

SUMMARY
Within the developing mammalian CNS, growth factors direct multipotent precursors to generate neurons versus glia, a process that if perturbed might lead to neural dysfunction. In this regard, genetic mutations resulting in constitutive activation of the protein tyrosine phosphatase SHP-2 cause Noonan Syndrome (NS), which is associated with learning disabilities and mental retardation. Here, we demonstrate that genetic knockdown of SHP-2 in cultured cortical precursors or in the embryonic cortex inhibited basal neurogenesis and caused enhanced and precocious astrocyte formation. Conversely, expression of a NS SHP-2 mutant promoted neurogenesis and inhibited astrogenesis. Neural cell fate decisions were similarly perturbed in a mouse knock-in model that phenocopies human NS. Thus, SHP-2 instructs precursors to make neurons and not astrocytes during the neurogenic period, and perturbations in the relative ratios of these two cell types upon constitutive SHP-2 activation may contribute to the cognitive impairments in NS patients.
INTRODUCTION
Development of the cerebral cortex involves a common pool of precursor cells that sequentially generate neurons and glia. Emerging evidence indicates that both intrinsic mechanisms and environmental growth factors are important for cortical cell fate decisions (Miller and Gauthier, 2007). In particular, appropriate early neurogenesis requires receptor tyrosine kinase (RTK) mediated activation of a MEK-ERK-C/EBP pathway (Ménard et al., 2002; Paquin et al., 2005), while the later onset of astrocyte formation requires activation of the gp130-JAK-STAT pathway (Bonni et al., 1997; Johe et al., 1996) by neuron-derived cardiotrophin-1 (Barnabé-Heider et al., 2005).
Implicit in any model where growth factors define neural cell fate is the assumption that multipotent precursors can respond to neurogenic and gliogenic factors even at times when those cell types are not normally generated. Support for this assumption comes from work showing that cortical precursors will inappropriately generate astrocytes during the neurogenic period if prematurely exposed to CNTF in vivo (Barnabé-Heider et al., 2005). One way to ensure that such inappropriate early gliogenesis does not occur is via intrinsic epigenetic mechanisms that make early cortical precursors relatively unresponsive to gliogenic cytokines (reviewed in Miller and Gauthier, 2007). Here, we have asked whether growth factor signaling provides another way to silence gliogenesis during the neurogenic period, and have focused on the protein tyrosine phosphatase SHP-2.
SHP-2 is a growth factor-regulated phosphatase that is widely-expressed and that modulates both the MEK-ERK and the gp130-JAK-STAT pathways (Neel et al., 2003; Ernst and Jenkins, 2004). SHP-2 is recruited to many receptor tyrosine kinases (RTKs) upon activation, and is essential for sustained MEK-ERK activation (Neel et al., 2003). SHP-2 is also recruited to the activated gp130 receptor, and negatively regulates the gp130-JAK-STAT pathway in some cells (Lehmann et al., 2003; Ernst and Jenkins, 2004). Thus, SHP-2 is an ideal candidate for promoting neurogenesis and inhibiting gliogenesis. Further support for the idea that SHP-2 may regulate neural development comes from the human genetic disorder Noonan Syndrome (NS) which occurs in 1 in 2500 live births. NS children present with cardiac defects, craniofacial abnormalities, and short stature (Noonan, 1994), and a large percentage (1/3 to 1/2) exhibit learning disabilities and mental retardation (Noonan, 1994; Yoshida et al., 2004; Lee et al., 2005). Approximately fifty percent of NS cases are caused by missense mutations in the human PTPN11 (SHP-2) gene (Tartaglia et al., 2001), and result in expression of a SHP-2 protein with increased basal or stimulated phosphatase activity (Fragale et al., 2004; Keilhack et al., 2005). NS has been modelled in the mouse by knocking-in a NS SHP-2 allele (Araki et al., 2004), but it is not yet known whether this mouse model displays neural and/or cognitive perturbations.
Here, we have provide evidence that SHP-2 is essential for normal cortical cell fate decisions, and that aberrant activation of SHP-2 in a NS mouse model decreases astrogenesis and enhances neurogenesis, suggesting that some cognitive impairments seen in this syndrome may be due to aberrant neural cell fate decisions during development.
RESULTS
SHP-2 is necessary for cultured cortical precursor cells to generate neurons
To elucidate mechanisms that regulate genesis of neurons versus astrocytes, we examined primary murine E12 cortical precursors, a system we characterized previously (Ménard et al., 2002; Barnabé-Heider and Miller, 2003; Barnabé-Heider et al., 2005). Upon plating in FGF2, these precursors are dividing, nestin-positive cells that generate neurons first at 1 day in vitro (DIV) and glia at 5–6 DIV. This increase in differentiated cells is accompanied by depletion of proliferating precursors.
We showed previously that cortical precursor neurogenesis requires activation of a MEK-C/EBP signalling pathway (Ménard et al., 2002; Barnabé-Heider and Miller, 2003; Paquin et al., 2005), while astrocyte formation requires cardiotrophin-1-mediated gp130-JAK-STAT activation (Barnabé-Heider et al., 2005). Because SHP-2 can regulate these two signalling cascades, we asked whether it regulates cell fate decisions in cortical precursors. Initially, we showed that SHP-2 is expressed in cortical precursors and newly-born cortical neurons by Western blots (Fig. 1A) and immunocytochemistry (Fig. 1B). We then characterized SHP-2 expression in the embryonic cortex by immunocytochemistry. During the neurogenic period, at E12 and E15, SHP-2 was enriched in nestin-positive precursors of the ventricular/subventricular zones (VZ/SVZ) relative to neurons in the cortical plate (Fig. 1C). By E17, when gliogenesis commences, the highest relative levels of SHP-2 were in neurons of the cortical mantle, with lower expression in the VZ/SVZ. A similar expression pattern was observed at P5 (Fig. 1C). Thus, SHP-2 is relatively high in neurogenic versus gliogenic cortical precursors.
Figure 1. SHP-2 expression is high in cortical precursors during the neurogenic, but not gliogenic period.
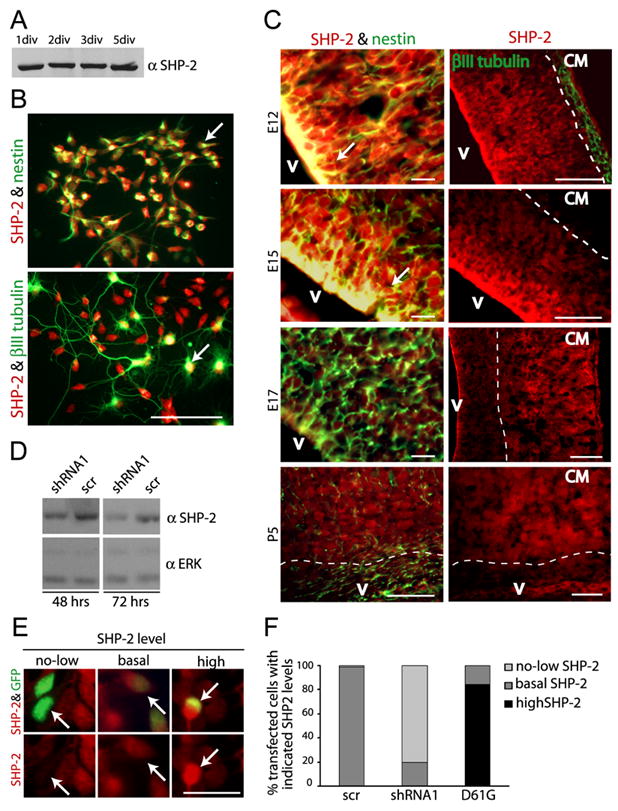
(A) Western blot for SHP-2 in equal amounts of lysate from precursors cultured for 1–5 DIV. (B) Immunocytochemistry for SHP-2 (red) and nestin (top panel, green) or βIII-tubulin (bottom panel, green) in precursors cultured for 2 DIV. Arrows denote double-labelled cells. Space bar = 50μm. (C) Immunocytochemistry for SHP-2 (all panels, red) and nestin (left panels, green) or βIII-tubulin (right top panel, green) in coronal cortical sections at E12, E15, E17 and P5. Arrows denote nestin-positive, SHP-2-positive precursors in the VZ/SVZ. The broken line demarcates the VZ/SVZ from the cortical mantle (CM). V = ventricle. Space bar = 50μm. (D) Western blot for SHP-2 in lysates from NIH 3T3 cells transfected with SHP-2 shRNA1 or a scrambled shRNA control (scr) for 48 or 72 hours. Blots were reprobed for ERK to ensure equal protein loading. (E) Immunocytochemistry for SHP-2 (red) in transfected precursors (green) indicating no or low (no-low), basal or high levels of SHP-2 expression. Arrows indicate the same transfected cell in the upper and lower panel. Space bar = 25μm. (F) Quantification of SHP-2 immunoreactivity as shown in (E) for precursors cotransfected with plasmids encoding EGFP and shRNA1, scrambled shRNA (scr) or, as a positive control, D61G SHP-2 for 2 DIV.
To ask if SHP-2 was necessary for neurogenesis, we used two different SHP-2 shRNAs. We determined the efficacy of these shRNAs by transfection into NIH-3T3 cells. Western blots showed that SHP-2 levels were reduced by both SHP-2 shRNAs at 48–72 hours posttransfection even though only ~ 30% of the 3T3 cells were transfected (Fig. 1D; data not shown). We then cotransfected cortical precursors with EGFP and the SHP-2 shRNAs and analyzed SHP-2 levels immunocytochemically at 2DIV; shRNA1 and shRNA2 both reduced SHP-2 levels, with shRNA1 being the more effective of the two (Fig. 1E,F; data not shown). We then performed similar experiments and asked if SHP-2 knockdown affected neurogenesis by immunostaining for the neuron-specific protein βIII-tubulin at 3DIV (Fig. 2A). Quantification revealed that SHP- 2 shRNA1 and shRNA2 both decreased the number of transfected neurons (Fig. 2B). To confirm that this was due to decreased SHP-2 levels, we performed a rescue experiment. Precursors were cotransfected with SHP-2 shRNA1 and HA-tagged human SHP-2 (which would not be targetted by the murine shRNA), and analyzed 3 days later for βIII-tubulin. The human SHP-2 completely rescued the decreased neurogenesis caused by murine SHP-2 shRNA (Fig. 2B).
Figure 2. Genetic knockdown of SHP-2 has no effect on cortical precursor cell survival or proliferation, but specifically inhibits neurogenesis.
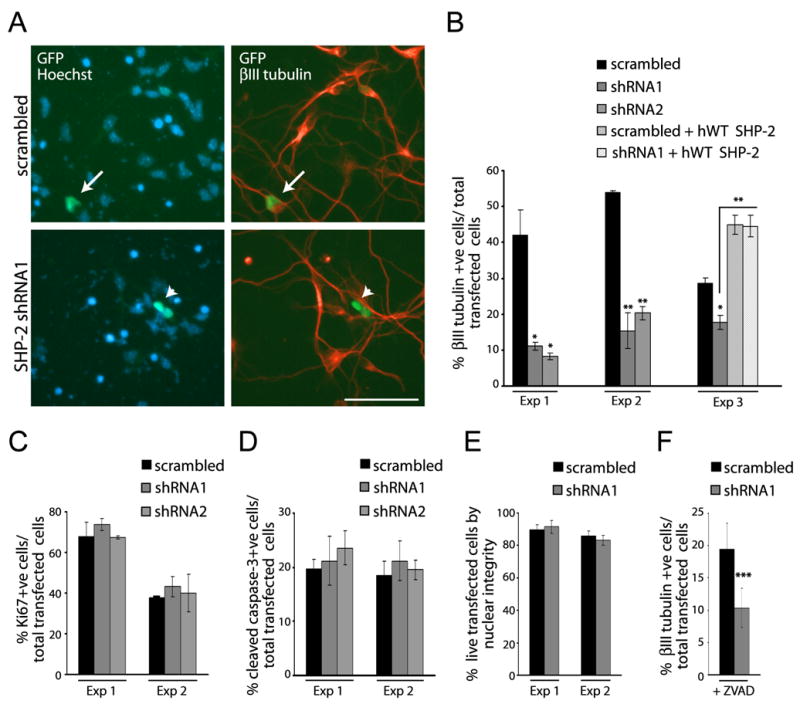
(A) Immunocytochemistry for EGFP (GFP; green) and βIII-tubulin (red) in precursors cotransfected with EGFP and SHP-2 shRNA1 or scrambled shRNA and cultured 3 DIV. Cells were counterstained with Hoechst 33258. Arrows denote double-labelled, transfected cells, and the arrowhead an EGFP-positive cell that is not double-labelled. Space bar = 50μm. (B) Quantification of transfected, βIII-tubulin-positive neurons in experiments similar to (A). Three experiments of 8 are shown. In experiment 3 (Exp 3), cultures were also cotransfected with murine SHP-2 shRNA1 and HA-tagged human SHP-2 (hWT SHP-2). *p<0.05, ** p<0.01, ANOVA. (C–E) Quantification of precursors transfected as in (A), cultured for 2 DIV, and stained for (C) Ki67, (D) caspase-3, or (E) with Hoechst 33258 to monitor nuclear morphology. In all cases, two experiments of three are shown. p>0.05, ANOVA (C,D). (F) Quantification of precursors transfected as in (A), cultured for 3 DIV in the presence of ZVAD, and immunostained for βIII tubulin. Results represent two pooled experiments. *** p<0.005.
While these data support the idea that SHP-2 is required for neurogenesis, alternative explanations are that SHP-2 is necessary for precursor proliferation or survival. To address these possibilities, precursors were cotransfected with EGFP and SHP-2 shRNA1, shRNA2, or scrambled shRNA, and were analyzed immunocytochemically at 2DIV for Ki67, to monitor proliferation, or cleaved caspase-3, to monitor apoptosis. Quantification revealed that the SHP-2 shRNAs had no significant effect on proliferation (Fig. 2C) or apoptosis (Fig. 2D) of transfected precursors. Counts of uncondensed Hoechst-positive nuclei confirmed that SHP-2 knock-down had no effect on survival (Fig. 2E). As a final control, we performed similar experiments in the presence of the pan-caspase inhibitor ZVAD, which completely inhibits apoptosis of cortical precursors (data not shown); SHP-2 shRNA still reduced the percentage of transfected neurons in the presence of ZVAD (Fig. 2F). Thus, SHP-2 is not essential for cortical precursor survival or proliferation, but is necessary for neurogenesis.
SHP-2 inhibits cytokine-mediated genesis of astrocytes in cortical precursor cultures
Since SHP-2 negatively regulates the JAK-STAT pathway in some cells, we asked whether it regulates astrocyte formation. To do this, we cotransfected precursors with EGFP and SHP-2 shRNA1, shRNA2, or scrambled shRNA, and then induced premature astrogenesis with exogenous CNTF (Johe et al., 1996; Bonni et al., 1997) at 2 DIV. Immunocytochemistry 4 days later (6 DIV) for GFAP (Fig. 3A) demonstrated that SHP-2 shRNA1 and shRNA2 robustly increased the number of transfected astrocytes (Fig. 3B). Similar results were obtained with the early astrocyte/astrocyte precursor marker CD44 (Fig. 3C) (Liu et al., 2004; Barnabé-Heider et al., 2005), indicating that SHP-2 normally functions to inhibit cytokine-mediated astrogenesis.
Figure 3. (A–E) Genetic knockdown of SHP-2 enhances cytokine-mediated gliogenesis.
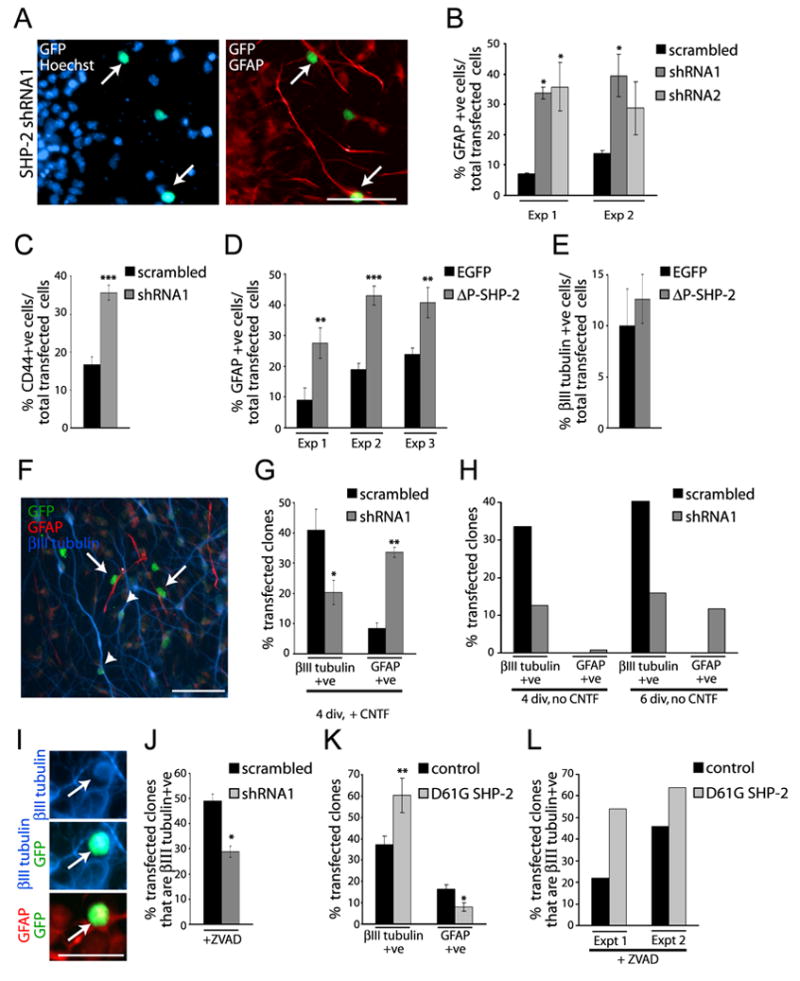
(A) Immunocytochemistry for EGFP (GFP; green) and GFAP (red) in precursors cotransfected with EGFP and SHP-2 shRNA1, exposed to 50 ng/ml CNTF at 2 DIV, and cultured for 4 more days. Cells were counterstained with Hoechst 33258. Arrows denote double-labelled, transfected cells. Space bar = 50μm. (B,C) Quantification of transfected cells expressing (B) GFAP or (C) CD44 in experiments similar to (A). *p<0.05, ***p<0.005, ANOVA (B). (D,E) Quantification of transfected cells expressing (D) GFAP or (F) βIII-tubulin in precursors cotransfected with EGFP and P-SHP-2. In (D) cells were exposed to CNTF at 2 DIV, and immunostained 3 days later, and in (E) cells were immunostained after 3 DIV. **p<0.01, ***p<0.005. (F–L) Clonal analysis demonstrates that SHP-2 instructs precursor cells to generate neurons at the expense of astrocytes. (F) Immunocytochemistry for EGFP (GFP, green), βIII tubulin (blue) and GFAP (red) in precursors cotransfected with EGFP and SHP-2 shRNA1, exposed to 50ng/ml CNTF at 2 DIV and cultured 4 more days. Arrows and arrowheads denote EGFP-positive astrocytes and neurons, respectively. Space bar = 50μm. (G) Quantification of three pooled experiments similar to (F) where precursors were exposed to CNTF at 2 DIV, and analyzed 2 days later. * p<0.05, **p<0.01. (H) Quantification of two experiments of three similar to (F), where cortical precursors were cultured 4 or 6 DIV in the absence of CNTF. (I) Immunocytochemistry of precursors cotransfected with EGFP (green) and SHP-2 shRNA1 for 2 days, exposed to CNTF for 2 days, and then stained for βIII-tubulin (blue) and GFAP (red). Arrows indicate the same transfected precursor coexpressing all three proteins. Scale bar = 25μm. (J) Quantification of 2 pooled experiments (of three) similar to (I), where cells were cultured in the presence of ZVAD from day 1 on. Cells were double-labelled for βIII-tubulin and EGFP. *p<0.05. (K) Quantification of the percentage of neurogenic and gliogenic clones in experiments similar to (F), where cells were cotransfected with EGFP and empty vector or D61G SHP-2, showing pooled data from 4 independent experiments. *p<0.05, **p<0.01. (L) Quantification of two experiments (of three) similar to (J) except that precursors were cotransfected with EGFP and scrambled vector or D61G SHP-2.
To ask whether this inhibition of astrogenesis required SHP-2 phosphatase activity, we used a dominant-negative SHP-2 mutant, P-SHP-2, that has a 30 nucleotide deletion in the phosphatase domain (Tang et al., 1995). Precursors were cotransfected with EGFP and P-SHP-2, exposed to CNTF at 2 DIV, and immunostained for GFAP at 6 DIV. This analysis revealed that, as seen with SHP-2 shRNAs, P-SHP-2 enhanced CNTF-mediated astrocyte formation (Fig. 3D). We therefore asked whether the neurogenic effects of SHP-2 also required phosphatase activity; precursors were transfected with P-SHP-2, and 3 days later were immunostained for βIII-tubulin. These experiments demonstrated that P-SHP-2 had no effect on the numbers of transfected neurons (Fig. 3E), suggesting that SHP-2 regulates neurogenesis via a non-phosphatase-dependent mechanism.
SHP-2 instructs cortical precursors to generate neurons versus astrocytes
To ask whether SHP-2 mediates these effects on cortical precursor differentiation by instructing precursors to generate neurons rather than astrocytes, we performed clonal analysis; individual precursors were transfected at very low densities with EGFP and SHP-2 shRNA1, CNTF was added at 2 DIV, and cultures were immunostained at 4 DIV for EGFP, βIII-tubulin and GFAP. Alternatively, experiments were performed in the absence of CNTF. In either case, isolated, EGFP-positive clones of cells were analyzed for their composition (Fig. 3F); clones containing at least one βIII-tubulin positive neuron but no astrocytes were classified as neuronal clones, those containing at least one GFAP-positive astrocyte but no neurons as astrocyte clones, and those containing both neurons and astrocytes as bipotent clones. Clones containing no cells expressing either of these markers were also observed. This analysis revealed that, in the presence of CNTF, SHP-2 knockdown decreased the number of neuronal clones and increased the number of astrocyte clones (Fig. 3G). Bipotent clone numbers were unchanged (4–5% with or without SHP-2 shRNA; p>0.05), and clone size was unaffected (p>0.05). In the absence of CNTF, SHP-2 knockdown reduced the number of neuronal clones, and caused premature genesis of astrocyte clones at 4 and 6 DIV (Fig. 3H). Thus, endogenous SHP-2 instructs precursors to generate neurons at the expense of glia.
This triple-label analysis also revealed one unexpected result; while control transfected cells never coexpressed neuronal and astrocytic proteins, SHP-2 knockdown caused the appearance of a small number of apparently “confused” cells that expressed both βIII-tubulin and GFAP in the presence of CNTF (Fig. 3I). We therefore ensured that the observed decrease in neuronal clones was not due to cell death by performing clonal experiments in the presence of ZVAD. Immunocytochemistry confirmed that SHP-2 shRNA1 decreased the number of neuronal clones in the presence or absence of ZVAD (Fig. 3J). Thus, endogenous SHP-2 ensures (i) that precursors become neurons rather than astrocytes, and (ii) that precursors do not attempt to adopt both fates in response to extrinsic cues.
A Noonan Syndrome SHP-2 mutant promotes neurogenesis and inhibits gliogenesis from cultured cortical precursors
In fifty percent of NS patients, the disorder is due to missense mutations that enhance the activity of SHP-2 (Tartaglia et al., 2001; Fragale et al., 2004; Yoshida et al., 2004). D61G SHP-2 is one such mutation seen in approximately 10% of NS patients (Yoshida et al., 2004). To ask if a NS SHP-2 mutant would alter cortical precursor cell fate decisions we cotransfected precursors with HA-tagged D61G SHP-2, HA-tagged WT SHP-2, or EGFP as a control. Immunocytochemistry and quantification three days later (Fig. 4A,B) revealed that both D61G and WT SHP-2 increased the percentage of transfected βIII-tubulin-positive cells (Fig. 4B). We then asked whether this increase was due to increased proliferation and/or survival of newly-born neurons as opposed to enhanced neurogenesis. Immunocytochemistry for Ki67 at 2DIV (Fig. 4C) revealed that D61G SHP-2 modestly decreased proliferation, consistent with enhanced neurogenesis. Analysis of Hoechst-positive apoptotic nuclei demonstrated that WT and D61G SHP-2 had no effect on neuronal survival, but promoted a small increase in survival of βIII tubulin-negative cortical precursors (Fig. 4D,E). This small survival effect did not contribute to the observed increase in neurogenesis, since D61G SHP-2 increased the percentage of neurons to a similar extent in the presence or absence of ZVAD (Fig. 4F).
Figure 4. A Noonan Syndrome activated SHP-2 mutant promotes neurogenesis and inhibits gliogenesis in cultured cortical precursors.
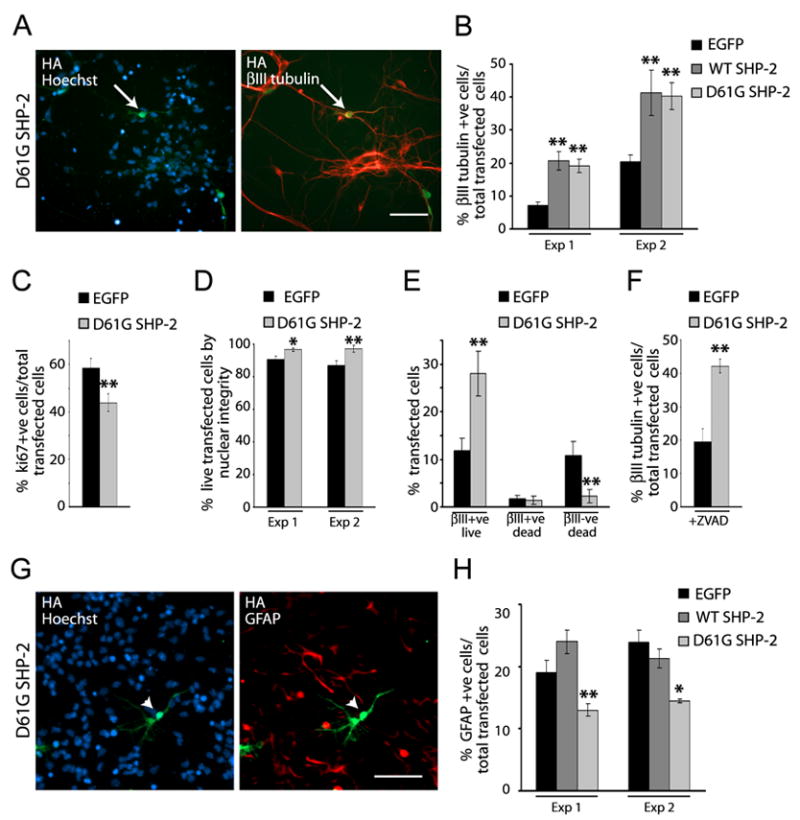
(A) Immunocytochemistry for HA (green) and βIII-tubulin (red) in precursors transfected with HA-tagged D61G SHP-2 and cultured 3 DIV. Cells were counterstained with Hoechst 33258. Arrow denotes a double-labelled, transfected cell. (B) Quantification of transfected, βIII-tubulin-positive neurons in experiments similar to (A) where precursors were cotransfected with EGFP and WT or D61G SHP-2. ** p<0.01, ANOVA. (C) Quantification of precursors transfected as in (A), cultured 2 DIV, and immunostained for Ki67. Three pooled experiments are shown. **p<0.01. (D,E) Quantification of precursors transfected as in (B), cultured 2 DIV, immunostained for EGFP and βIII-tubulin and counterstained with Hoechst 33258 to monitor nuclear morphology. (D) shows the percentage of live cells in 2 independent experiments, while (E) shows the percentage of βIII-tubulin-positive and negative cells with normal (live) versus apoptotic (dead) nuclear morphology within an individual experiment. *p<0.05, **p<0.01. (F) Quantification of one of two experiments as in (A), where precursors were cultured in the presence of ZVAD. ** p<0.01, Student’s t-test. (G) Immunocytochemitry for HA (green) and GFAP (red) in precursors transfected with EGFP or HA-tagged WT SHP-2 or D61G SHP-2, exposed to 50 ng/ml CNTF at 2 DIV, and cultured 4 more days. Cells were counterstained with Hoechst 33258. Arrowheads denote a transfected cell that is GFAP-negative. (H) Quantification of transfected, GFAP-positive astrocytes in two experiments (of 6) similar to (G). *p<0.05, ** p<0.01, ANOVA.
These data indicate that increased levels or activity of SHP-2 directly promote neurogenesis. We therefore asked whether the NS SHP-2 D61G mutant perturbed gliogenesis. Precursors were transfected as in the neurogenesis experiments, and exposed to CNTF at 2 DIV. Immunocytochemistry at 6 DIV revealed that D61G SHP-2 decreased the genesis of transfected, GFAP-positive astrocytes (Fig. 4G,H), while overexpression of WT SHP-2 had no effect (Fig. 4H). Thus, SHP-2 inhibits astrogenesis as it enhances neurogenesis.
Finally, we asked whether these changes were due to altered cell fate decisions by performing clonal analysis. Precursors were cotransfected at very low density with D61G SHP-2 and EGFP, exposed to CNTF at 2 DIV, and clones were then analyzed at 6 DIV (Fig. 3K). Immunocytochemistry revealed that D61G SHP-2 increased the number of neuronal clones and concomitantly decreased the number of astrocyte clones. D61G SHP-2 did not affect the number of bipotent clones (~ 3% in both cases, p=0.2), and clone size was unaffected (p>0.05). Similar results were obtained in the presence of ZVAD (Fig. 3L). Thus, D61G SHP-2 functions within individual cortical precursors to promote neurogenesis while inhibiting cytokine-induced astrocyte formation.
SHP-2 is necessary to promote neurogenesis during embryonic cortical development in vivo
To ask whether SHP-2 plays a similar role in vivo, we performed in utero electroporation to knock-down SHP-2 expression in precursors of the VZ/SVZ of the embryonic cortex (Barnabé-Heider et al., 2005; Paquin et al, 2005). We previously demonstrated that 1 day after in utero electroporation at E13/14, all of the transfected cells reside in the VZ/SVZ and approximately 85% of these cells are proliferating (Paquin et al., 2005). Many of these transfected cells differentiate into neurons over the next few days, and these neurons migrate out of the VZ/SVZ into the cortical plate. Some of the transfected cells also remain in the VZ/SVZ where they adopt an astrocyte fate during the early postnatal period.
We therefore performed in utero electroporation at E13/14 with plasmids encoding a nuclear EGFP and SHP-2 shRNA1 or scrambled shRNA, and analyzed the cortex 3-4 days later. Immunocytochemistry for EGFP revealed a robust difference between control and SHP-2 shRNA1-transfected brains, with approximately twice as many control transfected cells migrating out of the VZ/SVZ into the cortical mantle (which includes both the intermediate zone of migrating neurons and the cortical plate (Fig. 5A,B). Moreover, quantitative confocal analysis of sections immunostained for the neuronal protein HuD revealed that many more of the control, scrambled shRNA-transfected cells were HuD-positive neurons at this timepoint (Fig. 5C,D). In both SHP-2 shRNA1 and scrambled shRNA-transfected cells, these HuD positive neurons were located in the cortical mantle. Immunostaining for the early neuronal protein, βIII-tubulin demonstrated that only small numbers of newly-born neurons were present within the VZ/SVZ, and that these numbers were similar in both conditions (data not shown). Thus, SHP-2 knockdown decreased the genesis of cortical neurons in vivo as it did in culture.
Figure 5. Genetic knockdown of SHP-2 inhibits cortical neurogenesis in vivo.
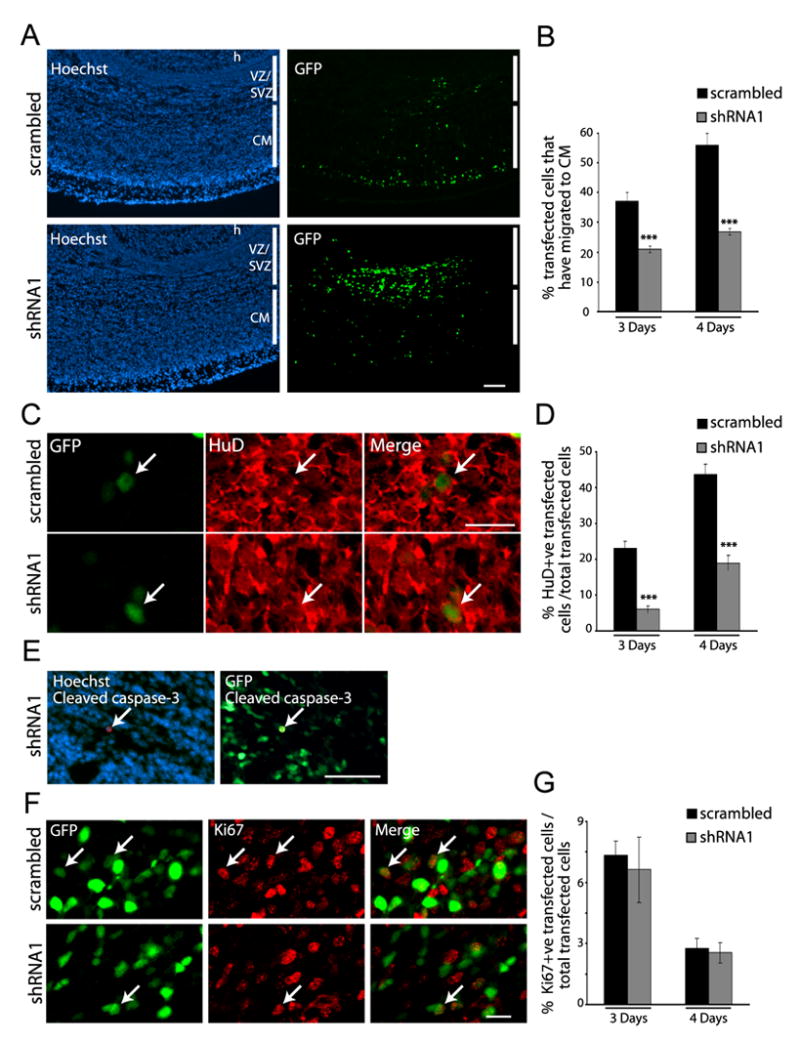
Precursor cells of the E13/E14 cortex were electroporated with EGFP and SHP-2 shRNA1 or scrambled shRNA, and analyzed four days later. (A) Immunocytochemistry for EGFP (GFP; green) in coronal cortical sections. Sections were counterstained with Hoechst 33258. h = hippocampus, VZ = ventricular zone, SVZ = subventricular zone, CM = cortical mantle. Scale bar = 100μm. (B) Quantification of GFP-positive cells that migrated to the cortical mantle in experiments similar to (A). For 4 days, n = 6 each. ***p<0.005. (C) Confocal images of a section through the cortical plate double-labelled for EGFP (GFP; green) and HuD (red). Arrows denote transfected, double-labelled cells. Scale bar = 20μm. (D) Quantification of transfected, HuD-positive cells in cortices electroporated with SHP-2 shRNA1 or scrambled shRNA in experiments similar to (C). For 4 days, n = 6 brains each. ***p<0.005. (E) Micrographs of a section double-labelled for EGFP (GFP) and cleaved caspase-3 (red), and counterstained with Hoechst 33258. Arrow denotes a rare, transfected caspase-3-positive cell. Scale bar = 50μm. (F) Confocal micrographs of the VZ/SVZ of sections double-labelled for EGFP (GFP; green) and Ki67 (red). Arrows denote double-labelled cells. Scale bar = 20μm. (G) Quantification of transfected, Ki67-positive cells in the VZ/SVZ of sections similar to (F). p>0.05.
To rule out the possibility that the decrease in neurons was due to enhanced cell death and/or altered proliferation, we analyzed adjacent sections for cleaved caspase-3 and Ki67. Analysis for cleaved caspase-3 revealed only rare double-labelled cells in control and experimental brains (Fig. 5E; less than 10 cells per brain in both cases). Similarly, the percentage of transfected Ki67-positive cells, which were located only in the VZ/SVZ, was similar in control and SHP-2 shRNA1-transfected brains at both 3 and 4 days post-electroporation (Fig. 5F,G). Thus, SHP-2 knockdown had no effect on survival or proliferation, but specifically inhibited neurogenesis.
SHP-2 knockdown leads to premature gliogenesis in the embryonic cortex
Since the culture data indicated that endogenous SHP-2 instructs precursors to make neurons rather than glia, we asked if it inhibited gliogenesis during the embryonic neurogenic period in vivo. To do this, embryos were electroporated with EGFP and SHP-2 shRNA1 at E13/E14, and analyzed 3 or 4 days later by double-labelling for GFP and GFAP. At E16/17 (3 days post-electroporation), no GFAP-positive cells were detected in cortices transfected with control, scrambled shRNA, but a small number of transfected astrocytes were present in cortices transfected with SHP-2 shRNA1 (Fig. 6A). By E17/18, some GFAP-positive astrocytes were observed in control cortices, but none of these derived from transfected precursors in the 3 brains that were analyzed (Fig. 6B). In contrast, SHP-2 shRNA-transfected, GFAP-positive astrocytes were observed at this timepoint (Fig. 6B). Thus, genetic knockdown of SHP-2 leads to precocious astrocyte formation during the late neurogenic period.
Figure 6. Inhibition of SHP-2 perturbs the timing and magnitude of gliogenesis in vivo.
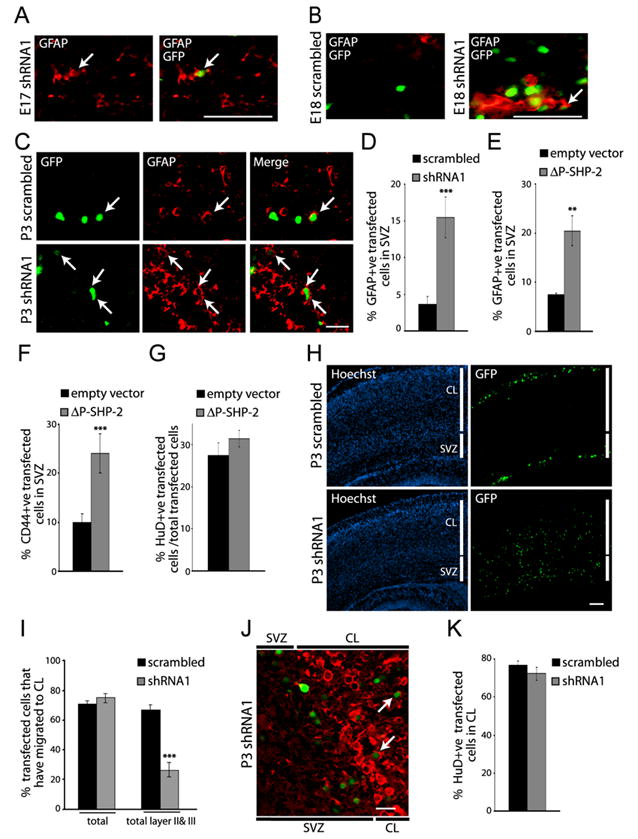
(A–D) Precursor cells of the E14 cortex were electroporated with EGFP and SHP-2 shRNA1 or scrambled shRNA, and analyzed at E17 (A), E18 (B), or P3 (C,D) by immunocytochemistry for EGFP (GFP; green) and GFAP (red). (A,B) Micrographs of coronal sections through the E17 (A) or E18 (B) SVZ transfected with SHP-2 shRNA1 or scrambled shRNA at E14. (C) Confocal images of the SVZ in cortices transfected at E14, and analyzed at P3. In (A–C), arrows indicate transfected, double-labelled cells. Scale bars = 50 (A,B) or 20 (C) μm. (D) Quantification of transfected, GFAP-positive cells in the SVZ in experiments similar to (C). n = 3 brains. ***p<0.005. (E,F) Quantification of experiments similar to that in (C) where embryos were electroporated at E14 with EGFP and ΔP-SHP-2 or empty vector, and analyzed immunocytochemically for EGFP and GFAP (E) or CD44 (F) at P3. In (E), n = 7 brains each. **p<0.01, ***p<0.001. (G) Quantification of experiments similar to (E,F) except that brains were analyzed 4 days after electroporation by immunostaining for EGFP and HuD. n = 2 each. p>0.05. (H) Coronal sections of cortices electroporated at E14 with EGFP and SHP-2 shRNA1 or scrambled shRNA, and immunolabelled at P3 for EGFP (GFP; green) and counterstained with Hoechst 33258. SVZ = subventricular zone, CL= cortical layers. Scale bar = 100μm. (I) Quantification of transfected cells within the cortical layers (total) or in layers II and III in experiments similar to (H). n = 3 each. ***p<0.001. (J) Confocal image of a section similar to those in (H) immunostained for EGFP (green) and HuD (red). The image shows both the SVZ and the deeper cortical layers (CL). Arrows denote transfected, double-labelled neurons. Scale bar = 20μm. (K) Quantification of transfected, HuD-positive cells within the cortical layers. n = 3 each. p>0.05.
To quantitatively assess this enhanced gliogenesis, we performed similar electroporations, and counted transfected GFAP-positive cells within the SVZ at P3 (Fig. 6C). This analysis (Fig. 6D) revealed that when SHP-2 was knocked-down, the number of transfected astrocytes increased by approximately 3-fold. Since our culture data also indicated that the phosphatase activity of SHP-2 was essential for its gliogenic effects, we performed similar experiments with P-SHP-2; P-SHP-2 increased the percentage of astrocytes that were generated by 2-3 fold, as monitored by immunocytochemistry for either GFAP or CD44 (Fig. 6E,F). We also analyzed neurogenesis in the P-SHP-2 transfected brains 4 days post-electroporation, and observed, as we had seen in culture, that it had no effect on the number of transfected, HuD-positive neurons (Fig. 6G). Thus, SHP-2 normally regulates the onset and extent of astrogenesis in vivo.
SHP-2 knockdown in the embryonic period leads to postnatal perturbations in cortical neurons
While the perturbations in neonatal gliogenesis following SHP-2 knockdown were localized to the SVZ, analysis of the remainder of the cortex revealed additional perturbations. In particular, while the percentage of transfected cells present in the SVZ versus the cortical layers was similar in scrambled shRNA and SHP-2 shRNA1-transfected cortices (Fig. 6H,I), cell location within the cortical layers differed. The majority (95%) of control transfected cells were present in cortical layers II and III, but approximately 65% of the SHP-2 shRNA1-transfected cells were scattered throughout layers IV-VI (Fig. 6I). These misplaced cells were not glia, since all of the transfected cells expressing GFAP or a marker for oligodendrocytes, O4, were in the SVZ (data not shown). Instead, the large majority of the SHP-2 shRNA1-transfected cells within the cortical layers were neurons, as defined by HuD immunostaining (Fig. 6J,K). Thus, by P3, the percentage of transfected cortical neurons was similar with scrambled shRNA versus SHP-2 shRNA1, but SHP-2 shRNA1-transfected neurons were inappropriately located. Since these experiments involved transient transfections, SHP-2 knockdown may be less efficient at later timepoints, with neurons ultimately being generated from “delayed” precursors between E17/18 and P3, and then migrating inappropriately due to their late birthdate. Alternatively, these data may indicate that SHP-2 plays a direct role in the migration of newly-born neurons.
A Noonan Syndrome SHP-2 mutant promotes neurogenesis and inhibits gliogenesis in the developing cortex
To ask whether the NS SHP-2 D61G mutation causes cell fate perturbation in the embryonic brain as it does in culture, we performed in utero electroporation at E13/14 with plasmids encoding nuclear EGFP and the D61G SHP-2 mutant or WT SHP-2. Immunocytochemistry and quantitative confocal microscopy for HuD three or four days later revealed an increase in the number of D61G SHP-2 transfected cells that were neurons four days post-electroporation (Fig. 7A). These neurons were all appropriately located within the cortical mantle. To ask whether WT SHP-2 had the same effect, we electroporated it into the cortex of E13/14 Tα1:nlacZ mice (Gloster et al., 1994; Paquin et al., 2005); these mice express nuclear β-galactosidase from the early neuron-specific promoter Tα1 α-tubulin. Analysis 3 days later revealed that WT SHP-2 robustly increased the number of newly-born, Tα1:nlacZ-positive neurons that were generated (Fig. 7B). Thus, as seen in culture, ectopic expression of either D61G or WT SHP-2 enhanced neurogenesis in vivo.
Figure 7. A Noonan Syndrome-associated SHP-2 mutant enhances neurogenesis and inhibits gliogenesis in vivo.
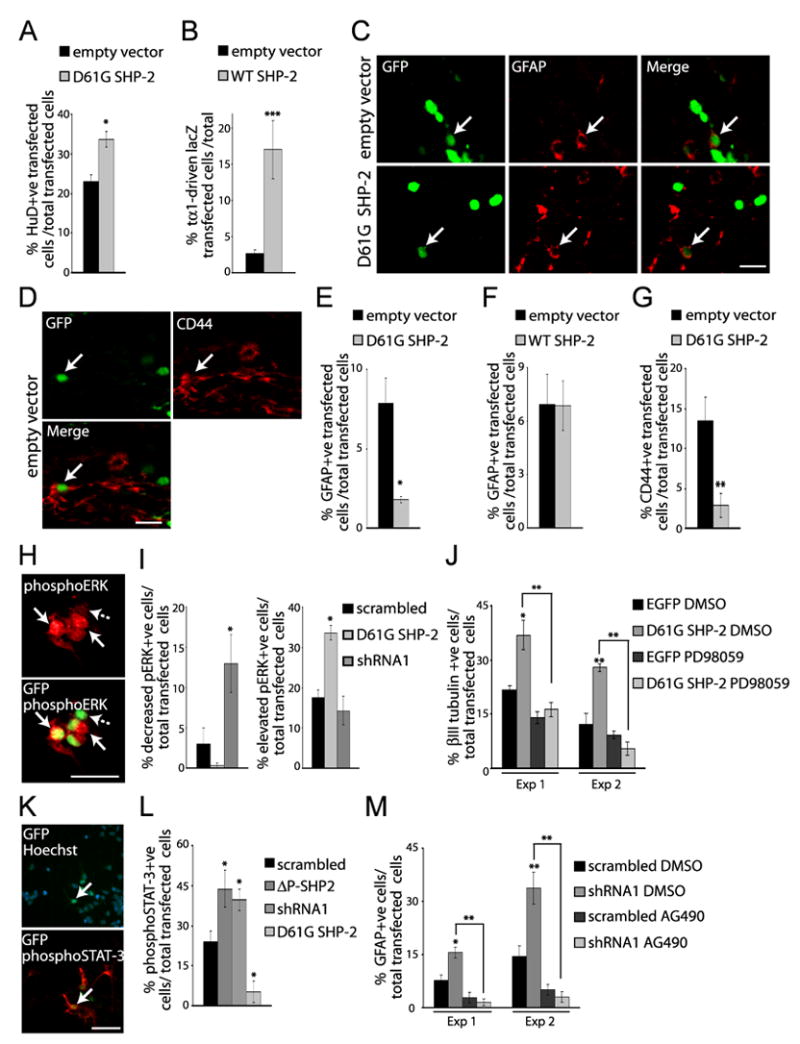
Precursor cells of the E13/14 cortex were electroporated with EGFP and D61G SHP-2, WT SHP-2 or the empty vector, and analyzed at E17/18 (A,B) or P3 (C–G). In panel (B), electroporations were performed with E13/14 Tα1:nlacZ mice. (A) Quantification of transfected, HuD-positive neurons in E17/18 coronal cortical sections immunostained for EGFP (GFP) and HuD. n = 3 control and 4 experimental brains. *p<0.05. (B) Quantification of transfected, Tα1:nlacZ-positive cells 3 days following electroporation, as detected by immunocytochemistry for EGFP and β-galactosidase. n = 2 each. ***p<0.005. (C,D) Confocal micrographs of coronal sections through the SVZ of P3 brains double-labelled for EGFP (GFP; green) and GFAP (C, red) or CD44 (D, red). Arrows denote double-labelled cells. Scale bar = 20μm. (E–G) Quantification of transfected, GFAP-positive (E,F) or CD44-positive (G) cells within the SVZ in experiments similar to (C). In (E), n = 7 and 6, in (F), n = 4 and 5, and in (G), n = 2 each experimental and control brains. *p<0.05, **p<0.01. (H–J) A Noonan syndrome-associated SHP-2 mutant enhances neurogenesis via MEK. (H) Immunocytochemistry for EGFP (GFP; green) and phosphorylated ERK (phosphoERK; red) in precursors cotransfected with EGFP (green) and D61G SHP-2 and cultured 3 DIV. Arrows and the dotted arrow denote transfected cells that express high or basal levels of phosphoERK, respectively. Scale bar = 25μm. (I) Quantification of transfected, phosphoERK-positive cells in one of two experiments where precursors were cotransfected with EGFP plus D61G SHP-2 or SHP-2 shRNA1. *p<0.05, ANOVA. (J) Quantification of transfected, βIII-tubulin-positive cells in three independent experiments where precursors were transfected with EGFP or HA-tagged D61G SHP-2, treated with PD98059 after 1 DIV, and analyzed 2 days later. *p<0.05, **p<0.01, ANOVA. (K–M) SHP-2 inhibits gliogenesis by negatively regulating gp130-JAK-STAT signaling. (K) Immunocytochemistry for EGFP (GFP; green) and phosphoSTAT-3 (red) in precursors cotransfected with EGFP and SHP-2 shRNA1, treated with 50ng/ml CNTF at 2 DIV and cultured one more day. Cultures were counterstained with Hoechst 33258. The arrow indicates a double-labelled cell. Scale bar = 50μm. (L) Quantification of transfected, phosphoSTAT-3-positive cells in one of 3 experiments similar to (K). Cultures were cotransfected with plasmids encoding EGFP and SHP-2 shRNA1, scrambled shRNA, ΔP-SHP-2, or D61G SHP-2. *p<0.05, ANOVA. (F) Quantification of transfected, GFAP-positive cells in 2 experiments where precursors were cotransfected with EGFP and SHP-2 shRNA1 or scrambled shRNA, and treated at 2 DIV with 50ng/ml CNTF with or without AG490 for 4 more days prior to immunostaining for EGFP and GFAP. *p<0.05,**p<0.01, ANOVA.
We also asked whether the D61G SHP-2 mutant inhibited astrocyte formation in vivo, as it did in culture. Cortices were electroporated at E14 with either D61G or WT SHP-2, and then analyzed immunocytochemically at P3 for expression of GFAP or CD44 (Fig. 7C,D). Quantitative analysis of these brains (Fig. 7E–G) revealed that D61G, but not WT, SHP-2 reduced astrocyte formation approximately 4-5-fold. As seen for neurons, D61G SHP-2 did not affect the localization of these cells, since these transfected astrocytes were located within the SVZ. Thus, the NS D61G SHP-2 mutant promotes the genesis of neurons and inhibits the genesis of astrocytes in the embryonic cortex.
SHP-2 promotes neurogenesis via activation of the MEK-ERK pathway, and inhibits gliogenesis by directly suppressing the gp130-JAK-STAT pathway
Our data indicate that endogenous SHP-2 promotes neurogenesis and inhibits gliogenesis. Since SHP-2 can promote RTK-mediated activation of the Ras-MEK-ERK pathway, and since we have previously shown that MEK activity is necessary and sufficient for neurogenesis (Ménard et al., 2002; Barnabé-Heider et al., 2003; Paquin et al., 2005), we asked whether SHP-2 promoted neurogenesis via MEK. Initially, we transfected precursors with EGFP and SHP-2 shRNA1 or the D61G SHP-2 mutant, immunostained them with an antibody specific to phosphorylated, activated ERK, and counted cells with basal, high or low levels of phosphoERK expression (Fig. 7H,I). This analysis revealed that SHP-2 shRNA1 reduced and D61G SHP-2 increased phosphoERK activation (Fig. 7I), indicating that SHP-2 is essential for basal levels of ERK activity in cortical precursors. We then asked if the increased MEK-ERK activation observed with D61G SHP-2 was responsible for its ability to promote neurogenesis; transfected precursors were treated with a pharmacological MEK inhibitor, PD98059, and then immunostained for βIII-tubulin 3 days post-transfection (Fig. 7J). We previously showed that PD98059 specifically inhibits MEK in cortical precursors, and that this has no effect on precursor cell survival or proliferation (Barnabé-Heider and Miller, 2003). These experiments demonstrated that PD98059 completely blocked the enhanced neurogenesis seen in cells transfected with D61G SHP-2. Thus, SHP-2 regulates neurogenesis via the MEK-ERK pathway.
Since gliogenesis in cortical precursor cultures depends upon the gp130-JAK-STAT pathway (Bonni et al., 1997; Johe et al., 1996; Barnabé-Heider et al., 2005), and since SHP-2 negatively regulates this signaling pathway in some cells (Ernst and Jenkins, 2004), we asked whether SHP-2 regulated gliogenesis via this pathway. Initially, we cotransfected precursors with EGFP and SHP-2 shRNA1, P-SHP-2, or D61G SHP-2, exposed them to CNTF at 2 DIV for one additional day, and then immunostained with an antibody specific for phosphorylated, activated STAT3 (Fig. 7K). This analysis revealed that transfection with SHP-2 shRNA1 or ΔP-SHP-2 increased, and D61G SHP-2 decreased, the number of precursors expressing detectable phosphoSTAT3 (Fig. 7L). To ask whether SHP-2 inhibited STAT activation further upstream at the level of JAK or gp130, we asked whether the enhanced gliogenesis seen upon SHP-2 knockdown was dependent upon JAK activation; precursors were cotransfected with EGFP and shRNA1, and were induced with CNTF at 2 DIV for an additional 4 days with or without the pharmacological JAK inhibitor AG490. We previously showed that this inhibitor specifically blocks JAK activity and astrogenesis in cortical precursors, with no effects on survival or neurogenesis (Barnabé-Heider et al., 2005). Immunocytochemistry for GFAP demonstrated that JAK inhibition completely blocked the increased gliogenesis observed with SHP-2 shRNA1 (Fig. 7M). Thus, endogenous SHP-2 inhibits astrocyte formation by negatively regulating the gp130-JAK-STAT pathway.
A Noonan Syndrome mouse model displays perturbed cell genesis in the neonatal forebrain and hippocampus
While our findings suggest that enhanced SHP-2 activity in NS could cause abnormal neural development, these were overexpression studies, whereas in NS patients only one copy of the mutant allele is present. We therefore examined the brains of a NS mouse model carrying one copy of D61G SHP-2 knocked into the endogenous SHP-2 locus. This mouse phenocopies several aspects of the human disorder (Araki et al., 2004), but its nervous system has not yet been examined. We therefore analyzed the cortex and hippocampus of newborn NS mice, at a time when astrogenesis had just started (Fig. 8A). Initially, we examined coronal sections through the dentate gyrus of SHP-2 D61G/+ mice versus their wildtype littermates and age-matched non-littermates (Fig. 8A, region 1). These sections were immunostained for the astrocyte marker GFAP (Fig. 8B) or the neuronal marker HuD, and counterstained with Hoechst 33258 (Fig. 8B) to allow quantitation of total neurons, astrocytes, cell numbers, and tissue area. These parameters were then quantitated in 3–4 coronal sections per brain at the same rostral-caudal level of the dentate gyrus (Figs. 8A,B). This analysis revealed significant differences between the hippocampus of wildtype versus NS mice. Counts of total Hoechst-positive nuclei revealed that cell density was significantly increased (~ 6%) in NS mice (Fig. 8C). Quantitation of HuD-positive neurons in the same area also revealed a small, but significant increase in both the density of neurons, and in the percentage of neurons (Fig. 8E). However, the most robust phenotype was revealed when GFAP-positive astrocytes were quantitated; both the density and the percentage of total cells that were astrocytes were decreased by 2.5-3 fold in the NS hippocampus (Fig. 8B,C).
Figure 8. A Noonan Syndrome mouse model shows enhanced neurogenesis and decreased gliogenesis in the neonatal hippocampus and cortex.
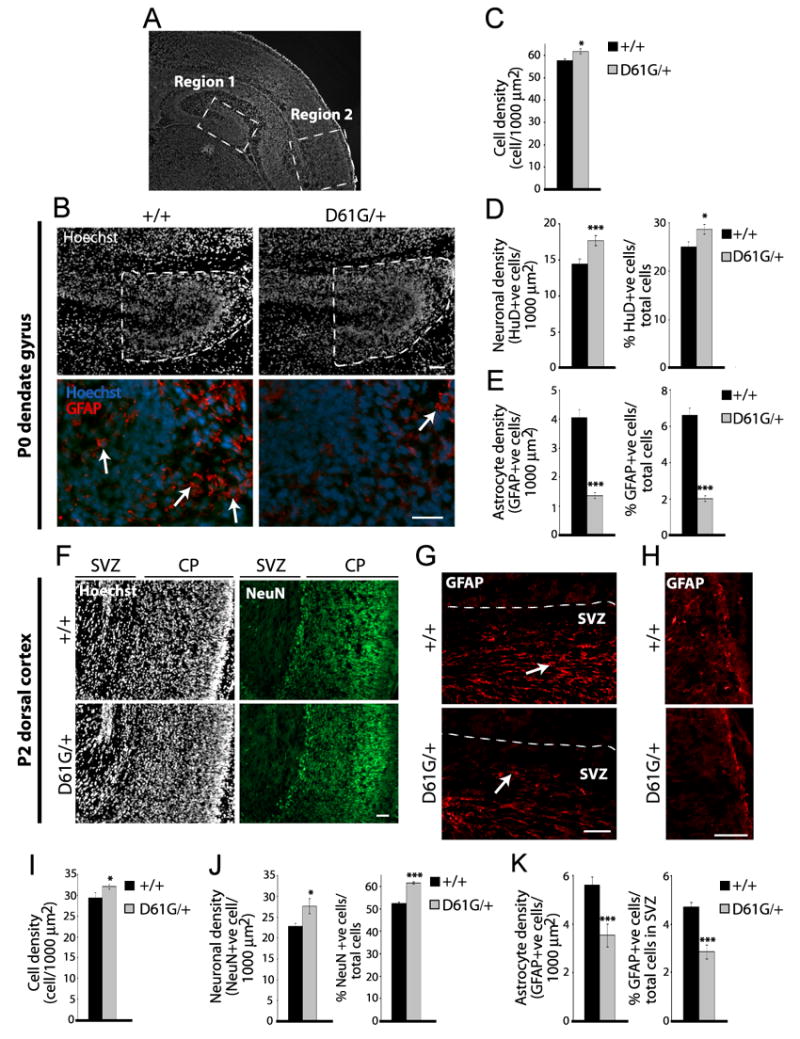
(A) Coronal section through the neonatal forebrain, depicting the regions that were analyzed in the hippocampus (region 1) and dorsal cortex (region 2). (B–E) Analysis of the P0 dentate gyrus (region 1). In all cases, at least 3 adjacent sections through this region were analyzed per brain. (B) Immunocytochemistry for GFAP (red) in coronal dentate gyrus sections at the same rostral-caudal level of the P0 NS (D61G/+) or wildtype (+/+) brain. Cell density was determined by counterstaining with Hoechst 33258 (white or blue). Dashed outline represents the area that was analyzed. Arrows denote GFAP-positive cells. Scale bar = 50μm (top) and 25μm (bottom). (C) Quantification of mean cell density in the area denoted in (B). n=6 and 5, control and NS brains. *p<0.05. (D) Quantification of mean HuD-positive cell density (left panel) and percentage of HuD-positive cells/total cells (right panel) in the area denoted in (B). n=6 and 5, control and NS brains. *p<0.05, ***p<0.005. (E) Quantification of mean GFAP-positive cell density (left panel) and the percentage of GFAP-positive cells/total cells (right panel) in the area denoted in (B). n=6 brains each. ***p<0.005. (F–K) Analysis of the postnatal day 2 (P2) dorsal cortex (region 2). In all cases, at least 4 adjacent sections through this region were analyzed per brain. (F) Immunocytochemistry for NeuN (green) in coronal sections at the same rostral-caudal level of the P2 NS (D61G/+) or wildtype (+/+) brain. Cell density was determined by counterstaining with Hoechst 33258 (white). CP = cortical plate. Scale bar = 100μm. (G,H) Immunocytochemistry for GFAP (red) in the SVZ (G) or glia limitans (H) in the dorsal cortex of P2 NS (D61G/+) versus wildtype (+/+) brains. Arrows denote GFAP-positive cells. Scale bar = 50μm. (I) Quantification of mean total cell density in sections similar to (F). n=2 brains each. *p<0.05. (J) Quantification of mean NeuN-positive cell density (left panel) and the percentage of NeuN-positive cells/total cells (right panel) in sections similar to (F). n=2 brains each. *p<0.05, ***p<0.005. (J) Quantification of mean GFAP-positive cell density (left panel) and the percentage of GFAP-positive cells/total cells (right panel) in the SVZ in sections similar to (G). n=3 and 2, control and NS brains. ***p<0.005.
Similar changes were seen in the dorsal cortex (Fig. 8A, region 2) of wildtype versus NS brains that were analyzed at postnatal day 2. The density of total cells displayed a small but significant increase (Fig. 8F,I), and both the density and percentage of cells expressing the neuron-specific protein NeuN-positive also showed a small but significant increase (Fig. 8F,J). In addition, quantitation of GFAP-positive cells within the SVZ demonstrated an approximately 2-fold decrease in the density and percentage of astrocytes in the NS versus wildtype cortex (Fig. 8G,K). The intensity of GFAP immunostaining in the glia limitans at this level was also reduced in the neonatal NS brains (Fig. 8H). Thus, in both the dorsal cortex and hippocampus of neonatal NS mice, there is a large decrease in astrocytes and a small but significant increase both in total cellular and neuronal density, alterations that may contribute to further perturbations during circuit formation, and that ultimately could lead to cognitive dysfunction.
DISCUSSION
The data presented here support three major conclusions. First, the experiments using SHP-2 shRNA indicate that endogenous SHP-2 is essential for the normal genesis of cortical neurons and astrocytes both in culture and within the environment of the embryonic cortex. Genetic knockdown of SHP-2 leads to a delay in neurogenesis and ultimately a perturbation in neuronal location. It also causes inappropriate precocious formation of astrocytes during the embryonic neurogenic period, and leads to a robust increase in the number of astrocytes generated. Second, as shown in the clonal experiments, SHP-2 mediates these effects by instructing neural precursors to generate neurons rather than astrocytes, thereby ensuring that precursors that are biased to a neuronal fate do not attempt to become glia even in the presence of cytokines. This occurs via two dissociable mechanisms; SHP-2 activates the neurogenic Ras-MEK-C/EBP pathway, and at the same time inhibits the gliogenic gp130-JAK-STAT pathway. Third, the constitutive activation of SHP-2 that occurs in NS causes a large reduction in astrocyte formation, and a coincident small increase in neuronal number in the forebrain and hippocampus of mice. These effects are seen both following ectopic expression of a NS SHP-2 allele and, importantly, in a NS mouse model carrying the same activated allele in place of the wildtype allele, as occurs in humans. Together these data indicate that SHP-2 regulates the timing and extent of neurogenesis versus gliogenesis by acting as a growth factor-regulated switch to bias cortical precursor cells towards one fate and against another. Perturbations in the ratio of neurons versus glia as a result of constitutive SHP-2 activation may contribute to the cognitive dysfunction observed in NS individuals.
During development of the mammalian nervous system, cell genesis is a timed event, with neurons generated first and glial cells second. Within the murine cortex, these neurogenic and gliogenic periods are temporally distinct; neurogenesis occurs from approximately E12-E17 while astrocyte formation is largely postnatal. Many of the previous studies examining how growth factors regulate this timer have focused upon positive, inductive mechanisms (reviewed in Miller and Gauthier, 2007). Here, we show that appropriately timed gliogenesis also depends upon a SHP-2-mediated suppressive signal that inhibits competent precursors from prematurely generating astrocytes. We propose that during the embryonic neurogenic period, SHP-2 is an integral upstream component of the neurogenic RTK-MEK-ERK-C/EBP pathway. At the same time, SHP-2 suppresses the low levels of gliogenic gp130-JAK-STAT signaling that may occur in response to cytokines such as neuropoietin 1 and cardiotrophin-like cytokine that are expressed in the embryonic brain (Uemura et al., 2002; Derouet et al., 2004). As development proceeds, newly-born cortical neurons synthesize and secrete cardiotrophin-1 (Barnabé-Heider et al., 2005), and cortical precursors become sensitized to cytokines via a variety of intrinsic mechanisms (reviewed in Miller and Gauthier, 2007). Under these new conditions, SHP-2 does not completely suppress gliogenesis, but instead regulates the number of astrocytes that are generated by attenuating gp130-JAK-STAT signaling. Thus, SHP-2 functions as an essential component of growth factor regulated signaling mechanisms that determine the timing and extent of neurogenesis versus gliogenesis in the developing cortex.
An equally important major conclusion of this study is that enhanced SHP-2 activity in NS causes perturbations in cortical cell fate decisions, leading to a small increase in neurogenesis and a robust decrease in gliogenesis. We observed these perturbations in a mouse model of NS that phenocopies the human condition with regard to all parameters so far examined (Araki et al., 2004). Moreover, our in utero electroporation studies argue that many of these effects are cell-autonomous, with the D61G SHP-2 allele directly perturbing precursor cell fate decisions. We suggest that these perturbations in cell genesis during early development could lead to subsequent alterations in cell survival and/or circuit formation, thereby substantively impacting cognitive function. As one example of such amplification, astrocyte development is perturbed in mice lacking the EGF receptor, and this ultimately causes dramatic degeneration of forebrain neurons (Wagner et al., 2006). Thus, genetic perturbations such as those seen in NS, or perhaps even in more commonly-studied disorders such as Rett Syndrome, might first perturb cell genesis, and then this might in turn alter many later aspects of neural development, ultimately resulting in impaired circuitry and cognitive dysfunction. Such a model does not preclude the importance of later perturbations that might occur in neuronal circuits as a direct consequence of activated SHP-2, it simply argues that earlier impairments such as altered cell genesis could have more widespread effects because they occur so early during development.
We provide strong evidence for perturbed neural development in a mouse model of NS. Do similar perturbations occur in the human syndrome? While it is clear that many of these individuals display learning disabilities and mental retardation (Noonan et al., 1994; Yoshida et al., 2004; Jongmans et al., 2005; Lee et al., 2005), there have only been a few anatomical/imaging studies reported. However, those few studies do indeed indicate that there are perturbations, including cortical dysplasia, cortical atrophy, and cerebellar malformations (Saito et al., 1997; Gorke, 1980; Peiris and Ball, 1982; Heye and Dunne, 1995). Since the mouse model that we studied here has been shown to phenocopy the human condition with regard to more well-studied aspects of the syndrome such as craniofacial abnormalities, cardiac defects and myeloproliferation, then we believe that neural defects similar to those documented here are likely to occur in the human condition. In this regard, Costello and cardio-facial-cutaneous (CFC) syndromes, which are in the same family of genetic disorders as NS and display mental retardation, have recently been shown to arise from activating mutations in the HRas-BRaf-MEK-ERK pathway (Bentires-Alj et al., 2006). Thus, we propose that the SHP-2-MEK-ERK-C/EBP pathway we have defined as important for neural cell fate determination is essential for normal human brain development.
Little is known about how genetic perturbations lead to mental retardation. Here, we have identified a signaling protein, SHP-2, that plays a key role in allowing environmental cues such as growth factors to instruct multipotent precursor cells to generate one cell type versus another. Moreover, we have shown that the same constitutively-activated SHP-2 that occurs in a subpopulation of NS patients causes aberrant neural development in mice, providing a potential explanation for the cognitive dysfunction and neuroanatomical perturbations observed in this disorder. Such a mechanism may well generalize to other syndromes where signaling pathways that are important for neural cell genesis are genetically perturbed.
EXPERIMENTAL PROCEDURES
Cell cultures, inhibitor treatments and transfections
E11.5–12.5 cortical precursors were cultured in 40ng/ml FGF2 as described (Barnabé-Heider et al., 2005) from CD1 mice at a density of 250,000 cells/ml. CNTF (Cedarlane Laboratories) was used at 50ng/ml. AG490 (Calbiochem) and PD98059 (BIOMOL Research Laboratories) were used at concentrations of 5 μM and 50 μM in 0.75% DMSO. ZVAD-FMK (Calbiochem ) was used at a concentration of 100 μM in 0.5% DMSO. Transfection conditions and plasmids are described in the supplementary materials. For clonal analysis, precursors were cotransfected at very low densities with EGFP and SHP-2 shRNA1 or D61G SHP-2 plasmids, and were immunostained at 4 or 6 DIV for EGFP, μIII-tubulin and GFAP. In all experiments, only clusters of isolated, EGFP-positive cells deriving from single transfected precursors were analyzed.
In utero electroporation
In utero electroporation was performed as described (Barnabé-Heider et al., 2005; Paquin et al., 2005) with E13/E14 CD1 or Tα1:nlacZ (Gloster et al., 1994) mice, injecting a nuclear EGFP expression plasmid driven from the EF1α promoter (pEF-EGFP) (as above) at a 1:3 ratio with pSUPER-shRNA1, pEF-D61GSHP-2 or pEF-ΔPSHP-2, or pEF-GM (empty vector), pSUPER-scrambled as a control (5 μg/μl) and 0.05% trypan blue as a tracer. The square electroporator CUY21 EDIT (TR Tech, Japan) was used to deliver five 50ms pulses of 50V with 950ms intervals per embryo. Brains were dissected in ice-cold HBSS, fixed in 4% PFA at 4ºC overnight, cryoprotected and cryosectioned coronally at 16 μm.
Immunocytochemistry and quantitation
Immunocytochemistry on cells was performed as described (Barnabé-Heider et al., 2005). For immunostaining of tissue, sections were post-fixed with 4% PFA, blocked and permeabilized with 10% BSA and 0.3% Triton-X, and then with the M.O.M. blocking kit (Vector Laboratories). Sections were incubated with primary antibodies at 4ºC overnight, with secondary antibodies at room temperature for 1 hour, counterstained with Hoechst 33258 (1:2000, Sigma-Aldrich) and mounted with GelTol (Fisher). Primary and secondary antibodies are described in the supplementary materials. For quantitation over 200 transfected cells in at least 8 randomly selected fields spanning the wells of precursor cultures were counted (per condition per experiment). In the clonal studies, 70 to 90 clones were analyzed (per condition per experiment). For electroporated brains, sections were analyzed using a Zeiss Pascal confocal microscope and the manufacturer’s software. 3–4 pictures per coronal section were taken with a 40X objective and a total of four sections per animal were analyzed, and compared to equivalent sections in littermate (or age-matched non-littermate where indicated) counterparts. For quantitation of total cells, neurons and astrocytes in SHP-2 D61G/+ mice (Araki et al., 2004), mice were bred on a 129J/B6 background, and brains harvested and processed as described above at postnatal day 0 and 2. In blind analyses, 3–4 coronal tissue sections were selected from the same rostral-caudal extent (based upon hippocampal and cortical morphology) in brains of D61G/+ and +/+ littermates and age-matched non-littermates, and cell counts were made from defined regions in the dorsal cortex and dentate gyrus. Digital image acquisition and analysis was performed using Northern Eclipse software (Empix Inc.) with a Sony XC-75CE CCD video camera. Statistics were performed using Students t-test, unless indicated otherwise. ANOVA indicates a one way ANOVA. Error bars denote SEM in all cases.
Western blot analysis and biochemistry
Cortical precursors and NIH 3T3 cells were lysed, 30–50 μg protein lysate was run on SDS-PAGE, and Western blots performed as described (Barnabé-Heider et al., 2005).
Supplementary Material
Acknowledgments
This work was supported by grants from the CIHR to F.D.M. and D.R.K., and N.I.H. grant R37CA49152 to B.G.N. F.D.M. and D.R.K. are CRC Senior Research Chairs, and F.D.M. is an H.H.M.I. International Research Scholar. A.S.G. was supported by NSERC, OGS, and in part by a HSC Foundation studentship award, and T.A. by the J.S.P.S. We thank Annie Paquin, Fanie Barnabé-Heider, Julie Wasylnka, Sagar Dugani and Karl Fernandes for their advice and assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Araki T, Mohi MG, Ismat FA, Bronson RT, Williams IR, Kutok JL, Yang W, Pao LI, Gilliland DG, Epstein JA, Neel BG. Mouse model of Noonan syndrome reveals cell type- and gene dosage-dependent effects of Ptpn11 mutation. Nat Med. 2004;10:849–857. doi: 10.1038/nm1084. [DOI] [PubMed] [Google Scholar]
- Barnabé-Heider F, Miller FD. Endogenously produced neurotrophins regulate survival and differentiation of cortical progenitors via distinct signaling pathways. J Neurosci. 2003;23:5149–5160. doi: 10.1523/JNEUROSCI.23-12-05149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnabé-Heider F, Wasylnka JA, Fernandes KJL, Porsche C, Sendtner M, Kaplan DR, Miller FD. Evidence that embryonic neurons regulate the onset of cortical gliogenesis via cardiotrophin-1. Neuron. 2005;4:253–265. doi: 10.1016/j.neuron.2005.08.037. [DOI] [PubMed] [Google Scholar]
- Bentires-Alj M, Kontaridis MI, Neel BG. Stops along the RAS pathway in human genetic disease. Nat Med. 2006;12:283–285. doi: 10.1038/nm0306-283. [DOI] [PubMed] [Google Scholar]
- Bonni A, Sun Y, Nadal-Vicens M, Bhatt A, Frank DA, Rozovsky I, Stahl N, Yancopoulos GD, Greenberg ME. Regulation of gliogenesis in the central nervous system by the JAK-STAT signaling pathway. Science. 1997;278:477–483. doi: 10.1126/science.278.5337.477. [DOI] [PubMed] [Google Scholar]
- Derouet D, Rousseau F, Alfonsi F, Froger J, Hermann J, Barbier F, Perret D, Diveu C, Guillet C, Preisser L, Dumont A, Barbado M, Morel A, Delapeyvrier O, Gascan H, Chevalier S. Neuropoietin, a new IL-6-related cytokine signaling through the ciliary neurotrophic factor receptor. Proc Natl Acad Sci. 2004;101:4827–4832. doi: 10.1073/pnas.0306178101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst M, Jenkins BJ. Acquiring signaling specificity from the cytokine receptor gp130. Trends Genet. 2004;20:23–31. doi: 10.1016/j.tig.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Fragale A, Tartaglia M, Wu J, Gelb BD. Noonan syndrome-associated SHP2/PTPN11 mutants cause EGF-dependent prolonged GAB1 binding an sustained ERK2/MAPK1 activation. Hum Mutat. 2004;23:267–277. doi: 10.1002/humu.20005. [DOI] [PubMed] [Google Scholar]
- Gloster A, Wu W, Speelman A, Weiss S, Causing C, Pozniak C, Reynolds B, Chang E, Toma JG, Miller FD. The T alpha 1 alpha-tubulin promoter specifies gene expression as a function of neuronal growth and regeneration in transgenic mice. J Neurosci. 1994;14:7319–7330. doi: 10.1523/JNEUROSCI.14-12-07319.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorke W. Cerebral defects in Noonan syndrome. Klin Padiatr. 1980;192:577–581. doi: 10.1055/s-2008-1035646. [DOI] [PubMed] [Google Scholar]
- Heye N, Dunne JW. Noonan's syndrome with hydrocephalus, hindbrain herniation, and upper cervical intracord cyst. J Neurol Neurosurg Psychiatry. 1995;59:338–339. doi: 10.1136/jnnp.59.3.338-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johe KK, Hazel TG, Muller T, Dugich-Djordjevic MM, McKay RD. Single factors direct the differentiation of stem cells from the fetal and adult central nervous system. Genes Dev. 1996;10:3129–3140. doi: 10.1101/gad.10.24.3129. [DOI] [PubMed] [Google Scholar]
- Jongmans M, Sisterman EA, Rikken A, Nillesen WM, Tamminga R, Patton M, Maier EM, Tartaglia M, Nooram K, van der Burgt I. Genotypic and phenotypic characterization of Noonan syndrome: New data and review of the literature. Amer J Med Genetics. 2005;134:165–170. doi: 10.1002/ajmg.a.30598. [DOI] [PubMed] [Google Scholar]
- Keilhack H, David FS, McGregor M, Cantley LC, Neel BG. Diverse biochemical properties of Shp2 mutants. J Biol Chem. 2005;280:30984–30993. doi: 10.1074/jbc.M504699200. [DOI] [PubMed] [Google Scholar]
- Lee DA, Portnoy S, Hill P, Gillberg C, Patton MA. Psychological profile of children with Noonan syndrome. Dev Med Child Neurol. 2005;47:35–38. doi: 10.1017/s001216220500006x. [DOI] [PubMed] [Google Scholar]
- Lehmann U, Schmitz J, Weissenbach M, Sobota RM, Hortner M, Friederichs K, Behrmann I, Tsiaris W, Sasaki A, Schneider-Mergener J, Yoshimura A, Neel BG, Heinrich PC, Schaper F. SHP-2 and SOCS3 contribute to Tyr-759-dependent attenuation of interleukin-6 signaling through gp130. J Biol Chem. 2003;278:661–671. doi: 10.1074/jbc.M210552200. [DOI] [PubMed] [Google Scholar]
- Liu Y, Han SS, Wu Y, Tuohy TM, Xu H, Cai J, Back SA, Sherman LS, Fischer I, Rao MS. CD44 epression identifies astrocyte-restricted precursor cells. Dev Biol. 2004;276:1–46. doi: 10.1016/j.ydbio.2004.08.018. [DOI] [PubMed] [Google Scholar]
- Ménard C, Hein P, Paquin A, Savelson A, Yang XM, Lederfein D, Barnabé-Heider F, Mir A, Sterneck E, Peterson AC, Johnson PF, Vinson C, Miller FD. An essential role for a MEK-C/EBP pathway during growth factor-regulated cortical neurogenesis. Neuron. 2002;36:597–610. doi: 10.1016/s0896-6273(02)01026-7. [DOI] [PubMed] [Google Scholar]
- Miller FD, Gauthier AS. Timing is everything: making neurons versus glia in the developing brain. 2007. (submitted) [DOI] [PubMed] [Google Scholar]
- Neel BG, Gu H, Pao L. The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Bioch Sci. 2003;28:284–293. doi: 10.1016/S0968-0004(03)00091-4. [DOI] [PubMed] [Google Scholar]
- Noonan JA. Noonan syndrome: an update and review for the primary pediatrician. Clin Ped. 1994;33:548–555. doi: 10.1177/000992289403300907. [DOI] [PubMed] [Google Scholar]
- Paquin A, Barnabé-Heider F, Kageyama R, Miller FD. CCAAT/enhancer-binding protein phosphorylation biases cortical precursors to generate neurons rather than astrocytes in vivo. J Neurosci. 2005;25:10747–10758. doi: 10.1523/JNEUROSCI.2662-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiris A, Ball MJ. Chiari (type 1) malformation and syringomyelia in a patient with Noonan's syndrome. J Neurol Neurosurg Psychiatry. 1982;45:753–754. doi: 10.1136/jnnp.45.8.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito Y, Sasaki M, Hanaoka S, Sugai K, Hashimoto T. A case of Noonan syndrome with cortical dysplasia. Pediatr Neurol. 1997;17:266–269. doi: 10.1016/s0887-8994(97)00094-5. [DOI] [PubMed] [Google Scholar]
- Tang TL, Freeman RM, Jr, O’Reilly AM, Neel BG, Sokol SY. The SH2-containing protein-tyrosine phosphatase SH-PTP2 is required upstream of MAP kinase for early Xenopus development. Cell. 1995;80:473–483. doi: 10.1016/0092-8674(95)90498-0. [DOI] [PubMed] [Google Scholar]
- Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, van der Burgt I, Crosby AH, Ion A, Jeffery S, Kalidas K, Patton MA, Kucherlapati RS, Gelb BD. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet. 2001;29:465–468. doi: 10.1038/ng772. [DOI] [PubMed] [Google Scholar]
- Uemura A, Takizawa T, Ochiai W, Yanagisawa M, Nakashima T, Taga T. Cardiotrophin-like cytokine induces astrocyte differentiation of fetal neuroepithelial cells via activation of STAT3. Cytokine. 2002;18:1–7. doi: 10.1006/cyto.2002.1006. [DOI] [PubMed] [Google Scholar]
- Wagner B, Natarajan A, Grunaug S, Kroismayr R, Wagner EF, Sibilia M. Neuronal survival depends on EGFR signaling in cortical but not midbrain astrocytes. EMBO J. 2006;25:752–762. doi: 10.1038/sj.emboj.7600988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida R, Hasegawa T, Hasegawa Y, Nagai T, Kinoshita E, Tanaka Y, Kanegane H, Ohyama K, Onishi T, Hanew K, Okuyama T, Horikawa R, Tanaka T, Ogata T. Protein-tyrosine phosphatase, nonreceptor type 11 mutation analysis and clinical assessment in 45 patients with Noonan Syndrome. J Clin EndoMetab. 2004;89:3359–3364. doi: 10.1210/jc.2003-032091. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.