. 21:113-134. [DOI] [PubMed] [Google Scholar]
- 92.Niederberger, P., G. Miozzari, and R. Hutter. 1981. Biological role of the general control of amino acid biosynthesis in Saccharomyces cerevisiae. Mol. Cell. Biol. 1:584-593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Normington, K., K. Kohno, Y. Kozutsumi, M. J. Gething, and J. Sambrook. 1989. S. cerevisiae encodes an essential protein homologous in sequence and function to mammalian BiP. Cell 57:1223-1236. [DOI] [PubMed] [Google Scholar]
- 94.Ogawa, N., J. DeRisi, and P. O. Brown. 2000. New components of a system for phosphate accumulation and polyphosphate metabolism in Saccharomyces cerevisiae revealed by genomic expression analysis. Mol. Biol. Cell 11:4309-4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Oliphant, A. R., C. J. Brandl, and K. Struhl. 1989. Defining the sequence specificity of DNA-binding proteins by selecting binding sites from random-sequence oligonucleotides: analysis of yeast GCN4 protein. Mol. Cell. Biol. 9:2944-2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Onda, M., K. Ota, T. Chiba, Y. Sakaki, and T. Ito. 2004. Analysis of gene network regulating yeast multidrug resistance by artificial activation of transcription factors: involvement of Pdr3 in salt tolerance. Gene 332:51-59. [DOI] [PubMed] [Google Scholar]
- 97.Oxelmark, E., A. Marchini, I. Malanchi, F. Magherini, L. Jaquet, M. A. Hajibagheri, K. J. Blight, J. C. Jauniaux, and M. Tommasino. 2000. Mmf1p, a novel yeast mitochondrial protein conserved throughout evolution and involved in maintenance of the mitochondrial genome. Mol. Cell. Biol. 20:7784-7797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pace, H. C., and C. Brenner. 2001. The nitrilase superfamily: classification, structure and function. Genome Biol. 2:REVIEWS0001.1-REVIEWS0001.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Patil, C., and P. Walter. 2001. Intracellular signaling from the endoplasmic reticulum to the nucleus: the unfolded protein response in yeast and mammals. Curr. Opin. Cell Biol. 13:349-355. [DOI] [PubMed] [Google Scholar]
- 100.Perpete, P., O. Duthoit, M. S. De, L. Imray, A. I. Lawton, K. E. Stavropoulos, V. W. Gitonga, M. J. Hewlins, and J. R. Dickinson. 2006. Methionine catabolism in Saccharomyces cerevisiae. FEMS Yeast Res. 6:48-56. [DOI] [PubMed] [Google Scholar]
- 101.Petersen, J. G., M. C. Kielland-Brandt, T. Nilsson-Tillgren, C. Bornaes, and S. Holmberg. 1988. Molecular genetics of serine and threonine catabolism in Saccharomyces cerevisiae. Genetics 119:527-534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Puig, S., E. Askeland, and D. J. Thiele. 2005. Coordinated remodeling of cellular metabolism during iron deficiency through targeted mRNA degradation. Cell 120:99-110. [DOI] [PubMed] [Google Scholar]
- 103.Ramos, F., E. Dubois, and A. Pierard. 1988. Control of enzyme synthesis in the lysine biosynthetic pathway of Saccharomyces cerevisiae. Evidence for a regulatory role of gene LYS14. Eur. J. Biochem. 171:171-176. [DOI] [PubMed] [Google Scholar]
- 104.Ramos, F., M. E. Guezzar, M. Grenson, and J. M. Wiame. 1985. Mutations affecting the enzymes involved in the utilization of 4-aminobutyric acid as nitrogen source by the yeast Saccharomyces cerevisiae. Eur. J. Biochem. 149:401-404. [DOI] [PubMed] [Google Scholar]
- 105.Reihl, P., and J. Stolz. 2005. The monocarboxylate transporter homolog Mch5p catalyzes riboflavin (vitamin B2) uptake in Saccharomyces cerevisiae. J. Biol. Chem. 280:39809-39817. [DOI] [PubMed] [Google Scholar]
- 106.Roberg, K. J., N. Rowley, and C. A. Kaiser. 1997. Physiological regulation of membrane protein sorting late in the secretory pathway of Saccharomyces cerevisiae. J. Cell Biol. 137:1469-1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rogers, B., A. Decottignies, M. Kolaczkowski, E. Carvajal, E. Balzi, and A. Goffeau. 2001. The pleiotropic drug ABC transporters from Saccharomyces cerevisiae. J. Mol. Microbiol. Biotechnol. 3:207-214. [PubMed] [Google Scholar]
- 108.Rose, M. D., L. M. Misra, and J. P. Vogel. 1989. KAR2, a karyogamy gene, is the yeast homolog of the mammalian BiP/GRP78 gene. Cell 57:1211-1221. [DOI] [PubMed] [Google Scholar]
- 109.Rowen, D. W., N. Esiobu, and B. Magasanik. 1997. Role of GATA factor Nil2p in nitrogen regulation of gene expression in Saccharomyces cerevisiae. J. Bacteriol. 179:3761-3766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ruepp, A., A. Zollner, D. Maier, K. Albermann, J. Hani, M. Mokrejs, I. Tetko, U. Guldener, G. Mannhaupt, M. Munsterkotter, and H. W. Mewes. 2004. The FunCat, a functional annotation scheme for systematic classification of proteins from whole genomes. Nucleic Acids Res. 32:5539-5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Saeed, A. I., V. Sharov, J. White, J. Li, W. Liang, N. Bhagabati, J. Braisted, M. Klapa, T. Currier, M. Thiagarajan, A. Sturn, M. Snuffin, A. Rezantsev, D. Popov, A. Ryltsov, E. Kostukovich, I. Borisovsky, Z. Liu, A. Vinsavich, V. Trush, and J. Quackenbush. 2003. TM4: a free, open-source system for microarray data management and analysis. BioTechniques 34:374-378. [DOI] [PubMed] [Google Scholar]
- 112.Scherens, B., A. Feller, F. Vierendeels, F. Messenguy, and E. Dubois. 2006. Identification of direct and indirect targets of the Gln3 and Gat1 activators by transcriptional profiling in response to nitrogen availability at short and long term. FEMS Yeast Res. 6:777-791. [DOI] [PubMed] [Google Scholar]
- 113.Schroder, M., J. S. Chang, and R. J. Kaufman. 2000. The unfolded protein response represses nitrogen-starvation induced developmental differentiation in yeast. Genes Dev. 14:2962-2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Schroder, M., R. Clark, C. Y. Liu, and R. J. Kaufman. 2004. The unfolded protein response represses differentiation through the RPD3-SIN3 histone deacetylase. EMBO J. 23:2281-2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sentheshanuganathan, S. 1960. The mechanism of the formation of higher alcohols from amino acids by Saccharomyces cerevisiae. Biochem. J. 74:568-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Shamji, A. F., F. G. Kuruvilla, and S. L. Schreiber. 2000. Partitioning the transcriptional program induced by rapamycin among the effectors of the Tor proteins. Curr. Biol. 10:1574-1581. [DOI] [PubMed] [Google Scholar]
- 117.Simonis, N., S. J. Wodak, G. N. Cohen, and J. van Helden. 2004. Combining pattern discovery and discriminant analysis to predict gene co-regulation. Bioinformatics 20:2370-2379. [DOI] [PubMed] [Google Scholar]
- 118.Sosa, E., C. Aranda, L. Riego, L. Valenzuela, A. DeLuna, J. M. Cantu, and A. Gonzalez. 2003. Gcn4 negatively regulates expression of genes subjected to nitrogen catabolite repression. Biochem. Biophys. Res. Commun. 310:1175-1180. [DOI] [PubMed] [Google Scholar]
- 119.Soussi-Boudekou, S., S. Vissers, A. Urrestarazu, J. C. Jauniaux, and B. Andre. 1997. Gzf3p, a fourth GATA factor involved in nitrogen-regulated transcription in Saccharomyces cerevisiae. Mol. Microbiol. 23:1157-1168. [DOI] [PubMed] [Google Scholar]
- 120.Spear, E., and D. T. Ng. 2001. The unfolded protein response: no longer just a special teams player. Traffic 2:515-523. [DOI] [PubMed] [Google Scholar]
- 121.Talibi, D., M. Grenson, and B. Andre. 1995. Cis- and trans-acting elements determining induction of the genes of the gamma-aminobutyrate (GABA) utilization pathway in Saccharomyces cerevisiae. Nucleic Acids Res. 23:550-557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Talla, E., F. Tekaia, L. Brino, and B. Dujon. 2003. A novel design of whole-genome microarray probes for Saccharomyces cerevisiae which minimizes cross-hybridization. BMC Genomics 4:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Thomas, D., A. Becker, and Y. Surdin-Kerjan. 2000. Reverse methionine biosynthesis from S-adenosylmethionine in eukaryotic cells. J. Biol. Chem. 275:40718-40724. [DOI] [PubMed] [Google Scholar]
- 124.Thomas, D., and Y. Surdin-Kerjan. 1997. Metabolism of sulfur amino acids in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 61:503-532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Travers, K. J., C. K. Patil, L. Wodicka, D. J. Lockhart, J. S. Weissman, and P. Walter. 2000. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 101:249-258. [DOI] [PubMed] [Google Scholar]
- 126.Turner, G. C., F. Du, and A. Varshavsky. 2000. Peptides accelerate their uptake by activating a ubiquitin-dependent proteolytic pathway. Nature 405:579-583. [DOI] [PubMed] [Google Scholar]
- 127.Urrestarazu, A., S. Vissers, I. Iraqui, and M. Grenson. 1998. Phenylalanine- and tyrosine-auxotrophic mutants of Saccharomyces cerevisiae impaired in transamination. Mol. Gen. Genet. 257:230-237. [DOI] [PubMed] [Google Scholar]
- 128.Valenzuela, L., C. Aranda, and A. Gonzalez. 2001. TOR modulates GCN4-dependent expression of genes turned on by nitrogen limitation. J. Bacteriol. 183:2331-2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Valenzuela, L., P. Ballario, C. Aranda, P. Filetici, and A. Gonzalez. 1998. Regulation of expression of GLT1, the gene encoding glutamate synthase in Saccharomyces cerevisiae. J. Bacteriol. 180:3533-3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Van Helden, J. 2003. Regulatory sequence analysis tools. Nucleic Acids Res. 31:3593-3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Van Helden, J., B. Andre, and J. Collado-Vides. 1998. Extracting regulatory sites from the upstream region of yeast genes by computational analysis of oligonucleotide frequencies. J. Mol. Biol. 281:827-842. [DOI] [PubMed] [Google Scholar]
- 132.Vuralhan, Z., M. A. Luttik, S. L. Tai, V. M. Boer, M. A. Morais, D. Schipper, M. J. Almering, P. Kotter, J. R. Dickinson, J. M. Daran, and J. T. Pronk. 2005. Physiological characterization of the ARO10-dependent, broad-substrate-specificity 2-oxo acid decarboxylase activity of Saccharomyces cerevisiae. Appl. Environ. Microbiol. 71:3276-3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Vuralhan, Z., M. A. Morais, S. L. Tai, M. D. Piper, and J. T. Pronk. 2003. Identification and characterization of phenylpyruvate decarboxylase genes in Saccharomyces cerevisiae. Appl. Environ. Microbiol. 69:4534-4541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wach, A. 1996. PCR-synthesis of marker cassettes with long flanking homology regions for gene disruptions in S. cerevisiae. Yeast 12:259-265. [DOI] [PubMed] [Google Scholar]
- 135.Webb, A. D., and J. L. Ingraham. 1963. Fusel oil. Adv. Appl. Microbiol. 5:317-353. [Google Scholar]
- 136.Wiame, J. M., M. Grenson, and H. N. Arst, Jr. 1985. Nitrogen catabolite repression in yeasts and filamentous fungi. Adv. Microb. Physiol. 26:1-88. [DOI] [PubMed] [Google Scholar]
- 137.Wong, D. T., H. A. Davis, K. Kuplent, S. Doares, J. A. Sorge, and R. Mullinax. 2000. Fairplay™ microarray labeling kit for preparing uniformly labeled cDNA. Strategies 13:131-132. [Google Scholar]
- 138.Wu, J., N. Zhang, A. Hayes, K. Panoutsopoulou, and S. G. Oliver. 2004. Global analysis of nutrient control of gene expression in Saccharomyces cerevisiae during growth and starvation. Proc. Natl. Acad. Sci. USA 101:3148-3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yang, R., S. A. Wek, and R. C. Wek. 2000. Glucose limitation induces GCN4 translation by activation of Gcn2 protein kinase. Mol. Cell. Biol. 20:2706-2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yang, Y. H., S. Dudoit, P. Luu, D. M. Lin, V. Peng, J. Ngai, and T. P. Speed. 2002. Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res. 30:e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.