

Abstract
p73, a member of the p53 family, expresses two classes of proteins: the full-length TAp73 and the N-terminally truncated ΔNp73. While TAp73 possesses many p53-like features, ΔNp73 is dominant negative towards TAp73 and p53 and appears to have distinct functions in tumorigenesis and neuronal development. Given its biological importance, we investigated the role of ΔNp73 in nerve growth factor (NGF)-mediated neuronal differentiation in PC12 cells. We show that overexpression of ΔNp73α or ΔNp73β inhibits NGF-mediated neuronal differentiation in both p53-dependent and -independent manners. In line with this, we showed that the level of endogenous ΔNp73 is progressively diminished in differentiating PC12 cells upon NGF treatment and knockdown of ΔNp73 promotes NGF-mediated neuronal differentiation. Interestingly, we found that the ability of ΔNp73 to suppress NGF-mediated neuronal differentiation is correlated with its ability to regulate the expression of TrkA, the high-affinity NGF receptor. Specifically, we found that ΔNp73 directly binds to the TrkA promoter and transcriptionally represses TrkA expression, which in turn attenuates the NGF-mediated mitogen-activated protein kinase pathway. Conversely, the steady-state level of TrkA is increased upon knockdown of ΔNp73. Furthermore, we found that histone deacetylase 1 (HDAC1) and HDAC2 are recruited by ΔNp73 to the TrkA promoter and act as corepressors to suppress TrkA expression, which can be relieved by trichostatin A, an HDAC inhibitor. Taken together, we conclude that ΔNp73 negatively regulates NGF-mediated neuronal differentiation by transrepressing TrkA.
p73 belongs to the p53 family, which also includes p53 and p63. These proteins share a significant degree of sequence similarity, especially in the DNA binding domain (20, 26, 27). However, p73 is not a classic Knudson-type tumor suppressor despite its remarkable structural and functional similarities to p53. For example, p73-deficient mice lack a tumor phenotype. Instead, these mice exhibit abnormalities in neuronal development, such as hippocampal dysgenesis, hydrocephalus, loss of Cajal-Retzius neurons, and defects in pheromone sensory pathways (53). In addition, wild-type p73 is frequently up-regulated, but not inactivated, in human primary tumors, suggesting that p73 may promote, rather than suppress, tumor formation (42).
These apparent contradictions of p73 activities might be due to the fact that p73 produces two classes of proteins, which are generated from two separate promoters and have distinct or even opposing functions. One, termed TAp73, is transcribed from the upstream promoter and contains an N-terminal activation domain with homology to that in p53. The other one, denoted ΔNp73, is transcribed from the downstream promoter in intron 3 and N-terminally truncated. Consequently, the TAp73 isoforms contain many p53-like properties, such as transactivation of a subset of p53 target genes necessary for induction of cell cycle arrest and apoptosis (4, 11, 15, 18, 24, 46, 49). In contrast, the ΔNp73 isoforms, because they lack the activation domain conserved in p53, are dominant negative towards TAp73 or p53 and thus are thought to have oncogenic potential (32). Consistent with this notion, ΔNp73 has been reported to be frequently up-regulated in human cancers, including breast cancer (54), lung cancer (45), and neuroblastoma (3). Moreover, overexpression of ΔNp73 promotes immortalization in primary cells and malignant transformation in NIH 3T3 fibroblasts (35, 43). In addition to its oncogenic potential, ΔNp73 plays a crucial role in neuronal survival through both p53-dependent and -independent mechanisms (29, 36, 37, 48). The underlying mechanism of these biological phenomena has been hypothesized as follows: ΔNp73, by forming a heterotetramer with TAp73 or competing with TAp73 or p53 to bind to the p53-responsive element (p53-RE), antagonizes TAp73 or p53 since it was thought not to contain an activation domain. However, we have reported recently that ΔNp73β, one of the ΔNp73 isoforms, is active in transactivation and suppression of cancer cell growth (30). In addition, ΔNp73α, the full-length ΔNp73, has been reported to specifically transactivate BTG2 in a p53-dependent manner in neuroblastoma cells but not in other cell types (4, 13). These findings suggest that ΔNp73 retains transcriptional activities, at least in some settings. On the other hand, these findings point out a possibility that in addition to antagonizing TAp73 and p53, ΔNp73 may regulate its own unique target genes for its biological activities.
Cellular differentiation, a highly coordinated process, is tightly regulated by a group of key transcriptional factors (25, 33). Precise regulation of this process is very important to ensure the production of properly differentiated cells. The transcriptional activity of p53 plays an important role in several types of cellular differentiation, such as epithelial differentiation, myogenic differentiation, and neuronal differentiation (1, 6, 28). Like p53, TAp73 also plays a critical role in cellular differentiation. Overexpression of TAp73 is sufficient to induce differentiation in neuroblastoma cells (5) and platelet-derived growth factor withdrawal- or thyroid hormone-induced differentiation in oligodendrocyte precursor cells (2). In contrast, overexpression of ΔNp73 inhibits differentiation in oligodendrocyte precursor cells (2) and interferes with multiple developmental programs (23). Moreover, ΔNp73 is likely to be responsible for the neuronal defects in p73-deficient mice since it is the most predominant isoform in the developing mouse brain (37). Such observations support a model whereby ΔNp73 might regulate cellular differentiation or development at least in part by antagonizing other p53 family members. However, whether these observations reflect the role of endogenous ΔNp73 remains unclear.
Nerve growth factor (NGF) plays a fundamental role in the neuronal development and maintenance of sympathetic and sensory neurons. The pheochromocytoma PC12 cell line is a well-established model for studying NGF-mediated neuronal differentiation (17). Upon NGF treatment, PC12 cells cease proliferation, exhibit somatic hypertrophy, and acquire neurites (47). The signal is initiated when NGF binds to its high-affinity receptor, TrkA, which is then phosphorylated and dimerized to activate its downstream cascades (22). Thus, the PC12 cell line is an ideal system with which to investigate the role of ΔNp73 in NGF-mediated neuronal differentiation. In this study, we show that overexpression of ΔNp73α or ΔNp73β inhibits, whereas knockdown of ΔNp73 promotes, NGF-mediated neuronal differentiation. Moreover, we show that endogenous ΔNp73 is gradually diminished in PC12 cells upon NGF treatment. To uncover the underlying mechanism, we show that ΔNp73, through binding to the p53-RE in the TrkA promoter, transrepresses TrkA expression, which then inhibits the NGF-mediated mitogen-activated protein kinase (MAPK) pathway. Conversely, we showed that the steady-state level of TrkA is increased in PC12 cells upon knockdown of ΔNp73. Finally, we showed that ΔNp73, by recruiting histone deacetylase 1 (HDAC1) and HDAC2 to the TrkA promoter, transcriptionally represses TrkA expression, which can be relieved by trichostatin A (TSA), an HDAC inhibitor.
MATERIALS AND METHODS
Reagents.
The dual-luciferase assay kit and nerve growth factor were purchased from Promega (Madison, WI). Anti-p21 and anti-TrkA were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antihemagglutinin (anti-HA) was purchased from Covance. TSA and antibodies against HDAC1/2, p-MEK1/2, and p-RSK90 were purchased from Upstate-Cell Signaling Biotechnology (Charlottesville, VA). Antiactin, anti-growth-associated protein 43 (anti-GAP-43), proteinase inhibitor cocktail, RNase A, and propidium iodide were purchased from Sigma (St. Louis, MO). p73-Ab1, p73-Ab2, and p73-Ab3 antibodies were purchased from Calbiochem (San Diego, CA). BL906 antibody was purchased from Bethyl Laboratories (Montgomery, TX). An expressed sequence tag (EST) clone (source ID 6826464; GenBank accession no. BC069182) was purchased from Open Biosystems (Huntsville, AL). Scrambled ΔNp73 small interfering RNA (siRNA) (GGCCGAUUGUCAAAUAAUU) was purchased from Dharmacon RNA Technologies. The iScript cDNA synthesis kit was purchased from Bio-Rad Laboratories.
Plasmids.
To generate HA-tagged murine ΔNp73α, a PCR fragment was amplified using an EST clone (ID 6826464) as a template. The following primers were used: upstream primer, 5′-ACCATGTACCCATACGATGTTCCAGATTACGCTCTTTACGTCGGTGACCCCATGAGA-3′; and downstream primer, 5′-CCGGGTCTCCAGGGTGATGATGACAAGGATGGGCCTCCGATTC-3′. This PCR product, which was then digested with SacII and SfiI, together with another DNA fragment, which was digested from an EST clone (ID 6826464) with SfiI and EcoRI, was cloned into pUHD10-3, a tetracycline-regulated expression vector (16), at SacII and EcoRI sites to generate pUHD10-3-HA-ΔNp73α. To generate HA-tagged murine ΔNp73β, a PCR fragment was amplified using an EST clone (ID 6826464) as a template with an upstream primer (5′-GGCCGCCGGTCTTTCGAGGGTCGCATCTGT-3′) and a downstream primer (5′-TTCAGAGCCCCAAGGTCCTGACGAGGCTGGGGT-3′). The fragment was then used to replace the region in pUHD10-3-HA-ΔNp73α between the KpnI and EcoRI sites. To generate a construct expressing siRNA targeting the rat ΔNp73 gene, two oligonucleotides were synthesized, which were annealed and then cloned into the pBabe-U6 expression vector (31) to generate pBabe-U6-Si-ΔNp73. The sequences of the oligonucleotides were as follows (lowercase letters indicate link sequences and restriction enzyme sites): forward, 5′-gatccccGCCACCAGTCCGCCCCTACttcaagagaGTAGGGGCGGACTGGTGGCtttttggaaa-3′; reverse, 5′-agcttttccaaaaaGCCACCAGTCCGCCCCTACtctcttgaaGTAGGGGCGGACTGGTGGCggg-3′. The nucleotides in boldface above represent the siRNA target region. pSuper-Si-p53 vector, which expresses an siRNA targeting the rat p53 coding region, pUHD10-3-HA-p53 vector, which expresses HA-tagged murine p53, and the luciferase reporters (the wild-type and mutant TrkA promoters) were described previously (55).
Cell culture.
The PC12 tet-off cells were maintained in DMEM (Dulbecco/Vogt modified Eagle's minimal essential medium) supplemented with 10% horse serum and 5% fetal bovine serum (FBS). H1299-HA-ΔNp73α-1 and H1299-HA-ΔNp73β cells were generated as described previously (29) and cultured in DMEM with 10% FBS. Selection drugs were added to the medium as needed. Cells were grown at 37°C with 5% CO2. For protein induction, cells were washed three times with DMEM and then cultured in DMEM with 10% tetracycline-free FBS. For NGF treatment, cells were uninduced or induced to express a protein of interest and then switched to DMEM with 2% tetracycline-free FBS plus 100 ng/ml of NGF for parental PC12 cells or 200 ng/ml of NGF for p53 knockdown PC12 cells.
Establishment of stable cell lines.
All transfections were achieved using Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen). To generate stable cell lines that inducibly express murine ΔNp73α or ΔNp73β, PC12 cells, which are capable of expressing Tet-VP16 for the tetracycline-repressible expression system (purchased from ClonTech), were transfected with a pUHD10-3 vector expressing HA-tagged murine ΔNp73α or ΔNp73β. To generate a PC12 cell line in which endogenous p53 is knocked down and an exogenous murine ΔNp73α is inducibly expressed, pSuper-Si-p53 and pUHD10-3-HA-ΔNp73α were cotransfected into PC12 cells. To generate ΔNp73 knockdown cell lines, pBabe-U6-Si-ΔNp73 was transfected into PC12 cells. After transfection, cells were selected with 1 μg/ml of puromycin or 1 μg/ml of puromycin and 100 μg/ml of G418 for 4 weeks. Individual clones were screened for inducible expression of ΔNp73α and ΔNp73β or lack of endogenous p53 or ΔNp73 by Western blot analysis.
Growth rate analysis.
To determine the rate of cell growth, 5 × 104 PC12 cells were seeded per 60-mm-diameter plate. Cells were uninduced or induced to express a protein of interest in the presence or absence of tetracycline (2 μg/ml). Culture media were replaced every 72 h. Each day over a 5-day period, three plates for each group were rinsed twice with phosphate-buffered saline (PBS) to remove dead cells. Live cells on the plates were trypsinized, and samples from each plate were counted three times using a Coulter cell counter (Coulter Corporation, Miami, FL). The average number of cells from each group was used for growth rate determination.
DNA histogram analysis.
Cells were seeded at 2 × 105 per 90-mm-diameter plate and uninduced or induced to express ΔNp73α or ΔNp73β in the presence or absence of tetracycline (2 μg/ml). After 48 h of induction, both floating and dead cells were collected and fixed with 10 ml of 75% ethanol overnight and resuspended in 300 μl of PBS solution containing 50 μg each of RNase A and propidium iodide per ml. The stained cells were analyzed in a fluorescence-activated cell sorter (FACS Calibur; Becton Dickinson) within 4 h. The percentage of cells in the sub-G1, G1, S, and G2/M phases was determined by the Cell Quest program.
Neurite outgrowth assay.
A total of 5 × 104 cells were plated in each well of a six-well plate. Cells were uninduced or induced as needed. After induction, cells were subject to NGF treatment and the differentiation medium was replaced every 2 days. Cells with at least one neurite longer than two body lengths were counted as neurite positive. At least 500 cells were counted for each group, with experiments performed in triplicate.
Luciferase assay.
A total of 1 × 104 PC12 cells were plated in each well of a 24-well plate for 16 h and then transfected with a luciferase reporter along with pUHD10-3 control vector or pUHD10-3 vector that expresses murine ΔNp73α, murine ΔNp73β, or wild-type murine p53. pRL-CMV, a Renilla luciferase assay vector, was also cotransfected as an internal control. Cell extracts were prepared 16 h after transfection and used for the dual-luciferase assay according to the manufacturer's instructions (Promega). The increase in relative luciferase activity is a product of the luciferase activity induced by activator divided by that induced by an empty vector. All experiments were repeated in triplicate.
Immunoprecipitation and Western blot analysis.
For immunoprecipitation, cells were lysed in 1% Triton lysis buffer (25 mM Tris [pH 7.4], 25 mM NaCl, 1% Triton X-100) supplemented with the proteinase inhibitor cocktail (100 μg/ml). Cell lysates were clarified by centrifugation at 14,000 rpm for 15 min at 4°C and then incubated with 1 μg antibody overnight. The immunocomplexes were brought down by protein A/G beads and subjected to Western blot analysis. For Western blot analysis, cell lysates were resuspended with 2× sample buffer and boiled for 5 min. Proteins were then resolved in a sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel and transferred to a nitrocellulose membrane. Membrane blocking, washing, antibody incubation, and detection by enhanced chemiluminescence were performed as previously described (7, 31).
RNA isolation and Northern blot analysis.
Total RNA was isolated with Trizol reagent and used for Northern blot analysis as described previously (7). The TrkA probe was made from a 650-bp cDNA fragment amplified by reverse transcription-PCR (RT-PCR) with an upstream primer (5′-ATGAGACCAGCTTCATC-3′) and a downstream primer (5′-GCTCCCACTTGAGAATG-3′). The glyceraldehyde-3-phosphate dehydrogenase (GADPH) probe was made from a 1.25-kbp PstI-PstI cDNA fragment of the rat GAPDH cDNA (12).
RT-PCR analysis.
RT-PCR was performed with the the iScript cDNA synthesis kit (Bio-Rad Laboratories) according to the manufacturer's instructions. The PCR program used for amplification was (i) 94°C for 5 min, (ii) 94°C for 45 s, (iii) 55°C for actin or 58°C for ΔNp73 for 45 s and (iv) 72°C for 1 min, and (v) 72°C for 10 min. From steps 2 to 4, the cycle was repeated 20 times for actin or 35 times for ΔNp73. The primers used to amplify actin were upstream primer 5′-GTGGGCCGCTCTAGGCACCAA-3′ and downstream primer ′5-CTCTTTGATGTCACGCACGAT-3′. The primers for ΔNp73 were upstream primer 5′-TCCAACACATCTCCGGGCAA-3′ and downstream primer 5′-TGGGACGAGGCATGAGTCTG-3′.
ChIP assay.
Cells were seeded at 2 × 107 per 150-mm plate and uninduced or induced to express a protein of interest. After induction, ΔNp73-DNA complexes were cross-linked by formaldehyde and used for chromatin immunoprecipitation (ChIP) assay as described previously (19, 50). The primers used to detect the TrkA promoter were TrkA-RE-5 (5′-CTCCTGGGCTTTCAGACTTC-3′) and TrkA-RE-3 (5′-CCCTAGTTGACAGGCTAAATA-3′). The primers used to detect the p21 promoter were p21-RE-5 (5′-GCCTCCTGAGTGCTGGG-3′) and p21-RE-3 (5′-AGGGTGGGGGGTGGTAT-3′).
RESULTS
Overexpression of murine ΔNp73β, but not ΔNp73α, suppresses cell proliferation in PC12 cells.
To investigate the role of ΔNp73 in neuronal differentiation, we generated stable PC12 cell lines that inducibly express HA-tagged murine ΔNp73α or ΔNp73β under the control of tetracycline-regulated promoter. The results are presented in Fig. 1A and B. Upon induction, ΔNp73α and ΔNp73β were detectable by anti-HA (Fig. 1A, ΔNp73α; and B, ΔNp73β), and their levels were comparable. We also examined the induction of p21, a common target of the p53 family. We found that p21 was induced by ΔNp73β (Fig. 1B, p21; compare lanes 1 and 3 with 2 and 4, respectively) but not by ΔNp73α (Fig. 1A, p21). To characterize if ΔNp73α or ΔNp73β has an effect on cell proliferation, growth curve analysis was performed over a 5-day period. We found that cell proliferation was markedly inhibited by ΔNp73β (Fig. 1D) but little if any by ΔNp73α (Fig. 1C). This is consistent with an earlier report (30). To further examine the effect of ΔNp73 on cell proliferation, DNA histogram analysis was performed. We found that overexpression of ΔNp73α had no effect on the cell cycle (Fig. 1E). However, apoptosis was induced by ΔNp73β as the percentage of cells with sub-G1 DNA content was increased from 2.85% to 13.37% (Fig. 1F). We would like note that a low level of ΔNp73β can induce only cell cycle arrest, but not apoptosis (data not shown). Together, we showed that ΔNp73β, but not ΔNp73α, is capable of inhibiting cell proliferation in PC12 cells.
FIG. 1.

Overexpression of ΔNp73β, but not ΔNp73α, suppresses cell proliferation in PC12 cells. (A and B) Establishment of stable PC12 cell lines inducibly expressing HA-tagged murine ΔNp73α (A) or ΔNp73β (B). PC12 cells were uninduced or induced to express ΔNp73α (A) or ΔNp73β (B) for 18 h. Cell extracts were collected for Western blot analysis. The levels of HA-tagged ΔNp73α (A) or ΔNp73β (B), p21, and actin were analyzed by Western blot analysis with antibodies against HA (α-HA), p21, and actin, respectively. Actin was used as a loading control. (C and D) Growth rates of cells with or without ΔNp73α (C) or ΔNp73β (D) expression over a 5-day period. A total of 5 × 104 cells were seeded per 60-mm-diameter plate, which were then uninduced or induced to express ΔNp73α (C) or ΔNp73β (D). At the time points indicated, live cells were collected and counted by the Coulter cell counter. The average numbers of cells from three plates were used for growth rate determination. (E and F) DNA histogram analysis of PC12 cells in the presence or absence of ΔNp73α (E) or ΔNp73β (F). PC12 cells were uninduced or induced to express ΔNp73α (E) or ΔNp73β (F) for 48 h, and the percentage of cells in each phase of the cell cycle was determined by DNA histogram analysis. DNA content was quantified using the data from at least three independent experiments.
Overexpression of murine ΔNp73α or ΔNp73β inhibits NGF-mediated neuronal differentiation in PC12 cells.
To determine if ΔNp73α or ΔNp73β plays a role in NGF-mediated neuronal differentiation, PC12 cells were uninduced or induced to express ΔNp73α or ΔNp73β for 18 h followed by treatment with NGF or no NGF treatment for 3 or 6 days. Phase-contrast microscopy was utilized to examine the morphology of PC12 cells. We showed that in the absence of NGF, no neurite outgrowth was observed in PC12 cells, regardless of expression of ΔNp73α (Fig. 2A, a and b) or ΔNp73β (Fig. 2B, a and b). However, in the presence of NGF, much less and shorter neurite outgrowth was observed in PC12 cells expressing ΔNp73α than in control cells (Fig. 2A, compare c and e with d and f, respectively). Surprisingly, neurites were also found to be fewer and shorter in PC12 cells expressing ΔNp73β than those in control cells (Fig. 2B, compare c and e with d and f, respectively), given that ΔNp73β has a strong transcriptional activity (Fig. 1). The neurite outgrowth was then quantified by counting cells with neurites longer than two body lengths. We found that the percentage of cells with neurites was decreased by ΔNp73α from 23% to 9% after 3 days of treatment and from 53% to 24.7% after 6 days of treatment (Fig. 2C). Likewise, ΔNp73β reduced NGF-mediated neurite outgrowth from 30.9% to 16.9% after 3 days of NGF treatment and from 58.3% to 33.6% after 6 days of NGF treatment (Fig. 2C). To confirm this, the induction of growth-associated protein 43 (GAP-43) and p21 was examined by Western blot analysis. GAP-43 is a neuronal differentiation marker whose expression directly correlates with the extent of differentiation, while p21 is required for the cell cycle arrest mediated by NGF during differentiation (14, 41). We showed that GAP-43 was induced in an NGF-dependent manner (Fig. 2D and E, GAP-43, compare lanes 1 and 2 with 3 and 4). However, the induction of GAP-43 was significantly decreased upon expression of ΔNp73α (Fig. 2D, GAP-43, compare lanes 3 and 4) or ΔNp73β (Fig. 2E, GAP-43, compare lanes 3 and 4). Interestingly, we found that while ΔNp73α attenuated NGF-mediated activation of p21 (Fig. 2D, p21, compare lanes 3 and 4), ΔNp73β induced p21 expression regardless of NGF treatment (Fig. 2E, p21, compare lanes 1 and 3 with 2 and 4, respectively). We also determined the level of p53, which was found not to be consistently up- or down-regulated by ΔNp73α or ΔNp73β regardless of NGF treatment (Fig. 2D and E, p53, compare lanes 1 and 3 with 2 and 4, respectively). Furthermore, we determined if the induction of p21 has an effect on the ability of ΔNp73β to inhibit NGF-mediated neuronal differentiation. To address this, two clones, PC12-HA-ΔNp73β-2 and PC12-HA-ΔNp73β-1, which express various levels of ΔNp73β (Fig. 2F, ΔNp73β, compare lanes 2 and 4), were selected for NGF treatment. We found that the extent of p21 induction was higher in PC12-HA-ΔNp73β-2 cells than that in PC12-HA-ΔNp73β-1 cells (Fig. 2F, p21, compare lanes 2 and 4). However, NGF-mediated expression of GAP-43 and neuronal differentiation were suppressed by ΔNp73β in both clones (Fig. 2F, GAP-43, compare lanes 1 and 3 with 2 and 4, respectively) (data not shown). This suggests that p21 induced by ΔNp73β does not impinge on the ability of ΔNp73β to suppress NGF-mediated neuronal differentiation. Taken together, our data indicate that ΔNp73α and ΔNp73β are capable of inhibiting NGF-mediated neuronal differentiation in PC12 cells.
FIG. 2.

Overexpression of ΔNp73α or ΔNp73β inhibits NGF-mediated neuronal differentiation in PC12 cells. (A and B) Representative microscopy images of PC12 cells with or without ΔNp73α (A) or ΔNp73β (B) expression in the presence or absence of NGF. PC12 cells were uninduced (a, c, and e) or induced (b, d, and f) to express ΔNp73α (A) or ΔNp73 (B) for 18 h followed by treatment with 100 ng/ml NGF (c, d, e, and f) or no NGF treatment (a and b) for 3 (c and d) or 6 (e and f) days. (C) Quantitative analysis of PC12 cells bearing neurites. Cells were treated as described for panels A and B. Neurite-positive cells are the ones with at least one neurite longer than two body lengths. The percentage of neurite outgrowth is calculated by dividing the neurite-positive cells by the total number of cells counted. Approximately 500 cells were counted for each sample. The error bars represent the standard deviations of triplicate results. (D, E, and F) Overexpression of ΔNp73α (D) or ΔNp73β (E and F) attenuates the induction of GAP-43. PC12 cells were uninduced or induced to express ΔNp73α (D) or ΔNp73β (E and F) for 18 h followed by treatment with NGF or no NGF treatment for 3 days. Cell lysates were examined by Western blot analysis with antibodies against HA (α-HA), p21, GAP-43, p53, and actin, respectively. Long Exp., long expression; Short Exp., short expression.
ΔNp73α inhibits NGF-mediated neuronal differentiation in p53 knockdown PC12 cells.
Previously, we and others have reported that p53 plays an important role in NGF-mediated neuronal differentiation in PC12 cells (9, 55). Since ΔNp73 has been found to be dominant negative towards p53 (37), it is important to determine whether ΔNp73 would inhibit NGF-mediated neuronal differentiation in the absence of p53. To this end, we generated stable PC12 cell lines in which endogenous p53 is targeted by siRNA and an HA-tagged murine ΔNp73α is inducibly expressed under a tetracycline-regulated promoter. Multiple clones were selected, and one representative clone, PC12(p53KD)-HA-ΔNp73α, is shown in Fig. 3A. Upon induction, ΔNp73α was detectable by anti-HA (Fig. 3A, ΔNp73α). Moreover, upon NGF treatment, endogenous p53 was accumulated in PC12 cells but not in p53 knockdown PC12 cells (Fig. 3A, p53, compare lanes 3 and 4 with lanes 7 and 8), suggesting that p53 is silenced by siRNA in PC12(p53KD) cells.
FIG. 3.
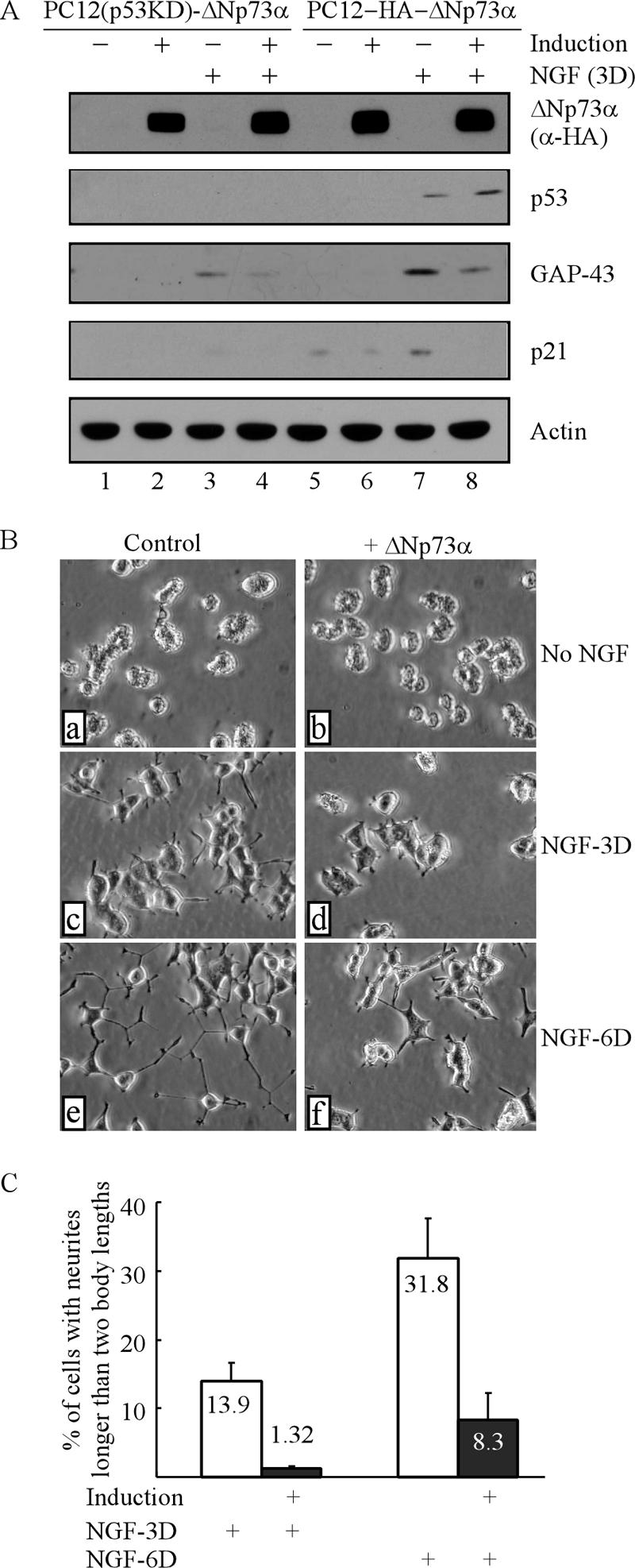
ΔNp73α inhibits NGF-mediated neuronal differentiation in p53 knockdown PC12 cells. (A) PC12(p53KD)-HA-ΔNp73α and PC12-HA-ΔNp73α cells were uninduced or induced to express ΔNp73α for 18 h followed by treatment with NGF or no NGF treatment for 3 days. Cell lysates were collected for Western blot analysis with antibodies against HA, p53, p21, GAP-43, and actin, respectively. (B) Representative microscopy images of p53 knockdown PC12 cells with or without ΔNp73α expression in the presence or absence of NGF. PC12 cells were uninduced (a, c, and e) or induced (b, d, and f) to express ΔNp73α for 18 h followed by treatment with 200 ng/ml NGT (c, d, e, and f) or no NGF treatment (a and b) for 3 (c and d) or 6 (e and f) days. (C) Quantitative analysis of PC12 cells bearing neurites shown in panel B.
Next, to determine if ΔNp73α inhibits NGF-mediated neuronal differentiation in the absence of p53, PC12(p53KD) cells were uninduced or induced to express ΔNp73α for 18 h followed by treatment with NGF or no NGF treatment for 3 or 6 days. It should be mentioned that a higher dosage of NGF (200 ng/ml instead of 100 ng/ml) was used since p53 knockdown renders PC12 cells less sensitive to NGF (55). As shown in Fig. 3B, in the absence of NGF, no morphological change was found in PC12(p53KD) cells regardless of ΔNp73α induction (Fig. 3B, a and b). In the presence of NGF, control PC12(p53KD) cells extended neurites in a time-dependent manner (Fig. 3C, c and e). However, the neurites were much fewer and shorter in ΔNp73α-expressing PC12(p53KD) cells than in control cells (Fig. 3B, compare d and f with c and e, respectively), suggesting that ΔNp73α is capable of inhibiting NGF-mediated neurite outgrowth in the absence of p53. By counting cells with neurites, we showed that ΔNp73α decreased NGF-mediated neurite outgrowth in p53 knockdown PC12 cells from 13.9% to 1.32% after a 3-day treatment and from 31.8% to 8.3% after a 6-day treatment (Fig. 3C). To confirm this, induction of GAP-43 and p21 was examined by Western blot analysis. We showed that due to lack of p53, the extent of GAP-43 induction was less in p53 knockdown PC12 cells than that in parental PC12 cells (Fig. 3A, GAP-43, compare lanes 3 and 4 with 7 and 8). Nevertheless, a further reduction was found in p53 knockdown PC12 cells upon ΔNp73α expression (Fig. 3A, GAP-43, compare lanes 3 and 4). In addition, p21 was not induced by NGF in p53 knockdown PC12 cells due to lack of p53 (Fig. 3A, p21, lanes 3 and 4), consistent with a previous report (55). Together, these data suggest that ΔNp73α is capable of inhibiting NGF-mediated neuronal differentiation independent of p53.
ΔNp73 is the predominant form in undifferentiated PC12 cells and down-regulated upon NGF treatment.
To determine the role of endogenous ΔNp73 in NGF-mediated neuronal differentiation, we examined whether commercially available anti-p73 antibodies are able to recognize exogenous murine ΔNp73, which shares a high degree of sequence identity with rat ΔNp73. To do this, PC12 cells were uninduced or induced to express ΔNp73α or ΔNp73β for 18 h. Cell lysates were immunoprecipitated with BL906 antibody (Bethyl Laboratories, catalog no. A300-126A), which was raised against amino acids 1 to 62 of human TAp73 and expected to react with only the TAp73 isoforms, or p73-Ab2 antibody (Calbiochem, catalog no. 0P109), which was raised against amino acids 380 to 495 of human p73α and expected to detect both the α and β isoforms of TA/ΔNp73. The immunoprecipitates were then examined by Western blot analysis with p73-Ab1 and p73-Ab3 antibodies, which preferentially detect α (TA/ΔNp73α) and β (TA/ΔNp73β) isoforms of p73, respectively. We showed that murine ΔNp73α was detectable by p73-Ab1 in p73-Ab2, but not BL906, immunoprecipitates (Fig. 4A, compare lanes 6 and 4). Similarly, murine ΔNp73β was detected by p73-Ab3 in p73-Ab2, but not BL906, immunoprecipitates (Fig. 4B, compare lanes 6 and 4).
FIG. 4.

ΔNp73 is the predominant isoform in undifferentiated PC12 cells and is down-regulated upon NGF treatment. (A and B) Characterization of p73 antibodies that can recognize exogenous murine ΔNp73α (A) or ΔNp73β (B). PC12 cells were uninduced or induced to express ΔNp73α (A) or ΔNp73β (B) for 18 h. Cell lysates were immunoprecipitated (IP) with 1 μg of BL906 and p73Ab-2 antibodies, respectively, followed by Western blot (WB) analysis with p73-Ab1 (A) or p73-Ab3 (B). Approximately 2% of cell lysates was used as input. (C, D, and E) Characterization of endogenous p73 in PC12 and HCT116 cells. Cell lysates were immunoprecipitated with 1 μg of BL906 and p73-Ab2 antibodies, respectively, followed by Western blot analysis with p73-Ab1 (C), p73-Ab3 (D), or BL906 (E). The HA-tagged murine ΔNp73α (C, lane 2), ΔNp73β (C and D, lane 1), and p73β (E, lane 2) as well as human TAp73α (E, lane 1) were used as controls. The asterisk in panel E, lane 3, represents a nonspecific protein. (F) Endogenous ΔNp73 is down-regulated upon NGF treatment in PC12 cells. PC12 cells were treated with NGF or not treated with NGF for 0 to 72 h. Cell lysates were collected at each time point and analyzed by Western blot analysis with p73-Ab1, p73-Ab3, GAP-43, and actin antibodies, respectively.
Next, we determined the expression of p73 in PC12 cells. To this end, PC12 cell extracts were immunoprecipitated with BL906 or p73-Ab2 antibody followed by Western blot analysis with p73-Ab1 or p73-Ab3 antibody. We showed that two polypeptides, whose apparent molecular masses are close to that for HA-tagged murine ΔNp73α (Fig. 4C, lane 2), were detected by p73-Ab1 in p73-Ab2, but not BL906, immunoprecipitates (Fig. 4C, compare lanes 4 and 5). We would like to note that HA-tagged ΔNp73α and ΔNp73β expressed in PC12 cells exist as a doublet (Fig. 4C, lanes 1 and 2). Another polypeptide, whose apparent molecular mass is close to that for HA-tagged ΔNp73β (Fig. 4D, lane 1), was detected by p73-Ab3 in p73-Ab2, but not BL906, immunoprecipitates (Fig. 4D, compare lanes 3 and 4). Therefore, these polypeptides are likely to be ΔNp73 since they were pulled down by p73-Ab2, an antibody for both TA/ΔNp73 isoforms, but not BL906, an antibody specific for TAp73 isoforms. To further confirm this, PC12 and HCT116 cell lysates were immunoprecipitated with BL906 or p73-Ab2 antibody followed by Western blot analysis with BL906 antibody. HCT116 cells, which express endogenous TAp73α (46), were utilized as a positive control. We showed that a polypeptide whose molecular mass is close to that of human TAp73α (Fig. 4E, lane 1) was detected by BL906 antibody in HCT116 but not PC12 cells (Fig. 4E, compare lanes 4 and 5 with lanes 7 and 8). To rule out the possibility of species variation, we also showed that BL906 was able to detect HA-tagged TAp73β, a murine TAp73 (Fig. 4E, lane 2). Together, we concluded that these polypeptides detected by p73-Ab1 and p73-Ab3 in PC12 cells are likely to be ΔNp73α and ΔNp73β. Thus, ΔNp73 is the predominant p73 isoform in undifferentiated PC12 cells.
To determine if expression of endogenous ΔNp73 is altered during NGF-mediated neuronal differentiation, PC12 cells were treated with NGF for 0 to 72 h. Cell lysates were collected at each time point and analyzed by Western blot analysis with antibodies against ΔNp73α (p73-Ab1), ΔNp73β (p73-Ab3), GAP-43, and actin, respectively. We showed that upon NGF treatment, GAP-43 was accumulated in a time-dependent manner (Fig. 4F, GAP-43). However, unlike GAP-43, the level of ΔNp73α was rapidly diminished in the first 18 h upon NGF treatment and then came back and ΔNp73αwas maintained at low levels from 24 to 72 h (Fig. 4E, ΔNp73α). Similarly, upon NGF treatment, the level of ΔNp73β was also decreased in the first 18 h and then ΔNp73β was maintained at low levels in differentiating PC12 cells (Fig. 4E, ΔNp73β).
Knockdown of endogenous ΔNp73 promotes NGF-mediated neuronal differentiation in PC12 cells.
To determine if down-regulation of ΔNp73 in differentiating PC12 cells plays a role in NGF-mediated neuronal differentiation, we generated stable PC12 cell lines in which endogenous ΔNp73 is silenced by siRNA. Two representative clones, PC12-ΔNp73-KD-1 and PC12-ΔNp73-KD-4, are presented in Fig. 5A. Western blot analysis revealed that the levels of endogenous ΔNp73α and ΔNp73β were significantly decreased in PC12-ΔNp73-KD cells compared to those in parental PC12 cells (Fig. 5A, ΔNp73α and ΔNp73β, compare lane 1 with 2 and 3). In line with this, RT-PCR analysis also showed that the level of ΔNp73 transcripts was markedly reduced in PC12-ΔNp73-KD cells compared to that in parental PC12 cells (Fig. 5B, ΔNp73, compare lane 1 with 2 and 3). Next, to determine if knockdown of ΔNp73 affects NGF-mediated neuronal differentiation, PC12 and PC12-ΔNp73-KD cells were treated with NGF or without NGF for 3 or 6 days. As shown in Fig. 5C, neither PC12 nor PC12-ΔNp73-KD cells were able to extend neurites in the absence of NGF (Fig. 5C, a to c). However, upon NGF treatment for 3 days, PC12-ΔNp73-KD cells appeared to extend many more and longer neurites than those in parental PC12 cells (Fig. 5C, compare d with e and f). Similar patterns were found when cells were treated with NGF for 6 days (Fig. 5C, compare g with h and i). By counting cells with neurites, we found that knockdown of ΔNp73 in PC12 cells substantially increased NGF-mediated neurite outgrowth from 33.5% to 49.1 to 53.5% after 3 days of treatment and from 61.2% to 77.6 to 90.1% after 6 days of treatment (Fig. 5D). To confirm this, the levels of ΔNp73 and GAP-43 were analyzed by Western blot analysis. We showed that the steady-state levels of ΔNp73α and ΔNp73β were much lower in PC12-ΔNp73-KD cells than those in PC12 cells (Fig. 5E, ΔNp73α, compare lanes 1 to 4 with 5 and 6; and 5F, ΔNp73β, compare lanes 3 to 6 with 1 and 2). We also noticed that the levels of ΔNp73 in two PC12-KD-ΔNp73 cells were slightly increased upon NGF treatment for 3 days (Fig. 5F, compare lanes 3 and 5 with 4 and 6, respectively), although the levels were much lower than those in the parental PC12 cells (Fig. 5F, compare lane 1 with 4 and 6, respectively). This is likely due to the effect of NGF on ΔNp73 modifications since NGF is known to up-regulate p53 via posttranslational modifications, such as acetylation (51). Nevertheless, we show that upon NGF treatment, the induction of GAP-43 was greatly increased in ΔNp73 knockdown PC12 cells compared to that in parental PC12 cells (Fig. 5F, GAP-43, compare lane 2 with lanes 4 and 6). We would like to note that even in the absence of NGF treatment, GAP-43 was detectable in ΔNp73 knockdown PC12 cells (Fig. 5E, GAP-43 Long Exp, compare lane 1 with lanes 3 and 5). Furthermore, a scrambled siRNA was used as a negative control. We showed that the scrambled siRNA was unable to target ΔNp73 (Fig. 5G, ΔNp73, compare lanes 1 and 3) or enhance GAP-43 expression upon NGF treatment (Fig. 5G, GAP-43, compare lanes 1 and 2 with 3 and 4, respectively). Together, these data suggest that knockdown of ΔNp73 in PC12 cells promotes NGF-mediated neuronal differentiation.
FIG. 5.

Knockdown of ΔNp73 promotes NGF-mediated neuronal differentiation in PC12 cells. (A) Establishment of stable PC12 cell line with ΔNp73 knockdown. PC12 and PC12-ΔNp73-KD cell lysates were collected and examined by Western blot analysis with p73-Ab1, p73-Ab3, and actin antibodies, respectively. (B)Total RNAs from PC12 and PC12-ΔNp73-KD cells were purified and subjected to RT-PCR analysis with primers to amplify ΔNp73 and actin. (C) Representative microscopy images of PC12 cells with or without ΔNp73 knockdown in the absence or presence of NGF. PC12 and PC12-ΔNp73-KD cells were treated without (a to c) or with (d to i) NGF for 3 days (NGF-3D [d to f]) or 6 days (NGF-6D [h and i]). (D) Quantitative analysis of PC12 and PC12-ΔNp73-KD cells with neurites shown in panel C. (E) PC12 and PC12-ΔNp73-KD cells were treated with or without NGF for 3 days followed by Western blot analysis with p73-Ab1 and actin, respectively. (F) The expression of GAP-43 is increased in PC12 cells upon knockdown of ΔNp73. PC12 cells were treated as described in panel E and examined by Western blot analysis with antibodies against ΔNp73β (p73-Ab3), GAP-43, and actin, respectively. (G) PC12 cells were either mock transfected or transfected with 25 nM scrambled siRNA for 3 days, followed by NGF treatment or no NGF treatment for 3 days. Cell lysates were collected for Western blot analysis with antibodies against ΔNp73β (p73-Ab3), GAP-43, and actin, respectively.
ΔNp73 transcriptionally suppresses TrkA expression, which attenuates the NGF-mediated MAPK pathway.
Previously, we reported that p53 plays an important role in NGF-mediated neuronal differentiation in PC12 cells by up-regulating TrkA, the high-affinity NGF receptor (55). Here, we found that knockdown of ΔNp73 promotes, whereas overexpression of ΔNp73 inhibits, NGF-mediated neuronal differentiation, suggesting that ΔNp73 might be able to inhibit TrkA expression. To test this, PC12-HA-ΔNp73α or PC12-HA-ΔNp73β cells were uninduced or induced to express ΔNp73α or ΔNp73β for 12 h followed by NGF treatment for 24 h or no NGF treatment. We found that TrkA expression was increased upon NGF treatment (Fig. 6A and B, TrkA, compare lanes 1 and 3), consistent with an early report (8). Moreover, we showed that ΔNp73α was capable of suppressing TrkA expression regardless of NGF treatment (Fig. 6A, TrkA, compare lanes 1 and 3 with 2 and 4, respectively). Similarly, TrkA expression was suppressed by ΔNp73β (Fig. 6B, TrkA, compare lanes 1 and 3 with 2 and 4, respectively). Additionally, TrkA expression was suppressed by ΔNp73α in p53 knockdown PC12 cells (data not shown). Next, to determine whether the repression of TrkA by ΔNp73 is preceded by a decrease in TrkA transcript, Northern blot analysis was performed and showed that the level of TrkA mRNA was down-regulated upon induction of ΔNp73α or ΔNp73β (Fig. 6C, TrkA, compare lanes 1 and 3 with 2 and 4, respectively). To determine if ΔNp73 transcriptionally represses TrkA expression in the absence of p53, p53 knockdown PC12 cells were uninduced or induced to express ΔNp73α in the presence or absence of NGF. We found that the level of TrkA mRNA was decreased upon ΔNp73α induction regardless of NGF treatment (Fig. 6D, TrkA, compare lanes 1 and 3 with 2 and 4, respectively). This suggests that ΔNp73 can repress TrkA expression in a p53-independent manner. Moreover, to determine if endogenous ΔNp73 has an effect on TrkA expression, we analyzed the steady-state level of TrkA in the parental and ΔNp73 knockdown PC12 cells. We showed that the steady-state level of TrkA was increased in ΔNp73 knockdown PC12 cells compared to that in parental PC12 cells (Fig. 6E, TrkA, compare lane 1 with lanes 2 and 3). Finally, to rule out the possibility that the inhibition of TrkA by ΔNp73 may be cell-type specific, H1299 cells that can inducibly express HA-tagged human ΔNp73α or ΔNp73β were utilized. We showed that upon ΔNp73α/β induction, the expression of TrkA was repressed in H1299 cells (Fig. 6F, TrkA, compare lanes 1 and 3 with 2 and 4, respectively). Next, we determined if ΔNp73-mediated repression of TrkA has an effect on the NGF-mediated MAPK pathway, whose sustained activation is required for NGF-mediated neurite outgrowth in PC12 cells (38). To this end, PC12-HA-ΔNp73β cells were uninduced or induced to express ΔNp73β for 12 h followed by NGF treatment for 0, 5, 15, and 30 min. As shown in Fig. 6G, NGF-mediated activation of MEK1/2 and RSK90 was attenuated in PC12 cells upon ΔNp73β induction compared to that in control cells (Fig. 6G, p-MEK1/2 and p-RSK90, compare lanes 1, 3, 5, and 7 with 2, 4, 6, and 8, respectively).
FIG. 6.

ΔNp73 transcriptionally inhibits TrkA expression, which leads to repression of the NGF-mediated MAPK pathway. (A and B) Overexpression of ΔNp73α (A) or ΔNp73β (B) represses TrkA expression. PC12 cells were uninduced or induced to express ΔNp73α (A) or ΔNp73β (B) for 12 h followed by treatment with NGF or no NGF treatment for 24 h. Cell extracts were analyzed by Western blot analysis with antibodies against HA, TrkA, and actin, respectively. (C) The level of TrkA mRNA is down-regulated by ΔNp73α or ΔNp73β. PC12 cells were uninduced or induced to express ΔNp73α or ΔNp73β for 18 h. The total RNAs from these cells were isolated and prepared for Northern blots, which were sequentially probed with cDNAs derived from the TrkA and GAPDH genes, respectively. (D) ΔNp73α down-regulates TrkA expression independent of p53. p53 knockdown PC12 cells were uninduced or induced to express ΔNp73α for 12 h followed by NGF treatment for 12 h. Total RNAs were isolated for Northern blot analysis as described in panel C. (E) The steady-state level of TrkA protein is increased in PC12 cells upon knockdown of ΔNp73. Cell lysates were collected for Western blot analysis with antibodies against TrkA and actin, respectively. (F) Overexpression of ΔNp73α and ΔNp73β suppresses TrkA expression in H1299 cells. H1299 cells were uninduced or induced to express ΔNp73α or ΔNp73β for 24 h. Cell lysates were collected and analyzed by Western blot analysis with antibodies against HA (α-HA), TrkA, and actin, respectively. (G) ΔNp73 attenuates the NGF-mediated MAPK pathway. PC12 cells were uninduced or induced to express ΔNp73β for 18 h followed by NGF treatment for 0, 5, 15, or 30 min. Cell extracts were collected for Western blot analysis with antibodies against HA, p-MEK1/2, p-RSK90, and actin, respectively.
ΔNp73 directly represses the TrkA promoter.
To further investigate the underlying mechanism, we sought to determine if ΔNp73 inhibits the TrkA promoter, which contains a p53-RE (55). To this end, a luciferase reporter under the control of the TrkA promoter with or without p53-RE (Fig. 7A, bottom panel) was cotransfected into PC12 cells with a control vector or a vector that expresses murine p53, ΔNp73α, or ΔNp73β. We show that the TrkA promoter with p53-RE was responsive to p53 (Fig. 7A), consistent with an early report (55). In contrast, this promoter was repressed by ΔNp73α and ΔNp73β in a dosage-dependent manner (Fig. 7A). However, the TrkA promoter without p53-RE was inert to p53, ΔNp73α, and ΔNp73β (Fig. 7A). Next, to determine if ΔNp73 directly binds to the TrkA promoter in vivo, a ChIP assay was performed. The ΔNp73-DNA complexes were immunoprecipitated by anti-HA and used for identification of ΔNp73-binding regions with primers located in the TrkA and p21 promoters (Fig. 7B). Normal immunoglobulin G (IgG) was used as a control. We found that upon ΔNp73β induction, the fragments containing the p53-RE in the TrkA or p21 promoters were enriched by anti-HA (Fig. 7C, TrkA-p53-RE and p21-p53-RE, compare lanes 1 and 2). No DNA fragment was enriched by the control IgG (Fig. 7C, TrkA-p53-RE and p21-p53-RE, lanes 3 and 4). Similarly, ΔNp73α was found to bind to the p53-RE in the TrkA and p21 promoters (data not shown). We also show that ΔNp73α directly bound to the TrkA and p21 promoters in the absence of p53 (Fig. 7D, TrkA-p53-RE and p21-p53-RE, compare lanes 1 and 2). Furthermore, to determine if endogenous ΔNp73 is able to bind to the TrkA promoter in vivo, PC12 cells were used for a ChIP assay. P73-Ab1 and -3 antibodies were used to immunoprecipitate ΔNp73-DNA complexes, while normal IgG and BL906 antibodies were used as controls. We showed that the fragment containing the p53-RE was enriched in p73-Ab1 and -3, but not in BL906 or IgG, immunocomplexes (Fig. 7E, TrkA-p53-RE, lanes 1 to 3).
FIG. 7.

ΔNp73 directly represses the TrkA promoter. (A) Wild-type, but not mutant, TrkA promoter is suppressed by ΔNp73. A schematic diagram of wild-type and mutant TrkA promoters is presented in the bottom panel. The dual-luciferase assay was performed as described in Materials and Methods. The increase in relative luciferase activity for each construct was calculated using an empty vector as a control. (B) Schematic presentation of the TrkA and p21 promoters with the locations of the transcriptional start site, p53-RE, and primers used for ChIP assays. (C and D) ΔNp73 binds to the TrkA and p21 promoters in vivo. Cells were uninduced or induced to express ΔNp73β (C) or ΔNp73α (D) for 18 h followed by the ChIP assay as described in Materials and Methods. Anti-HA antibody (α-HA) was used to immunoprecipitate ΔNp73β- or ΔNp73α-DNA complexes. Normal IgG was used as a control. (E) Endogenous ΔNp73 binds to the TrkA promoter under a nonstressed condition in PC12 cells. p73-Ab1 and -3 antibodies were used to immunoprecipitate ΔNp73-DNA complexes. Rabbit IgG and BL906 antibodies were used as a control.
ΔNp73-mediated repression of TrkA requires histone deacetylases.
Many studies have shown that gene silencing/repression is a highly regulated process including coordinated histone modifications. Therefore, we examined if HDACs are involved in ΔNp73-mediated repression of TrkA. To this end, trichostatin A, a histone deactylase inhibitor, was utilized. If HDACs are involved, ΔNp73-mediated TrkA repression would be derepressed upon TSA treatment. To test this, PC12-HA-ΔNp73β or PC12(p53KD)-HA-ΔNp73α cells were uninduced or induced to express ΔNp73β or ΔNp73α for 12 h and then treated with 0, 15, and 50 ng/ml of TSA for an additional 12 h. We found that in the absence of TSA, the expression of TrkA was repressed by ΔNp73α or ΔNp73β (Fig. 8A and B, TrkA, compare lanes 1 and 2). However, in the presence of TSA, the repression of TrkA by ΔNp73α or ΔNp73β was attenuated (Fig. 8A and B, TrkA, compare lanes 3 and 5 with 4 and 6, respectively). Moreover, immunoprecipitation followed by Western blot analysis was performed to determine if ΔNp73 can physically interact with HDACs. Anti-HA was used to immunoprecipitate ΔNp73-containing complexes or to detect ΔNp73 by Western blot analysis. We found that endogenous HDAC1 and HDAC2 were detected in ΔNp73α- or ΔNp73β-containing immunoprecipitates (Fig. 8C and D). Conversely, ΔNp73α was detected in both HDAC1- and HDAC2-containing immunoprecipitates (Fig. 8E). These data suggest that ΔNp73 physically interacts with HDAC1 and HDAC2. Furthermore, we determined if ΔNp73 is able to recruit HDAC1 and -2 to the TrkA promoter. To this end, PC12 cells were uninduced or induced to express ΔNp73β for 12 h for a ChIP assay. Chromatin was immunoprecipitated with normal IgG, antibody against acetylated histone 3, or a mixture of HDAC1 and -2 antibodies. We showed that upon ΔNp73β induction, the fragment containing the p53-RE in the TrkA promoter was enriched in HDAC1/2 immunocomplexes (Fig. 8F, compare lanes 7 and 8). In addition, the level of acetylated histone 3 bound to the p53-RE in the TrkA promoter was markedly decreased upon ΔNp73β expression (Fig. 8F, compare lanes 5 and 6). No fragment was enriched by normal IgG (Fig. 8F, lanes 3 and 4). Taken together, these results suggest that HDAC1 and -2 are recruited to the TrkA promoter by ΔNp73, which leads to diminished acetylation of histones and, subsequently, repression of TrkA.
FIG. 8.

ΔNp73-mediated repression of TrkA requires histone deacetylases. (A and B) Trichostatin A derepresses ΔNp73-mediated repression of TrkA. PC12 cells were uninduced or induced to express ΔNp73β (A) or ΔNp73α (B) for 12 h and then treated with 0, 15, and 50 ng/ml of TSA for additional 12 h. Cell lysates were collected for Western blot analysis with antibodies against HA, TrkA, and actin, respectively. (C to E) ΔNp73 interacts with endogenous HDACs. PC12 cells were uninduced or induced for 18 h to express ΔNp73α (C and E) or ΔNp73β (D). Cell lysates were immunoprecipitated (IP) with 1 μg of HA (C and D), HDAC1 (E), or HDAC2 (E) antibodies overnight. The immunocomplexes were examined by Western blot (WB) analysis with a mixture of anti-HDAC1 and -2 (C and D, upper panel) or with anti-HA (E, upper panel). The blots were then stripped and reblotted with anti-HA (C and D, lower panel) or a mixture of anti-HDAC1 and -2 (E, lower panel). (F) HDAC1 and -2 are recruited to the TrkA promoter. PC12-HA-ΔNp73β cells were uninduced and induced to express ΔNp73β for ChIP assay. Antiacetylated H3 and anti-HDAC1/2 were used to enrich the fragment containing the p53-RE in the TrkA promoter. Rabbit IgG was used as a control.
DISCUSSION
Many studies have shown that ΔNp73 possesses multiple functions distinct from TAp73, the latter of which has many p53-like features (4, 18, 24, 44). ΔNp73 is frequently overexpressed in a variety of human cancers (54). In addition, overexpression of ΔNp73 directly correlates with the poor prognosis of neuroblastoma (3) and lung cancer (45). Furthermore, ΔNp73 plays a crucial role in neuronal development and is a prosurvival protein in the nerve system (37, 53). However, the mechanism by which ΔNp73 exerts these biological activities is not clear. In the present study, we have investigated this and shown that ΔNp73 is a negative regulator of NGF-mediated neuronal differentiation in PC12 cells with the following pieces of evidence: (i) overexpression of ΔNp73 inhibits NGF-mediated neuronal differentiation, (ii) the level of endogenous ΔNp73 is progressively diminished in differentiating PC12 cells upon treatment with NGF, and (iii) knockdown of ΔNp73 promotes NGF-mediated neuronal differentiation. Interestingly, we also found that ΔNp73, by recruiting HDAC corepressors to the TrkA promoter, transcriptionally inhibits TrkA expression and consequently inhibits NGF-mediated neuronal differentiation. In line with this, we have shown that knockdown of ΔNp73 increases the steady-state level of TrkA and leads to enhanced neurite outgrowth in PC12 cells upon NGF treatment. Based on these findings, we present a model for the role of ΔNp73 in NGF-mediated neuronal differentiation (Fig. 9).
FIG. 9.
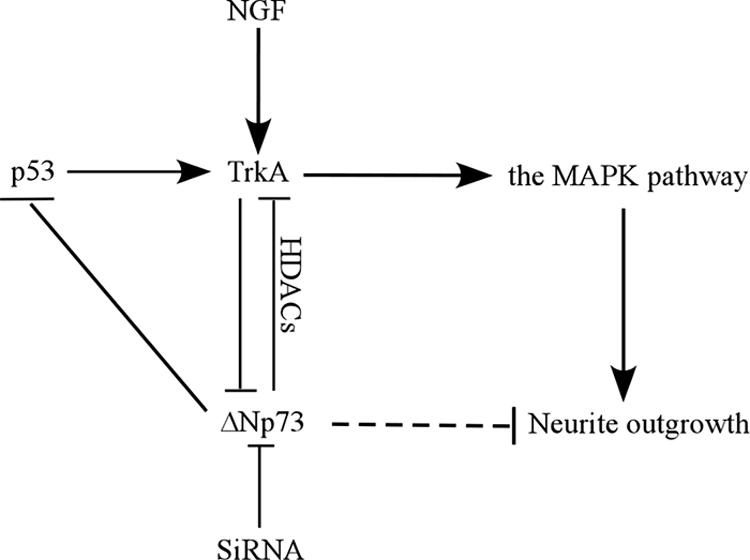
Model of the role of ΔNp73 in NGF-mediated neuronal differentiation in PC12 cells.
In this study, we found that while ΔNp73α and ΔNp73β are capable of inhibiting NGF-mediated neuronal differentiation via repression of TrkA, ΔNp73β exhibits several unique activities. Like its human counterpart, murine ΔNp73β is a strong transcriptional activator, as demonstrated by its ability to induce p21 and apoptosis (Fig. 1). Remarkably, these properties make ΔNp73β a p53/TAp73-like protein. However, unlike p53, which transactivates TrkA and promotes NGF-mediated neuronal differentiation (55), ΔNp73β has an opposing function. One possibility is that the activation domain in ΔNp73, which has no homology to that in p53, functions differently. As a result, p53, but not ΔNp73, can transactivate expression of certain genes, such as TrkA, which are required for neuronal differentiation. The other possibility is that ΔNp73 may regulate its own unique target genes to counter NGF-mediated neuronal differentiation, such as repression of GAP-43 induced by NGF (Fig. 2E). We would like to note that the induction of p21 by ΔNp73β is not sufficient, although p21 is known to be required, for NGF-mediated neuronal differentiation. This is consistent with a previous report that overexpression of p21 is not sufficient to induce PC12 cells to undergo differentiation (10).
Here, we found that ΔNp73 is the predominant p73 isoform in undifferentiated PC12 cells. Upon NGF treatment, endogenous ΔNp73 is down-regulated and maintained at low levels in differentiated PC12 cells. Similar to our findings, other studies showed that ΔNp73 is preferentially expressed in proliferating nephron precursors, while TAp73 is predominantly expressed in the differentiation domain of renal cortex during nephrogenesis (39, 40). Moreover, ΔNp63, another member of the p53 family, is also preferentially expressed in undifferentiated basal epithelium cells and its expression is repressed in differentiated cells (52). These observations suggest that different p53 family members have opposing functions during differentiation and that inhibition of cellular differentiation by ΔNp73 or ΔNp63 may contribute to tumor formation since lack of proper differentiation and unbalanced proliferation are two of the hallmarks of many types of cancer cells.
The finding that ΔNp73 represses TrkA expression may provide an insight into the mechanism of neuroblastoma pathogenesis. Both up-regulation of ΔNp73 and down-regulation of TrkA have been reported to be associated with the poor prognosis in neuroblastoma patients (3, 21, 34). Therefore, it would be interesting to find out if these two prognosis makers are directly correlated. In addition, we have shown that upon knockdown of ΔNp73, TrkA expression is up-regulated, leading to enhanced NGF-mediated neuronal differentiation. Furthermore, we have shown that upon treatment with TSA, an HDAC inhibitor, ΔNp73-mediated repression of TrkA is at least partially derepressed. These data point out the possibility that ΔNp73 might be explored as a therapeutic target for neuroblastoma and that HDAC inhibitors and others that target ΔNp73 can be used as agents for managing neuroblastoma.
Acknowledgments
This work was supported in part by NIH grants CA081237 and CA102188.
Footnotes
Published ahead of print on 12 March 2007.
REFERENCES
- 1.Almog, N., and V. Rotter. 1997. Involvement of p53 in cell differentiation and development. Biochim. Biophys. Acta 1333:F1-F27. [DOI] [PubMed] [Google Scholar]
- 2.Billon, N., A. Terrinoni, C. Jolicoeur, A. McCarthy, W. D. Richardson, G. Melino, and M. Raff. 2004. Roles for p53 and p73 during oligodendrocyte development. Development 131:1211-1220. [DOI] [PubMed] [Google Scholar]
- 3.Casciano, I., K. Mazzocco, L. Boni, G. Pagnan, B. Banelli, G. Allemanni, M. Ponzoni, G. P. Tonini, and M. Romani. 2002. Expression of DeltaNp73 is a molecular marker for adverse outcome in neuroblastoma patients. Cell Death Differ. 9:246-251. [DOI] [PubMed] [Google Scholar]
- 4.Cui, R., T. T. Nguyen, J. H. Taube, S. A. Stratton, M. H. Feuerman, and M. C. Barton. 2005. Family members p53 and p73 act together in chromatin modification and direct repression of alpha-fetoprotein transcription. J. Biol. Chem. 280:39152-39160. [DOI] [PubMed] [Google Scholar]
- 5.De Laurenzi, V., G. Raschella, D. Barcaroli, M. Annicchiarico-Petruzzelli, M. Ranalli, M. V. Catani, B. Tanno, A. Costanzo, M. Levrero, and G. Melino. 2000. Induction of neuronal differentiation by p73 in a neuroblastoma cell line. J. Biol. Chem. 275:15226-15231. [DOI] [PubMed] [Google Scholar]
- 6.De Laurenzi, V., A. Rossi, A. Terrinoni, D. Barcaroli, M. Levrero, A. Costanzo, R. A. Knight, P. Guerrieri, and G. Melino. 2000. p63 and p73 transactivate differentiation gene promoters in human keratinocytes. Biochem. Biophys. Res. Commun. 273:342-346. [DOI] [PubMed] [Google Scholar]
- 7.Dohn, M., S. Zhang, and X. Chen. 2001. p63alpha and DeltaNp63alpha can induce cell cycle arrest and apoptosis and differentially regulate p53 target genes. Oncogene 20:3193-3205. [DOI] [PubMed] [Google Scholar]
- 8.Eggert, A., N. Ikegaki, X. Liu, T. T. Chou, V. M. Lee, J. Q. Trojanowski, and G. M. Brodeur. 2000. Molecular dissection of TrkA signal transduction pathways mediating differentiation in human neuroblastoma cells. Oncogene 19:2043-2051. [DOI] [PubMed] [Google Scholar]
- 9.Eizenberg, O., A. Faber-Elman, E. Gottlieb, M. Oren, V. Rotter, and M. Schwartz. 1996. p53 plays a regulatory role in differentiation and apoptosis of central nervous system-associated cells. Mol. Cell. Biol. 16:5178-5185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Erhardt, J. A., and R. N. Pittman. 1998. Ectopic p21(WAF1) expression induces differentiation-specific cell cycle changes in PC12 cells characteristic of nerve growth factor treatment. J. Biol. Chem. 273:23517-23523. [DOI] [PubMed] [Google Scholar]
- 11.Fang, L., S. W. Lee, and S. A. Aaronson. 1999. Comparative analysis of p73 and p53 regulation and effector functions. J. Cell Biol. 147:823-830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fort, P., L. Marty, M. Piechaczyk, S. el Sabrouty, C. Dani, P. Jeanteur, and J. M. Blanchard. 1985. Various rat adult tissues express only one major mRNA species from the glyceraldehyde-3-phosphate-dehydrogenase multigenic family. Nucleic Acids Res. 13:1431-1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goldschneider, D., K. Million, A. Meiller, H. Haddada, A. Puisieux, J. Benard, E. May, and S. Douc-Rasy. 2005. The neurogene BTG2TIS21/PC3 is transactivated by DeltaNp73alpha via p53 specifically in neuroblastoma cells. J. Cell Sci. 118:1245-1253. [DOI] [PubMed] [Google Scholar]
- 14.Gollapudi, L., and K. E. Neet. 1997. Different mechanisms for inhibition of cell proliferation via cell cycle proteins in PC12 cells by nerve growth factor and staurosporine. J. Neurosci. Res. 49:461-474. [PubMed] [Google Scholar]
- 15.Gonzalez, S., C. Prives, and C. Cordon-Cardo. 2003. p73α regulation by Chk1 in response to DNA damage. Mol. Cell. Biol. 23:8161-8171. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16.Gossen, M., and H. Bujard. 1992. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. USA 89:5547-5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Greene, L. A., and A. S. Tischler. 1976. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc. Natl. Acad. Sci. USA 73:2424-2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harms, K., S. Nozell, and X. Chen. 2004. The common and distinct target genes of the p53 family transcription factors. Cell Mol. Life Sci. 61:822-842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harms, K. L., and X. Chen. 2005. The C terminus of p53 family proteins is a cell fate determinant. Mol. Cell. Biol. 25:2014-2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harms, K. L., and X. Chen. 2006. The functional domains in p53 family proteins exhibit both common and distinct properties. Cell Death Differ. 13:890-897. [DOI] [PubMed] [Google Scholar]
- 21.Hoehner, J. C., L. Olsen, B. Sandstedt, D. R. Kaplan, and S. Pahlman. 1995. Association of neurotrophin receptor expression and differentiation in human neuroblastoma. Am. J. Pathol. 147:102-113. [PMC free article] [PubMed] [Google Scholar]
- 22.Huang, E. J., and L. F. Reichardt. 2003. Trk receptors: roles in neuronal signal transduction. Annu. Rev. Biochem. 72:609-642. [DOI] [PubMed] [Google Scholar]
- 23.Huttinger-Kirchhof, N., H. Cam, H. Griesmann, L. Hofmann, M. Beitzinger, and T. Stiewe. 2006. The p53 family inhibitor DeltaNp73 interferes with multiple developmental programs. Cell Death Differ 13:174-177. [DOI] [PubMed] [Google Scholar]
- 24.Inoue, T., J. Stuart, R. Leno, and C. G. Maki. 2002. Nuclear import and export signals in control of the p53-related protein p73. J. Biol. Chem. 277:15053-15060. [DOI] [PubMed] [Google Scholar]
- 25.Johnson, K., and K. Calame. 2003. Transcription factors controlling the beginning and end of B-cell differentiation. Curr. Opin. Genet. Dev. 13:522-528. [DOI] [PubMed] [Google Scholar]
- 26.Kaghad, M., H. Bonnet, A. Yang, L. Creancier, J. C. Biscan, A. Valent, A. Minty, P. Chalon, J. M. Lelias, X. Dumont, P. Ferrara, F. McKeon, and D. Caput. 1997. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell 90:809-819. [DOI] [PubMed] [Google Scholar]
- 27.Kartasheva, N. N., A. Contente, C. Lenz-Stoppler, J. Roth, and M. Dobbelstein. 2002. p53 induces the expression of its antagonist p73 Delta N, establishing an autoregulatory feedback loop. Oncogene 21:4715-4727. [DOI] [PubMed] [Google Scholar]
- 28.Koster, M. I., and D. R. Roop. 2004. The role of p63 in development and differentiation of the epidermis. J. Dermatol. Sci. 34:3-9. [DOI] [PubMed] [Google Scholar]
- 29.Lee, A. F., D. K. Ho, P. Zanassi, G. S. Walsh, D. R. Kaplan, and F. D. Miller. 2004. Evidence that DeltaNp73 promotes neuronal survival by p53-dependent and p53-independent mechanisms. J. Neurosci. 24:9174-9184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu, G., S. Nozell, H. Xiao, and X. Chen. 2004. ΔNp73β is active in transactivation and growth suppression. Mol. Cell. Biol. 24:487-501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu, G., T. Xia, and X. Chen. 2003. The activation domains, the proline-rich domain, and the C-terminal basic domain in p53 are necessary for acetylation of histones on the proximal p21 promoter and interaction with p300/CREB-binding protein. J. Biol. Chem. 278:17557-17565. [DOI] [PubMed] [Google Scholar]
- 32.Moll, U. M., and N. Slade. 2004. p63 and p73: roles in development and tumor formation. Mol. Cancer Res. 2:371-386. [PubMed] [Google Scholar]
- 33.Nagamura-Inoue, T., T. Tamura, and K. Ozato. 2001. Transcription factors that regulate growth and differentiation of myeloid cells. Int. Rev. Immunol. 20:83-105. [DOI] [PubMed] [Google Scholar]
- 34.Nakagawara, A., M. Arima-Nakagawara, N. J. Scavarda, C. G. Azar, A. B. Cantor, and G. M. Brodeur. 1993. Association between high levels of expression of the TRK gene and favorable outcome in human neuroblastoma. N. Engl. J. Med. 328:847-854. [DOI] [PubMed] [Google Scholar]
- 35.Petrenko, O., A. Zaika, and U. M. Moll. 2003. ΔNp73 facilitates cell immortalization and cooperates with oncogenic Ras in cellular transformation in vivo. Mol. Cell. Biol. 23:5540-5555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pozniak, C. D., F. Barnabe-Heider, V. V. Rymar, A. F. Lee, A. F. Sadikot, and F. D. Miller. 2002. p73 is required for survival and maintenance of CNS neurons. J. Neurosci. 22:9800-9809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pozniak, C. D., S. Radinovic, A. Yang, F. McKeon, D. R. Kaplan, and F. D. Miller. 2000. An anti-apoptotic role for the p53 family member, p73, during developmental neuron death. Science 289:304-306. [DOI] [PubMed] [Google Scholar]
- 38.Qui, M. S., and S. H. Green. 1992. PC12 cell neuronal differentiation is associated with prolonged p21ras activity and consequent prolonged ERK activity. Neuron 9:705-717. [DOI] [PubMed] [Google Scholar]
- 39.Saifudeen, Z., V. Diavolitsis, J. Stefkova, S. Dipp, H. Fan, and S. S. El-Dahr. 2005. Spatiotemporal switch from DeltaNp73 to TAp73 isoforms during nephrogenesis: impact on differentiation gene expression. J. Biol. Chem. 280:23094-23102. [DOI] [PubMed] [Google Scholar]
- 40.Saifudeen, Z., S. Dipp, and S. S. El-Dahr. 2002. A role for p53 in terminal epithelial cell differentiation. J. Clin. Investig. 109:1021-1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schrama, L. H., G. Lepperdinger, A. Moritz, N. K. van den Engel, A. Marquart, A. B. Oestreicher, B. J. Eggen, W. J. Hage, K. Richter, and O. H. Destree. 1997. B-50/growth-associated protein-43, a marker of neural development in Xenopus laevis. Neuroscience 76:635-652. [DOI] [PubMed] [Google Scholar]
- 42.Stiewe, T., and B. M. Putzer. 2002. Role of p73 in malignancy: tumor suppressor or oncogene? Cell Death Differ. 9:237-245. [DOI] [PubMed] [Google Scholar]
- 43.Stiewe, T., S. Zimmermann, A. Frilling, H. Esche, and B. M. Putzer. 2002. Transactivation-deficient DeltaTA-p73 acts as an oncogene. Cancer Res. 62:3598-3602. [PubMed] [Google Scholar]
- 44.Tsai, K. K., and Z. M. Yuan. 2003. c-Abl stabilizes p73 by a phosphorylation-augmented interaction. Cancer Res. 63:3418-3424. [PubMed] [Google Scholar]
- 45.Uramoto, H., K. Sugio, T. Oyama, S. Nakata, K. Ono, M. Morita, K. Funa, and K. Yasumoto. 2004. Expression of deltaNp73 predicts poor prognosis in lung cancer. Clin. Cancer Res. 10:6905-6911. [DOI] [PubMed] [Google Scholar]
- 46.Urist, M., T. Tanaka, M. V. Poyurovsky, and C. Prives. 2004. p73 induction after DNA damage is regulated by checkpoint kinases Chk1 and Chk2. Genes Dev. 18:3041-3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vaudry, D., Y. Chen, C. M. Hsu, and L. E. Eiden. 2002. PC12 cells as a model to study the neurotrophic activities of PACAP. Ann. N. Y. Acad. Sci. 971:491-496. [DOI] [PubMed] [Google Scholar]
- 48.Walsh, G. S., N. Orike, D. R. Kaplan, and F. D. Miller. 2004. The invulnerability of adult neurons: a critical role for p73. J. Neurosci. 24:9638-9647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang, S., and W. S. El-Deiry. 2006. p73 or p53 directly regulates human p53 transcription to maintain cell cycle checkpoints. Cancer Res. 66:6982-6989. [DOI] [PubMed] [Google Scholar]
- 50.Willis, A., E. J. Jung, T. Wakefield, and X. Chen. 2004. Mutant p53 exerts a dominant negative effect by preventing wild-type p53 from binding to the promoter of its target genes. Oncogene 23:2330-2338. [DOI] [PubMed] [Google Scholar]
- 51.Wong, K., J. Zhang, S. Awasthi, A. Sharma, L. Rogers, E. F. Matlock, C. Van Lint, T. Karpova, J. McNally, and R. Harrod. 2004. Nerve growth factor receptor signaling induces histone acetyltransferase domain-dependent nuclear translocation of p300/CREB-binding protein-associated factor and hGCN5 acetyltransferases. J. Biol. Chem. 279:55667-55674. [DOI] [PubMed] [Google Scholar]
- 52.Yang, A., R. Schweitzer, D. Sun, M. Kaghad, N. Walker, R. T. Bronson, C. Tabin, A. Sharpe, D. Caput, C. Crum, and F. McKeon. 1999. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 398:714-718. [DOI] [PubMed] [Google Scholar]
- 53.Yang, A., N. Walker, R. Bronson, M. Kaghad, M. Oosterwegel, J. Bonnin, C. Vagner, H. Bonnet, P. Dikkes, A. Sharpe, F. McKeon, and D. Caput. 2000. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature 404:99-103. [DOI] [PubMed] [Google Scholar]
- 54.Zaika, A. I., N. Slade, S. H. Erster, C. Sansome, T. W. Joseph, M. Pearl, E. Chalas, and U. M. Moll. 2002. DeltaNp73, a dominant-negative inhibitor of wild-type p53 and TAp73, is up-regulated in human tumors. J. Exp. Med. 196:765-780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang, J., W. Yan, and X. Chen. 2006. p53 is required for nerve growth factor-mediated differentiation of PC12 cells via regulation of TrkA levels. Cell Death Differ. 13:2118-2128. [Epub ahead of print 26 May 2006.] [DOI] [PubMed] [Google Scholar]