

Abstract
Recent studies have established that DNA-dependent protein kinase (DNA-PK) undergoes a series of autophosphorylation events that facilitate successful completion of nonhomologous DNA end joining. Autophosphorylation at sites in two distinct clusters regulates DNA end access to DNA end-processing factors and to other DNA repair pathways. Autophosphorylation within the kinase's activation loop regulates kinase activity. Additional autophosphorylation events (as yet undefined) occur that mediate kinase dissociation. Here we provide the first evidence that autophosphorylation within the two major clusters (regulating end access) occurs in trans. Further, both UV-induced and double-strand break (DSB)-induced phosphorylation in the two major clusters is predominately autophosphorylation. Finally, we show that while autophosphorylation in trans on one of two synapsed DNA-PK complexes facilitates appropriate end processing, this is not sufficient to promote efficient end joining. This suggests that end joining in living cells requires additional phosphorylation events that either occur in cis or that occur on both sides of the DNA-PK synapse. These data support an emerging consensus that, via a series of autophosphorylation events, DNA-PK undergoes a sequence of conformational changes that promote efficient and appropriate repair of DSBs.
Rejoining of a single double-strand break (DSB) by the nonhomologous DNA end-joining (NHEJ) pathway requires multiple distinct steps (reviewed in references 15 and 16). The DNA-dependent protein kinase (DNA-PK) is central in NHEJ because it initially binds DNA breaks or discontinuities and targets other factors to the site of damage. To assemble the DNA-PK complex, two Ku heterodimers must bind the two free DNA ends and recruit two catalytic subunits of the kinase (DNA-PKcs). The two separate DNA-PK complexes must interact, or synapse, to facilitate alignment of the two DNA ends for repair. At this synapse, DNA-PK then regulates DNA end access not only to other NHEJ factors (Artemis, polλ, polμ, XRCC4/ligaseIV, and XLF) but also to other repair pathways. Elegant structural studies have provided a low-resolution structure of a DNA-PK synapse (21). The synapse provides an accessible platform on which the two DNA ends rest. The current challenge is to decipher how DNA-PK regulates end access and promotes end joining at this synapse.
Early work demonstrated that autophosphorylation of DNA-PK results in kinase inactivation and dissociation of the kinase's catalytic subunit (DNA-PKcs) from DNA end-bound Ku (4, 11, 17). Recent efforts in defining autophosphorylation sites within the large catalytic subunit of DNA-PK (DNA-PKcs) are providing insight into how DNA-PK autophosphorylation functions to regulate DSB repair (DSBR) (1, 3, 8, 9, 12, 18, 20). Initially, we defined a major cluster of six conserved sites between residues 2609 and 2647 (termed ABCDE). Functional assays revealed that while phosphorylation at any single site within the ABCDE cluster is not critical for DNA-PK's function, alanine substitution at all six sites virtually abolishes the ability of DNA-PK to function in NHEJ, suggesting that phosphorylation of any one of the sites could suffice functionally. DNA-PKcs with all six of these sites replaced with alanine is a fully functional protein kinase which interacts appropriately with other NHEJ factors (1, 9, 18). The specific NHEJ block in cells expressing the ABCDE mutant is at the level of end processing. This was determined both by sequencing rare VDJ coding joints and rejoined I-Sce1 breaks mediated by the mutant (8, 9), as well as by biochemical analyses of the mutant protein (1, 18). More recently, we defined a second major cluster of five conserved sites between residues 2023 and 2056 (termed PQR) (8). Blocking phosphorylation at PQR by substituting all five sites with alanine (PQR mutant) results in only a modest defect in NHEJ. As with the ABCDE mutant, the NHEJ defect in the PQR mutant can be attributed to a defect in end processing. However, whereas autophosphorylation of ABCDE promotes end processing, autophosphorylation of PQR inhibits end processing. Thus, the ABCDE and PQR sites function reciprocally to regulate DNA end access (8).
Recently, we have reported an additional autophosphorylation site within the activation loop of the kinase (threonine 3950, termed T) (10). Whereas mimicking phosphorylation at the T site inactivates the kinase, phospho-mimicking does not reduce affinity of the catalytic subunit for DNA-bound Ku. This suggests that an additional autophosphorylation event(s) is responsible for kinase dissociation.
Here we demonstrate that autophosphorylation events responsible for regulating end processing occur in trans. Further, although trans autophosphorylation of a kinase-inactive mutant can facilitate end processing in living cells, it does not substantially enhance end-joining rates. These data infer that although DNA-PK's ability to regulate end processing (via ABCDE and PQR autophosphorylation) is necessary during NHEJ, efficient end joining requires additional phosphorylation events on both sides of the DNA-PK synapse. Finally, both UV- and DSB-induced phosphorylation at these sites are predominately autophosphorylation events.
MATERIALS AND METHODS
Cells and transfectants.
Derivation of V3 DNA-PKcs transfectants has been described previously (9). The DNA-PKcs mutants utilized in this study include the following: ABCDE (all six sites in the ABCDE cluster replaced with alanine) (9); PQR (all five sites in the PQR cluster replaced with alanine) (8); ABCDE+PQR (all ABCDE and all PQR sites replaced with alanine) (8); K>R (a conserved lysine residue adjacent to the ATP binding site and lysine 3752 replaced with arginine) (14); T>D (the T loop site replaced with aspartic acid) (10); ACE>D (the A, C, and E sites replaced with aspartic acid); ACET>D (the A, C, E, and T sites replaced with aspartic acid); 3′ mutant (glycine 4122 replaced with glutamine, analogous to the mutation found in the XR-C2 DSBR mutant that dramatically reduces catalytic activity) (23); isoform II (DNA-PKcs kinase-inactive splice variant that lacks amino acids 3883 to 4128) (7); isoform III (DNA-PKcs kinase-inactive splice variant that lacks amino acids 3797 to 3828) (7). The positions of these mutants and splice variants are illustrated in Fig. 1A, and their characteristics are summarized in Table 1.
FIG. 1.

DNA DSBs and DNA cross-links induce ABCDE and PQR autophosphorylation in living cells. A. Diagrammatic representation of DNA-PKcs. Autophosphorylation sites are denoted with asterisks. Thirteen previously described autophosphorylation sites [A, B, C, D, E, M, P, Q, R, and T] are as follows: A = T2609, B = T2620 + S2624, C = T2638, D = T2647, E = S2612, M = S3205, P = S2023 + S2029; Q = S2041, R = S2056 + S2053, and T = T3950. LRR denotes the leucine-rich region (13). The position of splice variation (7) in the phosphatidylinositol 3-kinase (PI3K) domain is shown. Isoform II lacks residues 3883 to 4128; isoform III lacks residues 3797 to 3828. Conserved FAT and FAT-C domains denote regions in the C terminus conserved in ATM, ATR, DNA-PKcs, and related kinases. ATP denotes a lysine residue (K3752) adjacent to the proposed ATP binding site. B. Cell extracts (100 μg) from V3 transfectants expressing wild-type DNA-PKcs (lane 1), ABCDE mutant DNA-PKcs (lane 2), or PQR mutant DNA-PKcs (lane 3) were incubated under kinase-active conditions for 20 min at room temperature. Kinase reactions were then analyzed by immunoblotting with a phospho-specific antibody that recognizes phospho-A (2609, top panel), phospho-R (2056, middle panel), or total DNA-PKcs (bottom panel). C. V3 transfectants expressing wild-type DNA-PKcs were not treated (lanes 1 and 4) or treated with OA (lanes 2 and 5), zeocin (Zeo) and OA (lane 3), MMC (lane 6), or MMC and OA (lane 7) as indicated. Cells were harvested 1 hour later, and cell extracts were analyzed as described above.
TABLE 1.
DNA-PKcs mutantsa
| Mutant | Mutation | Kinase activityb | NHEJ activityc | Reference(s) |
|---|---|---|---|---|
| Wild type | Full-length human DNA-PKcs | + | Full | 14 |
| ABCDE | ABCDE sites to alanine | + | Very low; inhibits HR | 9 |
| PQR | PQR sites to alanine | + | Slightly decreased; enhances HR | 8 |
| ABCDE+PQR | ABCDE and PQR sites to alanine | + | Very low; enhances HR | 8 |
| K>R | Kinase-inactive mutant, K3752R | − | None | 14 |
| T>D | T loop site to aspartic acid | − | None | 10 |
| ACE>D | ACE sites to aspartic acid | + | Decreased | 9 |
| ACET>D | ACE and T sites to aspartic acid | − | None | 9, 10 |
| 3′mut | Residue 4122 G to glutamine | +/− | Very low | 23 |
| Isoform II | Deletion of residues 3883 to 4128 | − | None; inhibits HR | 7 |
| Isoform III | Deletion of residues 3797 to 3828 | − | None | 7 |
The characteristics of DNA-PKcs mutants utilized in this study are summarized here.
For kinase activation, + denotes full kinase activity, − denotes no detectable kinase activity, and +/− denotes weak kinase activity.
Based on function in both IR clonogenic survival assays and transient VDJ recombination assays.
To induce double-stranded breaks or DNA cross-links in living cells, 1 × 106 cells were incubated in 2 ml serum-free medium in the presence or absence of mitomycin C (MMC) or zeocin (a bleomycin analogue) for 1 h at 37°C. Cells were harvested and cell extracts analyzed by immunoblotting. To facilitate detection of these phosphorylation events, 1 μM okadaic acid (OA) was included (or not) to inhibit dephosphorylation by cellular protein phosphatases. To induce UV damage, 0.5 × 106 cells were plated in 100-cm2 dishes 18 h prior to irradiation; at this density, cells are actively dividing. Adherent cells were rinsed twice in phosphate-buffered saline (PBS) and exposed to 30-J/m2 UVC (with no medium or PBS). Cells were harvested 1 hour later and whole-cell lysates analyzed by immunoblotting.
To transiently coexpress green fluorescent protein (GFP)-tagged kinase-inactive or wild-type DNA-PKcs in the ABCDE+PQR mutant, 10 μg expression plasmid was transiently transfected using Fugene (Roche Molecular Biochemicals, Indianapolis, IN) according to the manufacturer's recommendations. After 48 h, cells were harvested and treated as indicated with DNA-damaging agents as described above.
Kinase assays.
Whole-cell extracts (WCE) were prepared as described previously (14). A 100-μg WCE aliquot was incubated for 20 min at room temperature under kinase-active conditions (50 mM HEPES [pH 7.5], 100 mM KCl, 10 mM MgCl2, 0.2 mM EGTA, 0.1 mM EDTA, 1 mM dithiothreitol, 20 μg/ml calf thymus DNA, 10 mM ATP). Reactions were stopped by the addition of sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) sample loading buffer, and samples were analyzed by immunoblotting.
Immunoblotting.
Kinase assay mixtures or cell extracts were analyzed after electrophoresis on 4.5% SDS-polyacrylamide gels and transferred to polyvinylidene difluoride membranes. To detect total DNA-PKcs, a monoclonal antibody raised against DNA-PKcs (42-27; a generous gift of Tim Carter, St. Johns University, New York, NY) was used as the primary antibody (1:1,000 dilution), and a goat anti-mouse immunoglobulin G (IgG) was used as the secondary antibody. To detect R (2056) phosphorylation, a phospho-specific antibody to this site (Abcam, Cambridge, MA) was utilized as the primary antibody (1:1,000 dilution), and a mouse anti-rabbit IgG was used as the secondary reagent. To detect A (2609) phosphorylation, two different phospho-specific antibodies to this site (generous gifts of Dale Ramsden, University of North Carolina, and Abcam, Cambridge, MA) were utilized as the primary antibody (1:1,000 dilution) and a mouse anti-rabbit IgG used as the secondary reagent. These two antibodies gave indistinguishable results.
VDJ assays.
Extrachromosomal VDJ recombination assays were performed essentially as described previously (9), except in these assays we used recombination-activating gene (RAG) expression vectors that utilize the elongation factor 1 promoter (EF1-α; generous gift of David Roth, New York University). All assays utilized full-length, untagged RAG1 and RAG2. Briefly, 1 μg of the substrate plasmid (pJH290) that detects coding joints was cotransfected with 2 μg of full-length RAG1, 2 μg RAG2, and 5 μg total DNA-PKcs plasmid when indicated. In mixing experiments, 2.5 μg of each DNA-PKcs plasmid was transfected. At 48 h after transfection, plasmids were recovered as alkaline lysates and transformed into bacteria.
Assessment of DNA-PKcs autophosphorylation in human cell lines.
HEK293 cells were grown in suspension as described previously (10). Approximately 2 liters of cells at a cell density of approximately 1 × 107 cells/ml was either harvested without further treatment, irradiated at 10 Gy and harvested after 1 h (IR), or incubated with either the ATM-specific inhibitor KU55933 (8 μM) or the DNA-specific inhibitor NU7441/KU57788 (5 μM) for 45 min prior to irradiation at 10 Gy and then harvested after 1 h. DNA-PKcs was partially purified in the presence of protein phosphatase inhibitors and protease inhibitors as described previously (10). Approximately 1 μg of DNA-PKcs was analyzed on SDS-PAGE followed by immunoblotting with phospho-specific antibodies to serine 2056, 2609, 2612, 2620, 2624, 2638, 2647, or 3950 or an antibody to total DNA-PKcs as described previously (10, 12).
Phosphorylation of ATM targets.
HEK293 cells were incubated with either dimethyl sulfoxide or a range of concentrations of the ATM inhibitor KU55933 as indicated. After 45 min, cells were irradiated at 10 Gy (where indicated) and harvested after 1 h. Cells were lysed in 1% NP-40-containing buffer in the presence of protein phosphatase, and protease inhibitors and whole-cell extracts were prepared as described previously (10). Fifty micrograms of total protein was run on SDS-PAGE and probed with antibodies to ATM (a kind gift from M. Lavin, Queensland Institute for Medical Research), ATM-serine 1981 (Rockland Immunochemicals), p53-serine 15, Chk2-threonine 68 (Cell Signaling Inc.), or actin (Sigma-Aldrich) as indicated below. In a similar experiment, extracts were probed for phosphorylation of DNA-PKcs on serine 2056, which we and others have previously shown is a DNA damage-inducible, DNA-PK-dependent site in vivo (6, 10).
RESULTS
DNA double-strand breaks and DNA cross-links induce ABCDE and PQR autophosphorylation in living cells.
We have utilized two phospho-specific DNA-PKcs antibodies to single sites in the ABCDE and PQR clusters to study autophosphorylation of DNA-PKcs mutants generated previously in our laboratory. The relative positions of these mutations are described in Fig. 1A; the characteristics of the mutants used in this study are summarized in Table 1. To demonstrate the appropriate specificity of the phospho-specific antibodies, in vitro kinase assays were performed using whole-cell extracts from V3 transfectants expressing either wild-type DNA-PKcs, ABCDE mutant (which has alanine substitutions at all six sites in the ABCDE cluster, 2609 to 2647), or the PQR mutant (which has alanine substitutions at all five sites within the PQR cluster, 2023 to 2056) (Fig. 1B). As can be seen, whereas A (2609) phosphorylation is readily detected in the PQR mutant, R (2056) phosphorylation is not. Similarly, R (2056) phosphorylation is easily detected in the ABCDE mutant, but A (2609) phosphorylation is not. In both cases, phosphorylation of the opposing cluster is somewhat reduced compared to wild-type DNA-PKcs, even though DNA-PKcs expression is similar (Fig. 1B, bottom panel) and enzymatic activity is indistinguishable (8). These data suggest cooperativity in these two autophosphorylation events.
Next, a V3 transfectant stably expressing wild-type DNA-PKcs was treated for 1 hour as indicated with combinations of OA (a protein phosphatase inhibitor), zeocin (which induces DSBs), or MMC (which induces DNA cross-links). Cell extracts were prepared and Western analyses performed. As can be seen, phosphorylation is detected at both R (2056) and A (2609) sites in cells treated with both zeocin and OA. Similarly, phosphorylation at both R (2056) and A (2609) is detected in cells treated with MMC and OA. In both cases, detection of A (2609) phosphorylation requires inhibition of phosphatases by okadaic acid (Fig. 1C and data not shown), suggesting that phosphorylated A (2609) is more susceptible to dephosphorylation than R (2056).
DNA-PKcs is primarily autophosphorylated in response to zeocin.
Although both the ABCDE and PQR sites can clearly be autophosphorylated in vitro (3, 12, 20), it has been reported that the ABCDE sites might also be the targets of other protein kinases (22), specifically ATR (24) and ATM (5). Yajima et al. reported that ATR-dependent A (2609) and D(2647) phosphorylation [but not R (2056) phosphorylation] is induced by UV irradiation in various human cell lines (24). Chen et al. reported that ATM specifically targets sites in the ABCDE cluster in response to DSBs (5). We examined A (2609) and R (2056) phosphorylation in cells expressing wild-type DNA-PKcs, the ABCDE+PQR mutant, or K>R mutant DNA-PKcs treated with UV irradiation in the presence of okadaic acid (Fig. 2A). These experiments were initially performed with cell clones expressing equivalent levels of DNA-PKcs; however, we also utilized clones expressing higher levels of the kinase-inactive mutant (compared to wild-type DNA-PKcs or the ABCDE+PQR mutant) to optimize detection. Although robust R (2056) and A (2609) autophosphorylation is detected in cells expressing wild-type DNA-PKcs treated with UV/OA, no K>R phosphorylation is detected on either A (2609) or R (2056) in response to UV damage. To address whether the ABCDE sites can be targeted by other protein kinases in response to zeocin-induced DSBs, V3 transfectants expressing either wild-type DNA-PKcs or the kinase-inactive K>R mutant DNA-PKcs were treated or not with zeocin and OA. Both R (2056) and A (2609) phosphorylations are readily detected in cells expressing wild-type DNA-PKcs. In some experiments (five of nine), minimal A (2609) and R (2056) phosphorylation was detected in zeocin- and OA-treated cells stably expressing the K>R mutant (Fig. 2B). We conclude (at least in the hamster cell lines studied here) that both UV- and DSB-induced phosphorylations of A (2609) and R (2056) are primarily autophosphorylations. Additionally, both sites are phosphorylated in response to both zeocin and UV.
FIG. 2.

DNA-PKcs primarily autophosphorylates A (2609) and R (2056) in response to DSBs. A. Stable V3 transfectants expressing wild-type DNA-PKcs (lanes 1 and 2), ABCDE+PQR mutant DNA-PKcs (lanes 3 and 4), or K>R mutant DNA-PKcs (lanes 5 and 6) were treated or not with 30 J UV and OA. After 1 h, cell extracts were prepared and analyzed by immunoblotting as in Fig. 1B. This is representative of five independent experiments. In UV experiments, low levels of A (2609) and R (2056) phosphorylation were detected in untreated cells and were presumably an artifact that resulted from mock UV treatment of cells in the absence of medium or PBS. B. Stable V3 transfectants expressing wild-type DNA-PKcs (lanes 1 and 2) or K>R mutant DNA-PKcs (lanes 3 and 4) were treated or not with zeocin (Z) and OA. After 1 h, cell extracts were prepared and analyzed by immunoblotting as described for Fig. 1B.
Since studies suggesting ATM- and ATR-mediated phosphorylation of these sites were done primarily in human cells (5, 24), we also examined IR-induced DNA-PKcs phosphorylation in HEK293 cells. Approximately 2 liters of cells at a cell density of approximately 1 × 107 cells/ml was either harvested without further treatment, irradiated at 10 Gy and harvested after 1 h, or incubated with either the ATM-specific inhibitor KU55933 (8 μM) or the DNA-specific inhibitor NU7441/KU57788 (5 μM) for 45 min prior to irradiation at 10 Gy (Fig. 3A). DNA-PKcs was partially purified, and phosphorylation was assessed by immunoblotting with a panel of phospho-specific antibodies. As can be seen, addition of 8 μM KU55933 (a highly specific ATM inhibitor) had no effect on IR-induced phosphorylation of DNA-PKcs. In contrast, 5 μM NU7441 (a highly specific DNA-PK inhibitor) completely ablated phosphorylation at all eight phosphorylation sites examined. To make sure the ATM inhibitor effectively inhibited the ATM kinase in the treated cells under the conditions utilized, phosphorylation of several known ATM targets was examined. The results showed that at 5 to 10 μM, KU55933 significantly inhibited autophosphorylation of ATM (serine 1981) and phosphorylation of p53 (serine 15) and Chk2 (threonine 68) while having a minimal effect on DNA-PK phosphorylation at R (2056) in vivo. From these data, we conclude that phosphorylation modifications within the ABCDE and PQR clusters are primarily autophosphorylation events.
FIG. 3.

In human cells, DNA-PKcs primarily autophosphorylates the ABCDE, PQR, and T sites in response to DSBs. A. Immunoblotting of DNA-PKcs partially purified from HEK293 cells treated or not with IR and with the ATM-specific inhibitor and the DNA-PKcs specific inhibitor as described in Materials and Methods. Phospho-specific antibodies recognized phosphorylated R (2056), A (2609), E (2612), B (2620 and 2624), C (2638), D (2647), and T (3950). B. Immunoblotting with ATM, p53, Chk2, actin, and DNA-PKcs from HEK293 cells treated or not with IR and with the ATM-specific inhibitor KU55933 as described in Materials and Methods. Phospho-specific antibodies recognized phosphorylated S1981 in ATM, phosphorylated S15 in p53, phosphorylated T68 in Chk2, and phosphorylated R (2056) in DNA-PKcs.
ABCDE and PQR autophosphorylation can occur in trans in vitro and in living cells.
We next utilized an extract “mixing” approach to determine whether R (2056) and A (2609) phosphorylation could occur in trans. R (2056) and A (2609) phosphorylations are not detected in kinase assays using extracts from the kinase-inactive K>R mutant or the ABCDE+PQR mutant that has alanine substitutions at both R (2056) and A (2609) sites (Fig. 4A, top and middle panels, lanes 2 and 4). However, when extracts from the ABCDE+PQR mutant and the K>R mutant are coincubated under kinase-active conditions, R (2056) and A (2609) phosphorylations are readily detected (Fig. 4A, top and middle panels, lane 3). We conclude that R (2056) and A (2609) can occur in trans in vitro. There are several explanations for the relatively low degree of R (2056) and A (2609) phosphorylation of the K>R mutant compared to wild-type DNA-PKcs. As noted above, autophosphorylation of R (2056) and A (2609) sites is less robust in the ABCDE and PQR mutants, respectively, than in wild-type DNA-PKcs; we suggested that this might reflect cooperativity in these phosphorylation events. These data suggest that this cooperativity may also be appreciated when autophosphorylation across the synapse is blocked, explaining less-robust phosphorylation of the K>R mutant by the ABCDE+PR mutant. Additionally, it is likely that the low expression level of the K>R mutant (compared to the wild-type control) in the transfectant utilized also contributes to the less-robust phosphorylation observed.
FIG. 4.

ABCDE and PQR autophosphorylation can occur in trans in vitro and in living cells. A. WCE from V3 transfectants expressing wild-type DNA-PKcs (lane 1), the ABCDE+PQR mutant (lane 2), the ATP binding site mutant K>R (lane 4), or both (lane 3) were incubated in the presence of ATP and calf thymus DNA as described in Materials and Methods. Phosphorylation at the R site (2056) or the A site (2609) was detected with phospho-specific antibodies, as indicated. B. Cells expressing wild-type DNA-PKcs (lanes 1 and 2), GFP-K>R (lanes 3 and 4), or the ABCDE+PQR combined mutant (lanes 5 to 10) were treated or not with zeocin (Zeo). The ABCDE+PQR mutant was either untransfected (lanes 5 and 6) or transiently transfected with plasmids encoding the GFP-K>R mutant DNA-PKcs (lanes 7 and 8) or GFP-tagged wild-type DNA-PKcs (lanes 9 and 10). After a 1-hour incubation with zeocin, cell extracts were analyzed by immunoblotting with the 2056 phospho-specific antibody (top panel) or a GFP-specific antibody (bottom panel). (Note: signal in untagged wild-type DNA-PKcs [lane 2, bottom panel] is residual phospho-R reactivity from the initial probing.)
To determine whether trans phosphorylation of a kinase-inactive molecule could also occur in living cells, the plasmid encoding a GFP-tagged K>R mutant was introduced transiently into cells stably expressing high levels of the ABCDE+PQR mutant. R (2056) phosphorylation was assessed by immunoblotting (Fig. 4B, top panel); subsequently, the same filter was reprobed with a GFP-specific antibody (Fig. 4B, bottom panel). Although zeocin induces robust phosphorylation at R (2056) in cells stably expressing wild-type DNA-PKcs (untagged), R (2056) phosphorylation is minimally detected in zeocin-treated cells stably expressing K>R-GFP and not detected in cells expressing ABCDE+PQR (Fig. 4B, lanes 4 and 6). Transient expression of GFP-K>R can be detected with GFP-specific antibodies in the stable ABCDE+PQR transfectant, although expression levels are much lower than in cells stably transfected with GFP-K>R (Fig. 4B, bottom panel, compare lanes 3 and 4 to lanes 7 and 8; in this panel, residual phospho R signal from the initial immunoblotting in panel A is still present on untagged wild-type DNA-PKcs). Although the relative level of transiently expressed GFP-K>R is minimal in the ABCDE+PQR transfectant, R (2056) phosphorylation is readily detected in cells treated with zeocin. In contrast, R (2056) phosphorylation is minimal in cells expressing much higher levels of GFP-K>R alone (either treated or not). These data demonstrate that R (2056) phosphorylation can occur in trans in vitro and in living cells.
The C terminus of DNA-PKcs may be important for DNA-PK synapsis.
We have previously studied several different DNA-PKcs isoforms and mutants that lack catalytic activity. Besides the K>R (kinase-inactive) mutant, we have studied two kinase-inactive splice variants that have deletions in the extreme C terminus of the molecule (DNA-PKcs isoforms II and III) (7): a mutation at residue 4122 (glycine to glutamine) in the FAT-C domain that explains the DNA-PKcs defect in the DSBR mutant cell line XR-C2 (23) (this mutant is termed 3′ mutant), and the T-loop autophosphorylation site mutant with aspartic acid substitution (10).
We utilized a similar extract mixing approach to assess trans phosphorylation of these proteins. As can be seen, neither A (2609) nor R (2056) phosphorylation was detected in the DNA-PKcs mutant with both the ABCDE and PQR clusters ablated (ABCDE+PQR) (Fig. 5A, lanes 2 and 12) or in any of the five kinase-inactive mutants studied (Fig. 5A, lanes 7 to 10 and 14). When extracts from the K>R mutant or the 3′ mutant were coincubated with extracts from the ABCDE+PQR mutant, both A (2609) and R (2056) phosphorylations were easily detected (Fig. 5A, lanes 3 and 6). However, only weak trans phosphorylation of DNA-PKcs isoform III (Fig. 5A, lane 4) and almost no trans phosphorylation of DNA-PKcs isoform II (Fig. 5A, lane 5) were detected. Although the expression level of the T>D mutant in this stable transfectant was low, trans phosphorylation on both A (2609) and R (2056) sites in the T>D mutant was still readily detected in vitro (lane 13).
FIG. 5.

The C terminus of DNA-PKcs may be important for DNA-PK synapsis. A. WCE from V3 transfectants expressing wild-type DNA-PKcs (lanes 1 and 11), the ABCDE+PQR mutant (lanes 2 and 12), the ATP binding site mutant K>R (lane 7), kinase-inactive isoform III (lane 8), kinase-inactive isoform II (lane 9), the kinase-impaired 3′ mutant (lane 10; this mutation is representative of the DNA-PKcs mutation in the DSBR mutant cell strain XR-C2), kinase-inactive mutant T>D (lane 14), or combinations of the ABCDE+PQR mutant with kinase-inactive mutants (lanes 3 to 6 and 13) as indicated were incubated in the presence of ATP and calf thymus DNA as described in Materials and Methods. Phosphorylation at the R site (2056) or the A site (2609) was detected with phospho-specific antibodies as indicated. B. The ABCDE+PQR mutant was either untransfected (lanes 3 and 4) or transiently transfected with plasmids encoding wild-type GFP-DNA-PKcs (lanes 1 and 2), GFP-K>R mutant DNA-PKcs (lanes 5 and 6), GFP-isoform III (lanes 7 and 8), untagged T>D mutant (lanes 9 and 10), FLAG-isoform II (lanes 11 and 12), or FLAG-wild-type DNA-PKcs (lanes 13 and 14). Cells were treated or not with zeocin (Z) as indicated. After 1 hour, cells were harvested and extracts analyzed by immunoblotting with a GFP-specific antibody (top panel) or the 2056 phospho-specific antibody (bottom panel).
We also tested trans phosphorylation of these kinase-inactive DNA-PK mutants and splice variants in living cells. V3 cells stably expressing high levels of the ABCDE+PQR mutant were transiently transfected with constructs encoding GFP-tagged or FLAG-tagged wild-type DNA-PKcs, GFP-K>R mutant, GFP-isoform III, FLAG-tagged isoform II, or untagged T>D DNA-PKcs (Fig. 5B). Consistent with the in vitro kinase assays, although GFP-tagged isoform III and FLAG-tagged isoform II are easily detected, R (2056) phosphorylation is only detected in the stable ABCDE+PQR transfectants coexpressing the GFP-tagged K>R mutant, T>D mutant, or wild-type DNA-PKcs. We conclude that isoforms II and III (which contain intact phosphorylation sites but lack regions of the phosphatidylinositol 3-kinase domain) are poor targets for trans autophosphorylation both in vitro and in living cells. In contrast, the K>R mutant, T>D mutant, and 3′ mutant, all harboring point mutations in the same regions, are good targets of trans autophosphorylation. Isoforms II and III lack residues in the extreme C terminus of the molecule. Whereas isoform III lacks 30 amino acids within the kinase domain, isoform II lacks the C-terminal 245 amino acids, including the ATP binding motif as well as the conserved FAT-C domain. We have shown previously that isoforms II and III both interact stably with DNA-bound Ku (7). The data presented here suggest that regions lacking in these isoforms may be important for interaction of the kinase domain with the ABCDE and PQR clusters or possibly initial kinase synapses. Structural studies from Llorca and colleagues (21) have shown that the C terminus of DNA-PKcs has a significantly different conformation when synapsed (i.e., with two complete DNA-PK complexes) than the conformation of DNA-PKcs bound to DNA (without Ku). The FAT-C domain rotates so that it assumes a conformation that bridges the head domain to Ku (in the same complex). In turn, Ku has extensive contacts with the arm domain of DNA-PKcs (in the same DNA-PK molecule). It is this interface that appears to synapse between the two DNA-PKs, forming a platform harboring the DNA ends.
trans phosphorylation of ABCDE or PQR on one of two synapsed DNA-PK complexes does not rescue the end-joining deficits associated with the ABCDE mutant.
We next considered whether coexpression of the inactive K>R mutant (with intact phosphorylation sites that can be phosphorylated in trans) with either the ABCDE or PQR mutant could correct the end-joining deficits of the two autophosphorylation site cluster mutants as measured by transient VDJ recombination assays. The strategy employed was to perform transient assays including kinase-inactive DNA-PKcs or kinase-active autophosphorylation site mutants either alone or in 1:1 ratios with each other. In our previous studies we employed RAG expression constructs that utilize the cytomegalovirus promoter to drive RAG expression. In these studies, we utilized RAG expression constructs that utilize the EF-1α promoter (generous gift of David Roth). These plasmids induced considerably higher VDJ recombination rates than in our previous studies, significantly increasing the sensitivity of the assay and facilitating the detection of low levels of recombination observed (for example, with the ABCDE mutant or the K>R mutant).
Robust VDJ recombination is induced in V3 cells transiently transfected with these RAG constructs and a wild-type DNA-PKcs construct (Fig. 6). Consistent with our previous reports, the K>R mutant does not support significantly more coding end joining than detectable in transfectants with no DNA-PKcs at all (14). The PQR mutant has only a modest coding end-joining deficit (approximately 2-fold decreased), whereas the ABCDE mutant has a major deficit in VDJ joining (approximately 15-fold decreased). Still, ABCDE supports significantly more coding end joining (sixfold higher) than the kinase-inactive K>R mutant.
FIG. 6.

trans autophosphorylation of ABCDE or PQR on one of two synapsed DNA-PK complexes cannot rescue the end-joining deficits associated with autophosphorylation blockade or kinase activity blockade. RAG expression from plasmid vectors initiates recombination in the V3 cells, as assessed by the plasmid substrate pJH290, which detects coding joints. DNA-PKcs-encoding plasmids were included in the transient assay as indicated (described in Materials and Methods). Recombination rates from four to nine independent assays were averaged. Error bars denote standard deviations.
As a proof of principle, mixing experiments were first performed with wild-type DNA-PKcs and the K>R mutant. In the mixing experiments, total DNA-PKcs plasmid transfected was constant. Thus, in wild-type-only transfections, 5 μg of wild-type plasmid was transfected compared to only 2.5 μg wild-type plasmid in the transfection of K>R plus wild type. However, DNA-PKcs was not limiting in these experiments, because recombination rates in transfections including either 2.5 μg or 5 μg of wild type alone were indistinguishable. The mixing experiments must be interpreted cautiously. One must assume that if equimolar amounts of each construct were expressed and if the mutant constructs could access DNA ends as well as wild-type DNA-PKcs, then 25% of the coding joints would be mediated by K>R plus K>R synapses, 25% by wild type plus wild type synapses, and 50% by a mixed synapse. If a mixed synapse were similarly efficient as a wild-type-alone synapse, one would expect a modest (∼25%) decrease in recombination rate, since the K>R plus K>R synapse cannot effectively mediate joining. If the mixed synapse were significantly less efficient in promoting end joining than wild-type DNA-PKcs, VDJ recombination rates would be altered by more than 25%. When the K>R mutant is coexpressed with wild-type DNA-PKcs, recombination rates are decreased to about 25% the level of wild-type DNA-PKcs. These data suggest that the K>R mutant can access the VDJ recombination intermediates and that mixed synapses (i.e., with wild type plus K>R mutant DNA-PKcs) are significantly less efficient than wild-type-alone synapses. To further establish that the kinase-inactive K>R mutant can act as a dominant negative inhibitor of wild-type DNA-PKcs, cell lines coexpressing GFP-tagged K>R mutant and wild-type DNA-PKcs were generated by introducing the GFP-K>R construct into a clonal transfectant expressing wild-type DNA-PKcs (Fig. 6B). Coexpression of the K>R mutant induces dramatic radiosensitization of the complemented V3 transfectant (Fig. 6C). These data are consistent with the conclusion that mixed synapses (i.e., with wild-type plus K>R mutant DNA-PKcs) are significantly less efficient in facilitating repair than wild-type-alone synapses.
We also tested whether isoform II that cannot be trans phosphorylated (possibly because of defective synapsis) could also inhibit end joining. In transfections including both wild-type DNA-PKcs and isoform II, recombination rates are about 60% of the level in transfections with wild-type DNA-PKcs alone. Thus, isoform II cannot inhibit end joining as well as the K>R mutant, perhaps because of an inability to synapse with a second DNA-PK complex.
We next tested whether trans phosphorylation of the intact sites in the K>R mutant could rescue end-joining defects in either the PQR or ABCDE mutants. Rescue of end joining would suggest that trans phosphorylation both relieves the end-processing deficits associated with the mutants and that autophosphorylation of ABCDE or PQR events are the only (or most) functionally relevant DNA-PK phosphorylation events. There are numerous explanations for lack of rescue. For example, both synapsed DNA-PK complexes might require ABCDE and PQR autophosphorylation, or additional phosphorylation events might be required that must occur on both sides of the synapsed complex.
In the PQR plus K>R mixing experiments, recombination rates were reduced ∼45% compared to PQR alone. This suggests that mixed synapses (K>R plus PQR) are less efficient than synapses with two PQR mutants, similar to the result obtained when mixing K>R plus wild-type DNA-PKcs. In any case, trans phosphorylation of PQR sites in a mixed synapse with the K>R mutant does not rescue the modest end-joining defect of the PQR mutant. In contrast, in transfections with both ABCDE and K>R present, recombination rates are similar to those observed with the ABCDE mutant alone. This suggests that the K>R plus ABCDE complexes are similarly efficient in end joining as synapses with two ABCDE mutants (or perhaps slightly better, since K>R-only complexes should reduce observed recombination rates by 25%). Still, recombination rates are 15-fold below wild-type levels; thus, mixed ABCDE plus K>R synapses are significantly impaired in mediating end joining. These data suggest that efficient end joining either requires ABCDE phosphorylation on both sides of the synapse or that additional phosphorylation events must occur on both sides of the synapse.
A candidate for such a site is the T loop site that we have recently characterized (10). Extract mixing experiments using a T3950 phospho-specific antibody showed no definitive trans phosphorylation, suggesting that T loop phosphorylation may occur in cis (data not shown). The T>D mutant lacks kinase activity, presumably because its T loop mimics the phosphorylated state that inactivates the kinase. Mutational analyses suggest that T loop phosphorylation is functionally important in NHEJ (10). VDJ assays were performed coexpressing the ABCDE mutant and the kinase-inactive T>D mutant; recombination rates in mixed transfections were somewhat reduced compared to those with the ABCDE mutant alone. Thus, if the T>D mutant accurately mimics phosphorylation, T loop phosphorylation is not sufficient to restore efficient end joining by an ABCDE plus kinase-inactive DNA-PK synapse. These data suggest that either both synapsed complexes require ABCDE phosphorylation or that additional phosphorylation events must occur on both sides of the complex.
If the ABCDE cluster and the T loop were the only DNA-PK targets that are absolutely required for end joining, we reasoned that combining phospho-mimicking mutations at these sites might bypass the requirement for DNA-PK's catalytic activity in end joining. Another combination mutant replacing the A, C, E, and T sites with aspartic acid (in the same molecule) was constructed. We have shown previously that an ABCDE>asp mutant partially mimics ABCDE phosphorylation by partially reversing the end-processing block associated with the ABCDE mutation. However, in transient assays, minimal recombination rates of the kinase-inactive T>D mutant were not altered at all by including A, C, and E aspartic acid substitutions in the same construct (ACET>asp). In sum, these data suggest that additional DNA-PK-mediated phosphorylation events are requisite for end joining and that some phosphorylation events must occur on both sides of the DNA-PK synapse. In fact, we have recently shown that a purified ABCDE+PQR+T mutant (harboring 13 alanine substitutions) can still substantially autophosphorylate in vitro and still undergoes phosphorylation-dependent dissociation as well as wild-type DNA-PKcs (10).
trans phosphorylation of the ABCDE sites in the K>R mutant substantially reverses the end-processing defects of both DNA ends in the synapse.
We considered that trans autophosphorylation in the mixed synapses might relieve the end-processing defects associated with the autophosphorylation mutants without affecting end joining rates. To address this possibility, DNA was prepared and sequenced from isolated coding joints (Table 2 and data available on request). Consistent with our previous reports (8, 9), coding joints mediated by the ABCDE mutant have minimal nucleotide loss compared to joints mediated by wild-type DNA-PKcs (1.9 bp/joint versus 6.5 bp/joint), whereas coding joints mediated by the PQR mutant have more extensive nucleotide loss than joints mediated by wild-type DNA-PKcs (10.2 bp/joint versus 6.5 bp/joint). Since joining rates with the K>R mutant are approximately 6-fold lower than with the ABCDE mutant and 40-fold lower than with the PQR mutant, one must assume that K>R-only-mediated joints will comprise a minimal fraction of recovered joints. If joining rates mediated by a mixed synapse (PQR+K>R) were lower than with PQR alone (as we concluded), then joints recovered in mixed transfections would include mostly those mediated by PQR alone. Joints recovered from transfections including PQR plus K>R compared to those recovered from PQR-alone transfections are indistinguishable, consistent with the hypothesis that most joints in the mixed transfection were mediated by the more efficient synapses with two PQR mutants.
TABLE 2.
trans phosphorylation of ABCDE on one of two synapsed DNA-PK complexes substantially relieves the end-processing defect associated with blocking ABCDE phosphorylationa
| Transfectant | No. of sequences | Avg nucleotide loss/joint | % Complete ends | Joints with SSH | % Complete ends with P segments | % P segments > 2 bp |
|---|---|---|---|---|---|---|
| Wild type | 50 | 6.5 | 22 (22/100) | 64 (32/50) | 55 (12/22) | 8 (1/12) |
| RAGs only | 42 | 22.2 | 37 (31/84) | 38 (16/42) | 87 (27/31) | 71 (20/28) |
| K>R | 45 | 9.5 | 28 (25/90) | 56 (25/45) | 56 (14/25) | 21 (3/14) |
| ABCDE | 50 | 1.9 | 58 (58/100) | 18 (9/50) | 36 (18/50) | 22 (4/18) |
| ABCDE+K>R | 59 | 5.7 | 26 (31/118) | 58 (34/59) | 16 (5/31) | 0 (0/5) |
| K>R+PQR | 41 | 10 | 12 (10/82) | 68 (28/41) | 20 (2/10) | 0 (0/2) |
| PQR | 45 | 10.2 | 13 (12/90) | 73 (33/45) | 25 (3/12) | 0 (0/3) |
Data were calculated from sequences of individual coding joints. SSH, short sequence homologies.
Since recombination rates in experiments including ABCDE and K>R were similar to recombination rates in experiments with ABCDE alone, we concluded that joints mediated by this mixed synapse were similarly effective in end joining compared to synapses with two ABCDE mutants. In this scenario, joints mediated by a mixed synapse would be twice as frequent as those mediated by ABCDE alone; joints mediated by K>R alone would be infrequent, because the K>R mutant cannot support substantial end joining. In fact, joints recovered from transfections that included ABCDE and K>R compared to those including only ABCDE were remarkably different. Joints recovered from mixed transfections not only have more nucleotide loss from each joint (5.7 bp/joint compared to 1.9 bp/joint), but also the percentage of complete ends is only 26% compared to 58% in joints mediated by ABCDE alone. In sum, by this analysis, end processing is very similar in joints recovered from transfections that include both ABCDE and the K>R mutant as with those mediated by wild-type DNA-PKcs, suggesting that trans phosphorylation of ABCDE sites in the K>R mutant of a mixed ABCDE plus K>R synapse substantially reverses the block in end processing.
In a mixed synapse, ABCDE sites will only be phosphorylated on one of the two DNA-PKcs molecules. We considered that ABCDE phosphorylation might be necessary on each side of the synapse to allow end processing of both DNA ends. If this were the case, one would expect to see asymmetric processing of the two ends in each joint. To address this question, joints that have one end with at least 3-bp loss were grouped and the degree of end processing was determined for the second end. This analysis was performed on wild-type, ABCDE, and mixed (ABCDE plus K>R) sequences (Fig. 7 and data available on request). In joints mediated by wild-type DNA-PKcs, where one end has lost three or more nucleotides, the second end does not necessarily display a similar degree of nucleotide loss, and the probabilities of deleting 0 to 1 bp versus 2 to 3 bp versus 4 bp or more are roughly equivalent. In contrast, in joints mediated by the ABCDE mutant, the majority of joints that have lost three or more nucleotides have minimal nucleotide loss from the second end (∼80% of the joints lose 0 to 1 bp). Joints mediated by mixed synapses that have lost three or more nucleotides are more similar to joints mediate by a wild-type synapse. The modest evidence of asymmetry is likely explained by joints mediated by ABCDE-alone synapses, which should comprise roughly one-third of joints recovered if mixed and ABCDE-alone synapses are similarly efficient. These data suggest that trans autophosphorylation of the K>R mutant in a mixed synapse can promote end processing of both synapsed ends. However, the mixed complex is still clearly impaired in mediating end joining.
FIG. 7.
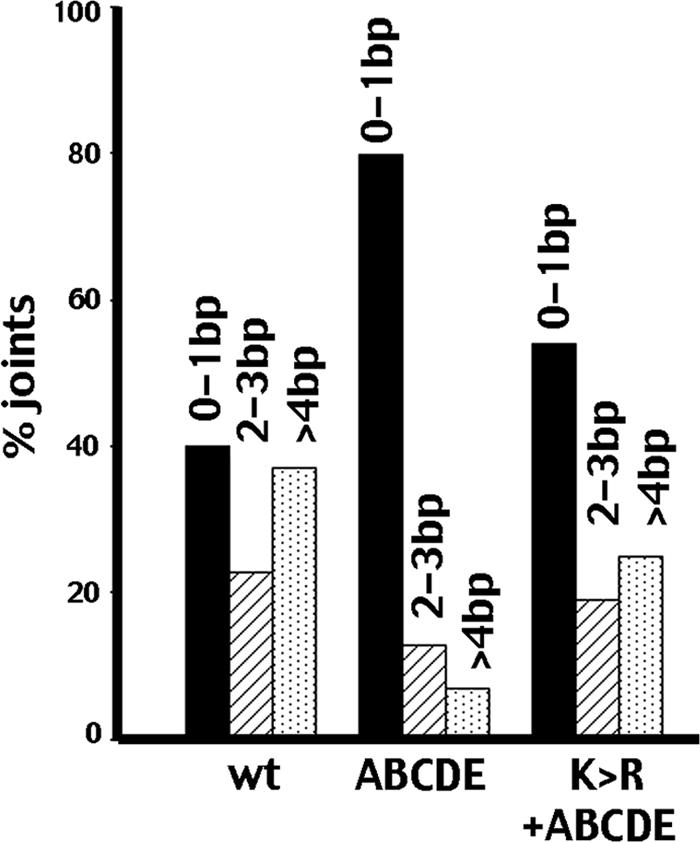
Nucleotide nibbling is not asymmetric in joints mediated by the ABCDE+K>R synapse. Sequences from wild-type-only, ABCDE-only, or ABCDE+K>R transfections (summarized in Table 1; data available on request) were analyzed as follows. All sequences that have one end with 3 bp or more deleted were grouped, and the number of the base pair missing from the second end was tabulated and grouped as follows: 0 to 1 bp (black bars); 2 to 3 bp (striped bars); 4 bp or more (stippled bars). Numbers presented are the percentages of joints with 3 bp or more deleted from one end.
Kinase-inactive DNA-PKcs protects coding ends from aberrant hairpin opening.
The higher recombination rates induced with these RAG expression vectors also allowed comparison of joints mediated by the K>R mutant compared to joints that occur in the complete absence of DNA-PKcs (i.e., classic SCID joints). SCID joints demonstrate excessive nucleotide loss and, in some joints, the presence of unusually long P segments (Table 2 and data available on request). In the RAG-only joints sequenced here, P segments are almost always present if a complete end is present (28/32 complete ends, 88%), and of those P segments, many are unusually long (20/28, 71%). Joints mediated in the presence of the K>R mutant display less nucleotide loss (9.5 versus 21.8). Moreover, P segments are much less frequent at complete ends (14/25 complete ends, 56%), and unusually long P segments are rare (3/14, 21%). This suggests that the K>R mutant prevents aberrant hairpin opening, and resulting long P segments, associated with coding end resolution in the absence of DNA-PKcs.
ABCDE phosphorylation on both sides of the synapse is required for efficient end joining.
The data presented so far demonstrate that ABCDE phosphorylation on one side of a DNA-PK synapse allows end processing to proceed fairly normally, at least in the characteristic nibbling expected during V(D)J recombination. (The rate of end processing cannot be established with these assays.) Clearly, autophosphorylation within the ABCDE cluster is requisite during NHEJ, since blocking phosphorylation at these sites dramatically disrupts NHEJ. We considered two possibilities regarding the importance of ABCDE autophosphorylation. First, ABCDE phosphorylation promotes end processing; one possibility is that blocking this function completely explains the severe NHEJ defect observed in cells expressing the ABCDE mutant. Alternatively, ABCDE phosphorylation may represent an initial event that induces a conformational change required to progress to further steps in NHEJ. With the first possibility, ABCDE coexpressed with wild-type DNA-PKcs should not act as a dominant negative, because the mutant would efficiently phosphorylate wild-type DNA-PKcs in trans, promoting end processing. Further steps in NHEJ should progress normally, since both the wild type and the ABCDE mutant are fully functional protein kinases that interact with other NHEJ factors and have all other autophosphorylation sites intact. If the second hypothesis were correct and ABCDE phosphorylation on both sides of a synapse were required to induce a conformational change that promotes subsequent steps in NHEJ, then the ABCDE mutant would functionally inhibit wild-type DNA-PKcs. This was tested in transient VDJ recombination assays (Fig. 6A). As can be seen, the ABCDE mutant inhibits coding joint formation similar to the kinase-inactive K>R mutant, suggesting that an ABCDE plus wild type synapse is just as defective in promoting end joining as a K>R plus wild type synapse. These data are consistent with a model whereby autophosphorylation within the ABCDE cluster induces a conformational change in the DNA-PK synapse that is required to progress to further steps in NHEJ.
DISCUSSION
Although it has been postulated that DNA-PK's autophosphorylation occurs in trans, this is the first direct demonstration of trans autophosphorylation of the DNA-PK complex. Llorca and colleagues surmised from cryo-electron microscopy studies that the ABCDE sites “without very substantial remodeling, would be inaccessible to the active site of the same molecule so that autophosphorylation by DNA-PKcs occurs in trans via interaction with a second molecule” (2, 19). However, in later elegant studies of the synapsed structure from the same authors, the catalytic domain is quite distant from the probable locations of both the ABCDE and PQR clusters of the opposing molecule (21). Certainly, the DNA-PK complex must assume several conformations during end joining. From an examination of the available structure, it is possible that a rotation around the structural interface of the synapse might accommodate interaction of the catalytic domain with the opposing phosphorylation sites (O. Llorca, personal communication). Alternatively, although synapsis has been linked to kinase activation, it is possible that trans phosphorylation occurs during kinase assembly prior to formation of a stable complex. While our data support a trans mechanism for phosphorylation within the major two clusters that regulate DNA end processing, it is still unclear whether T loop autophosphorylation and autophosphorylation events that mediate kinase dissociation occur in cis or trans.
Work presented here and reported previously (1, 4, 7-12, 17, 18) suggests the following model for DNA-PK's function during NHEJ. After initial binding of two Ku molecules and two DNA-PKcs subunits to the two DNA ends at a single DSB, the assembled DNA-PK complexes mediate DNA end synapsis. Because of the relative abundance of the DNA-PK subunits, it is likely that most DSBs are initially bound by DNA-PK. We have shown previously that DNA-PK's autophosphorylation status affects whether NHEJ or homologous recombination (HR) repairs a particular DSB (7, 8); it follows that every DSB initially bound by DNA-PK is not necessarily destined to be repaired by NHEJ. In the initial bound state (prior to ABCDE phosphorylation), DNA ends are protected from exonuclease activities. trans ABCDE phosphorylation on one side of a DNA-PK synapse promotes end processing of both DNA ends; synapsed ends are processed concurrently perhaps in the same vicinity, implying coupling of end processing with end pairing. PQR phosphorylation (in trans) may function to limit further end processing and to specifically promote NHEJ (and not HR) once end processing generates ligatable ends. This hypothesis is supported by three findings. First, PQR phospho-mimics allow minimal end processing while supporting completely wild-type levels of end joining. Second, PQR phosphorylation is absolutely required to block DNA end access to the HR pathway, regardless of the status of the ABCDE sites. Finally, the ABCDE mutant has a mild dissociation defect (autophosphorylation induced); however, this deficit is reversed by blocking PQR phosphorylation (10), suggesting that PQR phosphorylation helps maintain DNA end binding, consistent with a role in promoting subsequent steps in NHEJ. Although ABCDE autophosphorylation on one side of a DNA-PK synapse promotes end processing, the ABCDE cluster must be phosphorylated on both sides of the synapse to promote efficient end joining. We propose that ABCDE phosphorylation induces a conformational change that is required to progress to further steps in NHEJ. In contrast, although PQR phosphorylation protects DNA ends from excessive end processing, end joining is fairly efficient even when PQR sites cannot be phosphorylated. Activation (T) loop autophosphorylation results in kinase inactivation (but not dissociation from Ku bound to DNA) and is likely a late event during NHEJ; T phosphorylation is also required for efficient end joining (10). Finally, mutational analyses (presented here and previously) strongly suggest that additional (as-yet-undefined) phosphorylation events are required to promote efficient end joining. Work ongoing in our laboratories is focused at defining these events. In sum, these data reiterate an emerging consensus in this field: DNA-PK must undergo a series of autophosphorylation events that induce distinct conformations that allow DNA-PK to orchestrate the many distinct steps required to facilitate DNA double-strand break repair.
Acknowledgments
This work was supported by Public Health Service grant AI048758 (K.M.) and CIHR grant 13969 to S.P.L.-M.
We thank David Roth for RAG expression vectors and Dale Ramsden for his anti-phospho-2609 reagent. We thank Graeme Smith (KuDos Pharmaceuticals) for the kind gift of KU59933. We thank the MRC Protein Phosphorylation Unit, University of Dundee, for assistance in growing the HEK293 cells. We are especially grateful to Oscar Llorca for his input in considering structural implications of these data.
Footnotes
Published ahead of print on 12 March 2007.
REFERENCES
- 1.Block, W., Y. Yu, D. Merkle, J. Gifford, Q. Ding, K. Meek, and S. P. Lees-Miller. 2004. Autophosphorylation-dependent remodeling of the DNA-dependent protein kinase catalytic subunit (DNA-PKcs) regulates ligation of DNA ends. Nucleic Acids Res. 32:4351-4357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boskovic, J., A. Rivera-Calzada, J. D. Maman, P. Chacon, K. R. Willison, L. H. Pearl, and O. Llorca. 2003. Visualization of DNA-induced conformational changes in the DNA repair kinase DNA-PKcs. EMBO J. 22:5875-5882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chan, D. W., B. P. Chen, S. Prithivirajsingh, A. Kurimasa, M. D. Story, J. Qin, and D. J. Chen. 2002. Autophosphorylation of the DNA-dependent protein kinase catalytic subunit is required for rejoining of DNA double-strand breaks. Genes Dev. 16:2333-2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan, D. W., and S. P. Lees-Miller. 1996. The DNA-dependent protein kinase is inactivated by autophosphorylation of the catalytic subunit. J. Biol. Chem. 271:8936-8941. [DOI] [PubMed] [Google Scholar]
- 5.Chen, B. P., N. Uematsu, J. Kobayashi, Y. Lerenthal, A. Krempler, H. Yajima, M. Lobrich, Y. Shiloh, and D. J. Chen. 2006. Ataxia telangiectasia mutated (ATM) is essential for DNA-PKcs phosphorylations at the Thr-2609 cluster upon DNA double strand break. J. Biol. Chem. 282:6582-6587. [DOI] [PubMed] [Google Scholar]
- 6.Chen, B. P., D. W. Chan, J. Kobayashi, S. Burma, A. Asaithamby, K. Morotomi-Yano, E. Botvinick, J. Qin, and D. J. Chen. 2005. Cell cycle dependence of DNA-dependent protein kinase phosphorylation in response to DNA double strand breaks. J. Biol. Chem. 280:14709-14715. [DOI] [PubMed] [Google Scholar]
- 7.Convery, E., E. K. Shin, Q. Ding, W. Wang, P. Douglas, L. S. Davis, J. A. Nickoloff, S. P. Lees-Miller, and K. Meek. 2005. Inhibition of homologous recombination by variants of the catalytic subunit of the DNA-dependent protein kinase (DNA-PKcs). Proc. Natl. Acad. Sci. USA 102:1345-1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cui, X., T. Yu, S. Gupta, Y. M. Cho, S. P. Lees-Miller, and K. Meek. 2005. Autophosphorylation of DNA-dependent protein kinase regulates DNA end processing and may also alter double-strand break repair pathway choice. Mol. Cell. Biol. 25:10842-10852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ding, Q., Y. V. Reddy, W. Wang, T. Woods, P. Douglas, D. A. Ramsden, S. P. Lees-Miller, and K. Meek. 2003. Autophosphorylation of the catalytic subunit of the DNA-dependent protein kinase is required for efficient end processing during DNA double-strand break repair. Mol. Cell. Biol. 23:5836-5848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Douglas, P., W. D. Block, X. Cui, Y. Yu, Q. Ding, R. Ye, S. Gupta, N. Morrice, S. P. Lees-Miller, and K. Meek. 2007. The DNA-dependent protein kinase catalytic subunit (DNA-PKcs) is phosphorylated in vivo on threonine 3950, a highly conserved amino acid in the protein kinase domain. Mol. Cell. Biol. 27:1581-1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Douglas, P., G. B. Moorhead, R. Ye, and S. P. Lees-Miller. 2001. Protein phosphatases regulate DNA-dependent protein kinase activity. J. Biol. Chem. 276:18992-18998. [DOI] [PubMed] [Google Scholar]
- 12.Douglas, P., G. P. Sapkota, N. Morrice, Y. Yu, A. A. Goodarzi, D. Merkle, K. Meek, D. R. Alessi, and S. P. Lees-Miller. 2002. Identification of in vitro and in vivo phosphorylation sites in the catalytic subunit of the DNA-dependent protein kinase. Biochem. J. 368:243-251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gupta, S., and K. Meek. 2005. The leucine rich region of DNA-PKcs contributes to its innate DNA affinity. Nucleic Acids Res. 33:6972-6981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kienker, L. J., E. K. Shin, and K. Meek. 2000. Both V(D)J recombination and radioresistance require DNA-PK kinase activity, though minimal levels suffice for V(D)J recombination. Nucleic Acids Res. 28:2752-2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lees-Miller, S. P., and K. Meek. 2003. Repair of DNA double strand breaks by non-homologous end joining. Biochemie 85:1161-1173. [DOI] [PubMed] [Google Scholar]
- 16.Meek, K., S. Gupta, D. A. Ramsden, and S. P. Lees-Miller. 2004. DNA-PK: activity director at the end. Immunol. Rev. 200:132-141. [DOI] [PubMed] [Google Scholar]
- 17.Merkle, D., P. Douglas, G. B. Moorhead, Z. Leonenko, Y. Yu, D. Cramb, D. P. Bazett-Jones, and S. P. Lees-Miller. 2002. The DNA dependent protein kinase interacts with DNA to form a protein DNA complex that is disrupted by phosphorylation. Biochemistry 41:12706-12714. [DOI] [PubMed] [Google Scholar]
- 18.Reddy, V., Q. Ding, S. P. Lees-Miller, K. Meek, and D. A. Ramsden. 2004. Nonhomologous end-joining requires the DNA-PK complex to undergo remodeling at DNA ends. J. Biol. Chem. 279:39408-39413. [DOI] [PubMed] [Google Scholar]
- 19.Rivera-Calzada, A., J. D. Maman, L. Spagnolo, L. H. Pearl, and O. Llorca. 2005. Three-dimensional structure and regulation of the DNA-dependent protein kinase catalytic subunit (DNA-PKcs). Structure 13:243-255. [DOI] [PubMed] [Google Scholar]
- 20.Soubeyrand, S., L. Pope, B. Pakuts, and R. J. Hache. 2003. Threonines 2638/2647 in DNA-PK are essential for cellular resistance to ionizing radiation. Cancer Res. 63:1198-1201. [PubMed] [Google Scholar]
- 21.Spagnolo, L., A. Rivera-Calzada, L. H. Pearl, and O. Llorca. 2006. Three-dimensional structure of the human DNA-PKcs/Ku70/Ku80 complex assembled on DNA and its implications for DNA DSB repair. Mol. Cell 22:511-519. [DOI] [PubMed] [Google Scholar]
- 22.Wechsler, T., B. P. Chen, R. Harper, K. Morotomi-Yano, B. C. Huang, K. Meek, J. E. Cleaver, D. J. Chen, and M. Wabl. 2004. DNA-PKcs function regulated specifically by protein phosphatase 5. Proc. Natl. Acad. Sci. USA 101:1247-1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woods, T., W. Wang, E. Convery, A. Errami, M. Z. Zdzienicka, and K. Meek. 2002. A single amino acid substitution in DNA-PKcs explains the novel phenotype of the CHO mutant, XR-C2. Nucleic Acids Res. 30:5120-5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yajima, H., K. J. Lee, and B. P. Chen. 2006. ATR-dependent DNA-PKcs phosphorylation in response to UV-induced replication stress. Mol. Cell. Biol. 26:7520-7528. [DOI] [PMC free article] [PubMed] [Google Scholar]