

Abstract
Adiponectin is a secretory protein abundantly secreted from adipocytes. It assembles into a number of different higher-order complexes. Adipocytes maintain tight control over circulating plasma levels, suggesting the existence of a complex, highly regulated biosynthetic pathway. However, the critical mediators of adiponectin maturation within the secretory pathway have not been elucidated. Previously, we found that a significant portion of de novo-synthesized adiponectin is not secreted and retained in adipocytes. Here, we show that there is an abundant pool of properly folded adiponectin in the secretory pathway that is retained through thiol-mediated retention, as judged by the release of adiponectin in response to treatment of adipocytes with reducing agents. Adiponectin is covalently bound to the ER chaperone ERp44. An adiponectin mutant lacking cysteine 39 fails to stably interact with ERp44, demonstrating that this residue is the primary site mediating the covalent interaction. Another ER chaperone, Ero1-Lα, plays a critical role in the release of adiponectin from ERp44. Levels of both of these proteins are highly regulated in adipocytes and are influenced by the metabolic state of the cell. While less critical for the secretion of trimers, these chaperones play a major role in the assembly of higher-order adiponectin complexes. Our data highlight the importance of posttranslational events controlling adiponectin levels and the release of adiponectin from adipocytes. One mechanism for increasing circulating levels of specific adiponectin complexes by peroxisome proliferator-activated receptor gamma agonists may be selective upregulation of rate-limiting chaperones.
Adiponectin, a secretory protein expressed at high levels in plasma, plays a beneficial role in diabetes and cardiovascular disease (5, 21, 25). Recently, adiponectin has been implicated as a critical mediator of peroxisome proliferator-activated receptor gamma (PPARγ) agonist-induced insulin sensitization (13, 22, 27). The receptors for adiponectin that may mediate some aspects of adiponectin signaling in liver and muscle were recently described (20, 46). The 247-amino-acid mouse adiponectin contains three domains, which include a short signal sequence followed by a hypervariable region, an N-terminal collagenous domain, and a C-terminal globular domain (30). Circulating adiponectin forms at least three complexes, which are referred to as the high-molecular-weight (HMW), low-molecular-weight (LMW), and trimeric complexes (30, 43). Trimeric adiponectin is formed by interactions within the globular domain, further stabilized by a collagenous coiled coil that also supports formation of disulfide bonds between two of the three subunits mediated through cysteine 39 (Cys39). Different forms of adiponectin exert distinct functions. Absolute HMW levels and ratios of HMW to total levels of adiponectin in circulation change under different metabolic conditions (23, 31, 43, 45). HMW levels are regulated in diabetic and cardiovascular disease states with reduced HMW levels in circulation (31). Several recent papers have highlighted the usefulness of the different adiponectin complexes under a variety of conditions and demonstrated that HMW measurements as opposed to total adiponectin levels significantly improve the correlations under some conditions (6, 23, 41). Despite the ample literature on physiological functions of adiponectin, very little is known about the processes that govern adiponectin production and release from the adipocyte.
The formation of inter- and intramolecular disulfide bonds is essential for the maturation of cysteine-containing secretory proteins. The oxidizing environment within the lumen of the endoplasmic reticulum (ER) favors disulfide bond formation. However, the process is highly regulated and critically dependent on a number of oxidoreductases that catalyze disulfide bond exchange. Protein disulfide isomerase (PDI) is one of the critical players in this process. It transfers oxidizing equivalents to substrates and catalyzes disulfide bond formation. At the same time, PDI undergoes reduction by accepting two electrons. The regeneration of PDI is mediated by Ero1, an ER oxidoreductase that forms mixed disulfide bonds with PDI and transfers oxidizing equivalents to PDI. Ero1 itself is reduced in this process. Finally, Ero1 is reoxidized by transferring electrons to electron acceptors. Disulfide bond formation is catalyzed by sequential transfer of oxidizing equivalents between proteins in the thiol-disulfide exchange reaction.
In mammalian cells, two Ero1 genes have been identified. The two genes are differentially regulated and show distinct expression profiles in different tissues (9, 15). Ero1-Lα is ubiquitously expressed and is thought to be the rate-limiting step in disulfide bond formation in most cell types (9, 16). Ero1-Lβ is more abundantly expressed in professional secretory cells and may be involved in the unfolded protein response (15, 18). Unlike Saccharomyces cerevisiae Ero1p, which is a transmembrane protein in the ER, mammalian Ero1s do not contain membrane-spanning domains but are localized to the ER as well. However, they lack the characteristic ER retrieval signal found at the C termini of most soluble resident ER proteins. This suggests that a stable interaction with another resident ER protein may be responsible for the luminal retention of Ero1s (9). Such a molecule has indeed been reported by Anelli and colleagues, who identified ERp44, a resident ER protein which is a binding partner for Ero1s (3). ERp44 interacts with both Ero1-Lα and -β and is responsible for the retention of Ero1s in the ER. ERp44 has also been implicated in other critical processes. It has been shown to covalently interact with unassembled subunits of higher-order protein complexes in a process referred to as “thiol-mediated protein retention” (2).
Thiol-mediated retention was first discovered in the context of assembly and secretion of immunoglobulin M (IgM) complexes (1, 40). IgM secreted from plasma cells is almost always pentameric or hexameric. The monomer is synthesized, assembled, and retained intracellularly. Upon closer examination of the retention mechanism, Sitia and colleagues identified cysteine 575 in the tailpiece of the μ chain as the main residue mediating the interaction with a resident ER protein responsible for this retention (40). This is also the key residue responsible for the oligomerization of these IgM subunits into higher-order structures. Reducing agents, such as β-mercaptoethanol (BME), can relieve this retention at the whole-cell level (1). Thiol-mediated retention is not limited to IgM in plasma cells. Additional molecules in different cell types are found to be regulated this way (19, 35). Despite the importance of this thiol-mediated retention mechanism for the regulation and assembly of a number of important secretory factors, little was known about the ER proteins responsible for retention until recently (8). ERp44, an ER resident protein, was initially identified as a molecule involved in the control of ER redox potential (3). Later studies identified it as the key mediator of retention for IgM molecules (2).
Here, we show that the maturation and release of adiponectin from adipocytes are subject to thiol-mediated retention. ERp44 plays an important role in this process as well, forming a mixed disulfide bond with adiponectin via Cys39 on the adiponectin backbone. Ero1-Lα, an oxidoreductase in the ER lumen, is critically involved in the release of adiponectin from ERp44. The levels of both ERp44 and Ero1-Lα are differentially regulated in adipocytes under different metabolic conditions. As critical players for the assembly of adiponectin in the secretory pathway, both proteins represent major sites of regulation for the release of adiponectin from adipocytes and are therefore targets for pharmacological intervention.
MATERIALS AND METHODS
Animals.
Mice (FVB and C57/BL6) were maintained on a 12-hour light/dark cycle and on a standard chow diet. All animal experimental protocols were approved by the Institute for Animal Studies of the Albert Einstein College of Medicine.
Reagents and antibodies.
Dulbecco's modified Eagle's medium (DMEM) was purchased from Mediatech Inc. (Herndon, VA). BME was purchased from Sigma (St. Louis, MO). N-Ethylmaleimide (NEM) and dimethyl pimelimidate dihydrochloride (DMP) were obtained from Pierce (Rockford, IL). The rabbit polyclonal antibody to the guanine nucleotide dissociation inhibitor (GDI) was a generous gift from Perry Bickel (Washington University, St. Louis, MO). The antiadiponectin and antiresistin antibodies were described elsewhere (30, 33). Anti-green fluorescent protein (anti-GFP) antibody was purchased from Rockland (Gilbertsville, PA) and was conjugated to IRDye800. Antihemagglutinin (anti-HA; 1:200; Roche, Indianapolis, IN) and anti-myc (1:500; Cell Signaling, Beverly, MA) antibodies were commercially available.
Plasmid construction.
The ERp44 cDNA was amplified from a mouse fetal brain cDNA library (Clontech, Mountain View, CA) with primers P93 and P54. The PCR fragment was cloned into the pCR4TA cloning vector (Invitrogen, Carlsbad, CA) and verified by sequencing. An HA tag (YPYDVPDYA) was inserted between the leader sequence cleavage site and the beginning of the mature protein by sequential PCR-based reactions, similar to the strategy employed for human ERp44 cloning (3). Briefly, P93 and P96 were used for the first reaction and P97 and P54 were used for the second. The PCR products were used as templates for PCR with primers P93 and P54. Finally, the fragment was cleaved with EcoRV and XhoI and inserted into the pRA-GFP vector for mammalian expression (24). The following primers were used: P93, 5′ GATATCGCCACCATGAATCCTGCGGTCTTCCTG 3′; P96, 5′ GCGTAGTCAGGCACATCATACGGATATTCAGCTGTTATAGGTGTAAAAAT 3′; P97, 5′ TATCCGTATGATGTGCCTGACTACGCCGAAATAGCAAGTCTTGATTCAGA 3′; P54, 5′ AGTCTCGAGCACGTAAGCCTCTGCTGGTGGTGTGG 3′. The Ero1-Lα cDNA was obtained by PCR with P119 and P120, and a myc tag was included in frame in the C terminus. The PCR product was cloned into the pCR4TA vector (Invitrogen) and sequenced. A fragment obtained by EcoRV and XhoI digestion was inserted into the pRA-GFP vector for mammalian expression. The primers used are as follows: P119, 5′ GATATCTGGGCGGCGAGCGGCGATGG 3′; p120, 5′ CTCGAGTCACAGATCCTCTTCTGAGATGAGTTTTTGTTCCCCGTGAACATTCTGTAACAAGTGC 3′. The construction of adiponectin, resistin, and Cys39Ser adiponectin-expressing plasmids was described elsewhere (30, 33).
Generation of antibodies.
For the construction of fusion protein expression plasmid used for antibody generation, the sequence coding for residues 253 to 406 of mouse ERp44 was cloned into the BamHI and XhoI sites of pGEX-4T1 (GE Healthcare Bio-Science Corp., Piscataway, NJ) and made in Escherichia coli. The glutathione S-transferase-ERp44 fusion protein was purified and used to immunize rabbits to generate polyclonal antibodies (Covance, Denver, PA). To generate antibodies against Ero1-Lα, the sequence encoding residues 52 to 464 was cloned into pGEX-4T1. The glutathione S-transferase-Ero1-Lα fusion protein was purified and used to immunize rabbits to generate polyclonal antibodies (Covance). Monoclonal antibodies against adiponectin were raised in adiponectin-null mice with full-length mouse adiponectin produced and purified from HEK-293T cells. Two hybridomas, producing mab14 and mab45, were selected for further processing. Monoclonal antibodies were generated in collaboration with the AECOM Cancer Center Hybridoma Facility.
Cross-linking of adiponectin antibody.
We first determined the capacity of GammaBind G Sepharose (GE Healthcare Bio-Sciences Corp.) for binding to adiponectin monoclonal antibody mab14 and control mouse IgG (Sigma, St. Louis, MI). Subsequently, the beads were incubated with antibody overnight at 4°C. HEPES buffer (200 mM, pH 8.5) was used to wash the beads. Cross-linking was performed with 1 ml of 50 mM DMP at 25°C for 30 min with end-on rotation. This process was repeated twice, with fresh DMP used each time. The cross-linking was terminated with 50 mM Tris, pH 8.5. Un-cross-linked antibodies were removed with 200 mM glycine, 150 mM NaCl, pH 2.5. The cross-linking efficiency was checked by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Coomassie staining for heavy and light chains with the beads before and after the cross-linking. The beads were stored at 4°C in 25 mM HEPES buffer, 150 mM NaCl, 0.025% Triton X-100, pH 8.0.
Transfection.
The HEK-293T cells were maintained in DMEM with 10% fetal calf serum (FCS; JRH Biosciences, Lenexa, KA). The day before transfection, 3 × 105 cells were seeded in each well of six-well plates. On the day of transfection, TransIT-293 reagent was used according to the manufacturer's recommendations (Mirus, Madison, MI). After 36 h, the medium was changed to 1 ml DMEM containing 1% serum and collected for 12 h. Cells were lysed in HNET buffer (25 mM HEPES, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, pH 8.0) with 1× complete protease inhibitor cocktail (Roche). The extract was cleared by centrifugation at 20,000 × g at 4°C for 15 min. All samples were stored at −20°C with Laemmli sample buffer (2×) until further analysis.
For transfection in 3T3-L1 fibroblast cells, 106 cells were seeded in 10-cm plates. Two days later, transfection was done with SUPERFECT reagents (QIAGEN, Valencia, CA). Following transfection for 24 h, the cells were washed twice with 1× phosphate-buffered saline (PBS) and 5 ml fresh DMEM was added. After 8 h, the culture medium was collected and subjected to immunoprecipitation with antiadiponectin-coupled resin. The protein was then eluted off the beads with 1× sample buffer and used for immunoanalysis. Cells were lysed with HNET, and cell lysates were used for immunoblotting with various antibodies.
Adipocyte differentiation in cell culture.
3T3-L1 murine fibroblasts (a generous gift from Charles Rubin, Department of Molecular Pharmacology, Albert Einstein College of Medicine, Bronx, NY) were propagated and differentiated to adipocytes. The cells were maintained in “FCS” (DMEM containing 10% FCS supplemented with penicillin-streptomycin at 100 U/ml each) and allowed to reach confluence (day −2). After 2 days (day 0), the medium was changed to “DM1” (containing “FCS” and 160 nM insulin, 250 nM dexamethasone, and 0.5 mM 3-isobutyl-1-methylxanthine). Two days later (day 2), the medium was switched to “DM2” (“FCS” containing 160 nM insulin). After another 2 days, the cells were switched back to “FCS.” Cells were used between days 8 and 12 after induction of differentiation.
Immunoblot analysis.
The culture medium collected from transfected cells was resolved by SDS-PAGE on 10 to 20% Tris-tricine gels, 10% Bis-Tris Nupage gels, or 12% Tris-glycine gels (Invitrogen), followed by transfer to BA83 nitrocellulose (Schleicher and Schuell, Keene, NH). Blots were probed with various antibodies as indicated in the text. Primary and secondary antibodies were diluted in Tris-buffered saline with 0.1% Tween 20 and 4% nonfat milk. Bound antibodies were detected with IRDye800-conjugated anti-rabbit (Rockland) or Alexa Fluor 680-conjugated anti-mouse (Invitrogen) secondary antibodies. The membrane was then scanned by the LI-COR Odyssey infrared imaging system at the 700-nm and 800-nm channels simultaneously, and band intensity was quantified with Odyssey v1.2 software (LI-COR Biotechnology, Lincoln, NE).
Immunofluorescence microscopy.
Day 4 3T3-L1 adipocytes were trypsinized and seeded into six-well plates with sterile coverslips. After 2 days of recovery, the cells were washed with PBS and fixed in 4% paraformaldehyde for 20 min at 25°C. The fixation was terminated with 50 mM NH4Cl, and cells were permeabilized with 0.1% Triton X-100 in PBS for 5 min. After being washed three times for 5 min each, the cells were incubated with primary antibodies in dilution buffer (1% bovine serum albumin [BSA], 0.1% cold fish skin gelatin, 0.05% sodium azide, 1× PBS, pH 7.5) for 5 h at 25°C. To detect adiponectin, monoclonal antiadiponectin antibody mab14 was used at 2 μg/ml, and for detection of ERp44, the rabbit polyclonal anti-ERp44 antibodies were used at 10 μg/ml. After three washes performed as described above, the coverslips were placed in donkey anti-mouse Cy3-coupled (Jackson Immunoresearch Laboratories, West Grove, PA) and goat anti-rabbit Alexa Fluor 488-coupled (Invitrogen) secondary antibodies at a 1:500 dilution in PBS for 1 h. The cells were counterstained with DAPI (4′,6′-diamidino-2-phenylindole) at 100 ng/ml in PBS for 5 min (Sigma). Coverslips were then mounted on glass slides with ProLong Gold antifade (Invitrogen). All images were acquired with an Olympus IX81 electronically motorized microscope (Olympus, Melville, NY).
Co-IP.
The interaction between ERp44 and adiponectin may be disulfide bonded. In order to prevent the reshuffling of disulfide bonds during cell lysis, 10 mM NEM was included in the lysis buffer. For HEK-293T transfection experiments, 36 h after transfection, cells in six-well plates were washed for 5 min with 25 mM HEPES buffer, pH 8.0, containing 150 mM NaCl and 10 mM NEM. Then, 500 μl HNET lysis buffer was added to each well. After being centrifuged at 20,000 × g, the extracts were precleared by incubating them with 100 μl GammaBind protein G beads (GE Healthcare Bio-Sciences Corp.) for 2 h at 4°C. The supernatant was then incubated with either control IgG or anti-adiponectin antibody-coupled resin overnight at 4°C with end-on rotation. The material was washed six times with lysis buffer and dissolved in 2× sample buffer, and proteins were separated on a Bis-Tris Nupage gel and transferred to a membrane before Western blotting. Ten percent starting material prior to immunoprecipitation was taken as input. For experiments involving 3T3-L1 adipocytes, cells were grown in a 10-cm plate and were washed once with 10 ml HEPES buffer containing 150 mM NaCl and 10 mM NEM for 5 min at 4°C. Cells were then lysed in 1 ml HNET lysis buffer. The lysates were cleared at 20,000 × g at 4°C for 15 min. For coimmunoprecipitation (co-IP) experiments, 500 μg to 1 mg total protein was used.
Cycloheximide and insulin treatments.
Day 8 3T3-L1 adipocytes were washed twice with DMEM containing 1% BSA and then incubated with vehicle or 5 μM cycloheximide in 3 ml DMEM containing 1% BSA for 4 h. Supernatant (200 μl) was removed for gel filtration analysis of adiponectin. The cell extracts were immunoprecipitated with monoclonal antibody mab14-conjugated beads. For insulin stimulation, adipocytes in 10-cm plates were adapted in DMEM containing 1% BSA for 2 h. Insulin (160 nM) was used to treat the cells for 30 min. The culture medium was taken for immunoprecipitation with mab14-coupled beads and later used for immunoblotting with rabbit polyclonal antiadiponectin antibody. The cellular extracts were subjected to co-IP to examine the interaction between adiponectin and ERp44.
shRNA knockdown of ERp44 and Ero1-Lα.
Short hairpin RNA (shRNA) sequences for RNA interference of ERp44 and Ero1-Lα were designed and synthesized by Oligoengine (Seattle, WA). A total of three oligonucleotides for each gene were chosen and cloned into the pSUPER-hygromycin vector. The fragment containing shRNA and the H1 promoter was excised and cloned into the lentiviral transfer construct pCCL.PPT.hPGK.GFP.Wpre. The knockdown efficiency of each shRNA was tested by transient transfection of these constructs combined with the ERp44 or Ero1-Lα expression construct. The most efficient shRNA was used to produce lentivirus. The third-generation packaging plasmids pMD2.VSVG, pMDLg/pRRE, and pRSV-REV were used to produce the lentivirus. After titration with 293T cells, we obtained lentiviral preparations containing 2 × 107 to 4 × 107 transducing units/ml. To achieve overexpression of shRNA, we used a multiplicity of infection (MOI) of 5 for ERp44 and Ero1-Lα.
PPARγ agonist gavage and tissue collection.
Male FVB mice (10 weeks old) were used. The PPARγ agonist 2-(2-[4-phenoxy-2-propylphenoxy]ethyl)indole-5-acetic acid (COOH) was a kind gift from Merck Research Laboratories (Rahway, NJ) (10). The COOH and vehicle were gavaged daily at 12 noon for 14 days at 10 mg/kg body weight. The mice were subjected to tail bleeding 4 h after the last gavage, and 30 μl serum was collected. The mice were then sacrificed within 6 h after the last gavage. Liver and stomach tissues were exposed to HNET buffer and homogenized. The solution was cleared by centrifugation at 20,000 × g for 15 min at 4°C. Adipose tissues were first put into HNET buffer without Triton X-100 and homogenized. After low-speed centrifugation (2,600 × g at 4°C), the fat cake was removed from the top of the tube and Triton X-100 was added to a final concentration of 1%. After incubation at 4°C for 30 min, the extract was cleared at 20,000 × g for 15 min at 4°C and mixed with 2× Laemmli sample buffer. Tissue extracts were loaded on 10% Bis-Tris Nupage gels (1.5 mm) for analysis with indicated antibodies.
Reverse transcription-PCR and mouse tissue extract preparation.
Wild-type (WT) C57/B6 mice (10 weeks old) were anesthetized with isoflurane during tissue collection. Half of each tissue was used to prepare total RNA with TRIzol reagent (Invitrogen), and the other half was used for protein extraction as indicated above. Total RNA (1 μg) from each tissue was taken for first-strand cDNA synthesis with SuperScript III reverse transcriptase (Invitrogen). The primers used for Ero1-Lα amplification were 5′CGATATACAGTCCCCCGATG3′ and 5′ACTTTTTCCTCGCCCAGAAG3′. The primers used for GAPDH (glyceraldehyde-3-phosphate dehydrogenase) were 5′ AACTTTGGCATTGTGGAAGG 3′ and 5′ ACACATTGGGGGTAGGAACA 3′.
Adiponectin complex distribution analysis.
The complex distribution of adiponectin was determined by separating 15 μl of mouse serum over a Superdex 200 10/300 GL column (GE Healthcare Bio-Sciences Corp.) using a Biologic Workstation fast-performance liquid chromatography system (Bio-Rad, Hercules, CA). The column was equilibrated in column buffer (25 mM HEPES, 150 mM NaCl, 1 mM CaCl2, pH 8.0), and 0.22-ml fractions were collected. Samples (40 μl) were collected over the entire elution of adiponectin and incubated with 10 μl of 5× Laemmli sample buffer, followed by boiling for 5 min. Samples were loaded on a Criterion precast 26-well gel (Bio-Rad) and subjected to immunoblotting using 1:500 polyclonal antiadiponectin antibody, followed by incubation with IR-Dye 800-coupled goat anti-rabbit secondary antibody (Rockland). The fluorescence signal obtained at 30 kDa was quantified as described above.
Statistical analysis.
The results are shown as means ± standard errors of the means. Statistical analysis was performed by the Student t test with SigmaPlot 9.0 (Systat Software, Point Richmond, CA). Significance was accepted at P values of <0.05.
RESULTS
Thiol-mediated retention of adiponectin in 3T3-L1 adipocytes.
To determine the fate of newly synthesized adiponectin, we had previously performed pulse-chase studies with radiolabeled 35S-Cys/Met (12). We had demonstrated that within 3 h postsynthesis, about 50% of newly synthesized adiponectin is secreted. This was consistent with previous analysis which examined the secretion kinetics in a different context (37). However, upon extending the analysis to 24 h, it is apparent that the remaining intracellular protein is retained and, under the conditions used, fails to be secreted even after a prolonged chase period. To test whether this intracellular pool of adiponectin is misfolded and fails to be secreted due to aggregation in the secretory pathway or whether it is retained more specifically and potentially released under some conditions, we tested the effects of reducing agents on the maturation of intracellular adiponectin. Surprisingly, treatment of 3T3-L1 adipocytes with 5 mM BME allows intracellular adiponectin to be released from cells into the tissue culture supernatant (Fig. 1A). This is not a generalized phenomenon affecting all proteins in the secretory pathway. Other proteins that contain disulfide bonds tend to either be unaffected or in fact have reduced secretion in the presence of reducing agents. As a typical example, the secretion of the highly cysteine-rich protein resistin is reduced under the same conditions (Fig. 1A). Quantitatively, BME stimulates the secretion of adiponectin as much as seven- to eightfold (Fig. 1B). When we examined the intracellular material, we found that adiponectin is decreased after BME treatment, consistent with the increased release of adiponectin into the supernatant (Fig. 1C). This unusual behavior has been described previously only for proteins that undergo a process termed thiol-mediated protein retention, suggesting that a similar mechanism may be in place for adiponectin. This phenomenon was originally described by Alberini and colleagues as an important component of the maturation of IgM complexes (1).
FIG. 1.

Thiol-mediated retention of adiponectin. (A) Day 8 3T3-L1 adipocytes (∼107) were incubated with or without 5 mM BME in 3 ml DMEM. The medium was removed at indicated time points and resolved by SDS-PAGE. Upon transfer to nitrocellulose, the membrane was decorated with antiadiponectin (anti-APN) and antiresistin antibodies. (B) Three hours after BME treatment, culture medium was taken and subjected to immunoblotting with antiadiponectin antibody. Band intensity was quantified and plotted. Control, n = 4; BME, n = 4. (C) A representative result for intracellular adiponectin after treatment is shown. Actin was used as protein loading control. WB, Western blot.
ERp44 covalently interacts with adiponectin.
ERp44 was first discovered as a protein associated with Ero1-Lα (3). It was shown that ERp44 is a critical mediator of thiol-mediated retention of IgM subunits (2). We therefore wanted to test whether ERp44 is also involved in retention of adiponectin. As an initial step, we tested whether there is in fact an interaction between ERp44 and adiponectin and assessed this by co-IP experiments. We generated a series of monoclonal antibodies against the mouse full-length adiponectin expressed and purified from HEK-293T cells. Two hybridomas were chosen for further characterization. One of the monoclonal antibodies, mab14, was chosen for co-IP experiments. Since the molecular weight of ERp44 is close to the IgG heavy chain and adiponectin runs near the light chain on SDS-PAGE, we chemically cross-linked the monoclonal antiadiponectin antibody to GammaBind G resin to minimize leakage of IgGs. Upon cotransfection of expression plasmids encoding both ERp44 tagged with a HA epitope and adiponectin in HEK-293T cells, ERp44 and adiponectin form a stable complex, as judged by the ability of the antiadiponectin antibody to effectively immunoprecipitate ERp44 protein (Fig. 2A, lane 4). This is in fact a very stable interaction that involves a covalent bond between adiponectin and ERp44. The immunoprecipitated material was run under reducing conditions and blotted with antiadiponectin and anti-HA antibodies (Fig. 2B, left panel). Upon separation of the same material under nonreducing conditions, both anti-HA and antiadiponectin antibodies recognize the same band of around 75 kDa, which is consistent with the molecular mass of an ERp44-adiponectin complex (Fig. 2B, right panel). This indicates that the interaction between ERp44 and adiponectin is covalent and mediated by an intermolecular disulfide bond. In previous studies, we have described a mutation in cysteine residue 39 of adiponectin (30). Adiponectin carrying this mutation is secreted much more efficiently than WT molecules. The more rapid secretion kinetics suggest that the mutant protein may bypass this thiol-mediated retention mechanism. Indeed, this Cys39Ser mutant fails to form a complex with ERp44, as judged by a failure to immunoprecipitate ERp44 in transfected HEK-293T cells (Fig. 2C, compare lanes 8 and 9). Furthermore, this suggests that this mutant protein should also be unaffected by the presence of reducing agents during secretion. This is indeed the case. For this purpose, we generated cell lines that stably overexpress this adiponectin variant. The kinetics of secretion of the Cys39Ser mutant are completely unaffected by the presence of BME in these cells (Fig. 2D). In contrast, the secretion of WT adiponectin in stably transfected HEK-293T cells is increased, similar to what is observed in 3T3-L1 adipocytes.
FIG. 2.

Adiponectin interacts covalently with ERp44. (A) Extracts from HEK-293T cells transfected with adiponectin (APN) and HA-tagged ERp44 were subjected to co-IP with control IgG or monoclonal mab14-coupled beads. After co-IP, 10% of the supernatant was loaded to confirm complete removal of adiponectin. The beads were boiled in 2× sample buffer and loaded on a 10% Bis-Tris Nupage gel. After transference, the membrane was decorated with antiadiponectin and anti-HA antibodies. (B) The eluate of co-IP from panel A was resolved under reducing (with 100 mM dithiothreitol) and nonreducing conditions and subjected to immunoblotting with antiadiponectin and anti-HA antibodies. Arrows indicate the same band of about 75 kDa, which is recognized by both antiadiponectin and anti-HA antibodies. (C) The Cys39Ser mutant does not interact with ERp44. Cell extracts from transfected HEK-293T cells were subjected to co-IP with control IgG and mab14 antibody. Proteins were separated on a 12% Tris-glycine gel, and Western blotting (WB) was performed with antiadiponectin and anti-HA antibodies. (D) The WT adiponectin and Cys39Ser mutant-expressing cell lines were described previously (30). The same number of cells were plated in 10-cm plates and treated with 5 mM BME. At different time points, supernatant was removed and assayed for adiponectin levels by immunoblotting.
ERp44 is relevant for the maturation of adiponectin in 3T3-L1 adipocytes.
In the previous experiments, we demonstrated an interaction of a tagged ERp44 version with adiponectin in the heterologous HEK-293T system. In order to detect endogenous ERp44, we generated polyclonal antibodies against ERp44 in rabbits. These antibodies recognize a discrete band upon transient transfection of HEK-293T cells with a plasmid encoding HA-tagged mouse ERp44 (see Fig. S1, middle panel, in the supplemental material). The same membrane was decorated with anti-HA antibodies, and they recognized the same band as anti-ERp44 antibody (see Fig. S1, left panel, in the supplemental material). In 3T3-L1 adipocytes, the anti-ERp44 antibody recognized a single band of about 50 kDa (see Fig. S1, right panel, in the supplemental material). While the interaction between ERp44 and adiponectin can easily be detected in a heterologous expression system in HEK-293T cells, we wondered whether these two proteins also interact in the more native environment of 3T3-L1 adipocytes. Cross-linked antiadiponectin antibody-conjugated beads were used for incubation with adipocyte extracts. We found that adiponectin and ERp44 also interact in mature adipocytes (Fig. 3A, lane 4). Approximately 10% of total ERp44 can be recovered with antiadiponectin antibody, suggesting that this is a quantitatively significant interaction between these two proteins that is likely to be functionally important. We wanted to test the expression of ERp44 during adipocyte differentiation since adiponectin is dramatically induced in this process. We found that ERp44 is expressed at high levels throughout differentiation of 3T3-L1 fibroblasts to adipocytes and increases approximately two- to threefold during this process (Fig. 3B).
FIG. 3.

Adiponectin interacts with ERp44 in 3T3-L1 adipocytes. (A) About 750 μg adipocyte protein extract was used for co-IP with control (Ctl) IgG or mab14-coupled resin. The immunoblotting was done with antiadiponectin (anti-APN) and anti-ERp44 antibodies. (B) Extracts from 3T3-L1 adipocytes at various stages of differentiation were separated on a 10% Bis-Tris Nupage gel and then transferred for immunoblotting with various antibodies. GDI was used as an internal protein loading control. (C) Immunofluorescence staining for adiponectin and ERp44 in 3T3-L1 adipocytes as indicated in the picture. Adiponectin is shown in red, ERp44 is shown in green, and nuclei (DAPI) are shown in blue. Arrows indicate the fibroblast in the field, which does not show staining for adiponectin. Bar, 10 μm. WB, Western blot.
To see whether ERp44 and adiponectin overlap to any significant extent with respect to intracellular localization, we performed immunofluorescent staining with a mixed population of 3T3-L1 adipocytes and fibroblasts. Similar to the results reported by Bogan and Lodish (7), we found the bulk of the adiponectin signal in a punctate cytoplasmic pattern, consistent with an ER-based localization (Fig. 3C, top left panel). As expected, ERp44 localizes in a similar punctate cytoplasmic pattern. Surprisingly, a significant portion of ERp44 signal can be found in an asymmetric perinuclear distribution, highly characteristic of Golgi localization (Fig. 3C, top right panel). This is particularly striking in the case of the staining in fibroblast (which is completely devoid of adiponectin expression), where essentially all of the ERp44 signal can be found in the Golgi apparatus. The ER-based signals for adiponectin and ERp44 completely overlap in the 3T3-L1 adipocyte (Fig. 3C, bottom right panel), whereas there is no such overlap for the ERp44 localized to the Golgi stack. Combined, this suggests that the bulk of ERp44 is localized to the Golgi in 3T3-L1 fibroblasts. As these cells differentiate into adipocytes, a much higher proportion of ERp44 redistributes to the ER, leading to the high degree of colocalization with adiponectin in the ER.
The covalent intracellular interaction between ERp44 and adiponectin can be regulated.
At steady state, a significant pool of intracellular adiponectin is covalently bound to ERp44. We were interested in the fate of this pool. We used 5 μM cycloheximide to inhibit protein synthesis and examined the secretion dynamics of adiponectin. We found that after 2 h, the secretion of adiponectin is significantly reduced in the cycloheximide-treated group. After 4 h, the secretion rate is less affected by the presence of the protein synthesis inhibitor (Fig. 4A). After 6 h, the adipocytes under cycloheximide treatment start to overlap with the vehicle group. We then chose 4 h as the time point at which to analyze the multimerization of adiponectin. To examine whether the cycloheximide treatment had an effect on the relative distribution of adiponectin complexes released from the cells, the tissue culture supernatant was subjected to gel filtration analysis. We have developed conditions under which we can effectively separate the HMW, LMW, and trimeric forms of adiponectin (Fig. 4B). While a reduction of the overall signal can be observed as expected from the analysis of total adiponectin in Fig. 4A, the profile with adiponectin intensities can be seen in Fig. 4C, and a quantitation of the relative contributions of the different complexes compared to total adiponectin is shown in Fig. 4D. A clear bias toward the HMW and LMW forms at the expense of the trimer can be seen. This suggests that the favorable overall ratio of chaperone (ERp44) to passenger (adiponectin) encountered under cycloheximide treatment significantly affects the maturation and release of the HMW and LMW complexes. We then analyzed the interaction between ERp44 and adiponectin 4 h after initiation of this treatment. There is a >90% reduction in the ERp44-adiponectin interaction (Fig. 4E) in the cycloheximide treatment samples. This suggests that the population of adiponectin associated with ERp44 corresponds to the secretion-competent pool, and the binding and release of adiponectin to ERp44 may represent a critical regulatory step in adipocytes.
FIG. 4.

ERp44-adiponectin interaction is changed by cycloheximide treatment. (A) Day 8 3T3-L1 adipocytes were treated with 5 μM cycloheximide (CHX) or vehicle for up to 8 h. Supernatants from 0 h, 2 h, 4 h, 6 h, and 8 h were removed for immunoblotting with antiadiponectin antibody. The intensity for adiponectin was quantified and plotted. Vehicle, n = 5; cycloheximide, n = 5. (B) Supernatant (200 μl) from vehicle- and cycloheximide-treated adipocytes at 4 h was separated on a size fractionation column. Each fraction was run on a 26-well gel and later for immunoblotting of adiponectin. The ratio of each complex was expressed by the equation (area under curve for each complex)/(total area under curve). Vehicle, n = 3; cycloheximide, n = 3. (C) A representative complex distribution of adiponectin for vehicle- and cycloheximide-treated cells is shown. The immunoblotting was done with antiadiponectin antibody. (D) The complex distribution of the representative result for panel C is shown in the line plot. (E) Day 8 3T3-L1 adipocytes were treated with 5 μM cycloheximide or vehicle for 4 h. Protein extracts were used for co-IP with mab14-coupled beads. Five percent input before co-IP and 5% flowthrough after co-IP were loaded as controls. The Western analysis (WB) was performed with antiadiponectin and anti-ERp44 antibodies. A result representative of three independent experiments is shown here. Tri, trimeric form; SUP, supernatant.
The ERp44-adiponectin interaction can be manipulated in additional ways. In an acute setting, insulin has a stimulatory effect on adiponectin secretion from adipocytes in cell culture (7, 37). Consistent with these previous findings, treatment of 3T3-L1 adipocytes with insulin significantly increases the release of adiponectin into the tissue culture supernatant (Fig. 5A). Under the same conditions, the association of intracellular adiponectin with ERp44 is reduced compared to basal levels. A quantitation based on three experiments is shown in Fig. 5B, and a representative example is shown in Fig. 5C. Combined, this suggests that several treatments associated with altered levels of adiponectin secretion have a significant impact on the interaction of ERp44 with adiponectin. The disruption of the ERp44-adiponectin interaction may therefore represent one of the rate-limiting steps for adiponectin release from adipocytes.
FIG. 5.
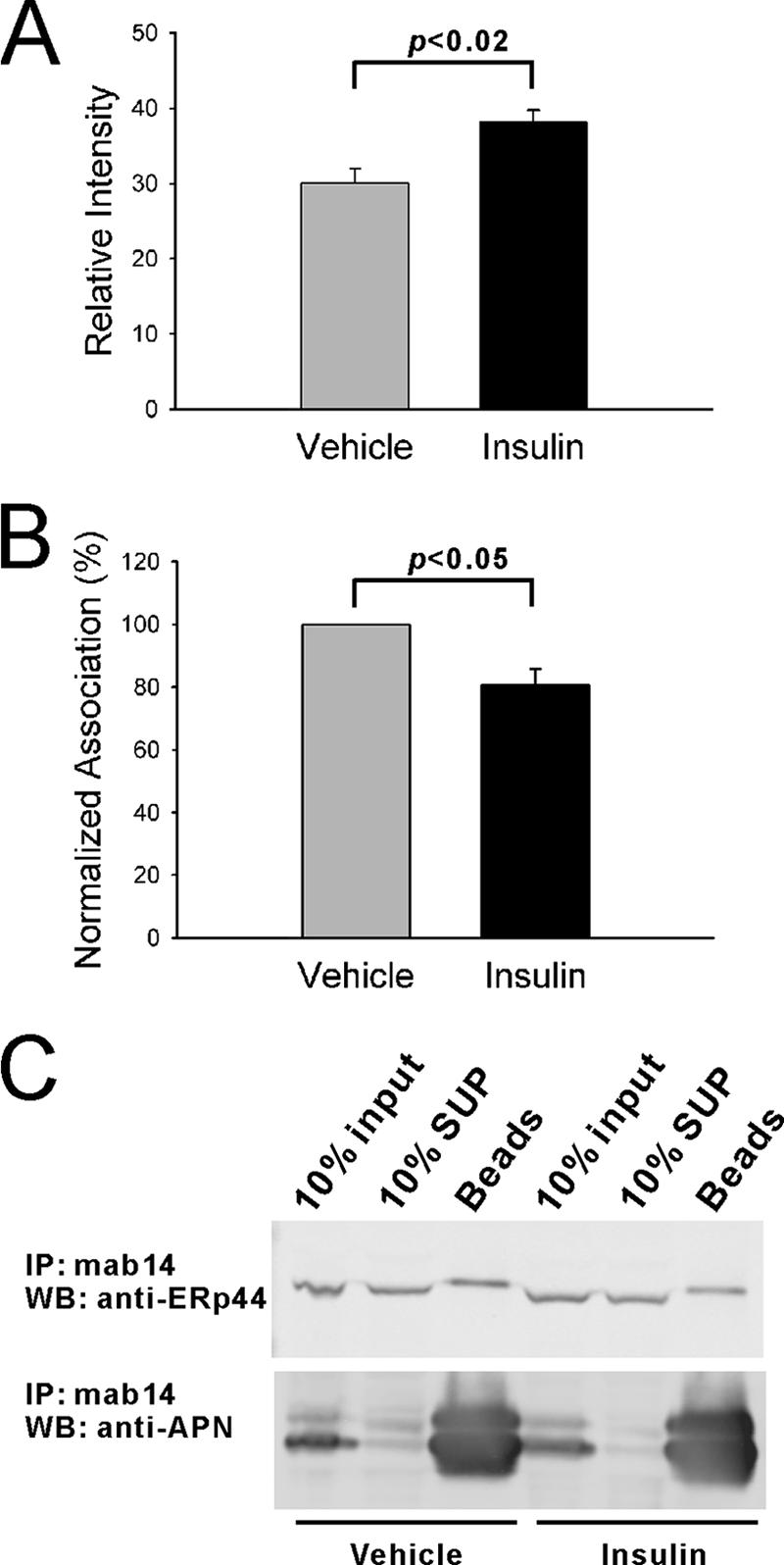
Interaction between ERp44 and adiponectin is decreased by insulin treatment. (A) Insulin (160 nM) was used to treat adipocytes. After 30 min, the culture medium was removed for immunoprecipitation and blotted with antiadiponectin antibody. Vehicle, n = 4; insulin, n = 4. (B) The cellular extracts from vehicle- and insulin-treated cells were used for co-IP with mab14 antibody-coupled beads. The beads were then resolved on a 10% Nupage gel for immunoanalysis with anti-ERp44 and antiadiponectin antibodies. The interaction between ERp44 and adiponectin was represented by the ratio of ERp44 over adiponectin. The ratio from vehicle-treated cells was taken as 100% and was used to normalize the ratio from insulin-treated samples. Vehicle, n = 3; insulin, n = 3. (C) A representative Western blot (WB) is shown for ERp44 and adiponectin immunoanalysis. SUP, supernatant.
The presence of ERp44 has a functional impact on adiponectin secretion.
If ERp44 is involved in the specific intracellular retention of adiponectin, overexpression of ERp44 is predicted to have a negative impact on the level of adiponectin secretion. Indeed, when ERp44 levels are progressively increased in HEK-293T cells, the degree of adiponectin release from the cells is dose dependently reduced (Fig. 6A). Overexpression of ERp44 has the potential to cause general cellular toxicity and may lead to an unspecific block of the secretory pathway. However, this is very unlikely because we found that the secretion of resistin is not affected. In addition, the secretion of the Cys39Ser mutant adiponectin is completely unaffected by ERp44 overexpression (Fig. 6A). WT adiponectin that fails to be secreted under conditions of high ERp44 levels can be efficiently released into the tissue culture supernatant upon a brief exposure to reducing agents, as judged by the increased time-dependent release of adiponectin into culture medium after treatment with BME (Fig. 6B). Examining in parallel the intracellular interaction between ERp44 and adiponectin, we found a significant reduction of adiponectin-associated ERp44 after BME treatment of intact cells (Fig. 6C). This corroborates that the interaction between ERp44 and adiponectin is susceptible to reducing agents and ERp44 is sufficient to effectively induce the highly specific retention of adiponectin intracellularly. A similar retention phenomenon was found when we cotransfected ERp44 and adiponectin in 3T3-L1 fibroblast cells. The reduced levels of secreted WT adiponectin can be accounted for by the increased intracellular retention of the protein (Fig. 6D).
FIG. 6.

ERp44 retains adiponectin intracellularly. (A) A fixed amount of either WT adiponectin (APN) or cys39ser (0.5 μg) mutant and increasing amount of ERp44 plasmid were cotransfected into HEK-293T cells. The amounts of HA-tagged ERp44 plasmid used were 0 μg, 0.025 μg, 0.05 μg, 0.075 μg, 0.1 μg, and 0.2 μg. The same amount of resistin-expressing plasmid (0.5 μg) was cotransfected as a control. The culture medium was removed and used for immunoblotting for adiponectin and resistin. Cell extracts were analyzed with immunoblotting for ERp44 to confirm the expression. (B) HEK-293T cells transiently transfected with ERp44 and adiponectin plasmids were treated with 5 mM BME or vehicle. The culture medium was collected and analyzed for adiponectin at different times. (C) The cell extracts from panel B were used for co-IP followed by immunoblotting with anti-ERp44 and antiadiponectin antibodies. (D) To repeat the cotransfection experiments in 3T3-L1 fibroblast cells, adiponectin and ERp44 plasmids were cotransfected into the cells. The culture medium and cell extracts were taken for Western blotting (WB) with antiadiponectin antibody.
Ero1-Lα disrupts the ERp44-adiponectin interaction upon cotransfection.
While ERp44 exerts potent effects on adiponectin retention, there has to be a mechanism in place within the lumen of the secretory pathway that facilitates the release of the passenger protein from ERp44. An important partner for ERp44 has recently been identified and shown to be Ero1-Lα (3). Ero1 is involved in the regeneration of PDI and hence involved in disulfide bond formation (17). Two closely related isoforms, encoding Ero1-Lα and Ero1-Lβ, exist in the human genome (9, 29). Since Ero1-Lα is critically involved in reducing the covalent disulfide bond between cargo and ERp44 (2), we postulated that Ero1-Lα can also release the retention of adiponectin by ERp44. The presence of increasing intracellular levels of Ero1-Lα alone does not have a strong impact on adiponectin secretion from HEK-293T cells (Fig. 7A). Interestingly, the disproportionate increase in Ero1-Lα leads to a decrease in the secretion of the highly sulfhydryl-rich protein resistin. When ERp44 is introduced at levels that lead to a highly effective intracellular retention of WT adiponectin, secretion of Cys39Ser adiponectin is not affected. Complementing the cells with increasing levels of Ero1-Lα leads to a dose-dependent release of the block on adiponectin secretion brought about by excessive ERp44 (Fig. 7B). This suggests that Ero1-Lα serves as a preferred partner of ERp44 and can release the retained cargo molecules from ERp44. In order to test this possibility directly, we immunoprecipitated the cellular extracts with anti-HA antibody, recognizing the tagged ERp44 in these cells. A significant amount of adiponectin can be recovered under these conditions. Upon cotransfection with Ero1-Lα, levels of ERp44-associated adiponectin were decreased. Instead, a significant portion of Ero1-Lα is recovered with ERp44. This is consistent with the role of Ero1-Lα as an effective competitor for ERp44 binding (Fig. 7C).
FIG. 7.

Ero1-Lα releases the retention by ERp44. (A) The conditions were the same as described for FIG. 6A for the cotransfection with Ero1-Lα and adiponectin (APN). The amounts of myc-tagged-Ero1-Lα used were 0 μg, 0.025 μg, 0.05 μg, 0.075 μg, 0.1 μg, and 0.2 μg. Intracellular materials were analyzed for Ero1-Lα to confirm the expression. (B) Ero1-Lα releases the retention of adiponectin by ERp44 in a dose-dependent manner. The same amount of adiponectin or Cys39Ser mutant plasmid (0.3 μg) was transfected to HEK-293T cells, together with different combinations of ERp44 and Ero1-Lα plasmids. The amount of ERp44 plasmid used was 0.06 μg, and the amounts of Ero1-Lα plasmids used were 0 μg, 0.01 μg, 0.02 μg, 0.05 μg, and 0.1 μg. (C) HEK-293T cells were transfected by adiponectin and HA-tagged ERp44, with or without Ero1-Lα. Immunoprecipitation (IP) was performed with anti-HA antibody. The recovered immunoprecipitate was analyzed for adiponectin, ERp44, and Ero1-Lα. Note that the presence of Ero1-Lα decreases the association of adiponectin with ERp44 while significant levels of Ero1-Lα are recovered with ERp44. WB, Western blot.
Reduction of the levels of either ERp44 or Ero1-Lα enhances the secretion of adiponectin.
To gain more insight into the relationship between ERp44 and adiponectin, we carried out an shRNA interference experiment for ERp44. We used a lentiviral system to get efficient delivery of the shRNA. We infected adipocytes with the same MOIs of control shRNA and shRNA for ERp44 as shown by GFP immunoblotting. With an MOI of 5, we achieved a reduction of ERp44 by more than 80% (Fig. 8A). A higher MOI did not result in a further reduction (data not shown). This may be due to the long half-lives of chaperones such as ERp44. Under these conditions, we found that the secretion of adiponectin is increased twofold, consistent with a function of ERp44 aimed at the retention of intracellular adiponectin (Fig. 8B).
FIG. 8.

Knockdown of ERp44 and Ero1-Lα increases the secretion of adiponectin. (A) Day 6 3T3-L1 adipocytes were infected by shRNA for ERp44 or control shRNA at an MOI of 5. After 4 days, the cell lysates were used for immunoblotting for ERp44, GFP, and actin. (B) On day 10, the medium was changed to serum-free DMEM for 8 h. Supernatants were run on Bis-Tris gel and later subjected to immunoanalysis for adiponectin. Relative adiponectin levels are shown. Control (Ctl) shRNA, n = 3; ERp44-shRNA, n = 3. (C) Day 6 3T3-L1 adipocytes were infected by shRNA for Ero1-Lα or control shRNA at an MOI of 5. After 4 days, the cell lysates were used for immunoblotting of Ero1-Lα, GFP, and actin. (D) On day 10, the medium was changed to serum-free DMEM for 8 h. Supernatants were run on Bis-Tris gel and later used for immunoblotting for adiponectin. Relative levels of adiponectin are shown. Ctl shRNA, n = 3; Ero1-Lα-shRNA, n = 3. (E) The supernatant (200 μl) from panel D was separated on a gel filtration column (as described in Materials and Methods). The relative levels of the HMW, LMW, and trimeric (Tri) forms were compared between control shRNA and Ero1-Lα-specific-shRNA-treated samples. Note that the levels of trimer increase, while the HMW and LMW levels decrease. WB, Western blot.
To study the endogenous Ero1-Lα in adipocytes at the protein level, we raised polyclonal antibodies to Ero1-Lα. This antibody specifically recognizes Ero1-Lα upon transfection into HEK-293T cells (see Fig. S2A, middle panel, in the supplemental material). The same membrane was decorated with anti-myc antibody, which recognized the same band (see Fig. S2A, left panel, in the supplemental material). This antibody recognized a single band in 3T3-L1 adipocyte extract (see Fig. S2A, right panel, in the supplemental material). During adipogenesis, we found that Ero1-Lα is dramatically induced as 3T3-L1 fibroblast cells differentiate into adipocytes (see Fig. S2B in the supplemental material). This is consistent with previous proteomic findings by Wilson-Fritch and colleagues (44). At the mRNA level, we find Ero1-Lα to be ubiquitously expressed, consistent with the published literature (29). We also generated extracts from a number of different tissues and found that Ero1-Lα protein is enriched in several tissues, such as pancreas, stomach, testis, and all five adipose tissue depots examined (see Fig. S2D in the supplemental material). The size of Ero1-Lα in adipose tissues is slightly different from the molecular weight observed in other tissues, which may be a reflection of differential posttranslational modifications. The protein levels of Ero1-Lα generally match relative mRNA levels (compare Fig. S2C and D in the supplemental material). Of interest is the fact that the tissues with higher than average expression of Ero1-Lα are all tissues rich in “professional” secretory cells.
To study the influence of Ero1-Lα on adiponectin secretion, we performed shRNA interference for Ero1-Lα with adipocytes. With an MOI of 5, we obtained reduction by 50% (Fig. 8C). Interestingly, when we analyzed the secretion of adiponectin, we found that the extracellular level of adiponectin is increased (Fig. 8D). This suggests that Ero1-Lα, like ERp44, may impose an additional layer of regulation on adiponectin secretion. Knockdown of Ero1-Lα thus releases the regulation and enhances the secretion. To see the role of Ero1-Lα in higher-order-form assembly and secretion, we analyzed the complex distribution of the secreted adiponectin. Compared to what is found for control shRNA, a reduction of Ero1-Lα levels significantly increases the secretion of trimeric adiponectin at the expense of decreased secretion of higher-order adiponectin complexes (Fig. 8E). Importantly, the reduction of ERp44 and Ero1-Lα did not have an effect on the secretion of other proteins released from adipocytes, such as complement factor 3B (data not shown), again supporting that the effects seen are highly specific for adiponectin.
ERp44 and Ero1-Lα show differential regulation under a number of conditions associated with altered adiponectin secretion.
We wanted to probe the relationship between ERp44, Ero1-Lα, and adiponectin levels under different physiological conditions in mice. When comparing levels of the chaperones in gonadal fat depots from ob/ob and WT littermates, significant reductions of ERp44 and Ero1-Lα levels in adipose depots can be seen (Fig. 9A). Adipose tissue-associated adiponectin is decreased in the ob/ob state. Upon analysis of the complex distribution of adiponectin in WT and ob/ob mouse sera, levels of the HMW form were found to be lower in ob/ob mice than in WT mice, while the LMW and trimer forms were higher (Fig. 9B). Therefore, lower HMW levels of adiponectin in ob/ob mice account for the overall decrease in total circulating levels of adiponectin.
FIG. 9.

Ero1-Lα and ERp44 are differentially regulated in different states in mice. (A) Tissue extracts from WT and ob/ob mice were used for immunoblotting with anti-Ero1-Lα, anti-ERp44, antiadiponectin, and anti-GDI antibodies on the same membrane. WT, n = 4; ob/ob, n = 3. (B) The sera from 16-week-old WT and ob/ob mice were separated, and the complex distribution of adiponectin is shown. WT, n = 3; ob/ob, n = 3. (C) Tissues from male and female mice were collected and analyzed for anti-Ero1-Lα, anti-ERp44, antiadiponectin, and anti-GDI antibodies. Male, n = 4; female, n = 4. (D) Sera from 8- to 12-week-old mice were separated as mentioned in Materials and Methods. Complex distribution of adiponectin was compared between males and females. Male, n = 3; female, n = 3. NS, not significant.
Adiponectin levels in circulation display a sexual dimorphism. Levels in females are higher than those in males (11). A similar dimorphism holds up for the chaperones. ERp44 and Ero1-Lα levels are higher in adipose tissue extracts isolated from female mice than in those from male mice (Fig. 9C). Again, this correlates well with the higher HMW levels of serum adiponectin in female mice than in male mice (Fig. 9D) (30).
Ero1-Lα is induced after PPARγ agonist treatment in mice.
Treatment of mice with the PPARγ agonist with a standard 14-day regimen results in a significant increase of circulating adiponectin in mice (Fig. 10A) (13). The HMW ratio is significantly increased while the LMW and trimer ratios are decreased after treatment (Fig. 10B). The increase of the HMW ratio is not due to decreases of the LMW and trimer ratios, as shown in Fig. 10C. We found that the absolute LMW and trimer levels are not dramatically changed, while the HMW level is increased. This suggests that the increase of the HMW level may account for the increased secretion of total adiponectin. Within adipose depots, the levels of Ero1-Lα and ERp44 are increased in gonadal (Fig. 10D) and visceral (Fig. 10E) fat tissues upon treatment with PPARγ agonist. Similarly, adipose tissue-associated adiponectin displays an increase. Intracellular steady-state levels of Ero1-Lα in the stomach remain relatively unchanged under these conditions. A representative set of Western blots is shown in Fig. 10F. Combined, this suggests that the two critical chaperones with a defined role for adiponectin maturation, Ero1-Lα and ERp44, are both targets for PPARγ agonist action.
FIG. 10.

ERp44 and Ero1-Lα are increased after PPARγ agonist (COOH) treatment in mice. (A) After COOH treatment, the levels of adiponectin level are increased twofold compared to those for the vehicle group. Serum (2 μl) from each mouse was separated on a 10% Bis-Tris Nupage gel and transferred to nitrocellulose. The membrane was blotted with antiadiponectin antibody, and the bands were quantified. Vehicle, n = 4; COOH, n = 5. (B) The complex distribution of adiponectin was determined, and the ratios for all three complexes were calculated. Vehicle, n = 3; COOH, n = 3. (C) A representative serum complex distribution of adiponectin from vehicle- and COOH-treated mice is shown overlapping on the same graph. (D) Gonadal adipose tissues were collected from vehicle and COOH groups, and extracts were separated on a 10% Nupage gel and blotted with anti-Ero1-Lα, anti-ERp44, antiadiponectin, and anti-GDI antibodies. Relative levels of Ero1-Lα, ERp44, and adiponectin were calculated by dividing the band intensity by the GDI intensity. For each protein in the vehicle and COOH groups, the lowest relative level was regarded as 100%. All other samples were represented by dividing their relative levels by the lowest relative level. Vehicle, n = 4; COOH, n = 4. (E) The same method was used to measure the relative levels of Ero1-Lα, ERp44, and adiponectin in visceral adipose tissues. Vehicle, n = 4; COOH, n = 4. (F) Tissues from representative vehicle- and COOH-treated mice were collected. The extract was loaded to a 10% Bis-Tris Nupage gel, and Western blotting was done with anti-Ero1-Lα, anti-ERp44, antiadiponectin, and anti-GDI antibodies. Tri, trimeric form.
DISCUSSION
Adiponectin circulates at relatively high levels in plasma. Its primary effect may be mediated by enhancing insulin sensitivity in liver and carbohydrate metabolism (4, 32, 47). In addition, adiponectin has been shown to have an impact on atherosclerosis, inflammation, and myocardial ischemia (28, 38, 39). Despite the wealth of knowledge on its impact on various tissues, very little is known about the regulation of adiponectin secretion by adipocytes.
There is no strong evidence to date to argue for the presence of a triggered secretory pathway in adipocytes. While numerous studies have focused on the mechanistic aspect of the insulin-stimulated translocation of Glut4 vesicles, these vesicles are not well suited for storing soluble components, due to the constant cycling of these vesicles to and from the plasma membrane, even in the unstimulated state. Using adiponectin as a marker protein, we have unveiled one of the mechanisms by which adipocytes can adapt to the need to release secretory proteins in a more regulated fashion. Thiol-mediated protein retention allows adipocytes to effectively retain adiponectin intracellularly and control the kinetics and efficiency of its assembly and release in a fashion similar to that observed for IgM molecules in B cells. The chaperone responsible for the retention is ERp44, an ER-resident molecule, which is also involved in the thiol-mediated retention of IgM. Equally important is Ero1-Lα, which is involved in the release of adiponectin from ERp44. The differential regulation of these chaperones under a variety of conditions imposes an additional important layer of control over the release of adiponectin from adipocytes beyond transcriptional regulation of the adiponectin gene.
Adiponectin has three functional domains, which include the N-terminal signal sequence followed by a hypervariable region, a collagenous domain, and the C-terminal globular domain. Cotranslationally, adiponectin monomers form trimers by initiation of strong hydrophobic interactions that occur in the globular domains. The collagenous domains act to establish close intersubunit interactions that result in a highly ordered and stable fiber. The trimer is further stabilized by an intratrimer disulfide bond mediated by cysteine 39. The third monomer with a free thiol group forms an intertrimer disulfide bond with the third monomer from another trimer. This trimer-dimer (hexamer) is also referred to as the LMW form. Several LMW forms may interact with each other and form higher-order complexes (HMW forms). All the three forms, trimer, LMW, and HMW, can easily be detected in circulation and can be biochemically purified. Several reports have highlighted differential functions for these complexes. In particular, levels of the HMW form correlate well with insulin sensitivity in mice and in humans (23, 31, 41). Since different oligomeric complexes of adiponectin may activate diverse signaling pathways in various cell types, the regulation of the secretion of different complexes must be tightly controlled. This is particularly important in light of the fact that to date, there is no evidence for an interchange between the different forms once they reach circulation (30).
In order to study the function of specific disulfide bonds in adiponectin complex formation, we generated the Cys39Ser mutant and stably transfected it to HEK-293T cells. When the secretion kinetics of the mutant are compared to those of WT adiponectin, it is apparent that the mutant is more rapidly secreted from these cells (30). This phenomenon prompted us to study the mechanism involved in the differential rate of secretion. We hypothesized that WT adiponectin is retained intracellularly via the Cys39 residue, while the Cys39Ser mutant can bypass this retention and is secreted more rapidly. The retention of proteins through specific cysteine residues is a hallmark of thiol-mediated retention. This phenomenon was first discovered by Sitia and colleagues (40). They found that plasma cells secrete only pentameric or hexameric forms of IgM. Cys575 in the tailpiece of the μ chain mediates this retention by interacting with ER resident protein(s). Reducing agents, such as BME, can destroy this interaction and release monomeric IgM (1). We treated 3T3-L1 adipocytes with 5 mM BME for 3 h and found that the secretion of adiponectin was increased seven- to eightfold while intracellular levels of adiponectin were reduced. This suggested that adiponectin secretion may be mediated by a process similar to that for IgM assembly. Anelli and colleagues identified ERp44 as a resident ER chaperone that mediates the retention of IgM (2). We show that ERp44 associates with adiponectin, which strongly suggests that it may play an important role in adiponectin multimerization. The interaction is covalent and mediated through residue Cys39 of adiponectin. The basic building block of adiponectin is a trimer. Two of the three Cys39 residues are forming a disulfide bond within a trimer. The third Cys39 residue is available for heterologous interactions. We have strong reasons to believe that it is this third Cys39 residue in a trimer that interacts with ERp44. During B-cell differentiation in vitro, ERp44 level is increased along with the increasing demand on the secretion machinery (42). Interestingly, we found that ERp44 levels during 3T3-L1 adipocyte differentiation increase as well, in line with the increased expression of adiponectin during differentiation.
The interaction between ERp44 and adiponectin is regulated under different conditions. The interaction is highly susceptible to protein synthesis inhibition. When we treated adipocytes with 5 μM cycloheximide, we found that the overall secretion of adiponectin is decreased due to the lack of de novo-synthesized adiponectin. However, there is a significant increase in the relative HMW level in culture medium. The HMW ratio is increased from 20% to 30% after cycloheximide treatment compared to results for vehicle treatment, while the trimer is reduced. Under these conditions, the level of ERp44 within the ER lumen is only minimally affected due to the long half-life that this protein displays. This may cause a shift in the chaperone-to-passenger-protein ratio, giving the cell more time and a higher capacity to assemble adiponectin complexes. Since trimeric adiponectin may be the primary form associated with ERp44, the folding machinery may assemble LMW and HMW forms from the trimer more efficiently when ER cargo input is decreased. The level of intracellular adiponectin is only marginally affected after short-term protein synthesis inhibition, yet the ERp44 interaction is completely lost and HMW secretion is increased, which suggests that the ERp44-associating adiponectin population may reflect the “assembly-competent” pool.
The interaction between adiponectin and ERp44 can also be regulated under acute insulin treatment. After 30 min of insulin stimulation, secretion of adiponectin is increased by 25% compared to results for vehicle treatment, as shown previously (7, 37). When we examined the intracellular material, we found that the interaction between ERp44 and adiponectin is decreased by 10% after insulin treatment. Since ERp44-adiponectin interaction is mediated by a disulfide bond, changes in the ER redox potential are therefore likely to have a profound impact on the stability of the ERp44-adiponectin interaction. Insulin can elicit the generation of H2O2 after 15 min of treatment, and insulin action was also found to be accompanied by sulfhydryl oxidation (14, 26). After short-term insulin stimulation, we reasoned that the increase of adiponectin secretion is the result of acute changes of the redox potential in the ER triggered by insulin signaling. Indeed, secretion of adiponectin directly from the ER was implied in immunofluorescence staining of 3T3-L1 adipocytes (7). So the mechanism for insulin stimulation of adiponectin may be different from cycloheximide treatment, which primarily affects adiponectin assembly and thus secretion of the HMW form. In contrast, insulin signaling may simply disrupt the linkage between ERp44 and adiponectin and stimulate the secretion of adiponectin directly from the ER as trimer. This suggests that the ERp44-associated adiponectin population reflects the “secretion-competent” pool. In this context, it will also be interesting to see what the effects on adiponectin secretion and complex distribution by other agents, such as H2O2 and ascorbic acid, are. We are currently focusing on these questions.
Ero1 is the rate-limiting enzyme in disulfide bond formation within the lumen of the ER. It is critically involved in recycling of PDI. The physical interaction between ERp44 and Ero1-Lα indicates that there may be some link between thiol-mediated retention and disulfide bond formation. Anelli and colleagues showed that Ero1-Lα is a privileged partner of ERp44 and has the ability to replace the ERp44-retained passenger protein (2). We found that Ero1-Lα is of similar importance for the ERp44-adiponectin interaction. Overexpression of ERp44 caused a decrease in the secretion of adiponectin. When Ero1-Lα is coexpressed, this inhibition is partially released in transient transfection studies in HEK-293T cells. Comparing females and males along with WT and ob/ob mice, we found that ERp44 and Ero1-Lα levels correlate very well with secreted adiponectin levels, especially those of HMW adiponectin.
In previous reports, we have implicated adiponectin as an important mediator of PPARγ agonist action (12, 13). PPARγ agonist treatment increases the secretion of adiponectin, especially the HMW form, leading to increased steady-state concentrations of adiponectin in plasma. In gonadal and visceral adipose depots, we found both ERp44 and Ero1-Lα to be significantly increased in response to PPARγ agonist treatment. This suggests that chaperones like ERp44 and Ero1-Lα are critical PPARγ targets, further stimulating the folding and secreting of adiponectin. There is evidence that the PPARγ agonist-mediated transcriptional induction of the adiponectin gene is relatively modest (13) or even completely absent (34), which suggests that chaperones in the secretory pathway may in fact represent the most relevant target genes for PPARγ agonists as they relate to improvements in adiponectin secretion. Therefore, we would like to propose that PPARγ agonists in adipocytes may mainly exert their effects by improving the efficiency of the secretory machinery through regulation of critical chaperones in the secretory pathway.
What could be the function of such a retention/release mechanism in the secretory pathway of adipocytes? The most straightforward explanation is that complex proteins such as adiponectin, which need significant posttranslational modifications and assembly, may inherently require more time to fold. Retention by chaperones within the ER may prolong the resident time for adiponectin in the secretory pathway and thus provide a better chance for proper folding of adiponectin into higher-order complexes. At the same time, ERp44 association may also be a way to differentiate assembly-/secretion-competent pools and degradation-prone pools of adiponectin. Another reason could be that this complex retention mechanism allows the fat cell to exert more control over secretion for a subset of secretory molecules. As pointed out by Roh and colleagues (36), there is very limited evidence that would support the existence of a regulated secretory pathway comparable to what can be found in neuroendocrine cells. While adipocytes have the ability to translocate vesicles containing Glut4 to the plasma membrane in an acute fashion, these Glut4-containing structures are not well suited for the trafficking of soluble constituents, such as adiponectin (7). Therefore, the adipocyte has very limited potential for posttranslational control over protein release from the cell. ER retention may add one layer of regulation for some secretory molecules. The kinetics are significantly slower than the kinetics for the release of neuroendocrine vesicles (10 to 15 min as opposed to milliseconds). Nevertheless, for the controlled secretion of proteins that do not critically rely on a very rapid release, an ER-based retention mechanism is highly suitable. Like adiponectin, lipoprotein lipase is shown to be mainly localized in the ER (36). It is not known whether lipoprotein lipase is subjected to thiol-mediated retention and whether ERp44 is involved. It is likely that additional proteins requiring a more controlled release from adipocytes interact with ERp44, while proteins in the constitutive pathway can bypass this interaction. We are currently addressing this point by performing additional large-scale co-IP experiments to determine which other passengers interact with ERp44 in adipocytes.
Clearly, the relationship between the levels of the two chaperones ERp44 and Ero1-Lα and adiponectin secretion is complex. In HEK-293T cells, cotransfection of ERp44 retains the majority of transfected adiponectin and the secretion of adiponectin is affected dramatically. Our shRNA experiments with 3T3-L1 adipocytes demonstrate that ERp44 can act as the rate-limiting step for adiponectin secretion. After an 80% knockdown of ERp44, the secretion of adiponectin is increased twofold. The adipocyte uses ERp44 to effectively control adiponectin retention within the secretory pathway. This is a “postfolding” phenomenon, since WT trimers can effectively be released from cells with reducing agents and the Cys39Ser mutant effectively escapes this mechanism.
Ero1-Lα offers an additional layer of regulation, such that the combined upregulation of both chaperones (such as that during PPARγ agonist treatment) leads to an increase in the release of the HMW form. The relative ratio of the two chaperones to each other as well as the overall ratio of chaperones to cargo (adiponectin) is a critical determinant of the total release of adiponectin from the cell as well as the extents of release of the different complexes. In HEK-293T cells, complementing with Ero1-Lα partially releases the retention by ERp44, which suggests that Ero1-Lα can serve as the preferred partner of ERp44 and replaces the associated cargo molecule. In 3T3-L1 adipocytes, adiponectin expression and higher-order-oligomer formation are complex and physiologically more relevant. The lowering of cellular Ero-1α levels by shRNA leads to an increased release of adiponectin due to an increase in the release of the trimeric adiponectin complex, which primarily is responsible for the overall increase in adiponectin secretion under these conditions. At the same time, the HMW and LMW forms are released at dramatically reduced levels. This strongly suggests a model in which the lowering of Ero1-Lα levels below a critical threshold causes trimeric adiponectin complexes to accumulate intracellularly to a level that exceeds the endogenous retention capacity and the trimer then escapes and is released.
A complete elimination of Ero1-Lα may display a much more severe and less specific phenotype. While the formation of the adiponectin's intratrimer disulfide bond is likely to require a low level of oxidoreductase activity in the secretory pathway as well, none of our experiments were performed under these conditions. We achieved a partial, 50 to 70% knockdown of Ero1-Lα in vitro. This is nevertheless physiologically relevant and reflects the reduction of Ero1-Lα in an ob/ob model or the difference between male and female mice, with the concomitant difference seen in the complex distribution in these models.
Supplementary Material
Acknowledgments
This work was supported by grants R01-DK55758, R24-DK071030, and R21-DK075887-01 (to P.E.S.). T.D.S. was supported by T32 HL007675. T.K. was supported by T32 DK007513, and M.W.R. was supported by Medical Scientist Training Grant T32-GM07288. P.E.S. is a recipient of an Irma T. Hirschl Career Scientist Award.
We thank Yuan Xin and Aisha Cordero for technical assistance and Susan Buehl in the Cancer Center Hybridoma Facility for expert help during the generation of hybridomas and production of monoclonal antibodies. We also thank the entire Scherer laboratory for advice and discussions.
Footnotes
Published ahead of print on 12 March 2007.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Alberini, C. M., P. Bet, C. Milstein, and R. Sitia. 1990. Secretion of immunoglobulin M assembly intermediates in the presence of reducing agents. Nature 347:485-487. [DOI] [PubMed] [Google Scholar]
- 2.Anelli, T., M. Alessio, A. Bachi, L. Bergamelli, G. Bertoli, S. Camerini, A. Mezghrani, E. Ruffato, T. Simmen, and R. Sitia. 2003. Thiol-mediated protein retention in the endoplasmic reticulum: the role of ERp44. EMBO J. 22:5015-5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anelli, T., M. Alessio, A. Mezghrani, T. Simmen, F. Talamo, A. Bachi, and R. Sitia. 2002. ERp44, a novel endoplasmic reticulum folding assistant of the thioredoxin family. EMBO J. 21:835-844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berg, A. H., T. P. Combs, X. Du, M. Brownlee, and P. E. Scherer. 2001. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat. Med. 7:947-953. [DOI] [PubMed] [Google Scholar]
- 5.Berg, A. H., T. P. Combs, and P. E. Scherer. 2002. ACRP30/adiponectin: an adipokine regulating glucose and lipid metabolism. Trends Endocrinol. Metab. 13:84-89. [DOI] [PubMed] [Google Scholar]
- 6.Bobbert, T., H. Rochlitz, U. Wegewitz, S. Akpulat, K. Mai, M. O. Weickert, M. Mohlig, A. F. Pfeiffer, and J. Spranger. 2005. Changes of adiponectin oligomer composition by moderate weight reduction. Diabetes 54:2712-2719. [DOI] [PubMed] [Google Scholar]
- 7.Bogan, J. S., and H. F. Lodish. 1999. Two compartments for insulin-stimulated exocytosis in 3T3-L1 adipocytes defined by endogenous ACRP30 and GLUT4. J. Cell Biol. 146:609-620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brewer, J. W., and R. B. Corley. 1996. Quality control in protein biogenesis: thiol-mediated retention monitors the redox state of proteins in the endoplasmic reticulum. J. Cell Sci. 109:2383-2392. [DOI] [PubMed] [Google Scholar]
- 9.Cabibbo, A., M. Pagani, M. Fabbri, M. Rocchi, M. R. Farmery, N. J. Bulleid, and R. Sitia. 2000. ERO1-L, a human protein that favors disulfide bond formation in the endoplasmic reticulum. J. Biol. Chem. 275:4827-4833. [DOI] [PubMed] [Google Scholar]
- 10.Carley, A. N., L. M. Semeniuk, Y. Shimoni, E. Aasum, T. S. Larsen, J. P. Berger, and D. L. Severson. 2004. Treatment of type 2 diabetic db/db mice with a novel PPARgamma agonist improves cardiac metabolism but not contractile function. Am. J. Physiol. Endocrinol. Metab. 286:E449-E455. [DOI] [PubMed] [Google Scholar]
- 11.Combs, T. P., A. H. Berg, M. W. Rajala, S. Klebanov, P. Iyengar, J. C. Jimenez-Chillaron, M. E. Patti, S. L. Klein, R. S. Weinstein, and P. E. Scherer. 2003. Sexual differentiation, pregnancy, calorie restriction, and aging affect the adipocyte-specific secretory protein adiponectin. Diabetes 52:268-276. [DOI] [PubMed] [Google Scholar]
- 12.Combs, T. P., U. B. Pajvani, A. H. Berg, Y. Lin, L. A. Jelicks, M. Laplante, A. R. Nawrocki, M. W. Rajala, A. F. Parlow, L. Cheeseboro, Y. Y. Ding, R. G. Russell, D. Lindemann, A. Hartley, G. R. Baker, S. Obici, Y. Deshaies, M. Ludgate, L. Rossetti, and P. E. Scherer. 2004. A transgenic mouse with a deletion in the collagenous domain of adiponectin displays elevated circulating adiponectin and improved insulin sensitivity. Endocrinology 145:367-383. [DOI] [PubMed] [Google Scholar]
- 13.Combs, T. P., J. A. Wagner, J. Berger, T. Doebber, W. J. Wang, B. B. Zhang, M. Tanen, A. H. Berg, S. O'Rahilly, D. B. Savage, K. Chatterjee, S. Weiss, P. J. Larson, K. M. Gottesdiener, B. J. Gertz, M. J. Charron, P. E. Scherer, and D. E. Moller. 2002. Induction of adipocyte complement-related protein of 30 kilodaltons by PPARgamma agonists: a potential mechanism of insulin sensitization. Endocrinology 143:998-1007. [DOI] [PubMed] [Google Scholar]
- 14.Czech, M. P., J. C. Lawrence, Jr., and W. S. Lynn. 1974. Evidence for the involvement of sulfhydryl oxidation in the regulation of fat cell hexose transport by insulin. Proc. Natl. Acad. Sci. USA 71:4173-4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dias-Gunasekara, S., J. Gubbens, M. van Lith, C. Dunne, J. A. Williams, R. Kataky, D. Scoones, A. Lapthorn, N. J. Bulleid, and A. M. Benham. 2005. Tissue-specific expression and dimerization of the endoplasmic reticulum oxidoreductase Ero1beta. J. Biol. Chem. 280:33066-33075. [DOI] [PubMed] [Google Scholar]
- 16.Fassio, A., and R. Sitia. 2002. Formation, isomerisation and reduction of disulphide bonds during protein quality control in the endoplasmic reticulum. Histochem. Cell Biol. 117:151-157. [DOI] [PubMed] [Google Scholar]
- 17.Frand, A. R., and C. A. Kaiser. 1999. Ero1p oxidizes protein disulfide isomerase in a pathway for disulfide bond formation in the endoplasmic reticulum. Mol. Cell 4:469-477. [DOI] [PubMed] [Google Scholar]
- 18.Gess, B., K. H. Hofbauer, R. H. Wenger, C. Lohaus, H. E. Meyer, and A. Kurtz. 2003. The cellular oxygen tension regulates expression of the endoplasmic oxidoreductase ERO1-Lalpha. Eur. J. Biochem. 270:2228-2235. [DOI] [PubMed] [Google Scholar]
- 19.Guenzi, S., A. M. Fra, A. Sparvoli, P. Bet, M. Rocco, and R. Sitia. 1994. The efficiency of cysteine-mediated intracellular retention determines the differential fate of secretory IgA and IgM in B and plasma cells. Eur. J. Immunol. 24:2477-2482. [DOI] [PubMed] [Google Scholar]
- 20.Hug, C., J. Wang, N. S. Ahmad, J. S. Bogan, T. S. Tsao, and H. F. Lodish. 2004. T-cadherin is a receptor for hexameric and high-molecular-weight forms of Acrp30/adiponectin. Proc. Natl. Acad. Sci. USA 101:10308-10313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kadowaki, T., and T. Yamauchi. 2005. Adiponectin and adiponectin receptors. Endocr. Rev. 26:439-451. [DOI] [PubMed] [Google Scholar]
- 22.Kubota, N., Y. Terauchi, T. Kubota, H. Kumagai, S. Itoh, H. Satoh, W. Yano, H. Ogata, K. Tokuyama, I. Takamoto, T. Mineyama, M. Ishikawa, M. Moroi, K. Sugi, T. Yamauchi, K. Ueki, K. Tobe, T. Noda, R. Nagai, and T. Kadowaki. 2006. Pioglitazone ameliorates insulin resistance and diabetes by both adiponectin-dependent and -independent pathways. J. Biol. Chem. 281:8748-8755. [DOI] [PubMed] [Google Scholar]
- 23.Lara-Castro, C., N. Luo, P. Wallace, R. L. Klein, and W. T. Garvey. 2006. Adiponectin multimeric complexes and the metabolic syndrome trait cluster. Diabetes 55:249-259. [PubMed] [Google Scholar]
- 24.Mancia, F., S. D. Patel, M. W. Rajala, P. E. Scherer, A. Nemes, I. Schieren, W. A. Hendrickson, and L. Shapiro. 2004. Optimization of protein production in mammalian cells with a coexpressed fluorescent marker. Structure 12:1355-1360. [DOI] [PubMed] [Google Scholar]
- 25.Matsuzawa, Y. 2005. Adiponectin: identification, physiology and clinical relevance in metabolic and vascular disease. Atheroscler. Suppl. 6:7-14. [DOI] [PubMed] [Google Scholar]
- 26.May, J. M., and C. de Haen. 1979. Insulin-stimulated intracellular hydrogen peroxide production in rat epididymal fat cells. J. Biol. Chem. 254:2214-2220. [PubMed] [Google Scholar]
- 27.Nawrocki, A. R., M. W. Rajala, E. Tomas, U. B. Pajvani, A. K. Saha, M. E. Trumbauer, Z. Pang, A. S. Chen, N. B. Ruderman, H. Chen, L. Rossetti, and P. E. Scherer. 2006. Mice lacking adiponectin show decreased hepatic insulin sensitivity and reduced responsiveness to peroxisome proliferator-activated receptor gamma agonists. J. Biol. Chem. 281:2654-2660. [DOI] [PubMed] [Google Scholar]
- 28.Okamoto, Y., S. Kihara, N. Ouchi, M. Nishida, Y. Arita, M. Kumada, K. Ohashi, N. Sakai, I. Shimomura, H. Kobayashi, N. Terasaka, T. Inaba, T. Funahashi, and Y. Matsuzawa. 2002. Adiponectin reduces atherosclerosis in apolipoprotein E-deficient mice. Circulation 106:2767-2770. [DOI] [PubMed] [Google Scholar]
- 29.Pagani, M., M. Fabbri, C. Benedetti, A. Fassio, S. Pilati, N. J. Bulleid, A. Cabibbo, and R. Sitia. 2000. Endoplasmic reticulum oxidoreductin 1-lbeta (ERO1-Lbeta), a human gene induced in the course of the unfolded protein response. J. Biol. Chem. 275:23685-23692. [DOI] [PubMed] [Google Scholar]
- 30.Pajvani, U. B., X. Du, T. P. Combs, A. H. Berg, M. W. Rajala, T. Schulthess, J. Engel, M. Brownlee, and P. E. Scherer. 2003. Structure-function studies of the adipocyte-secreted hormone Acrp30/adiponectin. Implications for metabolic regulation and bioactivity. J. Biol. Chem. 278:9073-9085. [DOI] [PubMed] [Google Scholar]
- 31.Pajvani, U. B., M. Hawkins, T. P. Combs, M. W. Rajala, T. Doebber, J. P. Berger, J. A. Wagner, M. Wu, A. Knopps, A. H. Xiang, K. M. Utzschneider, S. E. Kahn, J. M. Olefsky, T. A. Buchanan, and P. E. Scherer. 2004. Complex distribution, not absolute amount of adiponectin, correlates with thiazolidinedione-mediated improvement in insulin sensitivity. J. Biol. Chem. 279:12152-12162. [DOI] [PubMed] [Google Scholar]
- 32.Qi, Y., N. Takahashi, S. M. Hileman, H. R. Patel, A. H. Berg, U. B. Pajvani, P. E. Scherer, and R. S. Ahima. 2004. Adiponectin acts in the brain to decrease body weight. Nat. Med. 10:524-529. [DOI] [PubMed] [Google Scholar]
- 33.Rajala, M. W., Y. Lin, M. Ranalletta, X. M. Yang, H. Qian, R. Gingerich, N. Barzilai, and P. E. Scherer. 2002. Cell type-specific expression and coregulation of murine resistin and resistin-like molecule-alpha in adipose tissue. Mol. Endocrinol. 16:1920-1930. [DOI] [PubMed] [Google Scholar]
- 34.Rasouli, N., A. Yao-Borengasser, L. M. Miles, S. C. Elbein, and P. A. Kern. 2006. Increased plasma adiponectin in response to pioglitazone does not result from increased gene expression. Am. J. Physiol. Endocrinol. Metab. 290:E42-E46. [DOI] [PubMed] [Google Scholar]
- 35.Reddy, P., A. Sparvoli, C. Fagioli, G. Fassina, and R. Sitia. 1996. Formation of reversible disulfide bonds with the protein matrix of the endoplasmic reticulum correlates with the retention of unassembled Ig light chains. EMBO J. 15:2077-2085. [PMC free article] [PubMed] [Google Scholar]
- 36.Roh, C., R. Roduit, B. Thorens, S. Fried, and K. V. Kandror. 2001. Lipoprotein lipase and leptin are accumulated in different secretory compartments in rat adipocytes. J. Biol. Chem. 276:35990-35994. [DOI] [PubMed] [Google Scholar]
- 37.Scherer, P. E., S. Williams, M. Fogliano, G. Baldini, and H. F. Lodish. 1995. A novel serum protein similar to C1q, produced exclusively in adipocytes. J. Biol. Chem. 270:26746-26749. [DOI] [PubMed] [Google Scholar]
- 38.Shibata, R., N. Ouchi, M. Ito, S. Kihara, I. Shiojima, D. R. Pimentel, M. Kumada, K. Sato, S. Schiekofer, K. Ohashi, T. Funahashi, W. S. Colucci, and K. Walsh. 2004. Adiponectin-mediated modulation of hypertrophic signals in the heart. Nat. Med. 10:1384-1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shibata, R., K. Sato, D. R. Pimentel, Y. Takemura, S. Kihara, K. Ohashi, T. Funahashi, N. Ouchi, and K. Walsh. 2005. Adiponectin protects against myocardial ischemia-reperfusion injury through AMPK- and COX-2-dependent mechanisms. Nat. Med. 11:1096-1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sitia, R., M. Neuberger, C. Alberini, P. Bet, A. Fra, C. Valetti, G. Williams, and C. Milstein. 1990. Developmental regulation of IgM secretion: the role of the carboxy-terminal cysteine. Cell 60:781-790. [DOI] [PubMed] [Google Scholar]
- 41.Tonelli, J., W. Li, P. Kishore, U. B. Pajvani, E. Kwon, C. Weaver, P. E. Scherer, and M. Hawkins. 2004. Mechanisms of early insulin-sensitizing effects of thiazolidinediones in type 2 diabetes. Diabetes 53:1621-1629. [DOI] [PubMed] [Google Scholar]
- 42.van Anken, E., E. P. Romijn, C. Maggioni, A. Mezghrani, R. Sitia, I. Braakman, and A. J. Heck. 2003. Sequential waves of functionally related proteins are expressed when B cells prepare for antibody secretion. Immunity 18:243-253. [DOI] [PubMed] [Google Scholar]
- 43.Waki, H., T. Yamauchi, J. Kamon, Y. Ito, S. Uchida, S. Kita, K. Hara, Y. Hada, F. Vasseur, P. Froguel, S. Kimura, R. Nagai, and T. Kadowaki. 2003. Impaired multimerization of human adiponectin mutants associated with diabetes. Molecular structure and multimer formation of adiponectin. J. Biol. Chem. 278:40352-40363. [DOI] [PubMed] [Google Scholar]
- 44.Wilson-Fritch, L., A. Burkart, G. Bell, K. Mendelson, J. Leszyk, S. Nicoloro, M. Czech, and S. Corvera. 2003. Mitochondrial biogenesis and remodeling during adipogenesis and in response to the insulin sensitizer rosiglitazone. Mol. Cell. Biol. 23:1085-1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu, A., K. W. Chan, R. L. Hoo, Y. Wang, K. C. Tan, J. Zhang, B. Chen, M. C. Lam, C. Tse, G. J. Cooper, and K. S. Lam. 2005. Testosterone selectively reduces the high molecular weight form of adiponectin by inhibiting its secretion from adipocytes. J. Biol. Chem. 280:18073-18080. [DOI] [PubMed] [Google Scholar]
- 46.Yamauchi, T., J. Kamon, Y. Ito, A. Tsuchida, T. Yokomizo, S. Kita, T. Sugiyama, M. Miyagishi, K. Hara, M. Tsunoda, K. Murakami, T. Ohteki, S. Uchida, S. Takekawa, H. Waki, N. H. Tsuno, Y. Shibata, Y. Terauchi, P. Froguel, K. Tobe, S. Koyasu, K. Taira, T. Kitamura, T. Shimizu, R. Nagai, and T. Kadowaki. 2003. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 423:762-769. [DOI] [PubMed] [Google Scholar]
- 47.Yamauchi, T., J. Kamon, H. Waki, Y. Terauchi, N. Kubota, K. Hara, Y. Mori, T. Ide, K. Murakami, N. Tsuboyama-Kasaoka, O. Ezaki, Y. Akanuma, O. Gavrilova, C. Vinson, M. L. Reitman, H. Kagechika, K. Shudo, M. Yoda, Y. Nakano, K. Tobe, R. Nagai, S. Kimura, M. Tomita, P. Froguel, and T. Kadowaki. 2001. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat. Med. 7:941-946. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.