

Abstract
The persistent activation of signal transducer and activator of transcription 3 (Stat3) is a common feature of prostate cancer. However, little is known about the Stat3 targets that may mediate prostate tumorigenesis. The introduction of an activating mutant form of Stat3 (Stat3-C) into immortalized prostate epithelial cells resulted in tumorigenesis. Stat3-C-expressing cells had decreased E-cadherin levels, increased numbers of lamellipodia and stress fibers, and enhanced migratory capacities compared to vector control-expressing cells, with a concomitant increase in the expression of integrin β6 and its ligand, fibronectin (FN). Exogenously added FN increased cellular migration, with a concomitant loss of E-cadherin expression. The blockade of integrin αvβ6 in Stat3-C-expressing cells inhibited migration, increased E-cadherin levels, and reduced colony formation in soft agar. These results demonstrate the sufficiency of constitutively activated Stat3 in mediating prostate tumorigenesis and identify novel Stat3 targets that are involved in promoting cell migration and transformation.
Activated or tyrosine-phosphorylated Stat3 (pYStat3) protein is a critical signaling molecule in the regulation of tumorigenesis and the metastatic spread of cancer cells. In contrast to normal cells, which tightly control Stat3 activation, a large number of different tumor cell types exhibit constitutive Stat3 phosphorylation. The ramifications of persistent Stat3 phosphorylation include the constitutive expression of specific target genes which ultimately influence the phenotype of the cells. Tumorigenesis is a complex process involving a balance of factors, including the abolishment of contact-mediated growth arrest, the inhibition of apoptosis, angiogenesis, increased proliferation, and increased invasive and migratory capacities (23). Stat3 protein has been shown to mediate many of these factors both in vitro and in vivo (60).
Aberrant Stat3 activation has been implicated in the progression of prostate cancer. Activated Stat3 is found in both primary human prostate cancer samples and a number of prostate cancer-derived cell lines (15, 40, 43). The inhibition of Stat3 activity in these cell lines results in growth inhibition or the induction of apoptosis (20). The introduction of a constitutively active form of Stat3, Stat3-C, into a rat prostate epithelial cell line leads to growth in soft agar (27). Stat3-C expression in the hormone-responsive LnCaP cells results in androgen-independent growth (14). Interleukin-6 stimulation of prostate epithelial cells leads to Stat3 activation and the up-regulation of the androgen receptor, suggesting that this cytokine, through Stat3, may be involved in the development of androgen-independent prostate cancer (9). However, little is known about the Stat3 targets that are critical mediators of prostate tumorigenesis.
To further understand the role of activated Stat3 in prostate cancer, we introduced Stat3-C into immortalized human prostate epithelial cells and determined that Stat3-C is tumorigenic when expressed in these cells. Stat3-C expression altered the dynamics of the actin cytoskeleton and enhanced the ability of the prostate epithelial cells to migrate. The levels of a number of novel and relevant transcripts with respect to motility and the modulation of the cytoskeleton were increased as a consequence of the expression of either Stat3-C or activated Stat3. We identified integrin β6 (ITGβ6) and the extracellular matrix protein fibronectin (FN), a ligand for ITGαvβ6, as possible targets involved in mediating these changes (1, 10, 36, 51).
MATERIALS AND METHODS
Cell culture and plasmids.
RWPE-1 and DU145 cells were obtained from the American Type Culture Collection (Rockville, MD). RWPE-1 cells were maintained in K-SFM (GIBCO, Carlsbad, CA). DU145 cells were maintained in a mixture of Dulbecco's modified Eagle's medium and 10% heat-inactivated fetal bovine serum. pBabe-Stat3-C was generated as previously described, as were retroviral infections with the use of puromycin (2 μg/ml; Sigma, St. Louis, MO) (13). Two single cell clones and several independent polyclonal populations of both control and Stat3-C-expressing cells were isolated.
Antibodies.
Anti-Flag (M2; Sigma, St. Louis, MO) was used for Western blotting and electrophoretic mobility shift assay (EMSA) supershift analyses (2.5 μg). Anti-Stat3 and anti-Tyr705 Stat3 (Cell Signaling Technology, Beverly, MA) were used for chromatin immunoprecipitation, Western blotting (1:1,000), and immunohistochemistry (1:50). Fibronectin H-300 (1:200; Santa Cruz Biotechnology, Inc., Santa Cruz, CA) was used for immunofluorescence. Of the two ITGαvβ6 monoclonal antibodies (Biogen Idec, Inc., Cambridge, MA), 6.2E5 was used for Western blotting, immunofluorescence, and immunohistochemistry (1 μg/ml) and 6.3G9, a function-blocking antibody, was used for the migration and soft-agar assays (30 μg/ml). The E-cadherin monoclonal antibody (Zymed Laboratories, San Francisco, CA) was used at 1:1,000 for immunofluorescence and Western blotting.
Flow cytometry.
pBabe vector- or Stat3-C-expressing RWPE-1 cells were harvested by the addition of 5 mM EDTA in Ca2+-free phosphate-buffered saline (PBS) and washed twice with 1% bovine serum albumin (BSA)-PBS. Cells (106) were then incubated with or without the primary ITGαvβ6 antibody 6.3G9 (5 μg/ml; Biogen Idec, Inc., Cambridge, MA) for 30 min at 4°C, followed by incubation with or without the secondary goat anti-mouse Alexa 488 antibody (1:1,000; Molecular Probes Inc., Eugene, OR) for 30 min at 4°C. Between incubations, cells were washed twice with 1% BSA-PBS. Unstained and stained cells were resuspended in 500 μl of 1% BSA-PBS and then fixed in 4% paraformaldehyde for 30 min at room temperature and analyzed by using a Becton Dickinson FACScan.
Western blotting and EMSAs.
Western blot analyses were carried out as described previously (6). Cells were treated with or without 5-ng/ml oncostatin M (OSM; Chemicon International, Inc. Temecula, CA) for 1 h or with or without 1 μM pyridone 6 (Calbiochem, San Diego, CA) (52) for 24 h, and then cytoplasmic, nuclear, or radioimmunoprecipitation assay extracts were obtained as previously described (6). EMSAs were carried out as previously described (6) by using a radiolabeled high-affinity m67 DNA binding probe and the anti-Flag antibody for supershifting.
Luciferase assay.
Luciferase assays were carried out according to the instructions of the manufacturer of the assay kit (Promega, Fitchburg Center, WI) by cotransfecting cells with cDNA expression plasmids, reporter constructs, and Renilla luciferase (a dual luciferase reporter assay system) to normalize for transfection efficiency.
Soft-agar assay.
Anchorage-independent growth in triplicate 35-mm-diameter dishes was assessed as previously described (6). Cells (3 × 104) with or without 6.3G9 (30 μg/ml) or an isotype-matched control (immunoglobulin G [IgG]) antibody were plated. Colonies were stained with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (Sigma, St. Louis, MO) and counted after 3 weeks.
Migration assays.
The migration assays of RWPE-1 cells were performed as described previously (8). Cells (105 cells/well) with or without 6.3G9 (30 μg/ml) or an isotype-matched control (IgG) antibody were plated onto 8-μm cell culture inserts (Becton Dickinson Labware, Franklin Lakes, NJ) coated with or without 15 μg/cm2 human fibronectin (BD Biosciences, Bedford, MA) in K-SFM, and 0.5% serum was used as the chemoattractant. After 24 h of incubation, the cells were counted. Each condition was assayed in triplicate, experiments were performed independently at least three times, and the results were expressed as the number of cells per field. A one-way analysis of variance was used to determine significance.
Immunofluorescence microscopy.
Cells were grown on extracellular matrix 8-well BioCoat culture slides (Becton Dickinson Labware, Bedford, MA) coated with fibronectin or on plain glass culture slides with or without 6.3G9 (30 μg/ml) and then fixed in 4% paraformaldehyde and permeabilized in 0.1% Triton X (Sigma, St. Louis, MO). Slides were treated with Alexa Fluor 488 phalloidin (0.4 μg/μl; Molecular Probes Inc., Eugene, OR) for 20 min at room temperature, and fibronectin H-300, 6.2E5, or E-cadherin was added for overnight incubation at 4°C. If required, the secondary antibody goat anti-rabbit IgG-fluorescein isothiocyanate or goat anti-mouse IgG-rhodamine (Molecular Probes Inc., Eugene, OR) was added for 1 h at room temperature. Hoechst 33258 (Molecular Probes Inc., Eugene, OR) was added prior to mounting, and the cells were analyzed by confocal laser microscopy with a Zeiss LSM 510 instrument.
Semiquantitative PCR.
Reverse transcription-PCRs (RT-PCRs) with the various primer pairs were performed by preparing total RNA from cells treated with or without OSM for 4 h with or without 1 μM pyridone 6 for 24 h, followed by reverse transcription. Each PCR was carried out using a 32P-labeled deoxynucleoside triphosphate. Primer pairs were as follows: glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene forward primer, 5′-GTGAAGGTCGGAGTCAAC-3′, and reverse primer, 5′-TGGAATTTGCCATGGGTG-3′; fibronectin 1 gene forward primer, 5′-CCGAGGTTTTAACTGCGAAA-3′, and reverse primer, 5′-CCTCCAGGTGTCACCAATCT-3′; tenascin C (TNC) gene forward primer, 5′-AGAAAGTCATCCGGCACAAG-3′, and reverse primer, 5′-ACTCCAGATCCACCGAACAC-3′; and ITGβ6 forward primer, 5′-ACCTGTGAAGACTGCCTGCT-3′, and reverse primer, 5′-GGAGACAGGGTTTTCGATGA-3′.
Q-PCR.
For real-time PCR, total RNA was extracted as described above, followed by reverse transcription (100 ng of RNA) with the Thermoscript RT-PCR system (Invitrogen, Carlsbad, CA). Twenty nanograms of resultant cDNA was used in a quantitative PCR (Q-PCR) carried out with an iCycler (Bio-Rad Laboratories, Hercules, CA) and predesigned TaqMan gene expression assays (GenBank no. Mm00475156_m1; Applied Biosystems, Foster City, CA). Triplicate cycle threshold values were averaged, and the amounts of the target were interpolated from the standard curves and normalized to that of hypoxanthine guanine phosphoribosyltransferase.
Chromatin immunoprecipitation.
Chromatin immunoprecipitation assays of RWPE-1 cells expressing Stat3-C and the pBabe vector control were performed by using the chromatin immunoprecipitation assay kit (Upstate Biotechnology, Waltham, MA). Stat3-C-DNA complexes were precipitated by using anti-Stat3 antibody (9132; Cell Signaling). Polyclonal IgG antibody was used as a negative control, as was a Stat1 antibody (Santa Cruz, CA). Precipitated DNA was amplified by PCR using primers flanking Stat3 binding sites. Primers used for PCR were as follows, with positions numbered from the transcriptional start site given in parentheses: primer set 1/2, 5′-GAAGTCTATCTCCCTGCACTGCCC-3′ (−1669) and 5′-GAGAGAAACTAATTATAGACTACCTAGG-3′ (−1249), and primer set 5/6, 5′-CCTAGCCTTCCTTCTCATTTACTG-3′ (−455) and 5′-CAGAGGCTACCTGGACAGGTAAAGCAG-3′ (−18). The temperature of annealing was 56°C, and the procedure included 30 cycles. The input was 5% of the total.
Tumorigenicity assay.
RWPE-1 pBabe vector- or Stat3-C-expressing cells (107) were harvested and mixed with an equal volume of Matrigel (Becton Dickinson Labware, Bedford, MA), and 200-μl doses were injected into the flanks of 6- to 8-week-old male NCr athymic nude mice (NCI, Frederick, MD). Tumor sizes were measured once a week. Mice were sacrificed after 5 weeks of observation. Results are expressed as the number of tumors per injection. Nuclear extracts were isolated from the tumors and analyzed for the presence of Stat3-C by Western blotting with anti-Flag. All mice were housed under specific-pathogen-free conditions in facilities approved by the American Association of Accreditation of Laboratory Animal Care and in accordance with the present regulations and standards of the U.S. Department of Agriculture and the National Institutes of Health (protocol 00-11-091).
Immunohistochemistry.
Multitissue blocks of formalin-fixed, paraffin-embedded prostate cancer tissue were obtained from Imgenex (catalog no. IMH-303; San Diego, CA), and immunohistochemistry analysis for pYStat3 was performed as previously described (13). Immunohistochemistry analysis for ITGαvβ6 was performed as follows. Sections were deparaffinized and treated with 30% hydrogen peroxide in methanol, and antigen retrieval with pepsin (Zymed Laboratories, San Francisco, CA) at 37°C for 5 min ensued. Sections were sequentially blocked with avidin-biotin (Vector Laboratories, Burlingame, CA), incubated overnight at 4°C with the ITGαvβ6 antibody 6.2E5, and then washed in PBS and incubated with ABC reagents (ABC reagent kit; Vector Laboratories, Burlingame, CA). Sections were incubated with 3,3-diaminobenzidine and then stained with Mayer's hematoxylin. As a negative control, an isotype-matched control (IgG) antibody (Vector Laboratories, Burlingame, CA) was used to replace the primary antibody. Immunohistochemistry analyses of sequential sections of 40 primary prostate cancer and 9 benign prostate tissue samples were performed. To quantify the results, the level of immunostaining of tumor cells was graded as +3 (strong), +2 (moderate), +1 (weak), and 0 (no staining). Scoring of the tissue microarray was performed by two independent observers (H.A.-A. and J.B.), and the scores for staining intensity assigned by the two observers showed high levels of correlation for both pYStat3 and ITGαvβ6. Fisher's exact test was used to assess the correlation between pYStat3 and the corresponding pathological findings.
RESULTS
Stat3-C expression in RWPE-1 cells.
As Stat3-C was previously observed to mediate the transformation of immortalized breast epithelial cells, we wished to determine whether Stat3-C could mediate the transformation of immortalized prostate epithelial cells (13). For these studies, we used RWPE-1 cells which were derived from nonneoplastic human prostatic epithelial cells immortalized with human papillomavirus 18. This cell line expresses luminal cytokeratins and does not grow in soft agar or form tumors in nude mice (5, 56). Flag-tagged Stat3-C was introduced into the RWPE-1 cells by retroviral gene transfer, and polyclonal populations were isolated following puromycin selection. Single cell clones (2) and several independent polyclonal populations were analyzed and gave rise to similar results, of which we present representative examples. The expression of Stat3-C was demonstrated by anti-Flag and anti-Stat3 Western blot analysis (Fig. 1A). DNA binding activity assays revealed low levels of endogenous Stat3 binding in extracts derived from pBabe vector-expressing cells and robust constitutive binding in extracts derived from Stat3-C-expressing cells (Fig. 1B). Stat3-C binding specificity was confirmed by supershifting with an anti-Flag antibody (Fig. 1B).
FIG. 1.

Stat3-C induces tumorigenesis, morphological changes, and increased migration of RWPE-1 cells. (A) Anti-Flag Western blot showing the expression of Flag-tagged Stat3-C in RWPE-1 cells expressing the pBabe vector control (pB) and Stat3-C (3C). (B) EMSA performed with nuclear extracts from the cell lines described in the legend to panel A. Stat3-C DNA binding was supershifted with anti-Flag antibody. (C) Colony formation in soft agar by pBabe vector control- and Stat3-C-infected RWPE-1 cells. The numbers of colonies shown are the means ± standard deviations (SD) of triplicate results from three independent experiments. (D) Evaluation of tumor growth in nude mice injected with pBabe vector control- and Stat3-C-express-ing cells. Nuclear extracts from a Stat3-C-derived tumor (Tu) and the pBabe vector control- and Stat3-C-expressing cell lines were analyzed for the presence of Stat3-C by Flag immunoblotting. (E) pBabe vector control- and Stat3-C-expressing cells were stained with phalloidin (top panels) and anti-E-cadherin-phalloidin (bottom panels) and examined by confocal microscopy. E-Cad, E-cadherin. (F) Expression of E-cadherin as determined by Western blot analysis of whole-cell extracts isolated from pBabe vector control- and Stat3-C-expressing RWPE-1 cells. (G) pBabe vector control- and Stat3-C-expressing RWPE-1 cells were plated and subjected to a Boyden chamber assay, and cell migration was assessed after 24 h. Results are expressed as the number of cells per field and are the means ± SD of triplicate values from three independent experiments.
Stat3-C induces tumorigenesis, morphological changes, and increased migration of RWPE-1 cells.
The growth rate of Stat3-C-expressing cells was compared to that of pBabe vector-expressing cells, and no difference was observed, nor did Stat3-C expression relieve a requirement for epidermal growth factor (data not shown). Cellular transformation was determined by anchorage-independent growth. pBabe vector- and Stat3-C-expressing RWPE-1 cells were plated onto soft agar, and robust colony formation by the Stat3-C-expressing cells compared to that by the pBabe vector control cell line was observed (Fig. 1C). Similarly, the induction of tumorigenesis was demonstrated by injecting Stat3-C-expressing cells into the flanks of athymic nude mice. Six out of eight animals injected with the Stat3-C-expressing cells developed tumors, but none of those injected with the pBabe vector control cells did (Fig. 1D). The presence of Stat3-C within the tumor cells was confirmed via Western blot analysis using anti-Flag antibody (Fig. 1D). An examination of the morphological features of the RWPE-1 cells expressing Stat3-C versus the vector control revealed a striking difference in the abundance of filipodia (found predominantly in pBabe vector control cells) versus that of lamellipodia and an increase in stress fibers in the Stat3-C-expressing cells, suggestive of an epithelial cell-to-mesenchymal cell transition (EMT) (Fig. 1E). By immunocytochemistry analysis, equivalent levels of vimentin in both cell lines were detected, but a marked decrease in E-cadherin in the Stat3-C-expressing cells was observed (Fig. 1E and F and data not shown). Cells undergoing an EMT have an increased capacity for migration (25, 47). We therefore compared the abilities of pBabe vector- and Stat3-C-expressing cells to migrate in a Boyden chamber assay and a wound-healing assay and determined that Stat3-C-expressing cells migrated more than the pBabe vector-expressing cells (Fig. 1G and data not shown). Thus, Stat3-C can mediate tumorigenesis, morphological changes suggestive of an EMT, and increased migration of a nontumorigenic epithelial prostate-derived cell line.
Stat3-C-induced gene expression.
Gene expression profiling was used to identify potential Stat3-C-regulated transcripts that may contribute to the Stat3-C-mediated transformation and increased migration of prostate epithelial cells. We examined levels of mRNA corresponding to previously identified Stat3 target genes, including those for cyclin D1, vascular endothelial growth factor, survivin, Bcl-xL, a subfamily of Zrt/Irt-like protein zinc transporters (LIV-1), and myc, in the Stat3-C- versus pBabe vector-expressing cells and found no significant differences (data not shown). An Affymetrix analysis of RNA isolated from RWPE-1 Stat3-C-containing cells was performed, and the results were compared with those for pBabe vector control cells. Levels of mRNAs corresponding to 163 genes were up-regulated and those of mRNAs corresponding to 25 genes were down-regulated in RWPE-1 Stat3-C-expressing cells compared to those in the pBabe vector-expressing cells (change, twofold; P < 0.001) (see Table S1 in the supplemental material). A number of potentially interesting transcripts were identified in this screen, and the levels of these transcripts were confirmed to be increased in the Stat3-C-expressing cells compared to those in the pBabe vector-expressing cells by RT-PCR and Q-PCR (Fig. 2A and data not shown). To determine whether any of these differentially expressed genes could also be regulated by pYStat3, we treated RWPE-1 cells with OSM, a ligand leading to the transient activation of Stat3 as determined by Western blotting with pYStat3 (Fig. 2B). An Affymetrix analysis of RNA isolated from pBabe vector control cells treated with or without OSM was performed. Transcripts which were commonly up- or down-regulated in both Stat3-C-expressing and OSM-treated RWPE-1 cells were identified (see Table S2 in the supplemental material). The levels of mRNAs corresponding to 29 genes were up-regulated, and those of mRNAs corresponding to 4 genes were down-regulated. We examined three of the up-regulated transcripts, including those for ITGβ6 and its ligands, FN and TNC, as they have recently been implicated in colorectal tumorigenesis and tumor cell migration (4). We confirmed that OSM treatment of RWPE-1 cells led to an increase in the expression of ITGβ6, FN, and TNC transcripts as determined by RT-PCR and Q-PCR, suggesting that the corresponding genes may be transcriptionally regulated by activated Stat3 (Fig. 2C and D).
FIG. 2.
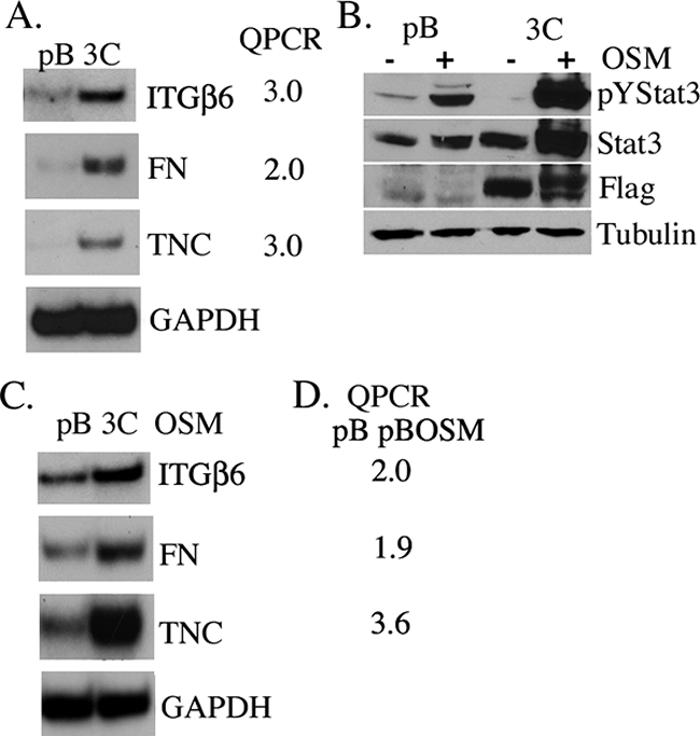
Stat3-C- and activated-Stat3-induced transcription. (A) Induction of ITGβ6, FN, and TNC mRNAs in pBabe vector control (pB)- and Stat3-C (3C)-expressing RWPE-1 cells as determined by RT-PCR and real-time Q-PCR and normalized to GAPDH mRNA expression. Results are expressed as the increase (n-fold) in levels in Stat3-C-expressing cells relative to those in pBabe control cells. (B) Western blot analysis showing pYStat3 in extracts from pBabe vector control- and Stat3-C-expressing cells stimulated with (+) or without (−) OSM for 1 h and reprobed with anti-Stat3, anti-Flag, and antitubulin. (C) Induction of ITGβ6, FN, and TNC mRNAs in pBabe vector control- and Stat3-C-expressing RWPE-1 cells stimulated with OSM for 4 h as determined by RT-PCR and normalized to GAPDH mRNA expression. (D) Real-time Q-PCR values for ITGβ6, FN, and TNC mRNAs from the pBabe vector control cells stimulated with and without OSM for 4 h.
Fibronectin expression is increased in Stat3-C-expressing cells and can mediate both morphological and migratory changes in pBabe vector control cells.
Fibronectin is an extracellular matrix protein secreted by cells, is involved in cell migration, and is a ligand for ITGαvβ6 (7). By immunocytochemistry analysis, we observed increased FN staining of the Stat3-C-expressing cells compared to that of the pBabe vector control cells (Fig. 3A). We asked whether the morphological differences observed between pBabe vector- and Stat3-C-expressing cells (Fig. 1E) could be conferred by the addition of FN. The F-actin filament distribution patterns in cells plated onto glass slides versus those plated onto FN-coated culture slides were examined. The morphology of pBabe vector control cells plated onto FN approximated that of Stat3-C-expressing cells plated onto glass, with relative decreases in the numbers and distribution of filipodia and increases in the numbers of lamellipodia and stress fibers as well as a marked diminution in E-cadherin levels (Fig. 3B and C and data not shown). The morphological changes could be observed within 4 h of plating of the cells onto fibronectin. These data suggest that exogenously added FN can alter the morphology of the RWPE-1 cells and potentiate an EMT by down-modulating E-cadherin expression. The effect of FN on cell migration was examined by both a Boyden chamber assay and a wound-healing assay (Fig. 3D and data not shown). In the Boyden chamber assay, FN potently enhanced the migration of pBabe vector control cells but not to the observed levels of migration of Stat3-C-expressing cells (Fig. 3D). Furthermore, the addition of FN enhanced the migration of Stat3-C-expressing cells (Fig. 3D). Thus, the expression of FN participates in mediating RWPE-1 cell migration, but additional molecules are likely to be involved in potentiating the phenotype of increased migration of Stat3-C-expressing cells.
FIG. 3.
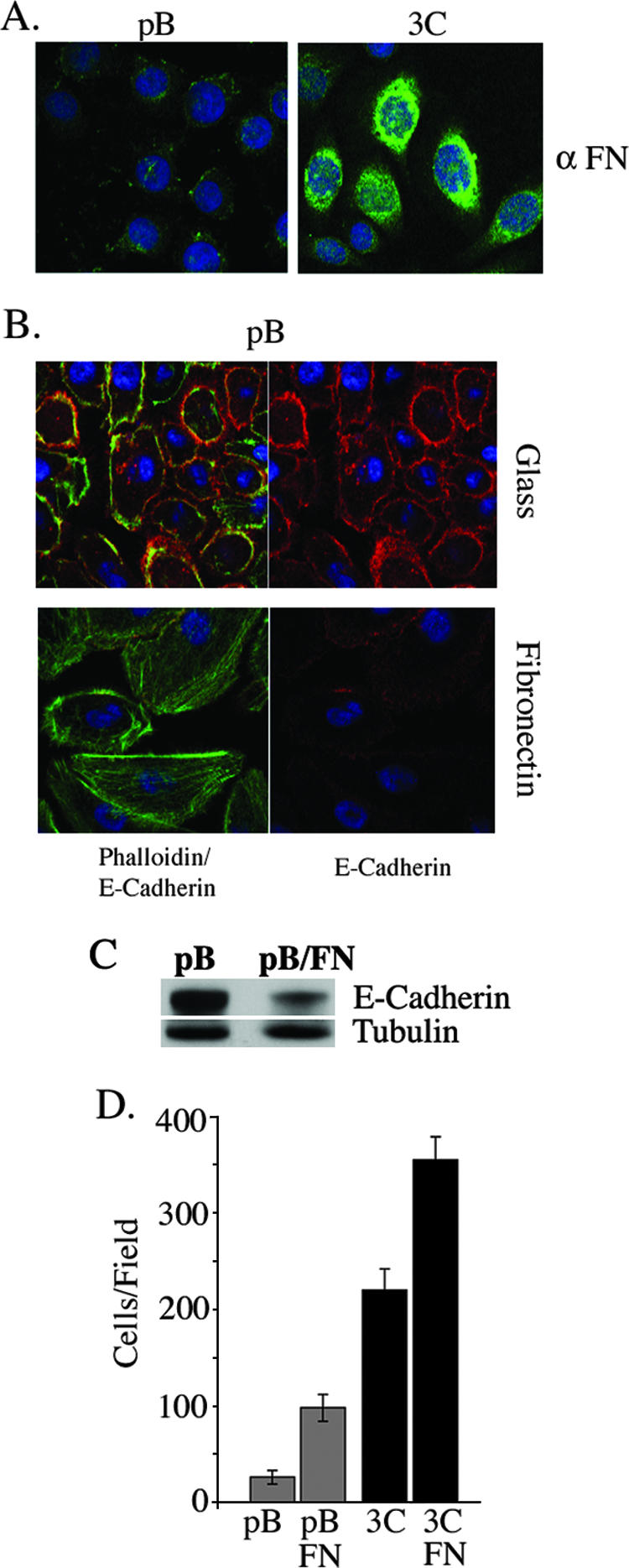
Fibronectin expression is increased in Stat3-C-expressing cells and can mediate both morphological and migratory changes in pBabe cells. (A) Expression of fibronectin in pBabe vector control (pB)- and Stat3-C (3C)-expressing RWPE-1 cells as shown by immunofluorescence. α FN, anti-FN. (B) pBabe vector control-expressing RWPE-1 cells were plated onto glass or fibronectin-coated culture slides, stained with phalloidin and anti-E-cadherin, and examined by confocal microscopy. (C) Extracts from pBabe vector control-expressing cells and pBabe vector control-expressing cells plated onto fibronectin-coated culture dishes were examined by Western blot analysis for E-cadherin expression. (D) Migration of pBabe vector control- and Stat3-C-expressing RWPE-1 cells in a Boyden chamber assay in the presence or absence of fibronectin. Results are expressed as the number of cells per field and are the means ± SD of triplicate values from three independent experiments.
ITGβ6 levels are increased in Stat3-C-expressing cells, and Stat3-C mediates the transcriptional activation of ITGβ6.
ITGαvβ6 is overexpressed in a variety of epithelial tumors, binds to a number of extracellular matrix proteins, including FN and TNC, and has been demonstrated to potentiate an EMT of colorectal cells (4). Here, we examined ITGαvβ6 protein levels by immunocytochemistry, Western blot, and fluorescence-activated cell sorter (FACS) analyses and observed a significant increase in the expression of ITGαvβ6 in the Stat3-C-expressing cells compared to that in pBabe vector control cells (Fig. 4A, B, and C). FACS analysis did not reveal any significant differences in the levels of expression of some other FN binding integrins, including ITGβ1, ITGαvβ3, ITGα4, and ITGα3 (data not shown). To investigate the role of Stat3 in the transcriptional regulation of the ITGβ6 gene, 293T cells were cotransfected with a human ITGβ6 promoter-luciferase gene construct and a Stat3-C or v-Src (which phosphorylates endogenous Stat3) expression plasmid. Both Stat3-C and v-Src could enhance luciferase activity, suggesting a role for Stat3-dependent regulation of the ITGβ6 gene (Fig. 4D). An examination of the ITGβ6 promoter revealed numerous Stat3 binding recognition sites (Fig. 4D). Cross-linking chromatin immunoprecipitation assays were carried out to determine whether the potential Stat binding sites within the ITGβ6 promoter bound Stat3. Primer sets were utilized to target the regions of the ITGβ6 promoter corresponding to Stat binding sites found between −1560 and −1288 and between −500 and the start site of transcription. Extracts from Stat3-C-expressing RWPE-1 cells were subjected to chromatin immunoprecipitation analysis, and the amplification by PCR of both promoter targets was observed when anti-Stat3 antisera was used to immunoprecipitate the protein-DNA complexes (Fig. 4E). We found no association between Stat1 and the ITGβ6 promoter, nor was Stat3 found on this promoter in pBabe vector control cells (see the supplemental material). These data indicate that activated Stat3 can be found associated with the ITGβ6 promoter and is likely to be a transcriptional regulator of ITGβ6.
FIG. 4.

ITGβ6 levels are increased in Stat3-C-expressing cells, and Stat3-C mediates the transcriptional activation of ITGβ6. (A) Expression of ITGαvβ6 in pBabe vector control (pB)- and Stat3-C (3C)-expressing RWPE-1 cells as shown by immunofluorescence. α ITGβ6, anti-ITGβ6. (B) Anti-ITGβ6 Western blot showing the increased expression of ITGαvβ6 in Stat3-C-expressing cells compared to that in pBabe vector control cells, as normalized to tubulin expression. (C) FACS analysis showing increased ITGαvβ6 surface expression in Stat3-C cells (yellow) compared to that in the pBabe vector control cells (green). Secondary antibodies alone corresponding to the pBabe vector control and Stat3-C are indicated by the red and blue curves. (D) 293T cells were transiently transfected with an ITGβ6 promoter-luciferase gene construct (start site of transcription, −926) together with the internal control Renilla luciferase reporter (a dual luciferase reporter system) and the pBabe vector control (β6/pB), Stat3-C (β6/3C), or v-Src (β6/vsrc). Luciferase activities were normalized against the internal control. Columns indicate the means of results from three experiments; errors bars indicate SD. Potential Stat3 binding sites are indicated as inverted triangles; those marked by dark triangles are optimal (TTN5AA) and those marked by light triangles have either a TTN4AA or a TTN6AA sequence or an AT-rich region at the N positions on the ITGβ6 promoter. Arrows indicate the directions of transcription, and nucleotide positions are shown below the diagram. (E) RWPE-1 cells expressing Stat3-C were subjected to a chromatin immunoprecipitation assay using antibodies to Stat3 (S3) or IgG as a negative control. Coprecipitated DNA was amplified by PCR using primers flanking the Stat3 binding sites at −1669 and −1249 (1/2) and primers surrounding the Stat3 binding sites at −455 and −18 (5/6). The input was 5% of the total.
ITGαvβ6 activity in Stat3-C-expressing cells potentiates cell migration.
In order to determine whether the enhanced ITGαvβ6 expression observed in the Stat3-C-expressing cells participated in the phenotype of increased migration, migration assays were performed in the presence of a function-blocking antibody to ITGαvβ6 (6.3G9) (57). The migration of Stat3-C-expressing cells was significantly inhibited in the presence of the 6.3G9 blocking antibody but not in the presence of the control antibody (Fig. 5A). The addition of FN enhanced the migration of Stat3-C-expressing cells, and this enhanced migration was also attenuated by the 6.3G9 blocking antibody (Fig. 5A). Control pBabe vector cells also express ITGβ6, albeit to a lesser degree, and thus the migration of these cells on FN was also blocked by the 6.3G9 antibody (Fig. 5A). When Stat3-C-expressing RWPE-1 cells were plated in the presence of the 6.3G9 blocking antibody, E-cadherin levels were restored to those observed in pBabe vector control cells (Fig. 5B, bottom panels). pBabe vector control cells plated onto fibronectin demonstrated a loss of E-cadherin expression and an increase in the numbers of actin stress fibers, which were both reversed when the cells were plated in the presence of the 6.3G9 blocking antibody (Fig. 5B, bottom panels) in contrast to the control antibody (top panel). These data suggest that ITGαvβ6, in part through its interactions with FN, plays a significant role in the phenotype of increased migration of Stat3-C-expressing RWPE-1 cells, possibly through the modulation of E-cadherin levels. A mechanism of ITGαvβ6-mediated EMT proceeds through the activation of latent transforming growth factor β (TGF-β), which in turn can lead to the up-regulation of FN and TNC (4, 41, 51). We explored this process as a possible mechanism explaining the morphological and migratory changes seen in our test cells. A TGF-β blocking antibody had no effect on the morphological phenotype of the Stat3-C-expressing cells plated in the presence or absence of FN (data not shown). These data suggest that ITGβ6 expression possibly mediates its effects through a region different from that involved in TGF-β activation (1, 41). We also determined that Stat3-C-mediated growth in soft agar could be partially inhibited by the 6.3G9 blocking antibody (Fig. 5C), suggesting that ITGαvβ6 can affect the anchorage-independent growth of these cells (1).
FIG. 5.

ITGαvβ6 expression potentiates cell migration. The inhibition of Stat3 phosphorylation in DU145 prostate cancer cells leads to a decrease in ITGβ6 mRNA levels. (A) Migration of pBabe vector control (pB)- and Stat3-C (3C)-expressing RWPE-1 cells incubated with 6.3G9 (3G9) or an isotype-matched control (IgG) antibody in a Boyden chamber assay in the presence or absence of FN. Results are expressed as the number of cells per field and are the means ± SD of triplicate values from three independent experiments. (B) pBabe vector control- and Stat3-C-expressing RWPE-1 cells were plated onto glass- or fibronectin-coated culture slides in the presence of 6.3G9 antibody (bottom panels) or control IgG antibody (top panels), stained with phalloidin and anti-E-cadherin, and examined by confocal microscopy. (C) Colony formation in soft agar by pBabe vector control- and Stat3-C-infected RWPE-1 cells plated in the presence of 6.3G9 antibody or IgG control antibody. The numbers of colonies shown are the means ± SD of triplicate results from three independent experiments. (D) Western blot analysis showing the expression of pYStat3 and total Stat3 (Stat3), normalized to tubulin expression, in extracts from DU145 cells after treatment with pyridone 6 (P6) or the dimethyl sulfoxide control (D) for 24 h. By RT-PCR, ITGβ6 mRNA levels were determined in parallel and normalized to those of GAPDH mRNA.
Inhibition of Stat3 phosphorylation in DU145 prostate cancer cells leads to a decrease in ITGβ6 mRNA levels.
Stat3 is persistently tyrosine phosphorylated in the prostate cancer-derived cell line DU145, due to the autocrine production of cytokines, while treatment with a Janus-activated kinase inhibitor leads to a decrease in phospho-Stat3 as determined by Western blot analysis (18, 40). Here, we used RT-PCR to examine ITGβ6 message levels in DU145 cells treated with or without a pan-Janus-activated kinase inhibitor and determined that a decrease in pYStat3 correlated with a reduction in the abundance of the ITGβ6 message (55) (Fig. 5D). Similarly, reducing Stat3 levels by using Stat3 short hairpin RNA resulted in a marked reduction in ITGβ6 mRNA expression (data not shown).
Immunohistochemical analysis of phosphorylated Stat3 and ITGβ6 in primary prostate tumors.
By immunohistochemistry analysis, we examined 40 primary prostate tumors for nuclear pYStat3 and determined that 19 (47%), 14 (40%), 4 (10%), and 1 (3%) showed staining patterns of 0, +1, +2, and +3, respectively (Fig. 6). In addition, nine samples of tissue that was histologically normal but derived from areas adjacent to tumors were examined for pYStat3, and five (56%), three (33%), and one (11%) showed staining patterns of 0, +1, and +2, respectively (data not shown). No significant correlation between the pYStat3 staining patterns and prostate-specific antigen levels, clinical stages, or Gleason scores was observed. We also examined serial sections of the same tumors for ITGαvβ6 levels and determined that 31 (77%), 5 (13%), 3 (7%), and 1 (3%) showed staining patterns of 0, +1, +2, and +3, respectively. No significant correlation between the ITGαvβ6 staining patterns and clinical stages, Gleason scores, or prostate-specific antigen levels was observed (data not shown). The strongest ITGαvβ6 staining was observed in the basal cells in benign tissue adjacent to a tumor or in ducts with high-grade intraepithelial neoplasia as seen in neoplastic samples 17 and 31 and in benign tissue sample 44. No significant correlation between pYStat3 and ITGαvβ6 levels in the tumor cells was observed (P = 0.43; Fisher's exact test), while a trend toward a positive correlation in basal cells was observed but was not statistically significant.
FIG. 6.

pYStat3 and ITGαvβ6 expression in prostate cancer cells. An association between tumor characteristics, ITGαvβ6 immunostaining, and pYStat3 immunostaining is displayed in the table. Microtissue arrays were stained with anti-phospho-Stat3 and anti-ITGαvβ6 antibodies. Examples of relative pYStat3 and ITGαvβ6 staining patterns are indicated by the large arrowheads showing cancer cells and the small arrowheads showing nontumorous basal cells. The number associated with each section corresponds to the Imgenex tissue microarray designation. Scores for pYStat3 staining patterns are as follows: tumor 13, +3; tumor 31, +1; tumor 11, 0; tumor 30, +2; tumor 17, +1; and benign tissue 44, +2. Scores for ITGαvβ6 staining patterns are as follows: tumor 13, +1; tumor 31, 0, tumor 11, +3; tumor 30, 0; tumor 17, 0; and benign tissue 44, +3. PSA, prostate-specific antigen.
DISCUSSION
Here, we examined the consequences of persistently activated Stat3 in prostate tumorigenesis and determined that the expression of Stat3-C is sufficient to mediate tumorigenesis and the migration of immortalized but nontransformed prostate epithelial cells. A comparison of the morphologies of the control and Stat3-C expressing cells revealed a shift in the distribution of filipodia toward a greater abundance of lamellipodia and, correspondingly, an increase in the migratory capacity of the cells (Fig. 1). Stat3 has been shown to play a critical role in cell migration through both transcriptional and nontranscriptional mechanisms (12, 35, 42, 48, 58, 59, 61).
Tumorigenesis involves the acquisition of genetic and epigenetic changes that cause the aberrant loss or gain of functions by cellular proteins. The consequences include the ability of tumor cells to proliferate, resist apoptosis, demonstrate angiogenic potential, migrate, and invade, as well as the ability of these cells to evade immune surveillance. Activated Stat3 has been linked to all of these processes in part through the transcriptional regulation of critical target genes, including those for c-myc, cyclin D1, matrix metalloproteinase 9 (MMP-9), MMP-2, vascular endothelial growth factor, Bcl-xL, Mcl-1, survivin, and LIV-1, as well as the repression of p53 and Fas (6, 11, 13, 16, 34, 44, 53, 58, 60). We and others previously demonstrated that Stat3-C can transform immortalized fibroblasts and breast epithelial cells, in part as a consequence of increased cyclin D1 and MMP-9 expression (6, 13, 37, 46). However, the levels of neither of these targets nor those of many of the above-mentioned targets (MMP-2, survivin, c-myc, and LIV-1) were increased in our Stat3-C-expressing RWPE-1 cells, suggesting that additional Stat3 targets which are tissue or tumor specific exist (data not shown).
In order to identify potential Stat3-C-regulated transcripts which may contribute to the Stat3-C-mediated transformation and increased migration of prostate epithelial cells, we found many potentially interesting up- or down-regulated transcripts in these cells by Affymetrix analysis (see Table S1 in the supplemental material). In addition to the levels of ITGβ6, FN, and TNC, we confirmed by RT-PCR that those of MMP-3, thrombospondin 1, S100 A9, nicotinamide N-methyltransferase, and brain-heart-protocadherin were also increased, while the levels of cyclin D1, Stat1, p73, and myeloid cell leukemia sequence 1 were unchanged in Stat3-C-containing cells compared to those in pBabe vector control cells (data not shown). Many of these proteins have known roles in EMT, migration, and tumorigenesis (3, 17, 22, 32, 37, 38, 49, 54).
We focused our attention on ITGβ6 and its ligand FN as possible targets, given their known roles in cell migration and tumorigenesis (1, 4, 10, 36, 51). TNC is also a ligand for ITGαvβ6, may modulate the affinity of FN for ITGαvβ6, and is implicated in tumorigenesis and the metastatic spread of cancer cells (28, 45). An increase in the expression of TNC in the Stat3-C-containing cells relative to that in the pBabe vector control cells was observed by Western blotting (data not shown). However, we did not examine the role of TNC in these processes given the lack of commercially available purified reagents and functional blocking antibodies. We attempted to decrease the levels of TNC by using small interfering RNA but did not see a significant decrease in mRNA levels. The expression of fibronectin has been associated with malignant transformation and resistance to the apoptosis of tumor cells (36). The accumulation of FN in a number of malignancies, including breast, lung, and pancreatic cancers, in particular by the peritumoral stromal cells, has been noted previously (21, 24, 30). The treatment of breast cancer-derived MCF-7 cells with OSM leads to a Stat3-dependent increase in cell migration, with a concomitant increase in FN production, after 72 h of OSM treatment (61). We observed a relatively rapid increase in FN mRNA levels after 4 h of OSM stimulation of RWPE-1 cells, suggesting that Stat3 may directly regulate the transcription of the FN gene in this cell type (Fig. 3). The addition of FN to our cultured prostate epithelial cells resulted in an increase in migration, with a change in the phenotype of the pBabe vector control cells to one approximating the phenotype of the Stat3-C-expressing cells, including a decrease in E-cadherin levels, which is a hallmark of an EMT (Fig. 3) (31). These data suggest that activated Stat3 is likely to be a regulator of FN production, which is partly responsible for both an EMT and enhanced cell migration.
Integrins are essential mediators of tumorigenesis, adhesion, and migration. ITGαvβ6 levels in particular have been found to be increased in many tumors, including colorectal tumors, squamous-cell carcinomas, and pancreatic and breast tumors (2, 4, 39, 52). Recently, it was determined that an EMT of colorectal cells also leads to an increase in the expression of ITGαvβ6 through the Ets-1 transcription factor. Furthermore, the migration of these cells on FN requires ITGαvβ6, and a correlation between ITGαvβ6 and aggressive colorectal tumors was determined (4). Here, we demonstrate that Stat3-C-containing prostate epithelial cells express high levels of ITGβ6 mRNA and protein. The activation of Stat3 by OSM led to an increase in the levels of ITGβ6 mRNA within 4 h of stimulation. Furthermore, both the inhibition of Stat3 activity and a decrease in the abundance of Stat3 by short hairpin RNA in DU145 cells (a cancer-derived human prostate epithelial cell line) resulted in a decrease in the ITGβ6 mRNA levels. Increased ITGβ6 expression in MCF10A cells (a human breast epithelial cell line) expressing Stat3-C was observed, suggesting that activated Stat3 may participate in regulating ITGβ6 expression in more than one tumor type (data not shown) (13). An examination of the ITGβ6 promoter revealed many optimal (TTN5AA, where N represents any nucleotide) Stat3 binding sites (Fig. 4). Activated Stat3 may bind to the ITGβ6 promoter and stimulate the expression of an ITGβ6 promoter-luciferase gene construct, suggesting that ITGβ6 is a direct transcriptional target of Stat3 (Fig. 4).
The engagement of integrins with extracellular matrix proteins results in the regulation of the small GTPases (19). The activity of the small GTPases (Rac, Rho, and Cdc42) regulates the dynamic organization of the actin cytoskeleton, which determines cell morphology and regulates cell migration (50). Here, we demonstrate that the addition of exogenous FN can enhance cell migration (Fig. 3). Huang et al. have shown that the ITGαvβ6-mediated migration of murine keratinocytes on FN is blocked by an anti-ITGαvβ6 antibody (29). Here, we show that the Stat3-C expressing cells migrated better than pBabe control cells and that this phenotype was enhanced in the presence of FN. Significantly, the enhanced migratory phenotype was inhibited by a function-blocking antibody, 6.3G9, to ITGαvβ6 (Fig. 5). Furthermore, culturing Stat3-C-expressing cells in the presence of 6.3G9 led to an increase in E-cadherin expression approximating the expression seen in the pBabe vector control cells (Fig. 5). We hypothesize that activated Stat3 may promote cell migration and an EMT of RWPE-1 cells as a function of the coordinated regulation of ITGαvβ6 and FN.
We examined primary prostate tissue samples for the expression of activated Stat3 and ITGαvβ6. We observed that a relatively small fraction of cancer samples expressed high levels of activated Stat3, and no significant correlation between activated Stat3 and clinical parameters was noted (Fig. 5). This finding is in contrast to observations which show that a high percentage of samples express activated Stat3 and that it correlates with a high Gleason score and an advanced pathological stage (15, 26, 40). A possible explanation is provided by the labile nature of the pYStat3 signal in primary cancer tissues (16). Specifically, we observed that the pYStat3 signal in cancer samples decreased within 30 min of procurement (data not shown). Thus, we may be underestimating the number of pYStat3-positive samples in our tissue microarrays. The incidence of strong expression (staining score, +2 to +3) of ITGαvβ6 in primary prostate cancer cells was low (10%), while the overall level of expression was 23%, and no significant correlation between high levels of pYStat3 and levels of ITGαvβ6 was observed. However, high levels of ITGαvβ6 expression as well as pYStat3 were found within basal cells of both nonneoplastic ducts and those with intraepithelial neoplasia, although the degree of positive correlation did not achieve statistical significance. The role these basal cells play in the pathogenesis of prostate cancer is unknown. Furthermore, it remains unclear whether the histologically benign-appearing ducts adjacent to cancer are not already in the process of transformation due to field effects. In summary, we have identified and characterized novel targets of activated Stat3 that contribute to Stat3-mediated migration and may play a role in prostate tumorigenesis.
Supplementary Material
Acknowledgments
This study was supported by National Institutes of Health grant R01 CA87637, a Breast Cancer Alliance award, and Holloway Foundation, Sussman Fund, and Lerner awards (J.B.) and a Cancer Research and Prevention Foundation and Intercultural Cancer Council fellowship and a National Cancer Institute Comprehensive Minority Biomedical Branch training award (J.A.).
We thank Peter Oettgen (Boston, MA) for the ITGβ6 luciferase construct and Sam Lee for technical assistance.
Footnotes
Published ahead of print on 16 April 2007.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Agrez, M., A. Chen, R. I. Cone, R. Pytela, and D. Sheppard. 1994. The alpha v beta 6 integrin promotes proliferation of colon carcinoma cells through a unique region of the beta 6 cytoplasmic domain. J. Cell Biol. 127:547-556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arihiro, K., M. Kaneko, S. Fujii, K. Inai, and Y. Yokosaki. 2000. Significance of alpha 9 beta 1 and alpha v beta 6 integrin expression in breast carcinoma. Breast Cancer 7:19-26. [DOI] [PubMed] [Google Scholar]
- 3.Bastian, M., M. Steiner, and P. Schuff-Werner. 2005. Expression of thrombospondin-1 in prostate-derived cell lines. Int. J. Mol. Med. 15:49-56. [PubMed] [Google Scholar]
- 4.Bates, R. C., D. I. Bellovin, C. Brown, E. Maynard, B. Wu, H. Kawakatsu, D. Sheppard, P. Oettgen, and A. M. Mercurio. 2005. Transcriptional activation of integrin beta6 during the epithelial-mesenchymal transition defines a novel prognostic indicator of aggressive colon carcinoma. J. Clin. Investig. 115:339-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bello, D., M. M. Webber, H. K. Kleinman, D. D. Wartinger, and J. S. Rhim. 1997. Androgen responsive adult human prostatic epithelial cell lines immortalized by human papillomavirus 18. Carcinogenesis 18:1215-1223. [DOI] [PubMed] [Google Scholar]
- 6.Bromberg, J. F., M. H. Wrzeszczynska, G. Devgan, Y. Zhao, R. G. Pestell, C. Albanese, and J. E. Darnell, Jr. 1999. Stat3 as an oncogene. Cell 98:295-303. [DOI] [PubMed] [Google Scholar]
- 7.Busk, M., R. Pytela, and D. Sheppard. 1992. Characterization of the integrin alpha v beta 6 as a fibronectin-binding protein. J. Biol. Chem. 267:5790-5796. [PubMed] [Google Scholar]
- 8.Chen, H. C. 2004. Boyden chamber assay. Methods Mol. Biol. 294:15-22. [DOI] [PubMed] [Google Scholar]
- 9.Chen, T., L. H. Wang, and W. L. Farrar. 2000. Interleukin 6 activates androgen receptor-mediated gene expression through a signal transducer and activator of transcription 3-dependent pathway in LNCaP prostate cancer cells. Cancer Res. 60:2132-2135. [PubMed] [Google Scholar]
- 10.Danen, E. H., and K. M. Yamada. 2001. Fibronectin, integrins, and growth control. J. Cell. Physiol. 189:1-13. [DOI] [PubMed] [Google Scholar]
- 11.Dauer, D. J., B. Ferraro, L. Song, B. Yu, L. Mora, R. Buettner, S. Enkemann, R. Jove, and E. B. Haura. 2005. Stat3 regulates genes common to both wound healing and cancer. Oncogene 24:3397-3408. [DOI] [PubMed] [Google Scholar]
- 12.Debidda, M., L. Wang, H. Zang, V. Poli, and Y. Zheng. 2005. A role of STAT3 in Rho GTPase-regulated cell migration and proliferation. J. Biol. Chem. 280:17275-17285. [DOI] [PubMed] [Google Scholar]
- 13.Dechow, T. N., L. Pedranzini, A. Leitch, K. Leslie, W. L. Gerald, I. Linkov, and J. F. Bromberg. 2004. Requirement of matrix metalloproteinase-9 for the transformation of human mammary epithelial cells by Stat3-C. Proc. Natl. Acad. Sci. USA 101:10602-10607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeMiguel, F., S. O. Lee, W. Lou, X. Xiao, B. R. Pflug, J. B. Nelson, and A. C. Gao. 2002. Stat3 enhances the growth of LNCaP human prostate cancer cells in intact and castrated male nude mice. Prostate 52:123-129. [DOI] [PubMed] [Google Scholar]
- 15.Dhir, R., Z. Ni, W. Lou, F. DeMiguel, J. R. Grandis, and A. C. Gao. 2002. Stat3 activation in prostatic carcinomas. Prostate 51:241-246. [DOI] [PubMed] [Google Scholar]
- 16.Diaz, N., S. Minton, C. Cox, T. Bowman, T. Gritsko, R. Garcia, I. Eweis, M. Wloch, S. Livingston, E. Seijo, A. Cantor, J. H. Lee, C. A. Beam, D. Sullivan, R. Jove, and C. A. Muro-Cacho. 2006. Activation of stat3 in primary tumors from high-risk breast cancer patients is associated with elevated levels of activated SRC and survivin expression. Clin. Cancer Res. 12:20-28. [DOI] [PubMed] [Google Scholar]
- 17.Epling-Burnette, P. K., J. H. Liu, R. Catlett-Falcone, J. Turkson, M. Oshiro, R. Kothapalli, Y. Li, J. M. Wang, H. F. Yang-Yen, J. Karras, R. Jove, and T. P. Loughran, Jr. 2001. Inhibition of STAT3 signaling leads to apoptosis of leukemic large granular lymphocytes and decreased Mcl-1 expression. J. Clin. Investig. 107:351-362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flowers, L. O., P. S. Subramaniam, and H. M. Johnson. 2005. A SOCS-1 peptide mimetic inhibits both constitutive and IL-6 induced activation of STAT3 in prostate cancer cells. Oncogene 24:2114-2120. [DOI] [PubMed] [Google Scholar]
- 19.Friedl, P. 2004. Prespecification and plasticity: shifting mechanisms of cell migration. Curr. Opin. Cell Biol. 16:14-23. [DOI] [PubMed] [Google Scholar]
- 20.Gao, L. F., D. Q. Xu, Y. T. Shao, D. Zhao, and X. J. Zhao. 2005. Knockdown of STAT3 expression using siRNA inhibits the growth of prostate cancer cell lines. Zhonghua Nan Ke Xue 11:29-33, 37. (In Chinese.) [PubMed] [Google Scholar]
- 21.Gress, T. M., F. Muller-Pillasch, M. M. Lerch, H. Friess, M. Buchler, and G. Adler. 1995. Expression and in-situ localization of genes coding for extracellular matrix proteins and extracellular matrix degrading proteases in pancreatic cancer. Int. J. Cancer 62:407-413. [DOI] [PubMed] [Google Scholar]
- 22.Guan, M., and Y. Chen. 2005. Aberrant expression of DeltaNp73 in benign and malignant tumours of the prostate: correlation with Gleason score. J. Clin. Pathol. 58:1175-1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hahn, W. C., and R. A. Weinberg. 2002. Rules for making human tumor cells. N. Engl. J. Med. 347:1593-1603. [DOI] [PubMed] [Google Scholar]
- 24.Han, S., N. Sidell, S. Roser-Page, and J. Roman. 2004. Fibronectin stimulates human lung carcinoma cell growth by inducing cyclooxygenase-2 (COX-2) expression. Int. J. Cancer 111:322-331. [DOI] [PubMed] [Google Scholar]
- 25.Hazan, R. B., G. R. Phillips, R. F. Qiao, L. Norton, and S. A. Aaronson. 2000. Exogenous expression of N-cadherin in breast cancer cells induces cell migration, invasion, and metastasis. J. Cell Biol. 148:779-790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Horinaga, M., H. Okita, J. Nakashima, K. Kanao, M. Sakamoto, and M. Murai. 2005. Clinical and pathologic significance of activation of signal transducer and activator of transcription 3 in prostate cancer. Urology 66:671-675. [DOI] [PubMed] [Google Scholar]
- 27.Huang, H. F., T. F. Murphy, P. Shu, A. B. Barton, and B. E. Barton. 2005. Stable expression of constitutively-activated STAT3 in benign prostatic epithelial cells changes their phenotype to that resembling malignant cells. Mol. Cancer 4:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang, W., R. Chiquet-Ehrismann, J. V. Moyano, A. Garcia-Pardo, and G. Orend. 2001. Interference of tenascin-C with syndecan-4 binding to fibronectin blocks cell adhesion and stimulates tumor cell proliferation. Cancer Res. 61:8586-8594. [PubMed] [Google Scholar]
- 29.Huang, X., J. Wu, S. Spong, and D. Sheppard. 1998. The integrin alphavbeta6 is critical for keratinocyte migration on both its known ligand, fibronectin, and on vitronectin. J. Cell Sci. 111:2189-2195. [DOI] [PubMed] [Google Scholar]
- 30.Jiang, Y., S. L. Harlocker, D. A. Molesh, D. C. Dillon, J. A. Stolk, R. L. Houghton, E. A. Repasky, R. Badaro, S. G. Reed, and J. Xu. 2002. Discovery of differentially expressed genes in human breast cancer using subtracted cDNA libraries and cDNA microarrays. Oncogene 21:2270-2282. [DOI] [PubMed] [Google Scholar]
- 31.Kang, Y., and J. Massague. 2004. Epithelial-mesenchymal transitions: twist in development and metastasis. Cell 118:277-279. [DOI] [PubMed] [Google Scholar]
- 32.Kaplan, D. H., V. Shankaran, A. S. Dighe, E. Stockert, M. Aguet, L. J. Old, and R. D. Schreiber. 1998. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc. Natl. Acad. Sci. USA 95:7556-7561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaplan, R. N., R. D. Riba, S. Zacharoulis, A. H. Bramley, L. Vincent, C. Costa, D. D. MacDonald, D. K. Jin, K. Shido, S. A. Kerns, Z. Zhu, D. Hicklin, Y. Wu, J. L. Port, N. Altorki, E. R. Port, D. Ruggero, S. V. Shmelkov, K. K. Jensen, S. Rafii, and D. Lyden. 2005. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 438:820-827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karni, R., R. Jove, and A. Levitzki. 1999. Inhibition of pp60c-Src reduces Bcl-XL expression and reverses the transformed phenotype of cells overexpressing EGF and HER-2 receptors. Oncogene 18:4654-4662. [DOI] [PubMed] [Google Scholar]
- 35.Kira, M., S. Sano, S. Takagi, K. Yoshikawa, J. Takeda, and S. Itami. 2002. STAT3 deficiency in keratinocytes leads to compromised cell migration through hyperphosphorylation of p130(cas). J. Biol. Chem. 277:12931-12936. [DOI] [PubMed] [Google Scholar]
- 36.Labat-Robert, J. 2002. Fibronectin in malignancy. Semin. Cancer Biol. 12:187-195. [DOI] [PubMed] [Google Scholar]
- 37.Leslie, K., C. Lang, G. Devgan, J. Azare, M. Berishaj, W. Gerald, Y. B. Kim, K. Paz, J. E. Darnell, C. Albanese, T. Sakamaki, R. Pestell, and J. Bromberg. 2006. Cyclin D1 is transcriptionally regulated by and required for transformation by activated signal transducer and activator of transcription 3. Cancer Res. 66:2544-2552. [DOI] [PubMed] [Google Scholar]
- 38.Li, C., F. Zhang, M. Lin, and J. Liu. 2004. Induction of S100A9 gene expression by cytokine oncostatin M in breast cancer cells through the STAT3 signaling cascade. Breast Cancer Res. Treat. 87:123-134. [DOI] [PubMed] [Google Scholar]
- 39.Li, X., Y. Yang, Y. Hu, D. Dang, J. Regezi, B. L. Schmidt, A. Atakilit, B. Chen, D. Ellis, and D. M. Ramos. 2003. Alphavbeta6-Fyn signaling promotes oral cancer progression. J. Biol. Chem. 278:41646-41653. [DOI] [PubMed] [Google Scholar]
- 40.Mora, L. B., R. Buettner, J. Seigne, J. Diaz, N. Ahmad, R. Garcia, T. Bowman, R. Falcone, R. Fairclough, A. Cantor, C. Muro-Cacho, S. Livingston, J. Karras, J. Pow-Sang, and R. Jove. 2002. Constitutive activation of Stat3 in human prostate tumors and cell lines: direct inhibition of Stat3 signaling induces apoptosis of prostate cancer cells. Cancer Res. 62:6659-6666. [PubMed] [Google Scholar]
- 41.Munger, J. S., X. Huang, H. Kawakatsu, M. J. Griffiths, S. L. Dalton, J. Wu, J. F. Pittet, N. Kaminski, C. Garat, M. A. Matthay, D. B. Rifkin, and D. Sheppard. 1999. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell 96:319-328. [DOI] [PubMed] [Google Scholar]
- 42.Ng, D. C., B. H. Lin, C. P. Lim, G. Huang, T. Zhang, V. Poli, and X. Cao. 2006. Stat3 regulates microtubules by antagonizing the depolymerization activity of stathmin. J. Cell Biol. 172:245-257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ni, Z., W. Lou, S. O. Lee, R. Dhir, F. DeMiguel, J. R. Grandis, and A. C. Gao. 2002. Selective activation of members of the signal transducers and activators of transcription family in prostate carcinoma. J. Urol. 167:1859-1862. [PubMed] [Google Scholar]
- 44.Niu, G., K. L. Wright, M. Huang, L. Song, E. Haura, J. Turkson, S. Zhang, T. Wang, D. Sinibaldi, D. Coppola, R. Heller, L. M. Ellis, J. Karras, J. Bromberg, D. Pardoll, R. Jove, and H. Yu. 2002. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene 21:2000-2008. [DOI] [PubMed] [Google Scholar]
- 45.Orend, G. 2005. Potential oncogenic action of tenascin-C in tumorigenesis. Int. J. Biochem. Cell Biol. 37:1066-1083. [DOI] [PubMed] [Google Scholar]
- 46.Paz, K., N. D. Socci, E. van Nimwegen, A. Viale, and J. E. Darnell. 2004. Transformation fingerprint: induced STAT3-C, v-Src and Ha-Ras cause small initial changes but similar established profiles in mRNA. Oncogene 23:8455-8463. [DOI] [PubMed] [Google Scholar]
- 47.Ridley, A. J., M. A. Schwartz, K. Burridge, R. A. Firtel, M. H. Ginsberg, G. Borisy, J. T. Parsons, and A. R. Horwitz. 2003. Cell migration: integrating signals from front to back. Science 302:1704-1709. [DOI] [PubMed] [Google Scholar]
- 48.Rivat, C., O. D. Wever, E. Bruyneel, M. Mareel, C. Gespach, and S. Attoub. 2004. Disruption of STAT3 signaling leads to tumor cell invasion through alterations of homotypic cell-cell adhesion complexes. Oncogene 23:3317-3327. [DOI] [PubMed] [Google Scholar]
- 49.Roessler, M., W. Rollinger, S. Palme, M. L. Hagmann, P. Berndt, A. M. Engel, B. Schneidinger, M. Pfeffer, H. Andres, J. Karl, H. Bodenmuller, J. Ruschoff, T. Henkel, G. Rohr, S. Rossol, W. Rosch, H. Langen, W. Zolg, and M. Tacke. 2005. Identification of nicotinamide N-methyltransferase as a novel serum tumor marker for colorectal cancer. Clin. Cancer Res. 11:6550-6557. [DOI] [PubMed] [Google Scholar]
- 50.Sahai, E., and C. J. Marshall. 2002. RHO-GTPases and cancer. Nat. Rev. Cancer 2:133-142. [DOI] [PubMed] [Google Scholar]
- 51.Sheppard, D. 2005. Integrin-mediated activation of latent transforming growth factor beta. Cancer Metastasis Rev. 24:395-402. [DOI] [PubMed] [Google Scholar]
- 52.Sipos, B., D. Hahn, A. Carceller, J. Piulats, J. Hedderich, H. Kalthoff, S. L. Goodman, M. Kosmahl, and G. Kloppel. 2004. Immunohistochemical screening for beta6-integrin subunit expression in adenocarcinomas using a novel monoclonal antibody reveals strong up-regulation in pancreatic ductal adenocarcinomas in vivo and in vitro. Histopathology 45:226-236. [DOI] [PubMed] [Google Scholar]
- 53.Taylor, K. M., S. Hiscox, and R. I. Nicholson. 2004. Zinc transporter LIV-1: a link between cellular development and cancer progression. Trends Endocrinol. Metab. 15:461-463. [DOI] [PubMed] [Google Scholar]
- 54.Thomasset, N., A. Lochter, C. J. Sympson, L. R. Lund, D. R. Williams, O. Behrendtsen, Z. Werb, and M. J. Bissell. 1998. Expression of autoactivated stromelysin-1 in mammary glands of transgenic mice leads to a reactive stroma during early development. Am. J. Pathol. 153:457-467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thompson, J., R. Cubbon, R. Cummings, L. Wicker, R. Frankshun, B. Cunningham, P. Cameron, P. Meinke, N. Liverton, Y. Weng, and J. DeMartino. 2002. Photochemical preparation of a pyridone containing tetracycle: a Jak protein kinase inhibitor. Bioorg. Med. Chem. Lett. 12:1219-1223. [DOI] [PubMed] [Google Scholar]
- 56.Webber, M. M., D. Bello, and S. Quader. 1997. Immortalized and tumorigenic adult human prostatic epithelial cell lines: characteristics and applications. Part 3. Oncogenes, suppressor genes, and applications. Prostate 30:136-142. [DOI] [PubMed] [Google Scholar]
- 57.Weinreb, P. H., K. J. Simon, P. Rayhorn, W. J. Yang, D. R. Leone, B. M. Dolinski, B. R. Pearse, Y. Yokota, H. Kawakatsu, A. Atakilit, D. Sheppard, and S. M. Violette. 2004. Function-blocking integrin alphavbeta6 monoclonal antibodies: distinct ligand-mimetic and nonligand-mimetic classes. J. Biol. Chem. 279:17875-17887. [DOI] [PubMed] [Google Scholar]
- 58.Xie, T. X., D. Wei, M. Liu, A. C. Gao, F. Ali-Osman, R. Sawaya, and S. Huang. 2004. Stat3 activation regulates the expression of matrix metalloproteinase-2 and tumor invasion and metastasis. Oncogene 23:3550-3560. [DOI] [PubMed] [Google Scholar]
- 59.Yamashita, S., C. Miyagi, T. Fukada, N. Kagara, Y. S. Che, and T. Hirano. 2004. Zinc transporter LIVI controls epithelial-mesenchymal transition in zebrafish gastrula organizer. Nature 429:298-302. [DOI] [PubMed] [Google Scholar]
- 60.Yu, H., and R. Jove. 2004. The STATs of cancer: new molecular targets come of age. Nat. Rev. Cancer 4:97-105. [DOI] [PubMed] [Google Scholar]
- 61.Zhang, F., C. Li, H. Halfter, and J. Liu. 2003. Delineating an oncostatin M-activated STAT3 signaling pathway that coordinates the expression of genes involved in cell cycle regulation and extracellular matrix deposition of MCF-7 cells. Oncogene 22:894-905. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.