

Abstract
A number of studies have identified cytosolic prostaglandin E2 synthase (cPGES)/p23 as a cytoplasmic protein capable of metabolism of prostaglandin E2 (PGE2) from the cyclooxygenase metabolite prostaglandin endoperoxide (PGH2). However, this protein has also been implicated in a number of other pathways, including stabilization of the glucocorticoid receptor (GR) complex. To define the importance of the functions assigned to this protein, mice lacking detectible cPGES/p23 expression were generated. cPGES/p23−/− pups die during the perinatal period and display retarded lung development reminiscent of the phenotype of GR-deficient neonates. Furthermore, GR-sensitive gluconeogenic enzymes are not induced in the prenatal period. However, unlike GR-deficient embryos, cPGES/p23−/− embryos are small and a proliferation defect is observed in cPGES/p23−/− fibroblasts. Analysis of arachidonic acid metabolites in embryonic tissues and primary fibroblasts failed to support a function for this protein in PGE2 biosynthesis. Thus, while the growth retardation of the cPGES/p23−/− pups and decreased proliferation of primary fibroblasts identify functions for this protein in addition to GR stabilization, it is unlikely that these functions include metabolism of PGH2 to PGE2.
Prostaglandin E2 (PGE2) is a lipid mediator involved in a wide range of normal biological functions as well as pathological processes such as chronic inflammation. Biosynthesis begins with the metabolism of arachidonic acid into prostaglandin endoperoxide (PGH2) by cyclooxygenase enzymes (COX-1 or COX-2). Three terminal synthase enzymes capable of producing PGE2 from COX-derived PGH2 in vitro have been reported. These include microsomal PGE2 synthase 1 (mPGES1), microsomal PGE2 synthase 2 (mPGES2), and the cytosolic PGE2 synthase (cPGES) (24, 59, 60). mPGES1 is a member of the MAPEG (membrane-associated proteins involved in eicosanoid and glutathione metabolism) protein superfamily, and its expression increases rapidly in many cells after exposure to proinflammatory cytokines and lipopolysaccharide (LPS) (24, 62, 65). Not surprisingly, in vivo studies of mice lacking this gene indicate that this enzyme is responsible for the increased levels of PGE2 measured in many inflammatory responses (62). However, PGE2 production can still be detected in the mPGES1-deficient animals, supporting the contention that additional pathways are present that, at least under some circumstances, can produce PGE2. One candidate is mPGES2. Similar to mPGES1, mPGES2 is a microsomal protein. However, as the expression of the gene coding for this protein is not induced by LPS treatment and increased levels are not observed in inflamed tissues, it has been suggested that this synthase contributes primarily to maintenance of basal PGE2 levels (38).
The third protein capable of in vitro PGE2 production was termed cPGES, reflecting the fact that, unlike the other two synthases, this protein was purified from rat brain cytosolic extracts. Similar to mPGES1, this synthase was shown to have glutathione-dependent PGE2 synthase activity (60). cPGES is expressed in many cell lines and in a wide range of tissues. Similar to mPGES2, expression of the gene is generally not sensitive to inflammatory stimuli, with one notable exception. Tanioka et al. demonstrated that LPS treatment increased levels of cPGES threefold in the brain (60). Cotransfection of human cPGES cDNA with COX-1, but not with COX-2, resulted in a large increase in PGE2 production (60). These findings led to the suggestion that in most cells COX-1 collaborates with cPGES in basal PGE2 production.
Peptide mapping demonstrated that cPGES was identical to human p23, a highly conserved protein that functions as a cochaperone for Hsp90 (25, 55, 60). The Hsp90 chaperone system has been shown to be critical for the function of a variety of hormone and growth factor receptors, including glucocorticoid receptor (GR), mineralocorticoid receptor, androgen receptor, epidermal growth factor receptor, and platelet-derived growth factor receptor (48). Steroid receptors, particularly GR, are used extensively to model the interactions of p23 with Hsp90 chaperone system and its client proteins. An important function of the Hsp90 complex is to facilitate folding of the hormone binding domain to a conformation capable of binding substrate. GR forms a heterocomplex with Hsp90, Hsp70, and p60, which are sufficient to carry out this folding change. p23 has been reported to increase the stability and hormone binding activity of the GR/Hsp90 complex (12). p23 binds directly to Hsp90 with bound ATP to stabilize the GR/Hsp90 complex from inactivation and disassembly.
GR is a ligand-activated transcription factor that mediates the biological effects of glucocorticoids. Upon steroid binding, GR translocates into the nucleus, dimerizes, and binds to glucocorticoid response elements (GREs) upstream of specific target genes. GR has also been shown to have DNA-binding-independent mechanisms of gene regulation through direct protein-protein interactions, or tethering, of transcription factors, such as AP-1 and NF-κB (40). Many of these glucocorticoid-regulated genes, such as the epithelial sodium channel subunit γ (ENaCγ) and the surfactant proteins (SP-A, SP-B, and SP-C), are important in the final stages of mouse lung development. These genes are responsible for the production of lung surfactant, which increases alveolar stability by decreasing surface tension, and the clearance of lung liquid to improve lung tissue compliance and gas exchange upon birth (33, 70). Glucocorticoids are also known to promote the differentiation of alveolar epithelial cells (AECs) and development of terminal alveoli required for the transition to air breathing (30, 56).
A multifunctional role has been proposed for cPGES/p23 as both a terminal PGE2 synthase and a cochaperone for the GR/Hsp90 complex. In addition, many other functions linked to its association with Hsp90 have been assigned to this protein. To date, the role of cPGES/p23 in these processes has been primarily characterized in vitro, so the in vivo role of cPGES/p23 remains unclear. We have generated a mouse line deficient in cPGES/p23 to better characterize this protein and elucidate its role in vivo. The cPGES/p23 deficiency causes perinatal lethality due to improper lung maturation. We demonstrate that cPGES/p23 is required for both DNA-binding-dependent and -independent GR functions. However, additional phenotypes of this animal suggest GR-independent functions for this protein in development. Characterization of primary embryonic fibroblasts derived from the cPGES/p23-deficient mice, as well as embryonic tissues, demonstrates that cPGES/p23 is not required for PGE2 synthesis.
MATERIALS AND METHODS
Experimental animals.
All studies were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (23a) as well as the Institutional Animal Care and Use Committee guidelines of the University of North Carolina at Chapel Hill.
Generation of cPGES/p23-deficient mice.
A mouse ES cell line (RST271) containing an insertional mutation in cPGES/p23 was obtained from BayGenomics, a gene-trapping resource (57). A gene-trap vector, pGTOTMpfs, was used that contained a splice-acceptor sequence upstream of a reporter gene, β-geo (a fusion of β-galactosidase and neomycin phosphotransferase II genes). This vector was inserted into the first intron of cPGES/p23, creating a fusion transcript containing sequences from the first exon joined to the β-geo marker. 5′ rapid amplification of cDNA ends followed by automated DNA sequencing was used to determine the identity of the trapped gene as cPGES/p23. Male chimeras generated from this ES cell line were bred to B6/D2 females to generate mice heterozygous for the mutated allele. These heterozygotes were intercrossed to produce mice homozygous for the mutation. Pregnant heterozygous females were euthanized with CO2, and embryos were removed by cesarean section. The genotypes of the offspring were determined by Southern blot analysis. A specific probe was designed 3′ of exon 2 and used to screen genomic DNA digested with PvuII. Insertion of the vector in the first intron results in a reduced fragment size for the mutant allele compared to the endogenous wild-type allele.
Northern blot analysis.
Total RNA was isolated from embryonic day 18.5 (E18.5) embryos with RNA-Bee (Tel-Test, Inc.) according to the manufacturer's instructions. Total RNA (20 μg) was separated by gel electrophoresis on a 1.2% agarose/formaldehyde gel and transferred to a nitrocellulose membrane (Immobilon NC; Amersham Biosciences) overnight. Northern blots were hybridized with a [32P]dCTP (Amersham Biosciences)-labeled cPGES cDNA probe prepared with a random primed labeling kit (Roche Diagnostics). The cPGES cDNA probe was generated from commercially available expressed sequence tag clones (Invitrogen Laboratories). Membranes were hybridized with a β-actin cDNA probe to verify similar RNA loading.
Prostanoid measurements in whole embryos and embryo lungs.
Tissues were flash frozen in liquid nitrogen and stored in the −80°C freezer until use. Samples were pulverized and then homogenized in 1× phosphate-buffered saline (PBS) with 1 mM EDTA (pH 7.4) and 10 mM indomethacin and sonicated. Lipids were purified through an Amprep octadecyl C18 mini column (Amersham Biosciences), and prostanoid content was determined by using enzyme immunoassay kits (Assay Designs) according to the manufacturer's instructions.
Generation of MEFs.
E13.5 embryos were used to generate mouse embryonic fibroblasts (MEFs). The embryos were washed with sterile PBS, finely minced, and trypsinized twice for 30 min in a 37°C H2O bath. Tissue debris was discarded, and the fibroblasts were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum and supplemented with 100 μg/ml streptomycin sulfate and 100 units/ml penicillin G sodium at 37°C with 5% CO2.
Prostanoid measurements in MEFs.
MEFs were seeded in nickel-well plates (105 cells/well) and grown to 80% confluence. Fibroblasts were washed and cultured in 1.5 ml serum-free medium for 1 h. Supernatant was collected, and the cells were trypsinized and pelleted for total protein determination by the bicinchoninic acid (BCA) method (Pierce). Enzyme immunoassay kits (Assay Designs) were used to determine the amount of PGE2 and thromboxane B2 (TxB2) in the supernatants. All assays were performed in triplicate, and cells treated with indomethacin were used as a negative control.
Proliferation assay.
Carboxyfluorescein diacetate succinimidyl ester (CFSE [CellTrace cell proliferation kit; Invitrogen]) was used to stain primary fibroblasts to monitor cell proliferation. Primary fibroblasts were plated in a six-well plate, and 16 h later, the cells were stained with 5 μM CFSE according to the manufacturer's instructions. Cells were harvested, counted, and analyzed with a FACScalibur flow cytometer (Becton Dickinson) and CellQuest software at the indicated time points. The doubling time has been calculated by using the equation Yt = Y(o)·2nt, where Yt = number of cells at time t, Y(o) = initial number of cells, and n = number of cell cycles/hour.
Light microscopy and histological analysis.
Tissues were removed from embryos, fixed overnight in 10% formalin, and embedded in paraffin. Sections of lung and liver tissues were stained with hematoxylin and eosin for routine histology. Liver sections were also stained with periodic acid-Schiff stain (PAS) for glycogen content. Sections were observed using a light microscope (Nikon FXA).
Transmission electron microscopy.
Lungs were dissected from E18.5 embryos and fixed in 2% paraformaldehyde-2.5% glutaraldehyde in 0.15 M sodium phosphate (pH 7.4). Following three rinses with phosphate buffer, the samples were postfixed for 1 h in 1% osmium tetroxide-1.25% potassium ferrocyanide in sodium phosphate buffer. The tissues were rinsed in deionized water and dehydrated through increasing concentrations of ethanol (30, 50, 75, 100, and 100% for 10 min each) and put through two changes of propylene oxide (15 min each). Tissue samples were infiltrated with a 1:1 mixture of propylene oxide-Polybed 812 epoxy resin (1A:2B formulation; Polysciences, Inc., Warrington, PA) for 3 h followed by an overnight infiltration in 100% resin. The tissue pieces were embedded in fresh epoxy resin and polymerized for 24 h at 60°C. The blocks were trimmed, and 1-μm sections of the tissue were mounted on glass slides and stained with 1% toluidine blue O in 1% sodium borate. Representative areas were selected by light microscopy, and 70-nm ultrathin sections were cut using a diamond knife. Ultrathin sections were collected on 200 mesh copper grids and stained with 4% aqueous uranyl acetate for 15 min, followed by Reynolds' lead citrate for 7 min. Sections were observed using a LEO EM910 transmission electron microscope at 80 kV (LEO Electron Microscopy, Thornwood, NY) and photographed using a Gatan Bioscan digital camera (Gatan, Inc., Pleasanton, CA).
Isolation of RNA and quantitative RT-PCR.
For quantitative real-time PCR (RT-PCR), lung tissue was removed from E18.5 embryos and total RNA was isolated by RNA-Bee (Tel-Test, Inc.) according to the manufacturer's instructions. The RNA concentration was determined by A260. RNA was purified using the RNeasy mini kit (QIAGEN), and 5 μg was used to generate cDNA with the high-capacity cDNA Archive kit (Applied Biosystems). cDNA (2.5 ng) was used in amplification reactions with Taqman PCR universal mastermix and commercially available primer and probe sets (Applied Biosystems) on an ABI Prism 7900 thermocycler. Expression levels were normalized to an endogenous control, β-actin. Quantification of samples was performed using the comparative cycle threshold (ΔΔCT) method, as described in the Assays-on-Demand Users Manual (Applied Biosystems, Foster City, CA). Relative gene expression was determined by the formula 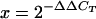 . Results were expressed as change (fold) relative to wild-type expression levels.
. Results were expressed as change (fold) relative to wild-type expression levels.
Western blot analysis.
Total protein was isolated from tissue by homogenizing frozen tissue in lysis buffer (50 mM HEPES, 0.15 M NaCl, 1.5 mM MgCl2, 1% Triton X-100, 10% glycerol, 10 μg/ml leupeptin, 500 μM Na3VO4, 0.1 M phenylmethlysulfonyl fluoride, Roche Complete mini protease inhibitor tablet). Homogenates were centrifuged at 14,000 rpm for 5 min at 4°C, and protein content of the supernatant was measured using the BCA assay (Pierce). A quantity of 75 μg of protein per sample was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and electrophoretically transferred onto polyvinylidene difluoride membranes (Hybond-P; Amersham Biosciences). Membranes were blocked in 5% nonfat milk in 1× TBST (Tris-buffered saline and 0.05% Tween 20) for 1 h at room temperature. Immunoblots were probed with primary antibody to GR (Abcam [ab3579, dilution of 2.5 μg/ml, incubated at 4°C overnight]), cPGES/p23 (Cayman [dilution of 1:500, incubated at room temperature for 1 h), and β-actin (Sigma [dilution of 1:5,000, incubated at room temperature for 1 h]) followed by goat anti-rabbit or goat anti-mouse secondary antibody (dilutions of 1:5,000 and 1:10,000, respectively) for 45 min at room temperature. Membranes were incubated with ECL enhanced chemiluminescence reagent, and signal was detected by autoradiography after exposure to Hyperfilm (Amersham Biosciences).
For the nuclear localization analysis, 1.5 × 106 fibroblasts were starved from serum for 24 h and activated with 10 nM dexamethasone for 0, 30, and 60 min. Cell pellet was resuspended in 200 μl of lysis buffer (Hank's balanced salt solution buffer, 1.26 mM calcium, 1 mM magnesium, 0.1% NP-40, 200 nM phenylmethylsulfonyl fluoride, and Roche Complete mini protease inhibitor tablet) and kept on ice for 20 min before centrifugation at 700 × g for 20 min at 4°C. Nuclear pellet was washed in 200 μl lysis buffer, centrifuged at 700 × g for 20 min at 4°C, resuspended in protein sample buffer, and processed for Western blotting. Cytoplasmic supernatant was recentrifuged at 12,000 × g for 20 min, and the supernatant was mixed with 80 μl of protein sample buffer and processed for Western blotting. GR was detected on Western blotting by M20 antibody (Santa Cruz Biotechnology). The blots were reprobed with antibodies to α-tubulin (Sigma) and histone H1 (Santa Cruz Biotechnology).
Transfection assay.
Primary fibroblasts were immortalized by transfection with pSV3-neo (ATCC), a plasmid expressing simian virus 40 large T antigen. Transformed fibroblasts (∼2 × 105 cells) were seeded in a six-well plate, and after 24 h, fibroblasts were transfected with either 3 μg of pGRE-luc (Clontech) and 0.05 μg of pCMVβ (Applied Biosystems) or 3 μg of pTAL-luc (Clontech) and 0.05 μg of pCMVβ using Lipofectamine Plus reagent (Invitrogen) according to the manufacturer's instructions. After transfection, the fibroblasts were incubated with dexamethasone (10 nM) for 24 h. Cell lysates were collected and analyzed for luciferase and β-galactosidase expression using the dual-light luciferase/β-galactosidase reporter gene assay kit (Applied Biosystems). Luciferase activity was normalized to β-galactosidase activity. Negative control values from the transformed fibroblasts transfected with pTAL-luc were subtracted from the experimental values.
Immunofluorescence staining of transformed MEFs for GR.
Coverslips (no. 1; 12-mm circle; thickness range, 0.13 to 0.17 mm [Fisher Scientific, Pittsburgh, PA]) were coated with 0.1% gelatin. Cells were resuspended in growth medium at a final concentration of 4 × 104 cells/ml. Five hundred or 1,000 μl of suspension was applied to a coverslip in a 24-well plate, and the cells were allowed to adhere for 3 h at 37°C. Cultivation medium was replaced by medium without serum, and cells were starved for 16 h. Cells were activated by 10 nM dexamethasone for 0, 30, and 60 min in medium without serum. Activation was stopped by ice-cold PBS, and cells were immediately fixed by methanol for 20 min at −20°C and permeabilized for 1 min by acetone at −20°C. Cells were blocked in blocking buffer (20% fetal bovine serum, 5% bovine serum albumin, 0.05% gelatin) for 30 min at room temperature and incubated with GR antibody (M-20; Santa Cruz Biotechnnology Inc., Santa Cruz, CA) diluted 1:150 in TBST buffer (10 mM Tris, 150 mM NaCl, 0.1% Tween) overnight at 4°C. Coverslips were washed three times in TBST buffer and incubated with fluorescein isothiocyanate-conjugated antirabbit antibody (Jackson ImmunoResearch, West Grove, PA) diluted 1:100 at room temperature for 1 h. Coverslips were washed three times with TBST buffer and two times with PBS, dried, mounted on slides (SuperFrost Plus microscope slides; Fisher Scientific) in mounting solution, and examined with a Nikon Microphot FXA upright fluorescence microscope (Nikon, Inc., Garden City, NY) using a ×60 objective. ImageJ software (NIH) was used to measure the mean pixel density in the nucleus.
Statistical analysis.
Values are presented as mean ± standard error of the mean and are analyzed by Student's two-tailed t test (Mann-Whitney or Tukey-Kramer test for multiple comparisons where indicated). A P value of <0.05 is considered statistically significant.
RESULTS
Generation of cPGES/p23−/− mice.
Mouse ES cells with a gene trap inserted in the first intron of cPGES/p23 were obtained from BayGenomics (57). The gene trap contains splice acceptors, which direct anomalous splicing of the primary transcript, resulting in loss of cPGES/p23 expression. Male chimeras generated from this ES cell line were bred to B6/D2 females to generate mice heterozygous for the mutated allele. No pups homozygous for the mutant allele were identified at weaning in litters obtained by intercross of these heterozygous animals. In contrast, Southern blot analysis of E18.5 embryos demonstrated the presence of homozygous fetuses at normal Mendelian frequency (Fig. 1A). Examination and monitoring of neonates indicated that at least some cPGES/p23−/− pups were alive at birth, but these died shortly thereafter, after taking several agonal breaths.
FIG. 1.
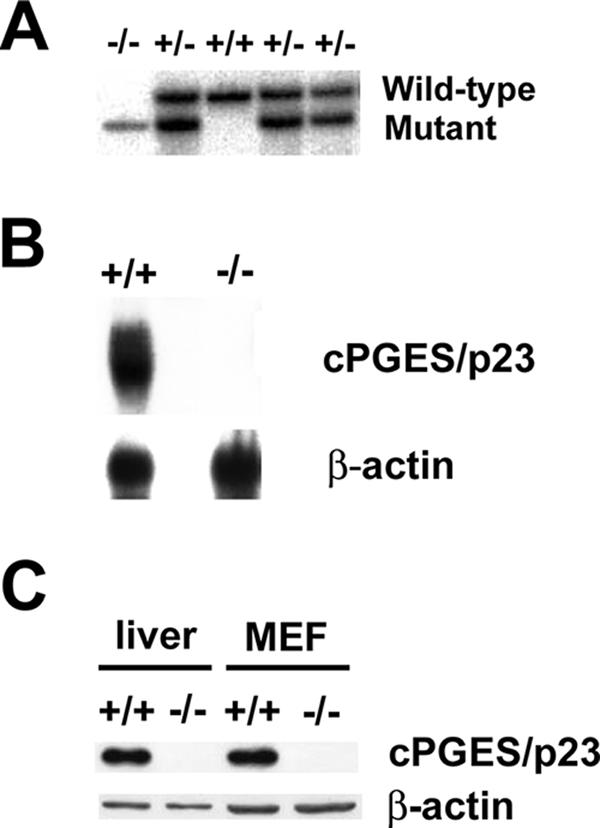
Generation of cPGES/p23−/− mice. (A) Southern blot analyses of DNA from E18.5 embryos generated by the intercross of mice heterozygous for the cPGES/p23 mutant allele. (B) Northern blot analyses of cPGES/p23 mRNA expression in whole E18.5 embryos. Analysis with a β-actin-specific probe verified equal RNA sample loading. (C) Western blot analysis of cPGES/p23 protein expression in E18.5 liver and MEFs. β-Actin expression is shown as a loading control.
To verify that the insertion of the gene trap into intron 1 of the cPGES/p23 gene resulted in loss of expression, RNA was prepared from cPGES/p23−/− and cPGES/p23+/+ embryos and expression of cPGES/p23 was determined by Northern blot analysis. No cPGES/p23 transcript was detected by this method (Fig. 1B). Similarly, cPGES/p23 could not be detected by Western blot analysis of protein extracts prepared from the livers and primary fibroblasts of cPGES/p23−/− embryos (Fig. 1C).
Mice lacking COX-2, as well as mice lacking the PGE2 EP4 receptor, die in the perinatal period due to a patent ductus arteriosus (31, 45). The majority of mice lacking both COX-1 and COX-2 die within 30 min of delivery. We therefore examined the possibility that cPGES/p23 was required for the PGE2 production essential for maturation of the vessel. Similar to mice lacking EP4 and COX-2, the ductus failed to close in the cPGES/p23-deficient mice; however, in contrast to the EP4−/− and COX-2−/− mice, death of the cPGES/p23-deficient neonates preceded the inflation of the lungs. Although this early death of the cPGES/p23-deficient mice excludes the assessment of this protein in PGE2 production under various physiological stresses in the mature animal, survival of these embryos to full term allowed us to assess prostanoid production both in fetal tissues and in cell lines derived from the cPGES/p23-deficient embryos.
Analysis of prostaglandin production in cPGES/p23−/− embryos.
To identify a role for cPGES/p23 in PGE2 production, we determined PGE2 levels in full-term embryos and E18.5 lungs. Consistent with the PGE2 synthase function assigned to this protein, PGE2 levels were reduced in the cPGES/p23−/− embryos and embryonic lungs compared to the wild-type tissues. However, a similar decrease in the levels of other COX-dependent metabolites, TxB2 and prostacyclin, in these tissues was also observed (Fig. 2A). This finding was not consistent with loss of expression of a PGE2 synthase, but rather suggested cPGES/p23 might have a pleiotropic function in prostaglandin synthesis.
FIG. 2.

cPGES/p23-deficient embryos have a significant decrease in prostanoid levels and a corresponding decrease in cPLA2 and COX expression. (A) PGE2, TxB2, and 6-keto-PGF1α (a stable metabolite of prostacyclin [PGI2]) levels in whole E18.5 embryos and lungs dissected from E18.5 embryos were measured by enzyme immunoassay. n = 4. *, P < 0.05, and #, P ≤ 0.09 by Student's t test or P ≤ 0.05 by Mann-Whitney test. (B) Determination of cPLA2, COX-2, and COX-1 expression in total RNA isolated from E18.5 lungs by RT-PCR. Expression levels were normalized to β-actin, an endogenous control, and the results were expressed as change (fold) relative to wild-type expression levels. cPGES/p23−/− lungs, n = 7; wild-type lungs, n = 9. *, P < 0.05; #, P = 0.06.
The first step in the synthesis of prostanoids is the release of arachidonic acid from the plasma membrane by cytosolic phospholipid A2 (cPLA2). A small decrease in the expression of this enzyme was detected by quantitative RT-PCR of RNA prepared from cPGES/p23−/− E18.5 lungs (Fig. 2B). However, this change did not achieve statistical significance. COX-1 and COX-2 are essential for the production of PGH2, the common substrate for all prostanoids. We, therefore, next determined whether loss of cPGES/p23 resulted in altered expression of these two enzymes. A 50% reduction in COX-2 and a 30% reduction in COX-1 transcripts were observed in RNA prepared from the cPGES/p23−/− lungs (Fig. 2B).
Increased basal PGE2 and TxB2 levels in MEFs.
To further examine the impact of decreased expression of cPGES/p23 on prostanoid biosynthesis, we examined the production of PGE2 in MEFs derived from E13.5 embryos. Since MEFs produce high levels of PGE2, even in the absence of stimuli (14), we measured constitutive production of PGE2 in cultures of cPGES/p23−/− and cPGES/p23+/+ fibroblasts. Equal numbers of cells were seeded and grown to confluence in 24-well plates. The cells were cultured in serum-free DMEM for 1 h, and the PGE2 released into the medium during this time period was measured. Surprisingly, basal levels of PGE2 were higher in the cPGES/p23−/− fibroblasts than in the wild-type fibroblasts, which again is not consistent with loss of a PGE2 synthase (Fig. 3). Parallel cultures treated with indomethacin, which inhibits the enzymatic activity of both COX-1 and COX-2, served as a negative control in this assay. Compared to these negative control indomethacin-treated cells, thromboxane levels were below the detectable limit in the wild-type fibroblasts. However, thromboxane was easily measured in the supernatant collected from the cPGES/p23−/− cells (Fig. 3); thus, similar to the in vivo measurements, the change in PGH2 metabolites was not limited to PGE2.
FIG. 3.

cPGES/p23-deficient primary fibroblasts have a significant increase in PGE2 and TxB2 levels. MEFs were isolated from cPGES/p23−/− and wild-type embryos. Cells were seeded in 24-well plates and grown to 80% confluence. After 1 h of incubation in serum-free medium, the supernatant was removed and PGE2 and TxB2 levels in the supernatant were determined by enzyme immunoassay. The cells in each well were harvested for total protein determination by the BCA method. Results are expressed as picograms of prostanoid per microgram of total cell protein per well. Parallel cultures treated with indomethacin, which inhibits the enzymatic activities of both COX-1 and COX-2, served as a negative control in this assay. n = 6. *, P < 0.01.
Growth retardation of the cPGES/p23-deficient embryos.
Our failure to define a role for cPGES/p23 in the direct regulation of PGE2 levels makes it unlikely that this previously assigned function contributes to the perinatal lethality of these mice. Further analysis of the mutant animals was carried out to determine whether the phenotype of the cPGES/p23−/− embryos would support other functions assigned to this protein. Embryos were removed at E15.5 and E18.5, each embryo was weighed, and the crown-to-rump length was determined. The weight and length of the E15.5 cPGES/p23-deficient pups tended to be less than those of the wild-type littermates, but this difference did not achieve statistical significance with this small group of animals. However, later in gestation, a significant decrease in both the weight and crown-to-rump length of the mice lacking cPGES/p23 became apparent (Fig. 4A).
FIG. 4.

Loss of cPGES/p23 results in growth defects in embryos and proliferation defects in the primary embryonic fibroblasts. (A) E15.5 and E18.5 embryos were removed by cesarean section, and weight and crown-to-rump length were determined. E15.5, cPGES/p23−/−, n = 6, and wild type, n = 4; E18.5, cPGES/p23−/−, n = 6, and wild type, n = 7. *, P < 0.05. (B) Growth curve for cPGES/p23−/− and wild-type primary embryonic fibroblasts. Equal numbers of cells of each genotype were plated in a six-well plate (three wells per time point for each genotype). At 24-h intervals, cells were harvested and counted. The supernatant was collected from each well, and the numbers of dead cells present in the supernatant did not differ between samples. In contrast, a marked reduction in the number of cells present in the wells seeded with cPGES/p23−/− cells was observed. These data are representative of two independent experiments, each performed with two different −/− and +/+ MEF cultures. (C) Proliferation assay comparing cPGES/p23−/− and wild-type primary fibroblasts. Cells were stained with CFSE and analyzed by flow cytometry 24 and 48 h after staining. The CFSE intensity at the time of labeling is shown at 0 h. A shift left at 24 and 48 h represents a decrease in CFSE intensity which corresponds to the rate of proliferation in the population. Shown are the representative results from three independent experiments. Black, wild type; gray, cPGES/p23−/−.
To determine whether the smaller size of the cPGES/p23 embryos reflected a role for cPGES/p23 in proliferation, embryonic fibroblast cultures were established from control and mutant E13.5 embryos. After isolation of these cells and establishment of the primary cultures, equal numbers of wild-type and cPGES/p23−/− cells were seeded and the growth was monitored daily. Attached viable cells, as well as the dead cells present in the culture media, were counted at 24, 48, and 72 h after plating. As can be seen in Fig. 4B, the growth of the cPGES/p23-deficient cells lagged behind that of the wild type. This did not appear to represent a loss of cells due to cell death, as no difference was observed in the number of dead cells present in the media. The average doubling time for the two wild-type primary cell cultures, derived from two different embryos, was calculated to be 18.3 h, while the average doubling time for two cPGES/p23−/− cell cultures was 25.4 h. An additional two cultures of each genotype were examined, and the average doubling times were 17.0 h and 33.1 h for the wild-type and cPGES/p23−/− cell cultures, respectively. To further examine this point, cells were labeled with CFSE. This dye is retained in the cells but diminishes in intensity with each round of proliferation. Cells were stained 24 h after plating, and flow cytometric analysis was performed to determine the proliferation rate at 24 and 48 h after staining. As shown in Fig. 4C, the cPGES/p23−/− fibroblasts were not proliferating as rapidly as the wild type to produce the next generations at both 24 and 48 h after addition of the dye. Overall, this supports a role of cPGES/p23 either directly or indirectly in cell cycle progression of primary fibroblasts.
cPGES/p23−/− mice have abnormal lung development.
The small decrease in growth of the cPGES/p23−/− pups alone is unlikely to account for the perinatal lethality of this mutation. As discussed above, necropsy of the pups indicated minimal inflation of the cPGES/p23−/− lungs after birth. In contrast to the impact of cPGES/p23 on the overall growth of the pup, the size of the cPGES/p23−/− lungs did not differ from that of wild-type embryos, and, in fact, measurement of total RNA content indicated an increase in the cellularity of the lungs at E18.5 (Fig. 5A). We, therefore, compared the architecture of the cPGES/p23−/− and wild-type lungs at various stages of development. Lung development can be divided into four stages (9). The embryonic stage is marked by the initial branching of the lung from the primitive foregut and the development of the bronchopulmonary segments. During the pseudoglandular stage (E12.5 to E16.0), there is further branching of the duct system generating the conducting portion of the respiratory system, including the terminal bronchioles. These early developmental events were not altered in the mice lacking cPGES/p23. As shown in Fig. 5B, no apparent differences are observed between the E15.5 wild-type and cPGES/p23−/− lung. During the next two stages of development, the canalicular phase (E16.0 to E17.5) and the saccular/alveolar phase (E17.5 to postnatal day 5), the terminal bronchioles undergo further branching to form the respiratory bronchioles. This is followed by the formation of terminal sacs and thinning of the stroma that separated these saccules from the developing vascular bed, thus forming the thin diffusible interface between the airways and the pulmonary circulation required after birth. It is during the canalicular stage that differences in the development of the cPGES/p23−/− lungs become apparent. At E18.5, the cPGES/p23−/− lungs still display morphology similar to the wild-type lungs early in the canalicular stage (Fig. 5B). Terminal sac development appears absent in the cPGES/p23−/− embryos, resulting in a more condensed lung architecture. Thus, the development of the cPGES/p23−/− lungs appears arrested in the canalicular stage (E16.0 to E17.5).
FIG. 5.

Histological analysis of lung tissue from E15.5 to E18.5 wild-type and cPGES/p23−/− embryos by light microscopy. (A) Total RNA was extracted from E18.5 embryo lungs. Quantitation of RNA revealed a significant increase in the cPGES/p23−/− lungs, suggesting hypercellularity. (B) Lungs were removed from embryos at E15.5, E16.5, E17.5, and E18.5 and fixed overnight in 10% formalin. Lung sections were stained with hematoxylin-eosin. The cPGES/p23−/− lung development appears arrested at the canalicular stage (E16.0 to 17.5). Notable is the failure of the development of terminal sacs. Original magnification, ×10.
Transmission electron microscopy was used to compare the epithelial cell profiles as well as other ultrastructural features of the lungs from cPGES/p23−/− and wild-type full-term embryos. In the wild-type embryonic lung, type II AECs, progenitors of type I alveolar epithelial cells (type I AECs), can easily be distinguished within each saccule by their cuboidal shape, apical microvilli, and the presence of the lamellar bodies, structures essential for the intracellular storage of surfactant. The saccules are largely lined by type I AECs (Fig. 6A). The thin cytoplasmic extensions of this cell type reduce the air-capillary interface. Thus, the differentiation of these cells from their type II AEC precursors is essential for survival. Marked differences are observed in the ultrastructure of the distal lungs obtained from cPGES/p23−/− embryos. Most notably, no type I AECs were observed in the cPGES/p23−/− lungs (Fig. 6B). Type II AECs could, however, be identified based on their unique apical microvilli and lamellar bodies. Consistent with differentiation of this cell type, surfactant can be identified in the airways of the cPGES/p23−/− lung. The distal airway of the cPGES/p23−/− embryos also contains a large number of cuboidal cells which cannot be classified, based on morphological characteristics. It is likely that these represent precursors of type II AECs.
FIG. 6.

Abnormal ultrastructure of the distal airway in cPGES/p23−/− embryos. Transmission electron microscopy was used to analyze ultrastructural morphology in E18.5 lung tissue from wild-type (A) and cPGES/p23−/− (B) embryos. The electron micrographs demonstrate squamous type I AECs (black arrowhead) and cuboidal type II AECs containing lamellar bodies (white arrows) and apical microvilli (black arrows) in the wild-type lung. In contrast, while type II AECs were detected in the cPGES/p23−/− lungs, type I AECs could not be identified. s, surfactant. (C) RT-PCR analysis was used to determine expression of a bronchial epithelial cell marker (NKCC1), a Clara cell-specific marker (TTF-1), type I AEC markers (aquaporin-5 and T1α), and type II AEC markers (DC-LAMP, SP-A, SP-B, and SP-C). Expression levels were normalized to β-actin, an endogenous control; the results are expressed as change (fold) relative to wild-type expression levels. cPGES/p23−/− lungs, n = 7; wild-type lungs, n = 9. *, P < 0.05.
To further examine the developmental changes in the cPGES/p23−/− lungs, we prepared RNA from E18.5 embryo lungs and quantitated the expression of genes which are commonly used as cell-specific markers for airway epithelia, type I AECs, and type II AECs. In the lung, especially in the neonatal period, the airway epithelia expresses high levels of the Na+-K+-2Cl− cotransporter (NKCC1) (53). The thyroid transcription factor (TTF-1) is expressed in both type II AECs and in Clara epithelial cells and has been used previously as a specific marker of lung epithelial cell demarcation (76, 79). No difference in expression of these two genes was observed between wild-type and cPGES/p23−/− animals, indicating normal proximal-distal epithelial cell patterning (Fig. 6C).
To quantitate the loss of type I AECs, quantitative RT-PCR was used to compare expression of type I cell-specific markers, T1α and aquaporin 5, in RNA prepared from wild-type and E18.5 cPGES/p23−/− lungs (72). A dramatic decrease in expression of T1α and aquaporin 5 was observed in the cPGES/p23−/− lungs, consistent with our failure to identify type I cells in these lung (Fig. 6C).
Analysis of a type II cell-specific marker, dendritic cell lysosomal-associated membrane protein (DC-LAMP) (54), showed a significant decrease in the cPGES/p23−/− lungs (Fig. 6C). Similarly, a dramatic decrease in the expression of surfactant genes (SP-A, SP-B, and SP-C) was observed (Fig. 6C). The remarkable changes in the expression of these genes were greater than predicted given our ability to identify type II AECs and surfactant in cPGES/p23-deficient lungs.
Expression of GR-sensitive genes in the cPGES/p23−/− lungs.
The severe lung phenotype of the cPGES/p23-deficient mice is similar to the lung phenotype of the GR-deficient mice (5, 8). We, therefore, next determined whether expression of GR-sensitive genes was altered in the cPGES/p23−/− lungs. The best studied of these genes are those encoding the surfactants. As described above, we observed a significant decrease in mRNA levels for these genes in the cPGES/p23−/− embryos. The expression of DC-LAMP has also been shown to be sensitive to glucocorticoids (32). Again, a significant decrease in the expression of this gene was observed in the mutant lungs. However, interpretation of these differences is complicated since these genes are expressed almost exclusively by type II AECs, and thus the paucity of mRNA may reflect both decreased numbers of mature type II AECs and regulation of these genes by GR. We therefore also examined the expression of ENaCγ, whose expression has also been shown to be sensitive to glucocorticoids (53, 68). mRNA levels of ENACγ were analyzed by quantitative RT-PCR, and a significant decrease in expression was measured in the cPGES/p23−/− lungs (Fig. 7A).
FIG. 7.

cPGES/p23−/− mice have alterations in expression of glucocorticoid-regulated genes in the lung. (A) RT-PCR analysis was used to determine expression levels of glucocorticoid-regulated genes, including ENaCγ and MK, in total RNA isolated from E18.5 lungs. Expression levels were normalized to β-actin, an endogenous control; the results are expressed as change (fold) relative to wild-type expression levels. cPGES/p23−/− lungs, n = 7; wild-type lungs, n = 9. *, P < 0.05. (B) Western blot analysis of GR expression in E18.5 lungs from wild-type and cPGES/p23−/− embryos demonstrates similar GR protein levels. β-Actin expression verifies equal sample loading.
Recently, glucocorticoids have been shown to be important in regulating the growth factor midkine (MK). From E16.5 to E17.5, a significant decrease in the expression of the MK gene was observed in wild-type mice but not embryos lacking GR. Thus, in these mutant animals, levels at birth are threefold higher than those in wild-type littermates (27). We reasoned that if cPGES/p23 is essential for GR function, we would expect a similar pattern of expression in cPGES/p23−/− lungs. Consistent with this, MK expression was threefold higher in the cPGES/p23-deficient mice than in the wild-type mice (Fig. 7A). Taken together, the decreased expression of ENACγ and the increased expression of MK indicated that cPGES/p23 is essential in the regulation of glucocorticoid-sensitive genes in the mouse lung. To determine whether this loss of GR function was the result of decreased levels of GR, protein extracts were prepared from wild-type and cPGES/p23−/− fetal lungs. No significant difference in the level of the steroid receptor was observed, suggesting that loss of cPGES/p23 primarily altered the activity of the receptor (Fig. 7B).
cPGES/p23 is necessary for the induction of gluconeogenic enzymes.
The analysis of expression of glucocorticoid-regulated genes in the lung is complicated by the dramatic alterations in the development of this organ in the cPGES/p23−/− animals. To further verify the relationship between cPGES/p23 and expression of GR-sensitive transcripts, we examined the expression of genes required for gluconeogenesis in the liver of the E18.5 wild-type and cPGES/p23−/− embryos. While gluconeogenesis in the mouse does not occur until after birth, the induction of some of the enzymes, including glucose-6-phosphatase and serine dehydratase, is observed late in gestation (2, 18). The final step of both gluconeogenic and glycogenolytic pathways is carried out by glucose-6-phosphatase, and the contribution of glucocorticoids to the regulation of the expression of this gene has been noted (66). The promoter contains a number of GREs, and the binding of GR together with other accessory factors forms the glucocorticoid response unit, which regulates gene expression. Levels of expression of both glucose-6-phosphatase and serine dehydratase were significantly decreased in the cPGES/p23−/− livers (Fig. 8A). A similar decrease has been reported for these genes in GR-deficient mice (7). To further examine the role of cPGES/p23 in maturation of the liver, we examined the accumulation of glycogen, which normally begins to accumulate late in gestation. Previous studies have suggested a role for glucocorticoids in this induction (67). The cPGES/p23-deficient mice have abnormal liver morphology compared to the wild-type mice at E17.5 (Fig. 8B and C). Analysis of the livers by PAS staining confirmed the failure of glycogen to accumulate in the cPGES/p23−/− livers (Fig. 8D and E).
FIG. 8.

cPGES/p23−/− mice display abnormal liver morphology and have alterations in expression of glucocorticoid-regulated genes in the liver. (A) RT-PCR analysis was used to determine expression levels of gluconeogenic enzymes, glucose-6-phosphatase (G6pc) and serine dehydratase (Sds), in total RNA isolated from E18.5 livers. Expression levels were normalized to β-actin, an endogenous control; the results are expressed as change (fold) relative to wild-type expression levels. n = 5. *, P < 0.01. (B to E) Livers were removed from embryos at E17.5 and fixed overnight in 10% formalin. Liver sections from wild-type (B) and cPGES/p23−/− (C) embryos were stained with hematoxylin and eosin for routine histological examination. Glycogen content in the liver was assessed by PAS staining. Note the intense staining in the wild-type liver (D) compared to the cPGES/p23−/− liver (E). Representative results for four embryo livers analyzed for each genotype are shown. Original magnification, ×10.
Delayed maturation of the skin in cPGES/p23−/− embryos.
Upon external examination, the skin of the cPGES/p23−/− mice appeared more translucent than that of their littermates. Histological examination of the dorsal stratified epithelium from one pair of E17.5 wild-type and cPGES/p23−/− embryos and three pairs of genetically matched E18.5 embryos showed that loss of cPGES/p23 had indeed altered the development of this organ. As expected, all of the structures of the skin were apparent on examination of the sections obtained from the E17.5 wild-type fetus (Fig. 9). A single basal layer of proliferating cells and the suprabasal layers of terminally differentiating cells transitioning as they move towards the surface of the skin were observed. Transcriptionally active spinous and granular cells, as well as several layers of stratum corneum, consisting of metabolically inert enucleated cells, are apparent at this embryonic stage. Similar to the wild-type pups, the basal epithelial cells were easily identified in the cPGES/p23−/− animals. A well-defined stratum spinosum was also observed. However, remarkable differences were noted in the terminal differentiation of the kerotinocytes. A well-defined stratum granulosum, characterized by darkly stained keratohyaline granules, was not present in the cPGES/p23−/− skin, although a few cells with some keratohyalin granules could be identified. A dramatic deficit in the stratum corneum was also observed. By E18.5, the stratum granulosum was more apparent in the mutant fetuses, although the development of this layer and the degree of granulation varied between the mutant pups. However, in all three animals, the lamellar organization of the granulocytic layer, the flattening of the cells, and the discarding of the nucleus had not progressed to the same degree as in the skin of the control animals. In fact, in many instances, numerous nucleated cells with some keratohyalin granules were present at the epithelial surface. In all of the cPGES/p23 mutant animals, deficiencies in the development of the stratum corneum were apparent.
FIG. 9.

Delayed maturation of the skin in cPGES/p23−/− embryos. Aberrations in differentiation and formation of the cornified envelope in cPGES/p23−/− stratified epithelia are shown. Skin from wild-type and cPGES/p23−/− E17.5 and E18.5 fetuses was fixed in formalin and embedded in paraffin, and sections were cut (5 μm) and stained with hematoxylin and eosin. Sections correspond to the dorsal skin above the scapular region of the embryo. SC, stratum corneum; GR, granular layer; KG, keratohyaline granules; SP, suprabasal layer; BL, basal layer; DE, dermis.
Loss of GR transactivation and tethering mechanisms in cPGES/p23−/− fibroblasts.
As normal levels of GR protein were measured in the cPGES/p23−/− fetal tissue, we next examined directly the ability of these receptors to activate gene transcription in cells lacking cPGES/p23. Transformed embryonic fibroblasts were transiently transfected with pGRE-luc, a reporter plasmid with three GRE enhancer elements. Binding of ligand-bound GR to the GREs drives luciferase expression. The fibroblasts were cotransfected with pCMVβ, and β-galactosidase expression was used to normalize for transfection efficiency. pTAL-luc, a reporter plasmid with no GRE enhancer elements, was independently transfected as a negative control, and these baseline expression values were subtracted from the experimental values. Upon incubation with dexamethasone, a synthetic glucocorticoid, the wild-type fibroblasts had a significant increase in luciferase expression. The cPGES/p23-deficient fibroblasts had significantly lower luciferase expression compared to the wild type, suggesting that loss of cPGES/p23 reduces GR transactivation (Fig. 10A).
FIG. 10.

Loss of cPGES/p23 results in defective GR transcriptional activation and protein-protein tethering mechanisms. (A) Transformed fibroblasts were transfected with the reporters pGRE-luc or pTAL-luc (negative control) and pCMV-β to normalize for transfection efficiency. The transfected fibroblasts were incubated in the presence or absence of dexamethasone (Dex [10 nM]) for 24 h. Luciferase expression and β-galactosidase expression were measured in the cell lysates. The luciferase values were normalized to the β-galactosidase values, and the values obtained from the fibroblasts transfected with the negative control (pTAL-luc) were subtracted from the values obtained from the fibroblasts transfected with pGRE-luc (three replicates for each treatment and genotype). As expected, dexamethasone treatment resulted in a significant increase in luciferase expression in the wild-type fibroblasts. This induction was significantly attenuated in the fibroblasts lacking cPGES/p23. *, P < 0.01. These data are representative of three independent experiments performed with two different cell cultures per genotype. (B) Treatment of embryonic fibroblasts with PMA (10−7 M for 6 h), which induces transcription of genes via AP-1, increased expression of collagenase-3 in both wild-type and cPGES/p23−/− cultures. However, subsequent treatment with dexamethasone (10−6 M for 6 h.) and PMA disrupts induction of this gene in wild-type but not in the cPGES/p23−/− fibroblasts. RT-PCR analysis was used to determine expression levels of collagenase-3 in total RNA isolated from the treated fibroblasts. Expression levels were normalized to β-actin, an endogenous control; the results are expressed as change (fold) relative to untreated controls. The data are representative of two independent experiments with different MEF cultures. n = 3. *, P < 0.05 compared to controls, and #, P < 0.05, compared to all groups by the Tukey-Kramer test for multiple comparisons.
GR has been shown to inhibit gene transcription through direct protein-protein interaction, or tethering, with transcription factors, such as AP-1. This interaction was first established for the AP-1-regulated gene coding for collagenase-3 (47). Relative expression of collagenase-3 was determined after activating AP-1 with phorbol 12-myristate 13-acetate (PMA). The cPGES/p23−/− fibroblasts had a similar induction of collagenase-3 compared to the wild-type fibroblasts, suggesting that the ability of AP-1 to induce gene transcription is not affected by loss of cPGES/p23. Wild-type fibroblasts treated with dexamethasone and PMA had a significant reduction in collagenase-3 expression (Fig. 10B). However, the AP-1-induced expression of collagenase-3 in the cPGES/p23−/− fibroblasts was not inhibited upon treatment with dexamethasone, suggesting that loss of cPGES/p23 reduces the ability of GR to negatively regulate transcription factors such as AP-1.
Defective nuclear translocation of GR in cPGES/p23−/− fibroblasts.
To determine the mechanism for the reduction in GR transactivation and tethering in the cPGES/p23−/− fibroblasts, we investigated the ability of GR to translocate to the nucleus after dexamethasone stimulation. Using immunofluorescent staining, the wild-type fibroblasts showed an increase in GR in the nucleus after 30 and 60 min of dexamethasone stimulation (Fig. 11A). cPGES/p23−/− fibroblasts showed no increase in GR in the nucleus after 30 and 60 min of dexamethasone stimulation. The nuclear localization of GR was quantitated by measuring mean pixel density of the nucleus. A slight increase in GR in the nucleus was measured at 60 min of dexamethasone stimulation in the cPGES/p23−/− fibroblasts; however, only the wild-type measurements taken at 30 and 60 min of dexamethasone stimulation had a statistically significant increase in pixel density in the nucleus compared to the nonstimulated cells.
FIG. 11.

Defective nuclear translocation of GR in cPGES/p23−/− fibroblasts. (A) Immunofluorescent staining of transformed fibroblasts for GR after treatment with dexamethasone demonstrates defective translocation in the cPGES/p23−/− fibroblasts. Fibroblasts were treated with dexamethasone (10 nM) for 0, 30, and 60 min in serum-free medium. Software (ImageJ, NIH) was used to determine the mean pixel density of the nucleus of representative cells in each culture. n = 12. *, P < 0.05 compared to all other groups by the Tukey-Kramer test for multiple comparisons. (B) Western blot analysis demonstrates increases in GR in the nucleus and decreases in GR in the cytoplasm after dexamethasone stimulation (10 nM) for 0, 30, and 60 min in the wild-type fibroblasts. No alterations in GR localization were observed in the cPGES/p23−/− fibroblasts after stimulation. Sample loading was verified by expression of α-tubulin for the cytoplasmic fraction and histone H1 for the nuclear fraction. Change was determined by densitometric analysis and normalized to the loading controls.
To further verify the translocation defect, Western blot analysis was performed on nuclear and cytosolic extracts from the fibroblasts after stimulation with dexamethasone. Similar to the immunofluorescence, the wild-type fibroblasts had an increase of GR in the nucleus and a decrease of GR in the cytosol at both 30 and 60 min of dexamethasone treatment. The cPGES/p23−/− fibroblasts had no change in GR in either the nuclear or the cytosolic fraction (Fig. 11B).
DISCUSSION
We have shown that cPGES/p23 is essential for progression of lung development from the early to the late canalicular stage and that pups lacking cPGES/p23 die in the perinatal period as a result of neonatal respiratory insufficiency. Loss of cPGES/p23 results in delayed maturation of the skin. cPGES/p23−/− pups also fail to induce high levels of gluconeogenic enzymes in the prenatal period. These phenotypes are consistent with the proposed function of cPGES/p23 as an Hsp90 cochaperone essential for stabilization of GR. However, the cPGES/p23−/− embryos are smaller in size, and primary cultures of embryonic fibroblast show decreased proliferation. As these phenotypes are not observed in the GR-deficient mice, they likely define cPGES/p23 functions that are independent of GR. Evaluation of prostaglandin production in the cPGES/p23−/− embryos and in the embryo-derived primary fibroblasts does not support a direct role for cPGES/p23 in PGE2 synthesis.
We noted differences in the terminal differentiation of the skin of the cPGES/p23−/− pups. In all mice, a severe deficiency in the cornification of the epithelium was noted at both 17.5 and 18.5 days of gestation. However, at 17.5 days, the skin of the cPGES/p23−/− mice was also deficient in granulocytes, while this stratum was easily identified in wild-type mice. Interestingly, previous studies have shown that injection of glucocorticoids accelerates the functional, morphological, and lipid biochemical maturation of the permeability barrier of the skin (1). Later studies of the glucocorticoid-deficient animals supported the role of this hormone in the differentiation of this organ (20). Similar to the cPGES/p23−/− mice, differences between the wild-type and mutant animals were more pronounced earlier in development. At E17.5, neither a well-defined granular layer nor a stratum corneum was evident by light microscopy in the glucocorticoid-deficient fetuses; however, these deficits diminished later in gestation. Taken together, this suggests that the alterations in differentiation of the skin of the cPGES/p23−/− mice are likely the consequence of loss of GR function.
Morphologically the lungs of the cPGES/p23 full-term embryos resemble those of the early canalicular stage of development (E16.5). Most notable is the absence of the terminal saccules characteristic of the full-term embryo. Examination of the ultrastructure of the lung and analysis of the expression of genes specific to distinct populations of airway epithelia indicate normal development of the airways, likely attenuated differentiation and/or maturation of type II AECs, and little to no differentiation of type I AECs from type II precursors. This phenotype is very similar to that described for GR-deficient mice. A GR hypomorphic (GRhypo) mouse line with extremely low levels of GR activity was generated from ES cells in which the neomycin marker gene was inserted into exon 2 by homologous recombination (8, 63). While 10% of theses hypomorphs survive on a mixed genetic background, the coisogenic 129 mice die of respiratory insufficiency in the immediate perinatal period. Similar to the cPGES/p23−/− pups, the lungs of these pups failed to complete the final stages of maturation, and their architecture was comparable to that of an E16.5 embryo. However, unlike the cPGES/p23−/− embryonic lung, some type I AECs were identified in the GRhypo lungs. Two possible explanations for these differences noted in the differentiation of the lungs and survival of the GR and cPGES/p23−/− mice are possible. First, this difference may reflect a GR-independent cPGES/p23 function in lung development. cPGES/p23 may play a role in stabilizing other Hsp90 clients, such as the retinoic acid receptor and the estrogen receptor, and many of these have documented roles in lung development (21, 28, 46, 74). An alternate possibility is that analysis of the GRhypo mice may reflect residual activity of the GR in these animals compared to the cPGES/p23−/− animals. Mice carrying a deletion of the second or third exon have a more severe phenotype, with no animals surviving the perinatal period (GRnull); however, a detailed description of the lung phenotype, as in the GRhypo mice, has not been described for these mice (5, 63). Final determination of whether GR-independent cPGES/p23 pathways contribute to lung development will require the direct comparison of congenic GRnull animals and cPGES/p23−/− animals under identical breeding and experimental conditions. However, the remarkable similarities in the phenotype of the GRhypo and cPGES/p23−/− animals are consistent with the hypothesis that cPGES/p23 is essential for GR actions during lung development and that the failure of the cPGES/p23−/− lungs to mature reflects primarily the loss of this function of cPGES/p23.
The GR-regulated pathways critical for lung development have not been identified. Interestingly, a recent study showed that GR-deficient mice fail to downregulate the expression of MK, a heparin-binding growth/differentiation factor, which plays a role in mesenchyme-epithelium interactions during organogenesis (26, 27, 35). The expression pattern of MK during lung development is complex, but expression is dramatically reduced during sacculation of the lung at day 17 of gestation. It has been suggested that the decrease in expression of MK signals the lung to initiate maturation, including the thinning of the mesenchyme critical to this stage of development (36). The high levels of MK in the lungs of E18.5 GR-deficient embryos are also observed in the cPGES/p23-deficient embryos. This continued expression of MK in the cPGES/p23−/− embryos supports the hypothesis that the failure to observe normal transition from the canalicular stage to the saccular stage of lung development is secondary to the loss of GR activity in the cPGES/p23−/− embryos.
The GR modulates gene expression through both DNA-binding-dependent and -independent mechanisms. The relative importance of these two pathways in development has been determined by generation of mice expressing a mutant GR (GRdim) (52). This mutation prevents dimerization of the receptor, binding of the receptor to GRE sites in promoters, and transactivation of genes. Study of these mice showed that lung maturation requires DNA-binding-independent regulatory functions of GR since GRdim mice survive into adulthood and have no severe respiratory defects. The cPGES/p23−/− mice die at birth, with a phenotype similar to that of the GRhypo mice, indicating that cPGES/p23 is important in regulating the GR in its DNA-binding-independent processes. In contrast, similar to the GR-deficient mice, gluconeogenic enzymes are not induced in the postnatal period in GRdim mice, demonstrating that expression of these genes is dependent on DNA binding and transactivation (40). The loss of expression of glucose-6-phosphatase and serine dehydratase in the livers from full-term cPGES/p23−/− pups indicates that cPGES/p23 is essential for the prenatal activation of enzymes involved in gluconeogenesis. Again, the most likely interpretation of this phenotype is that cPGES/p23 is required for the stabilization of the GR conformation required for ligand binding, subsequent nuclear translocation, and transactivation. Consistent with this, we have shown that the glucocorticoid dexamethasone failed to induce GRE-dependent expression of reporter genes in fibroblast lines established from the cPGES/p23-deficient mice. Our studies also indicate that cPGES/p23 is essential for the regulation of AP-1 by GR, a function that is not lost in the GRdim mice and thus is a DNA-binding-independent function. Furthermore, we have demonstrated that the loss of both DNA-binding-dependent and -independent mechanisms is due to the requirement of cPGES/p23 for the translocation of GR to the nucleus.
The cPGES/p23−/− pups are approximately 10% smaller in size than wild-type pups. No similar defect has been reported for GR-deficient pups—neither the GRhypo nor the GRnull lines—suggesting this growth retardation is not the result of decreased GR activity (5, 8). Primary fibroblasts derived from the cPGES/p23−/− embryos have a decreased rate of proliferation. Again, a similar defect has not been reported for the GR-deficient primary fibroblasts. In fact, addition of glucocorticoids to fibroblast cultures has been shown to inhibit proliferation (50, 71). cPGES/p23 together with Hsp90 has been implicated in a number of pathways which, if dysfunctional, could alter growth, cell cycle transition, and senescence of cells (3, 34, 39, 49). For example, the Saccharomyces cerevisiae homologue of p23, Sba1, and Wos2, the Schizosaccharomyces pombe homologue, together with the corresponding Hsp90 analogues have been shown to regulate Cdc2, a protein kinase involved in mitotic control (37). A possible role for cPGES/p23 and Hsp90 in the function of telomerase has also been reported (22). However, it is unlikely that dysfunction of this complex would manifest itself in the first generation of the cPGES/p23−/− pups. Mouse chromosomes possess much longer telomeres than human cells, and several generations of breeding are required before loss of telomerase activity results in altered growth of mice (6). Additional studies will be required to fully characterize the role of cPGES/p23 in growth and proliferation.
cPGES/p23 was independently identified as a cytosolic protein with glutathione-dependent PGE2 synthase activity. The identification was based not only on the activity of protein purified from cellular extracts but also on the increase in PGE2 production by cells cotransfected with a cDNA encoding this protein and COX-1 (60). This led to the hypothesis that the cPGES/COX1 pathway was responsible for the production of PGE2 detected in mice and cell lines lacking mPGES1 (62). Analysis of the cPGES/p23−/− mice fails to support the hypothesis that cPGES/p23 is a bona fide PGE2 synthase. While loss of cPGES/p23 altered PGE2 production, a corresponding alteration in the production of other COX-dependent prostanoids suggests a more complex and indirect role for cPGES/p23 in this pathway.
Our demonstration that cPGES/p23 is essential for the non-DNA-binding actions of GR and extensive studies documenting a role for GR in regulation of the arachidonic acid pathway provides an explanation for our results as well as the assignment of a synthase function to this protein. A number of negative regulatory targets for GR in the arachidonic acid pathway have been identified, including lipocortin 1 and p11, proteins capable of modifying the release of arachidonic acid from membrane phospholipids (11, 77). The primary enzyme responsible for the release of arachadonic acid from the membrane for metabolism by the prostanoid pathway is cPLA2 (4, 17). Glucocorticoids have been shown to inhibit the expression and activity of this protein during inflammatory responses (13, 23, 42). Glucocorticoids have also been shown to reduce COX-2 activity in cells by decreasing COX-2 transcription via tethering of transcription factors, such as NF-κB, essential for induction of this gene during inflammation (40-42, 51). Furthermore, posttranscriptional regulation and translational regulation of COX-2 activity have been reported (29, 43, 44). Consistent with our studies of primary embryonic fibroblasts, decreased GR activity secondary to loss of cPGES/p23 would result in loss of this inhibitory pathway, resulting in an increase in PGE2 and other COX-2-dependent metabolites. Transfection of cell lines with p23 expression vectors has been reported, similar to loss of cPGES/p23, to result in inhibition of GR activity in cells lines (16, 73). This raises the possibility that the increased PGE2 production noted in cells transfected with cPGES/p23 was the indirect consequence of decreased GR function in this line.
Interestingly, the impact of loss of cPGES/p23 on prostanoid production by fetal tissues was very different from that observed with cultured cells. Here loss of this protein resulted in decreased levels of PGE2 and other COX-dependent prostanoids. A number of possible explanations can be proposed. First, the ability of GR to stimulate the production of prostanoids in specific cell types has been reported, particularly in primary amniotic fibroblasts (15, 58, 78). These studies show that dexamethasone increases expression of both cPLA2 and COX-2 in these cells. Consistent with this finding, prenatal dexamethasone increased prostaglandin synthesis in rat fetal lung (64). Potential GREs have been defined in the COX-2 and cPLA2 promoters, which may play a role in the induction of gene transcription (61, 75). It is interesting to speculate that, in the mouse embryo, glucocorticoids, which increase in levels as gestation progresses, contribute to the increase in prostaglandin production observed late in gestation. Alternatively, the diminished prostanoid production might be the indirect consequence of alterations in development of the cPGES/p23−/− embryos. Finally, the decreases in prostanoids in the cPGES/p23−/− mice may reflect an as-yet-unidentified GR-independent function for cPGES/p23 in regulation of the arachidonic acid pathway. For example, cPGES/p23 has been shown to facilitate signaling of the aryl hydrocarbon receptor, which in turn modulates the COX-2 pathway (10, 69). While additional experiments will be necessary to address these questions, taken together, our data do not support the designation of cPGES/p23 as the third PGE2 synthase, cPGES, and we suggest that this protein simply be referred to as p23.
While the manuscript for this article was in preparation, Grad et al. reported the generation of mice from the identical ES cell clone carrying an insertional mutation in the cPGES/p23 gene (19). A similar defect in lung and skin development was noted, and cotransfection of a GR reporter construct and a GR-expressing construct in fibroblasts derived from the embryos demonstrated decreased GR-dependent transcription. In contrast to the results shown here, the growth retardation of the pups and decreased proliferation in the primary fibroblasts were not noted in these studies. We also have reported the defective induction of gluconeogenic enzymes and decreased glycogen accumulation in the fetal livers. In addition, we have shown that the ability of GR to regulate the activity of other transcription factors, namely AP-1, is dependent on expression of p23. These deficiencies, as well as decreased expression of genes involved in lung development, suggest a role for p23 in both the DNA-binding-dependent and -independent mechanisms of GR. Finally, our analysis of prostanoids in the p23-deficient animals and primary fibroblasts failed to support a role for this protein as a PGE2 synthase.
Acknowledgments
We thank Anne Latour, Leigh Jania, MyTrang Nguyen, and Subhashini Chandrashekaran for their technical assistance and helpful discussions; Jay Snouwaert for designing the probe for Southern blot analysis; and Bob Bagnell and Victoria Madden at UNC Microscopy Services.
This work was supported by National Institutes of Health grants HL-68141 (B. H. Koller) and DK-38108 (B. H. Koller and T. M. Coffman) and American Heart Association grant 0415427U (A. Kern Lovgren).
Footnotes
Published ahead of print on 16 April 2007.
REFERENCES
- 1.Aszterbaum, M., K. R. Feingold, G. K. Menon, and M. L. Williams. 1993. Glucocorticoids accelerate fetal maturation of the epidermal permeability barrier in the rat. J. Clin. Investig. 91:2703-2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beaudry, J.-B., C. E. Pierreux, G. P. Hayhurst, N. Plumb-Rudewiez, M. C. Weiss, G. G. Rousseau, and F. P. Lemaigre. 2006. Threshold levels of hepatocyte nuclear factor 6 (HNF-6) acting in synergy with HNF-4 and PGC-1α are required for time-specific gene expression during liver development. Mol. Cell. Biol. 26:6037-6046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Betsholtz, C., L. Karlsson, and P. Lindahl. 2001. Developmental roles of platelet-derived growth factors. BioEssays 23:494-507. [DOI] [PubMed] [Google Scholar]
- 4.Bonventre, J. V., Z. Huang, M. R. Taheri, E. O'Leary, E. Li, M. A. Moskowitz, and A. Sapirstein. 1997. Reduced fertility and postischaemic brain injury in mice deficient in cytosolic phospholipase A2. Nature 390:622-625. [DOI] [PubMed] [Google Scholar]
- 5.Brewer, J. A., O. Kanagawa, B. P. Sleckman, and L. J. Muglia. 2002. Thymocyte apoptosis induced by T cell activation is mediated by glucocorticoids in vivo. J. Immunol. 169:1837-1843. [DOI] [PubMed] [Google Scholar]
- 6.Chang, S. 2005. Modeling aging and cancer in the telomerase knockout mouse. Mutat. Res. 576:39-53. [DOI] [PubMed] [Google Scholar]
- 7.Cole, T. J., J. A. Blendy, A. P. Monaghan, K. Krieglstein, W. Schmid, A. Aguzzi, G. Fantuzzi, E. Hummler, K. Unsicker, and G. Schutz. 1995. Targeted disruption of the glucocorticoid receptor gene blocks adrenergic chromaffin cell development and severely retards lung maturation. Genes Dev. 9:1608-1621. [DOI] [PubMed] [Google Scholar]
- 8.Cole, T. J., N. M. Solomon, R. Van Driel, J. A. Monk, D. Bird, S. J. Richardson, R. J. Dilley, and S. B. Hooper. 2004. Altered epithelial cell proportions in the fetal lung of glucocorticoid receptor null mice. Am. J. Respir. Cell Mol. Biol. 30:613-619. [DOI] [PubMed] [Google Scholar]
- 9.Costa, R. H., V. V. Kalinichenko, and L. Lim. 2001. Transcription factors in mouse lung development and function. Am. J. Physiol. Lung Cell. Mol. Physiol. 280:L823-L838. [DOI] [PubMed] [Google Scholar]
- 10.Cox, M. B., and C. A. Miller III. 2002. The p23 co-chaperone facilitates dioxin receptor signaling in a yeast model system. Toxicol. Lett. 129:13-21. [DOI] [PubMed] [Google Scholar]
- 11.Croxtall, J. D., Q. Choudhury, H. Tokumoto, and R. J. Flower. 1995. Lipocortin-1 and the control of arachidonic acid release in cell signalling. Glucocorticoids (changed from glucorticoids) inhibit G protein-dependent activation of cPLA2 activity. Biochem. Pharmacol. 50:465-474. [DOI] [PubMed] [Google Scholar]
- 12.Dittmar, K. D., D. R. Demady, L. F. Stancato, P. Krishna, and W. B. Pratt. 1997. Folding of the glucocorticoid receptor by the heat shock protein (hsp) 90-based chaperone machinery. The role of p23 is to stabilize receptor.hsp90 heterocomplexes formed by hsp90.p60.hsp70. J. Biol. Chem. 272:21213-21220. [DOI] [PubMed] [Google Scholar]
- 13.Dolan-O'Keefe, M., and H. S. Nick. 1999. Inhibition of cytoplasmic phospholipase A2 expression by glucocorticoids in rat intestinal epithelial cells. Gastroenterology 116:855-864. [DOI] [PubMed] [Google Scholar]
- 14.Durant, S., D. Duval, and F. Homo-Delarche. 1988. Mouse embryo fibroblasts in culture: characteristics of arachidonic acid metabolism during early passages. Prostaglandins Leukot. Essent. Fatty Acids 32:129-137. [PubMed] [Google Scholar]
- 15.Economopoulos, P., M. Sun, B. Purgina, and W. Gibb. 1996. Glucocorticoids stimulate prostaglandin H synthase type-2 (PGHS-2) in the fibroblast cells in human amnion cultures. Mol. Cell. Endocrinol. 117:141-147. [DOI] [PubMed] [Google Scholar]
- 16.Freeman, B. C., and K. R. Yamamoto. 2002. Disassembly of transcriptional regulatory complexes by molecular chaperones. Science 296:2232-2235. [DOI] [PubMed] [Google Scholar]
- 17.Fujishima, H., R. O. Sanchez Mejia, C. O. Bingham III, B. K. Lam, A. Sapirstein, J. V. Bonventre, K. F. Austen, and J. P. Arm. 1999. Cytosolic phospholipase A2 is essential for both the immediate and the delayed phases of eicosanoid generation in mouse bone marrow-derived mast cells. Proc. Natl. Acad. Sci. USA 96:4803-4807.10220374 [Google Scholar]
- 18.Girard, J., P. Ferre, J. P. Pegorier, and P. H. Duee. 1992. Adaptations of glucose and fatty acid metabolism during perinatal period and suckling-weaning transition. Physiol. Rev. 72:507-562. [DOI] [PubMed] [Google Scholar]
- 19.Grad, I., T. A. McKee, S. M. Ludwig, G. W. Hoyle, P. Ruiz, W. Wurst, T. Floss, C. A. Miller III, and D. Picard. 2006. The Hsp90 cochaperone p23 is essential for perinatal survival. Mol. Cell. Biol. 26:8976-8983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hanley, K., K. R. Feingold, L. G. Komuves, P. M. Elias, L. J. Muglia, J. A. Majzoub, and M. L. Williams. 1998. Glucocorticoid deficiency delays stratum corneum maturation in the fetal mouse. J. Investig. Dermatol. 111:440-444. [DOI] [PubMed] [Google Scholar]
- 21.Holley, S. J., and K. R. Yamamoto. 1995. A role for Hsp90 in retinoid receptor signal transduction. Mol. Biol. Cell 6:1833-1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holt, S. E., D. L. Aisner, J. Baur, V. M. Tesmer, M. Dy, M. Ouellette, J. B. Trager, G. B. Morin, D. O. Toft, J. W. Shay, W. E. Wright, and M. A. White. 1999. Functional requirement of p23 and Hsp90 in telomerase complexes. Genes Dev. 13:817-826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hong, S. L., and L. Levine. 1976. Inhibition of arachidonic acid release from cells as the biochemical action of anti-inflammatory corticosteroids. Proc. Natl. Acad. Sci. USA 73:1730-1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23a.Institute of Laboratory Animal Resources Commission on Life Sciences. 1996. Guide for the care and use of laboratory animals. National Research Council, National Academy Press, Washington, DC.
- 24.Jakobsson, P. J., S. Thoren, R. Morgenstern, and B. Samuelsson. 1999. Identification of human prostaglandin E synthase: a microsomal, glutathione-dependent, inducible enzyme, constituting a potential novel drug target. Proc. Natl. Acad. Sci. USA 96:7220-7225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson, J. L., T. G. Beito, C. J. Krco, and D. O. Toft. 1994. Characterization of a novel 23-kilodalton protein of unactive progesterone receptor complexes. Mol. Cell. Biol. 14:1956-1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kadomatsu, K., R. P. Huang, T. Suganuma, F. Murata, and T. Muramatsu. 1990. A retinoic acid responsive gene MK found in the teratocarcinoma system is expressed in spatially and temporally controlled manner during mouse embryogenesis. J. Cell Biol. 110:607-616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaplan, F., J. Comber, R. Sladek, T. J. Hudson, L. J. Muglia, T. Macrae, S. Gagnon, M. Asada, J. A. Brewer, and N. B. Sweezey. 2003. The growth factor midkine is modulated by both glucocorticoid and retinoid in fetal lung development. Am. J. Respir. Cell Mol. Biol. 28:33-41. [DOI] [PubMed] [Google Scholar]
- 28.Knoblauch, R., and M. J. Garabedian. 1999. Role for Hsp90-associated cochaperone p23 in estrogen receptor signal transduction. Mol. Cell. Biol. 19:3748-3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lasa, M., M. Brook, J. Saklatvala, and A. R. Clark. 2001. Dexamethasone destabilizes cyclooxygenase 2 mRNA by inhibiting mitogen-activated protein kinase p38. Mol. Cell. Biol. 21:771-780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liggins, G. C. 1969. Premature delivery of foetal lambs infused with glucocorticoids. J. Endocrinol. 45:515-523. [DOI] [PubMed] [Google Scholar]
- 31.Loftin, C. D., D. B. Trivedi, H. F. Tiano, J. A. Clark, C. A. Lee, J. A. Epstein, S. G. Morham, M. D. Breyer, M. Nguyen, B. M. Hawkins, J. L. Goulet, O. Smithies, B. H. Koller, and R. Langenbach. 2001. Failure of ductus arteriosus closure and remodeling in neonatal mice deficient in cyclooxygenase-1 and cyclooxygenase-2. Proc. Natl. Acad. Sci. USA 98:1059-1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McDevitt, T. M., L. Gonzales, R. C. Savani, and P. L. Ballard. 2007. Role of endogenous TGF-β in glucocorticoid-induced lung type II cell differentiation. Am. J. Physiol. Lung Cell. Mol. Physiol. 292:L249-L257. [Epub 22 September 2006.] [DOI] [PubMed] [Google Scholar]
- 33.Mendelson, C. R. 2000. Role of transcription factors in fetal lung development and surfactant protein gene expression. Annu. Rev. Physiol. 62:875-915. [DOI] [PubMed] [Google Scholar]
- 34.Miettinen, P. J., J. E. Berger, J. Meneses, Y. Phung, R. A. Pedersen, Z. Werb, and R. Derynck. 1995. Epithelial immaturity and multiorgan failure in mice lacking epidermal growth factor receptor. Nature 376:337-341. [DOI] [PubMed] [Google Scholar]
- 35.Mitsiadis, T. A., T. Muramatsu, H. Muramatsu, and I. Thesleff. 1995. Midkine (MK), a heparin-binding growth/differentiation factor, is regulated by retinoic acid and epithelial-mesenchymal interactions in the developing mouse tooth, and affects cell proliferation and morphogenesis. J. Cell Biol. 129:267-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morrisey, E. E., and R. C. Savani. 2003. Midkine: a potential bridge between glucocorticoid and retinoid effects on lung vascular development. Am. J. Respir. Cell Mol. Biol. 28:5-8. [DOI] [PubMed] [Google Scholar]
- 37.Munoz, M. J., E. R. Bejarano, R. R. Daga, and J. Jimenez. 1999. The identification of Wos2, a p23 homologue that interacts with Wee1 and Cdc2 in the mitotic control of fission yeasts. Genetics 153:1561-1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murakami, M., K. Nakashima, D. Kamei, S. Masuda, Y. Ishikawa, T. Ishii, Y. Ohmiya, K. Watanabe, and I. Kudo. 2003. Cellular prostaglandin E2 production by membrane-bound prostaglandin E synthase-2 via both cyclooxygenases-1 and -2. J. Biol. Chem. 278:37937-37947. [DOI] [PubMed] [Google Scholar]
- 39.Nakamura, S., H. Watanabe, M. Miura, and T. Sasaki. 1997. Effect of the insulin-like growth factor I receptor on ionizing radiation-induced cell death in mouse embryo fibroblasts. Exp. Cell Res. 235:287-294. [DOI] [PubMed] [Google Scholar]
- 40.Newton, R. 2000. Molecular mechanisms of glucocorticoid action: what is important? Thorax 55:603-613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Newton, R., L. M. Kuitert, M. Bergmann, I. M. Adcock, and P. J. Barnes. 1997. Evidence for involvement of NF-kappaB in the transcriptional control of COX-2 gene expression by IL-1beta. Biochem. Biophys. Res. Commun. 237:28-32. [DOI] [PubMed] [Google Scholar]
- 42.Newton, R., L. M. Kuitert, D. M. Slater, I. M. Adcock, and P. J. Barnes. 1997. Cytokine induction of cytosolic phospholipase A2 and cyclooxygenase-2 mRNA is suppressed by glucocorticoids in human epithelial cells. Life Sci. 60:67-78. [DOI] [PubMed] [Google Scholar]
- 43.Newton, R., J. Seybold, L. M. Kuitert, M. Bergmann, and P. J. Barnes. 1998. Repression of cyclooxygenase-2 and prostaglandin E2 release by dexamethasone occurs by transcriptional and post-transcriptional mechanisms involving loss of polyadenylated mRNA. J. Biol. Chem. 273:32312-32321. [DOI] [PubMed] [Google Scholar]
- 44.Newton, R., J. Seybold, S. F. Liu, and P. J. Barnes. 1997. Alternate COX-2 transcripts are differentially regulated: implications for post-transcriptional control. Biochem. Biophys. Res. Commun. 234:85-89. [DOI] [PubMed] [Google Scholar]
- 45.Nguyen, M., T. Camenisch, J. N. Snouwaert, E. Hicks, T. M. Coffman, P. A. Anderson, N. N. Malouf, and B. H. Koller. 1997. The prostaglandin receptor EP4 triggers remodelling of the cardiovascular system at birth. Nature 390:78-81. [DOI] [PubMed] [Google Scholar]
- 46.Patrone, C., T. N. Cassel, K. Pettersson, Y.-S. Piao, G. Cheng, P. Ciana, A. Maggi, M. Warner, J. A. Gustafsson, and M. Nord. 2003. Regulation of postnatal lung development and homeostasis by estrogen receptor β. Mol. Cell. Biol. 23:8542-8552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pfahl, M. 1993. Nuclear receptor/AP-1 interaction. Endocr. Rev. 14:651-658. [DOI] [PubMed] [Google Scholar]
- 48.Pratt, W. B. 1998. The hsp90-based chaperone system: involvement in signal transduction from a variety of hormone and growth factor receptors. Proc. Soc Exp. Biol. Med. 217:420-434. [DOI] [PubMed] [Google Scholar]
- 49.Pratt, W. B., and D. O. Toft. 2003. Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp. Biol. Med. (Maywood) 228:111-133. [DOI] [PubMed] [Google Scholar]
- 50.Ramalingam, A., A. Hirai, and E. A. Thompson. 1997. Glucocorticoid inhibition of fibroblast proliferation and regulation of the cyclin kinase inhibitor p21Cip1. Mol. Endocrinol. 11:577-586. [DOI] [PubMed] [Google Scholar]
- 51.Ray, A., and K. E. Prefontaine. 1994. Physical association and functional antagonism between the p65 subunit of transcription factor NF-kappa B and the glucocorticoid receptor. Proc. Natl. Acad. Sci. USA 91:752-756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reichardt, H. M., K. H. Kaestner, J. Tuckermann, O. Kretz, O. Wessely, R. Bock, P. Gass, W. Schmid, P. Herrlich, P. Angel, and G. Schutz. 1998. DNA binding of the glucocorticoid receptor is not essential for survival. Cell 93:531-541. [DOI] [PubMed] [Google Scholar]
- 53.Rochelle, L. G., D. C. Li, H. Ye, E. Lee, C. R. Talbot, and R. C. Boucher. 2000. Distribution of ion transport mRNAs throughout murine nose and lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 279:L14-L24. [DOI] [PubMed] [Google Scholar]
- 54.Salaun, B., B. de Saint-Vis, N. Pacheco, Y. Pacheco, A. Riesler, S. Isaac, C. Leroux, V. Clair-Moninot, J. J. Pin, J. Griffith, I. Treilleux, S. Goddard, J. Davoust, M. Kleijmeer, and S. Lebecque. 2004. CD208/dendritic cell-lysosomal associated membrane protein is a marker of normal and transformed type II pneumocytes. Am. J. Pathol. 164:861-871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smith, D. F., L. E. Faber, and D. O. Toft. 1990. Purification of unactivated progesterone receptor and identification of novel receptor-associated proteins. J. Biol. Chem. 265:3996-4003. [PubMed] [Google Scholar]
- 56.Snyder, J. M., H. F. Rodgers, J. A. O'Brien, N. Mahli, S. A. Magliato, and P. L. Durham. 1992. Glucocorticoid effects on rabbit fetal lung maturation in vivo: an ultrastructural morphometric study. Anat. Rec. 232:133-140. [DOI] [PubMed] [Google Scholar]
- 57.Stryke, D., M. Kawamoto, C. C. Huang, S. J. Johns, L. A. King, C. A. Harper, E. C. Meng, R. E. Lee, A. Yee, L. L'Italien, P. T. Chuang, S. G. Young, W. C. Skarnes, P. C. Babbitt, and T. E. Ferrin. 2003. BayGenomics: a resource of insertional mutations in mouse embryonic stem cells. Nucleic Acids Res. 31:278-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sun, K., R. Ma, X. Cui, B. Campos, R. Webster, D. Brockman, and L. Myatt. 2003. Glucocorticoids induce cytosolic phospholipase A2 and prostaglandin H synthase type 2 but not microsomal prostaglandin E synthase (PGES) and cytosolic PGES expression in cultured primary human amnion cells. J. Clin. Endocrinol. Metab. 88:5564-5571. [DOI] [PubMed] [Google Scholar]
- 59.Tanikawa, N., Y. Ohmiya, H. Ohkubo, K. Hashimoto, K. Kangawa, M. Kojima, S. Ito, and K. Watanabe. 2002. Identification and characterization of a novel type of membrane-associated prostaglandin E synthase. Biochem. Biophys. Res. Commun. 291:884-889. [DOI] [PubMed] [Google Scholar]
- 60.Tanioka, T., Y. Nakatani, N. Semmyo, M. Murakami, and I. Kudo. 2000. Molecular identification of cytosolic prostaglandin E2 synthase that is functionally coupled with cyclooxygenase-1 in immediate prostaglandin E2 biosynthesis. J. Biol. Chem. 275:32775-32782. [DOI] [PubMed] [Google Scholar]
- 61.Tazawa, R., X. M. Xu, K. K. Wu, and L. H. Wang. 1994. Characterization of the genomic structure, chromosomal location and promoter of human prostaglandin H synthase-2 gene. Biochem. Biophys. Res. Commun. 203:190-199. [DOI] [PubMed] [Google Scholar]
- 62.Trebino, C. E., J. L. Stock, C. P. Gibbons, B. M. Naiman, T. S. Wachtmann, J. P. Umland, K. Pandher, J. M. Lapointe, S. Saha, M. L. Roach, D. Carter, N. A. Thomas, B. A. Durtschi, J. D. McNeish, J. E. Hambor, P. J. Jakobsson, T. J. Carty, J. R. Perez, and L. P. Audoly. 2003. Impaired inflammatory and pain responses in mice lacking an inducible prostaglandin E synthase. Proc. Natl. Acad. Sci. USA 100:9044-9049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tronche, F., C. Kellendonk, H. M. Reichardt, and G. Schutz. 1998. Genetic dissection of glucocorticoid receptor function in mice. Curr. Opin. Genet. Dev. 8:532-538. [DOI] [PubMed] [Google Scholar]
- 64.Tsai, M. Y., M. W. Josephson, B. Handschin, and D. M. Brown. 1983. The effect of prenatal dexamethasone on fetal rat lung prostaglandin synthesis. Prostaglandins Leukot. Med. 11:171-177. [DOI] [PubMed] [Google Scholar]
- 65.Uematsu, S., M. Matsumoto, K. Takeda, and S. Akira. 2002. Lipopolysaccharide-dependent prostaglandin E(2) production is regulated by the glutathione-dependent prostaglandin E(2) synthase gene induced by the Toll-like receptor 4/MyD88/NF-IL6 pathway. J. Immunol. 168:5811-5816. [DOI] [PubMed] [Google Scholar]
- 66.Vander Kooi, B. T., H. Onuma, J. K. Oeser, C. A. Svitek, S. R. Allen, C. W. Vander Kooi, W. J. Chazin, and R. M. O'Brien. 2005. The glucose-6-phosphatase catalytic subunit gene promoter contains both positive and negative glucocorticoid response elements. Mol. Endocrinol. 19:3001-3022. [DOI] [PubMed] [Google Scholar]
- 67.Vanstapel, F., F. Dopere, and W. Stalmans. 1980. The role of glycogen synthase phosphatase in the glucocorticoid-induced deposition of glycogen in foetal rat liver. Biochem. J. 192:607-612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Venkatesh, V. C., and H. D. Katzberg. 1997. Glucocorticoid regulation of epithelial sodium channel genes in human fetal lung. Am. J. Physiol. 273:L227-L233. [DOI] [PubMed] [Google Scholar]
- 69.Vogel, C., A. M. Boerboom, C. Baechle, C. El-Bahay, R. Kahl, G. H. Degen, and J. Abel. 2000. Regulation of prostaglandin endoperoxide H synthase-2 induction by dioxin in rat hepatocytes: possible c-Src-mediated pathway. Carcinogenesis 21:2267-2274. [DOI] [PubMed] [Google Scholar]
- 70.Wallace, M. J., S. B. Hooper, and R. Harding. 1995. Effects of elevated fetal cortisol concentrations on the volume, secretion, and reabsorption of lung liquid. Am. J. Physiol. 269:R881-R887. [DOI] [PubMed] [Google Scholar]
- 71.Wang, Z., and M. J. Garabedian. 2003. Modulation of glucocorticoid receptor transcriptional activation, phosphorylation, and growth inhibition by p27Kip1. J. Biol. Chem. 278:50897-50901. [DOI] [PubMed] [Google Scholar]
- 72.Williams, M. C. 2003. Alveolar type I cells: molecular phenotype and development. Annu. Rev. Physiol. 65:669-695. [DOI] [PubMed] [Google Scholar]
- 73.Wochnik, G. M., J. C. Young, U. Schmidt, F. Holsboer, F. U. Hartl, and T. Rein. 2004. Inhibition of GR-mediated transcription by p23 requires interaction with Hsp90. FEBS Lett. 560:35-38. [DOI] [PubMed] [Google Scholar]
- 74.Wongtrakool, C., S. Malpel, J. Gorenstein, J. Sedita, M. I. Ramirez, T. M. Underhill, and W. V. Cardoso. 2003. Down-regulation of retinoic acid receptor alpha signaling is required for sacculation and type I cell formation in the developing lung. J. Biol. Chem. 278:46911-46918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wu, T., T. Ikezono, C. W. Angus, and J. H. Shelhamer. 1994. Characterization of the promoter for the human 85 kDa cytosolic phospholipase A2 gene. Nucleic Acids Res. 22:5093-5098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yang, H., M. M. Lu, L. Zhang, J. A. Whitsett, and E. E. Morrisey. 2002. GATA6 regulates differentiation of distal lung epithelium. Development 129:2233-2246. [DOI] [PubMed] [Google Scholar]
- 77.Yao, X. L., M. J. Cowan, M. T. Gladwin, M. M. Lawrence, C. W. Angus, and J. H. Shelhamer. 1999. Dexamethasone alters arachidonate release from human epithelial cells by induction of p11 protein synthesis and inhibition of phospholipase A2 activity. J. Biol. Chem. 274:17202-17208. [DOI] [PubMed] [Google Scholar]
- 78.Zakar, T., J. J. Hirst, J. E. Mijovic, and D. M. Olson. 1995. Glucocorticoids stimulate the expression of prostaglandin endoperoxide H synthase-2 in amnion cells. Endocrinology 136:1610-1619. [DOI] [PubMed] [Google Scholar]
- 79.Zhou, L., L. Lim, R. H. Costa, and J. A. Whitsett. 1996. Thyroid transcription factor-1, hepatocyte nuclear factor-3beta, surfactant protein B, C, and Clara cell secretory protein in developing mouse lung. J. Histochem. Cytochem. 44:1183-1193. [DOI] [PubMed] [Google Scholar]