

Abstract
A series of 7-N-acyllavendamycins with zero, one or two substitutents at the C-2′, C-3′ and C-11′ were synthesized through short and efficient methods. Pictet-Spengler condensation of 7-N-acylamino-2-formylquinoline-5,8-diones with tryptamine or tryptophans produced the desired lavendamycins. Screening data on a panel of three ras oncogene transformed cell lines and the non-transformed parent cell line showed that a significant number of these analogues are potent antitumor agents and appear to be particularly active against K-ras transformed cells. Compared with the corresponding quinolinediones, these novel lavendamycins are much more inhibitory toward the transformed cells indicating that the β-carboline moiety of the lavendamycin analogues plays an important role in its potency and selective toxicity.
Keywords: Lavendamycins, Quinoline-5, 8-diones, Antitumor, ras
Introduction
Normal Ras proteins function as GTPases and serve as essential mediators in signaling pathways that convey extracellular signals from surface receptors to the interior of the cell, functioning as molelcular switches in processes governing cell proliferation, survival, and differentiation.1-3 In mammals, there are three functional ras genes located on different chromosomes that code for four highly homologous isoforms; all of which associate with the plasma membrane.4, 5 It is well established that mutations, amplifications or overexpression of the ras proto-oncogenes may lead to the development of transformation and malignancy in mice and humans.4,6–9 Indeed, single point mutations can lead to oncogenically transformed ras genes which have been detected in up to 35% of human tumors with K-ras mutations occurring most frequently.3,6,10–14 In these tumors, an activated Ras protein contributes to several aspects of the malignant phenotype, including the deregulation of tumor cell growth, programmed cell death and invasiveness as well as the ability to induce new blood vessel formation.11 Interestingly, although all three genes are expressed at least at some level in almost all tissue cell types14–16, different ras mutations have been found to be preferentially associated, although certainly not exclusively, with specific types of cancer. For instance, K-ras appears most commonly in pancreatic cancers, cholangiocarcinomas, colorectal malignancies and in adenocarcinomas of the lung whereas mutations in H-ras are rare but are found in certain squamous cell carcinomas and N-ras are found most frequently in acute leukemias and myelodysplastic syndromes.10 Only thyroid tumors often contain mutated ras genes having comparable frequencies of H-ras, K-ras and N-ras activations.17 Clearly, ras genes are of critical importance both for normal cell functioning and in the development of serious human or animal cancers.4,8,9
Although investigators first assumed that the different isoforms of Ras were functionally redundant, it is now becoming increasingly clear using a variety of approaches that each may have distinct biologic functions, involvement in different downstream pathways and cellular localizations.9,11,18–26 With the heavy involvement of ras oncogenes in human tumors there has been great interest in developing agents which target Ras and their signalling pathways.27 As the three normal Ras proteins have such different effector functions and localizations, it would not be surprising that some of these agents might have different effects on transformed cells and tumors carrying different ras-oncogenes. Indeed, in this study using H-ras, N-ras and K-ras oncogene transformed normal rat kidney epithelial cells as well as the 3 Lewis Lung (3LL) transformed cell line as a murine reference tumor, we show that certain analogues of lavendamycins have anti-ras inhibitory effects being particularly active toward K-ras transformed cells. We herein describe the syntheses of a number of these lavendamycins, their corresponding quinoline- 5,8-diones and the evaluation of their activities against several transformed cell lines and a normal rat kidney epithelial cell line. The antibiotic antitumor agent lavendamycin (1) was originally isolated from the fermentation broth of Streptomyces lavendulae in 1981.28 In biosynthesis and bioassays lavendamycin is similar to another potent antibiotic antitumor agent streptonigrin (2).28–32 The clinical use of both of these antibiotics, however, has been precluded because of their toxicity toward human cells.28,33–35 It was thought, however, that analogues of these naturally occurring compounds might have valuable therapeutic value, and thus efficient synthetic methods for these analogues were needed. In 1993 and 1996 our group reported two efficient 5-step syntheses for lavendamycin methyl ester (35) by which we could produce this compound in an overall yield of about 40%.36, 37 These methods were much superior to the previously reported 9- and 20- step syntheses, both of which produced 35 in overall yields of less than 2%. 38,39
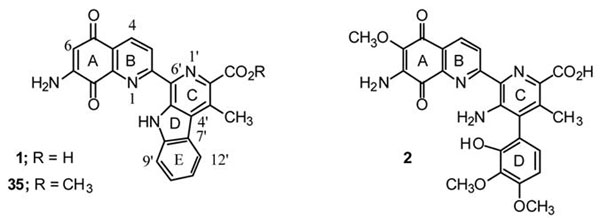
The efficiency of our methods, particularly those reported in 1996 37, enabled us to synthesize a large number of variously substituted lavendamycin analogues facilitating screening assays for biological activity and SAR studies. We have previously reported that a significant number of the lavendamycin analogues have potent biological activity including anti–HIV reverse transcriptase40 and antitumor activity.41,42 More importantly, compared to lavendamycin (1) and streptonigrin (2) most of the analogues have low cellular and animal toxicity.40 The present study extends these findings with the synthesis of several novel lavendamycin analogues that not only have antitumor effects but also appear to be most highly active against K-ras transformed cells.
Results and Discussion
Synthetic Chemistry
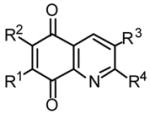
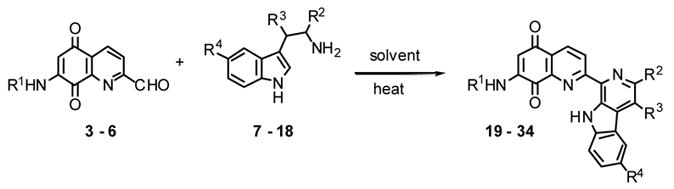
Tables 2 and 3 present the structures of the lavendamycins and quinioline-5,8-diones which were the subject of our biological studies in this report. As shown in Scheme 1 and Table 1, Pictet-Spengler (P-S) condensation of 7-N-acylamino-2-formylquinoline-5,8-diones 3–6 with β-methyltryptophan esters 9, 11, 14 or trytophan esters 7, 8, 10–13 and 15–18 yielded the corresponding lavendamycin derivatives 19–34.
Table 2.
Structures of Quninoline-5,8-diones and Their In Vitro Differential Cytotoxicity
 |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| LC50 (μM)a | |||||||||
| Cell Linesb |
|||||||||
| No. | R1 | R2 | R3 | R4 | NRK | K/1 | H/1.2 | N/4.2 | 3LL |
| 3 | CH3COHN | H | H | CHO | 1.3 | 2.6 | 2.5 | NT | 1.2 |
| 4 | ClCH2CONH | H | H | CHO | 3.6 | 5.8 | 3.4 | 3.2 | 2.1 |
| 5 | n-C3H7COHN | H | H | CHO | 3.0 | >3.3 | 3.3 | >3.3 | 2.5 |
| 6 | i-C3H7COHN | H | H | CHO | >3.3 | 2.15 | >3.3 | >3.3 | 1.0 |
| 47 | CH3COHN | H | H | CH3a | 2.4 | 5.9 | 4.4 | NT | 2.0 |
| 48 | ClCH2CONH | H | H | CH3 | 3.2 | 6.6 | 2.8 | 3.4 | 2.3 |
| 49 | n-C3H7COHN | H | H | CH3 | 2.0 | >3.3 | >3.3 | >3.3 | 2.5 |
| 50 | i-C3H7COHN | H | H | CH3 | 1.5 | 7.5 | 1.45 | 1.15 | 0.71 |
| 51 | CH3CONH | H | CH3 | H | 1.3 | 1.8 | 1.1 | NT | 1.0 |
| 52 | H | CH3COHN | H | CH3 | 21.5 | >33 | 20 | NT | >33 |
| 53 | CH3CONH | H | H | COOH | >33 | >33 | >31 | NT | 26 |
| 54 | H2N | H | H | CH3 | 0.8 | 1.3 | 0.6 | NT | 0.8 |
| 55 | H2N | H | H | CHO | 2.4 | 4.3 | 2.2 | NT | 2.3 |
| 56 | CH3O | H | H | CH3 | 6.9 | 11.8 | 4.2 | NT | 7.4 |
| 57 | HO(CH2)4NH | H | H | CH3 | 10.5 | 13.5 | 10.5 | NT | 3.8 |
LC50 is the concentration of analogue that afforded 50% reduction in colony number after a 5-day incubation. Most of the reported values are derived from representative experiments performed in triplicate and interpolated from at least eight different concentrations of drugs ranging from 0.01 to 33 μM. Those values reported as > 3.3 were determined in screening assays where only two concentrations of drug were used, i.e. 3.3 and 33 μM. All experiments were performed at least twice and the data presented for each compound is that obtained from one experiment. See Experimental Section for culture details. NT = Not tested.
Cell lines: NRK, normal rat kidney epithelial; K/1, K-ras transformed normal rat kidney epithelial; H/1.2, H-ras transformed normal rat kidney epithelial; N/4.2, N-ras transformed normal rat kidney epithelial; and 3LL, Lewis Lung carcinoma.
Table 3.
In Vitro Differential Cytotoxicity of Lavendamycins
 |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| LC50 (μ M)a | |||||||||
| Cell Linesb |
|||||||||
| No. | R1 | R2 | R3 | R4 | NRKE | K/1 | H/1.2 | N/4.2 | 3LL |
| 19 | CH3CO | CO2CH2CH3 | H | H | 0.30 | 0.04 | 0.36 | 0.20 | 3.2 |
| 20 | CH3CO | CO2CH(CH2)5 | H | H | 0.70 | 0.40 | 0.69 | 0.70 | 1.0 |
| 21 | CH3CO | CO2C5H11-i | CH3 | H | 4.16 | 5.0 | 4.3 | 3.89 | 7.4 |
| 22 | CH3CO | CO2CH2CH2N(CH3)2 | H | H | 0.37 | 0.14 | 0.24 | 0.29 | 0.50 |
| 23 | CH3CO | CO2CH2CH2N(CH3)2 | CH3 | H | 0.09 | 0.06 | 0.09 | 0.09 | 0.48 |
| 24 | CH3CO | CO2C4H9-n | H | OCH3 | 2.5 | 2.2 | 2.1 | 2.2 | >3.3 |
| 25 | CH3CO | CO2C4H9-n | H | F | 0.62 | 0.33 | 0.70 | 0.84 | 2.1 |
| 26 | ClCH2CO | CO2CH3 | CH3 | H | 0.58 | 0.79 | 0.58 | 0.70 | 2.0 |
| 27 | ClCH2CO | CO2C4H9-n | H | H | 0.96 | 0.52 | 0.74 | 1.05 | 2.3 |
| 28 | ClCH2CO | CO2C8H17-n | H | H | >33 | 15.0 | >33 | >33 | >33 |
| 29 | n-C3H7CO | CO2CH3 | H | H | 0.70 | 0.50 | 0.66 | 0.64 | 1.3 |
| 30 | n-C3H7CO | CO2CH3 | CH3 | H | 0.32 | 0.33 | 0.40 | 0.42 | 0.82 |
| 31 | n-C3H7CO | CO2C5H11-I | H | H | 1.00 | 0.60 | 0.37 | 0.65 | 2.4 |
| 32 | n-C3H7CO | CO2C8H17-n | H | H | >33 | 3.3 | 16.0 | 15.0 | 16.0 |
| 33 | i-C3H7CO | CO2CH3 | H | H | 0.70 | 0.50 | 0.66 | 0.64 | 1.3 |
| 34 | i-C3H7CO | CO2C4H9-n | H | H | 0.90 | 0.40 | 0.61 | 0.50 | 0.90 |
| 35 | H | H | H | H | 0.10 | 0.031 | 0.11 | 0.06 | 0.25 |
| 36 | CH3CO | H | H | H | 0.40 | 0.44 | 0.40 | 0.49 | 3.0 |
| 37 | CH3CO | CH2OH | H | H | 0.25 | 0.20 | 0.20 | 0.22 | 0.44 |
| 38 | CH3CO | CO2H | H | H | 0.15 | 0.07 | 0.11 | 0.10 | 0.40 |
| 39 | CH3CO | CONH2 | H | H | 0.90 | 0.35 | 0.23 | 0.22 | 0.21 |
| 40 | CH3CO | CO2CH3 | H | H | 0.50 | 0.11 | 0.51 | 0.52 | 0.84 |
| 41 | CH3CO | CO2C5H11-I | H | H | 1.62 | 0.09 | 1.42 | 1.5 | 1.69 |
| 42 | CH3CO | CO2C8H17-n | H | H | >33 | 0.25 | 7.6 | 9.0 | >33 |
| 43 | CH3CO | CO2CH2Ph | H | H | 9.5 | 9.5 | >10 | >33 | >33 |
| 44 | CH3CO | CO2CH3 | CH3 | H | 0.90 | 0.10 | 0.70 | NT | >33 |
| 45 | ClCH2CO | CO2C5H11-I | H | H | 1.35 | 0.56 | 1.32 | 1.2 | 3.2 |
| 46 | n-C3H7CO | CO2C4H9-n | H | H | 0.66 | 0.50 | 0.40 | 0.70 | 2.3 |
LC50 is the concentration of analogue that afforded 50% reduction in colony number after a 5-day incubation.With two exceptions, the reported values are derived from representative experiments performed in triplicate and interpolated from at least eight different concentrations of drug ranging from 0.01 to 33 μM. Those values reported as 3.3 or > 3.3 were determined in screening assays where only two concentrations of drug were used, i.e., 3.3 and 33 μM. All experiments were performed at least twice. NT = Not tested.
Scheme 1.
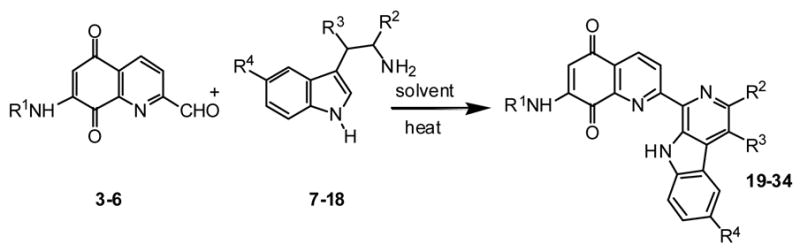
Table 1.
Synthesis and Structures of Lavendamycins
| No. | R1 | R2 | R3 | R4 | % yield | hrs (°C )a |
|---|---|---|---|---|---|---|
| 19 | CH3CO | CO2C2H5 | H | H | 55 | 3 (25-reflux), 19 (reflux) |
| 20 | CH3CO | CO2CH(CH2)5 | H | H | 59 | 3 (25-reflux), 1(reflux) |
| 21 | CH3CO | CO2C5H11-i | CH3 | H | 59 | 3 (<140), 1 (140) |
| 22 | CH3CO | CO2(CH2)2N(CH3)2 | H | H | 20 | 27 (100) |
| 23 | CH3CO | CO2(CH2)2N(CH3)2 | CH3 | H | 36 | 5.5 (100) |
| 24 | CH3CO | CO2C4H9-n | H | OCH3 | 66 | 6 (70-90) |
| 25 | CH3CO | CO2C4H9-n | H | F | 51 | 5 (25–150) |
| 26 | ClCH2CO | CO2CH3 | CH3 | H | 46 | 3 (25–135), 16 (135) |
| 27 | ClCH2CO | CO2C4H9-n | H | H | 20 | 3 (76), 5.75 (76) |
| 28 | ClCH2CO | CO2C8H17-n | H | H | 53 | 6 25–100), 5.5 (100) |
| 29 | n-C3H7CO | CO2CH3 | H | H | 59 | 3 (25–130), 22 (130) |
| 30 | n-C3H7CO | CO2CH3 | CH3 | H | 44 | 3 (25–130), 16 (130) |
| 31 | n-C3H7CO | CO2C5H11-i | H | H | 70 | 4 (25-reflux), 17 (reflux) |
| 32 | n-C3H7CO | CO2C8H17-n | H | H | 80 | 5 (25–85), 16 (85) |
| 33 | i-C3H7CO | CO2CH3 | H | H | 63 | 3 (25–130), 4 (130) |
| 34 | i-C3H7CO | CO2C4H9-n | H | H | 65 | 5(25-125), 3 (125) |
The reaction solvent used for producing 22-25, 27 and 29 was dry anisole and for the remainder dry xylene was used.
In certain instances the condensation did not directly produce the target lavendamycins but interestingly gave the corresponding dihydroxy derivative (Scheme 2; e.g. 27b) or a mixture of the dihydroxy and the target lavendamycin (e.g. 29). These products were then converted to lavendamycins 27 and 29 respectively by air or DDQ oxidation.43
Scheme 2.
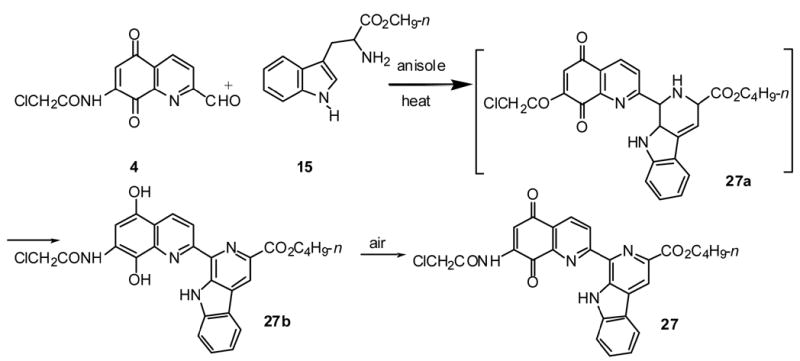
As presented in Scheme 2, we suggest that Pictet-Spengler condensation of aldehyde 4 with tryptophan 15 will yield the expected tetrahydro-β-carboline 27a which is then aromatized through the reduction of its quinolinedione portion to the corresponding dihydroxy derivative 27b. Tetrahydroderivative 27a, could not be isolated due to its immediate conversion to the more stable derivative 27b. The structure of 27b was confirmed by NMR, IR and Mass spectroscopic methods. IR and NMR spectra of 27b show signals for the phenoilic hydroxyl groups which were absent in the spectra of lavendamycin 27. Upon addition of a drop of D2O to the solution of 27b in DMSO-d6, the NMR signals at 9.76 and 9.93 (OH's) as well as those at 10.22 and 12.33 ppm (NH's) competely disappeared.
The formation of lavendamycin 29 also goes through this transformation. Considering the mild reaction conditions used for the production of lavendamycins (Table 1), it should be noted that this unique auto redox phenomenon may facilitate the ease of Pictet-Spengler condensations to produce a number of lavendamycins at significantly lower temperatures compared to the higher temperatures required for the P-S condensation of simple aldehydes with tryptophans.44,45
The syntheses of lavendamycin derivatives 35–46 (Table 3) have been described in our previous reports.36,37,40,42 Quinolinediones 3–5, 47–50, and 56 listed in Table 2 were prepared according to our reported methods.36,37,40,42,48 Isobtyramido-2-formylquinoline-5,8-dione (6) was prepared by the oxidation of dione 50 with selenium dioxide via a similar method to that used for aldehyde 3.36 7-Acetamidoquinoline-5,8-dione-2-carboxylic acid (53) was obtained by the oxidation of adehyde 3 with sodium perborate.
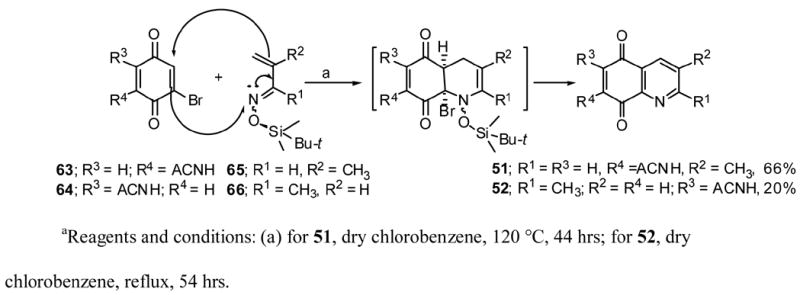
Quniolinediones 51 and 52 were prepared via the Diels-Alder reactions of the corresponding azadienes (E/Z mixtures) with the desired dienophiles as presented in Scheme 3.36,46
Scheme 3.
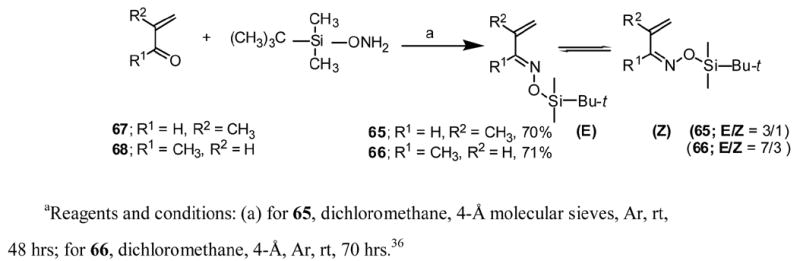
Azadienes 65 and 66 were prepared according to Scheme 4. The preparation of azadiene 66 was previously reported by us 36 and a similar method was used to prepare azadiene 65 as a mixture of E and Z isomers.46
Scheme 4.
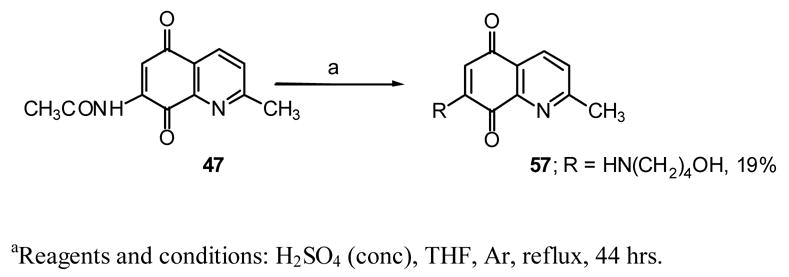
Dienophile 63 and 64 were prepared through our previously reported methods.36,47 Dione 56 was prepared by the methanolysis of 4748 and 57 was obtained by the reaction of tetrahydrofuran with 47 in the presence of acid (Scheme 5).
Scheme 5.
Based on present data and our previously reported results for the conversion of 47 to 7-methoxydione 56,48 the following mechanism (Fig. 1) is proposed for this novel transformation.
Fig 1.
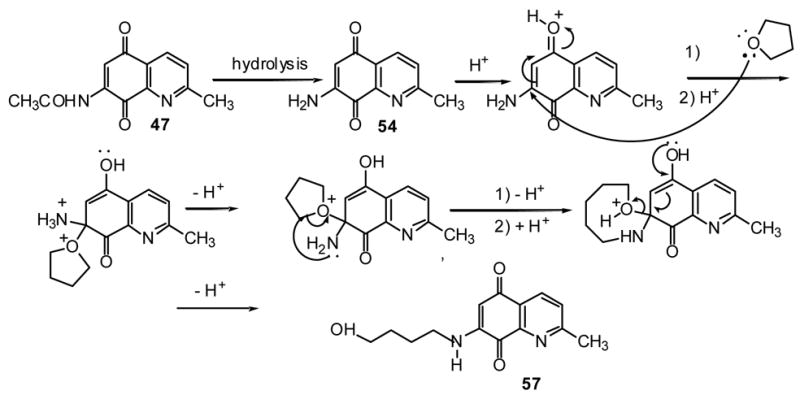
As experimentally observed, the first step of the reaction is the hydrolysis of 47 to the aminodione 54 followed by the conversion of the latter to the final product 57. Aminodione 55 was prepared by the hydrolysis of 3 with an aqueous solution of sodium bicarbonate at room temperature.
The preparations of tryptophans and their derivatives necessary for the synthesis of lavendamycins listed in Table 1 are described below. Tryptophan esters 7, 15 and 17 were obtained by the neutralization of their commercially available hydrochloride salts with 14% ammonium hydroxide solution and extraction with ethyl acetate. Tryptophan esters 8, 9, 12, 13, 16 and 18 were prepared by the Fischer esterification of the corresponding commercially available tryptophans with the desired alcohols in the presence of hydrogen chloride. (2R3S,2S3R)-β-Methyltryptophan was prepared according to the method of Snyder and Matteson.49 Esters 10 and 11 were obtained through the reactions shown in Scheme 6.
Scheme 6.
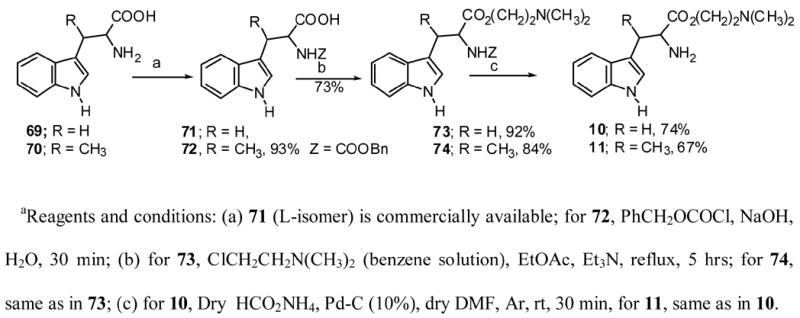
β-Dimethylaminoethyl chloride was prepared50 and then reacted with tryptophan and β-methyltryptophan 71 and 72 followed by the deprotection of the resulting 73 and 74 using modified methods of those previously reported.51,52
(2R3S,2S3R)-β-Methyltryptophan methyl ester (14) was prepared according to our own procedure.53
Activity of Quinolinediones and Lavendamycins Toward Transformed Cell Lines and SAR Study
As shown in Table 2, fifteen quinoline-5,8-diones were examined for their biological activity in vitro toward five cell lines; a normal rat kidney epithelial line (NRK-52E), three NRK lines transformed by the H-, K- and N-ras oncogenes as well as the unrelated 3LL carcinoma line. This differential cytotoxicty assay is particularly valuable in identifying compounds that interfere with the biochemical pathways activated or driven by oncogenes. The quinolinediones were frequently more or equally toxic to parent NRK cells than to the tumor lines tested. Regardless of the R group substituents, the lethal concentration needed to kill fifty percent of the cells (LC50) tested ranged between 0.8 μM and 33 μM with no particular pattern of sensitivity or selective toxicity toward any of the cell lines. In addition, representative diones such as 5, 47 and 49 were found to be highly toxic in vivo in mice with maximum tolerated doses in mice of <10 mg/kg. Previously, it has been suggested that the naphthoquinone or the quinolinedione moieties of the antitumor agents such as anthracyclines, streptonigrin and lavendamycin are essential for antimicrobial and antitumor activities of the parent compounds.34,54-63 Indeed, naphthoquinones have been shown64 to prolong the life of mice with the ascitic sarcoma 180 and quinones and quinolinediones have been shown to cleave DNA at rates which correlate with their molecular reduction potentials.65 In our work, however, the quinolinediones were in general no more active toward the tumors tested than they were to normal cells and no alterations in substituents seemed to significantly enhance their antitumor activity. Indeed, Boger et al, also found that the parent compounds, streptonigrin and lavendamycin, with their entire ring skeleton were considerably more active in their biologic studies than a complete set of partial structures.34
In contrast to the quinolinediones, several of the lavendamycins were toxic to some of the transformed cells at very low concentrations ( Tables 2 and 3). Five of the lavendamycins, 19, 23, 35, 38, and 41 had LC50's less than 0.1μM against the K-ras transformed cells with a range of 0.03–9.5 μM for all the lavendamycins and a median of 0.4 μM. The NRK cells were also more sensitive to the lavendamycins (median LC50 0.7μM) than they were to the quinolinediones (median LC50 3.2 μM), but these normal cells were, in several instances, considerably less sensitive to several of the lavendamycins than were the transformed cells, particularly the K-ras line (Tables 3). The complete pentacyclic skeleton of lavendamycin has a definite effect on its cytotoxicity toward the ras transformed cells. Thus, compared to their corresponding quinolinediones, the LC50’s of many of the lavendamycins toward the oncogene transformed cells are many fold lower (Tables 2 and 3). For example, this can be seen by a comparison of the data for lavendamycin 19 and 23 with quinolinedione 3; 45 with 48; 35 with 54; and 40 and 44 with that of 47. Most importantly, a large number of the lavendamycin analogues, but none of the quinolinediones, demonstrate significantly greater activity toward K/1-ras transformed cells in comparison to activity toward the parental NRK cells, the H-ras, N-ras transformed cell lines and the 3LL tumor line. This selective activity can be seen in Table 4 in which the differential indices of each of the analogues toward the four tumor lines is presented. The differential index for each analogue is calculated by dividing the LC50 toward the NRK cells by the LC50 toward the tumor cells. This value is an excellent measure of the selective toxicity of a compound toward tumor cells. Thirteen of the twenty-eight analogues tested had differential indices greater than 2 toward the K-ras transformed cells, whereas only three had indices over 2 toward either H-ras or N-ras transformed cells and only two were selectively toxic toward the 3LL cell line. As shown in Table 4, six of the lavendamycins were at least 4.5 times more toxic to the K-ras transformed cells than normal cells including compound 42, the octyl ester, with a remarkable differential index of 132.
Table 4.
Differential Indices of Lavendamycins
 |
||||||||
|---|---|---|---|---|---|---|---|---|
| Differential Indexa |
||||||||
| Tumor Cell Linesb |
||||||||
| No. | R1 | R2 | R3 | R4 | K/1 | H/1.2 | N/4.2 | 3LL |
| 19 | CH3CO | CO2CH2CH3 | H | H | 7.5 | 0.8 | 0.7 | 0.1 |
| 20 | CH3CO | CO2CH(CH2)5 | H | H | 1.7 | 1.0 | 1.0 | 0.7 |
| 21 | CH3CO | CO2C5H11-i | CH3 | H | 0.9 | 1.1 | 1.2 | 0.6 |
| 22 | CH3CO | CO2CH2CH2N(CH3)2 | H | H | 2.6 | 1.5 | 1.3 | 0.7 |
| 23 | CH3CO | CO2CH2CH2N(CH3)2 | CH3 | H | 1.5 | 1.0 | 1.0 | 0.2 |
| 24 | CH3CO | CO2C4H9-n | H | OCH3 | 1.1 | 1.2 | 1.1 | <0.7 |
| 25 | CH3CO | CO2C4H9-n | H | F | 1.9 | 0.9 | 0.8 | 0.3 |
| 26 | ClCH2CO | CO2CH3 | CH3 | H | 0.7 | 1.0 | 0.8 | 0.3 |
| 27 | CH3CO | CO2C4H9-n | H | OCH3 | 1.1 | 1.2 | 1.1 | <0.7 |
| 28 | ClCH2CO | CO2C8H17-n | H | H | >2.2 | 1.0 | 1.0 | 1.0 |
| 29 | n-C3H7CO | CO2CH3 | H | H | 1.4 | 1.1 | 1.1 | 0.5 |
| 30 | n-C3H7CO | CO2CH3 | CH3 | H | 1.0 | 0.8 | 0.8 | 0.4 |
| 31 | n-C3H7CO | CO2C5H11-i | H | H | 1.6 | 2.7 | 1.5 | 0.4 |
| 32 | n-C3H7CO | CO2C8H17-n | H | H | >10 | 2 | >2.2 | >2 |
| 33 | i-C3H7CO | CO2CH3 | H | H | 1.4 | 1.1 | 1.1 | 0.5 |
| 34 | i-C3H7CO | CO2C4H9-n | H | H | 2.3 | 1.5 | 1.8 | 1.0 |
| 35 | H | CO2CH3 | CH3 | H | 3.2 | 0.9 | 1.6 | 0.4 |
| 36 | CH3CO | H | H | H | 0.9 | 1.0 | 0.8 | 0.1 |
| 37 | CH3CO | CH2OH | H | H | 1.2 | 1.2 | 1.1 | 0.6 |
| 38 | CH3CO | CO2H | H | H | 2.1 | 1.4 | 1.5 | 0.4 |
| 39 | CH3CO | CONH2 | H | H | 2.5 | 1.5 | 4.1 | 4.3 |
| 40 | CH3CO | CO2CH3 | H | H | 4.5 | 1 | 1 | 0.6 |
| 41 | CH3CO | CO2C5H11-i | H | H | 18 | 1.1 | 1.1 | 1.0 |
| 42 | CH3CO | CO2C8H17-n | H | H | >132 | 4.3 | 3.6 | 1.0 |
| 43 | CH3CO | CO2CH2Ph | H | H | 1.0 | <0.9 | <0.3 | <0.3 |
| 44 | CH3CO | CO2CH3 | CH3 | H | 9.0 | 1.3 | N.T. | 0.03 |
| 45 | ClCH2CO | CO2C5H11-i | H | H | 2.4 | 1.0 | 1.1 | 0.4 |
| 46 | n-C3H7CO | CO2C4H9-n | H | H | 1.3 | 1.6 | 0.9 | 0.3 |
Differential index of cytotoxicity was determined by dividing the LC50 value for the normal rat kidney epithelial cells ( NRK) by the LC50 value for the tumor cells. Index values for analogues which reflected LC50 activity at concentrations at least two times lower toward tumor cells than to normal cells are shown in bold.
See footnotes for Table 2.
In addition to the need for the entire lavendamycin skeleton, the greatest selective toxicity is observed for analogues having a carboxylic acid function or its derivative at the C-2' position. This selective toxicity is reduced significantly when the oxidation state of the C-2' group is reduced as shown by comparing data for analogues with CH2OH (37) and H (36) in position C-2' with those of compounds 38-42 (Table 3 and 4). Among the 7-acetamido carboxylic acid derivatives, an ester group at the C-2' (19, 40-42) maximizes K/1-ras oncogene specificity and selective cytotoxicity compared to an acid (38) or an amide (39) function. Benzyl and cyclohexyl esters (43) and (20), however, do not show significant activity enhancement of their activity.
Of particular interest is that the K-ras specificity of the lavendamycin esters increases as the size of the ester chain increases; 5-fold for methyl (40), 8-fold for ethyl (19), 18-fold for isoamyl (41) and > 130- fold for n-octyl (42). This may be due to the enhanced lipophilicity of the molecules with longer ester chains and their consequent ease of passage through the cell membrane.
It is important to note that among the lavendamycins tested, an acetyl group on the C-7 NH2 enhances the compounds’ selective toxicities much more than other acyl groups. For example, analogue 44 is more active than 26 and 30; and 42 is much more active than 28 and 32. Likewise, the selective toxicity of the N-acetyl analogue 41 is greater than that of the corresponding analogues 31 and 45; and 40 has enhanced activity compared to 29 and 33..
In contrast to previous reports on quinones,64,65 it appears that the reduction potentials of the lavendamycin analogues have no influence on the toxicity or selective activity of these compounds toward the K-ras transformed cells. For example, compound 44 which was 9 times more toxic for the K-ras cells than for the NRKE cells had a redox potential almost identical to that of compound 36 which had a differential index of 0.9, i.e. V = −0.91 and V = −0.95, respectively.42 As the mechanism of action of these analogues toward the ras transformed cells is most likely directed toward the different Ras proteins or the metabolic pathways that are activated or driven by the ras oncogenes and not through DNA cleavage or interaction with the oncogenes themselves, it is not unexpected that redox potential plays little or no role in the activity of these compounds in this tumor system.
In Vivo Toxicity and Antitumor Activity
We have previously reported that the lavendamycin analogues have remarkably low toxicity in vivo, particularly in comparison to streptonigrin and lavendamycin itself which have maximum tolerated doses in mice of 0.4 and 12.8 mg/kg, respectively. 40,66,67 For example, the maximum tolerated dose in mice for analogues 35 and 39 was found to be 400 mg/kg. Analogues 41 and 42 were also well tolerated when given to nude mice at both 150 and 300 mg/kg/day for eight days with no deaths and only slight weight loss. In another trial, no signs of toxicity developed following treatment of nude mice with analogue 44 at 10, 50 and 100 mg/kg/day for seven days. Preliminary studies in nude mice transplanted with K/1-NRK tumors and treated with these analogues, i.e., 41, 42 and 44, demonstrated promising tumor inhibition with little or no toxicity 10 days following implantation. Analogue 41 inhibited tumor growth by 88 ± 5 percent at 150 mg/kg/day for 8 days compared to vehicle treated animals and 42 inhibited growth by 78 ± 13 percent at 300 mg/kg/day for 8 days. In these limited studies, compound 44 appeared to be the most potent in vivo with 69 ± 5 percent inhibition on day 10 in animals treated with only 100 mg/kg/day for seven days. Further in vivo studies with these promising analogues may reveal even greater tumor inhibition either at higher doses or different dosing regimens.
Conclusions
Short and efficient syntheses of a variety of novel acyllavendamycins as well as some of the corresponding quinoline-5,8-diones are described. An extensive SAR study of a large number of these lavendamycin analogues, shows these compounds, particularly those with large alkyl groups at the C-2' ester position, to be potent antitumor agents both in vitro and in vivo. These analogues appear to be much more active against K-ras oncogene transformed cells than either H- or N-ras transformed or 3LL cells. In addition, the lavendamycins are clearly much more potent with substantially higher selective toxicity toward the cancer cells compared to their corresponding quinoline-5,8-diones. This finding suggests the critical importance of the β-carboline moiety in the potency and selectivity of the lavendamycins. In contrast to the simpler quinolinediones, it appears that the antitumor activity does not correlate with the molecular reduction potentials or direct effects on the oncogenes themselves. It appears likely, using the differential cytotoxicity assay employed in this study, that the remarkable activity of these compounds toward K-ras transformed cells may be due to the inhibition of K-ras directed pathways or oncogene products. Indeed, such oncogene specific activity has been shown for a farnesyl transferase inhibitor which caused a selective, dose-dependent, reversible blockade in proliferation of H-Ras-transformed rat intestinal epithelial (RIE-1) cells whereas control cell lines, K-ras transformed cells, and activated raf-transfected RIE cells were unaffected.68 Apparently the loss of farnesylated H-Ras protein in particular led to a marked reduction in transforming growth factor-alpha with other inhibitory downstream effects. The apparent sensitivity of K-ras transformed cells to some of the lavendamycins in the present study may also serve to highlight other functional differences between the ras oncogenes and their products and strongly suggests that tumors caused by oncogenes even in the same family are, in fact, quite different targets which may require different drug treatments or formulations for effective treatment.27,69 The high degree of antitumor activity and the large differential indices of a significant number of the lavendamycin analogues suggest the potential use of these compounds as highly potent, nontoxic anticancer drugs for ras associated human tumors. Further study of these compounds may also lead to a better understanding of K-ras driven oncogenesis.
Experimental Section Chemistry. General Methods
For General Methods see ref. 40 (J. Med. Chem. 2003, 46, 5773).
General Procedure for the Synthesis of Lavendamycins
Unless otherwise stated, lavendamycin derivatives 19-34 were synthesized via a procedure similar to that used for analogue 19. For each lavendamycin, the required starting materials (Scheme 1, and Table 1), were mixed in the desired solvent and heated for several hours as shown in Table 1, until TLC of the reaction mixture indicated the absence of the starting materials. A brief experimental procedure, work-up and analytical data are given for each derivative
7-N-Acetyldemethyllavendamycin ethyl ester (19)
In a dry 250 mL round-bottomed two-necked flask, equipped with a magnetic bar, water cooled reflux condenser under argon, 7-acetamido-2-formylquinoli-5,8-dione (3, 151 mg, 0.62 mmol), L-tryptophan ethyl ester (7, 144 mg, 0.62 mmol) were added to 165 mL of dried distilled xylene and the mixture while being stirred was slowly heated to 167 °C and refluxed for 19 hrs. The solid product was filtered and dried under vacuum (154 mg, 55%): mp 230– 232 °C (dec.); Rf = 0.49 (0.05/5 MeOH/CH2Cl2); 1H NMR (CDCl3) Δ 1.55 (t, 3H, J = 7.1), 2.38 (s, 3H), 4.59 (q, 2H, J = 7.1), 7.38–7.45 (m, 1H), 7.62–7.70 (m, 1H), 7.71 (d, 1H, J = 7.7), 8.00 (s, 1H), 8.26 (d, 1H, J = 7.77), 8.43 (br s, 1H), 8.57 (d, 1H, J = 8.1), 8.98 (s, 1H), 9.21 (d, 1H, J = 8.1), 11.8 (br s, 1H); HRMS calculated for C25H18N4O5 454.1277, found 454.1270.
7-N-Acetyldemethyllavendamycin cyclohexyl ester (20)
Aldehyde 3 (58.6 mg, 0.24 mmol) and L-tryptophan cyclohexyl ester (8, 66 mg, 0.24 mmol) in 100 mL dry xylene was stirred and slowly heated to reflux over a course of 3 hrs. The reaction mixture was refluxed for another hr and then hot filtered to remove the solid impurity. The filtrate was evaporated to dryness in vacuo and the solid was washed with a small portion of acetone to give 72.2 mg (59%) of an orange product: mp > 280 °C; Rf = 0.62 (1/2 EtOAc/CHCl3); 1H NMR (CDCl3) Δ 1.66–2.17 (m, 10H), 2.37 (s, 3H), 5.10–5.30 (m, 1H), 7.41 (dd, 1H, J = 8.0, J = 8.0), 7.68 (dd, 1H, J = 8.0, 8.0), 7.75 (d, 1H, J = 8.0), 7.98 (s, 1H), 8.27 (d, 1H, J = 8.0), 8.42 (br s, 1H), 8.56 (d, 1H, J = 8.4), 8.95 (d, s, 1H), 9.22 (d, 1H, J = 8.4), 11.8 (br, s, 1H); HRMS calculated for C29H24N4O5 508.1746, found 508.1733.
7-N-Acetyllavendamycin isoamyl ester (21)
A stirred mixture of aldehyde 3 (44.8 mg, 0.2 mmol), (2RS,3SR)-β-methyltryptophan isoamyl ester (9, 57.6 mg, 0.2 mmol) in 67 mL of dry xylene was slowly heated to 140 °C over a 3 hr period and then maintained this temperature for 1 hr. The hot mixture was filtered to give an orange solid. The filtrate upon cooling gave more product. The total product weight was 60.2 mg (59%): mp 258–258.5 °C; Rf = 0.71 (0.5/5 MeOH/CH2Cl2); 1H NMR (CDCl3) Δ 1.06 (d, 6H, J = 6.3), 1.83–1.95 (m, 3H), 2.37 (s, 3H), 3.19 (s, 3H), 4.53 (t, 2H, J = 6.7), 7.36–7.44 (m, 1H), 7.75 (d, 1H, J = 8.0), 7.98(s, 1H), 8.36 (d, 1H, J = 8.0), 8.39 (br s, 1H), 8.53 (d, 1H, J = 8.4), 9.11 ( d, 1H, J = 8.4), 11.80 (br s, 1H); HRMS calculated for C29H26N4O5 510.1903, found 510.1878.
7-N-Acetyldemethyllavendamycin N,N-dimethlaminoethyl ester (22)
In a 50 mL two-necked round-bottomed flask a solution of aldehyde 3 (12.2 mg, 0.05 mmol) and L-tryptophan N,N-dimethylaminoethyl ester (10, 14 mg, 0.05 mmol) in 18 mL of dry anisole was heated at 100 °C for 27 hrs. The mixture was allowed to cool to room temperature and the solid impurity was filtered. The filtrate was evaporated under reduced pressure and the resulting residue was dissolved in 6 mL CHCl3, and purified by thick layer chromatography (alumina) using MeOH/CH2Cl2 2% as the eluting solvent. A pure orange solid (5 mg, 20% yield) was obtained: mp 242 °C (dec); Rf = 0.39 (0.5/5 MeOH/CH2Cl2, Al2O3); 1H NMR (CDCl3) Δ 2.36 (s, 3H), 2.43 (s, 3H), 2.87 (t, 2H, J = 5.9), 4.61 (t, 2H, J = 5.9), 7.40 (dd, 1H, J = 8.1, 7.3), 7.67 (dd, 1H, J = 8.1, 7.3), 7.74 (d, 1H, J = 8.1), 7.98 (s, 1H), 8.24 (d, 1H, J = 8.1), 8.55 (d, 1H, J = 8.4), 8.97 (s, 1H), 9.19 (d, 1H, J = 8.4), 11.82 (br s, 1H); HRMS calculated for C27H23N5O5 497.1694, found 497.1680.
N-Acetyllavendamycin N,N-dimethylaminoethyl ester (23)
The procedure for the synthesis of 23 was similar to that of 22. Aldehyde 3 (31.7 mg, 0.13 mmol), was added to 16 mL of dry anisole and was heated to 80 °C. (2RS,3SR)-β-Methltryptophan N,N-dimethylaminoethyl ester (11, 57.6 mg, 0.2 mmol) was added and the stirred mixture was heated at 100 °C for 5.5 hrs. The reaction mixture was allowed to cool to room temperature and the solid was filtered. The filtrate was distilled under vacuum to dryness. Ethyl acetate (3 mL) was added and the mixture was stirred and then filtered to yield an orange solid (24.5 mg, 36%): mp 181.5 °C (dec); Rf = 0.60 (0.2/5 MeOH/CH2Cl2); 1H NMR (CDCl3) Δ 2.37 (s, 3H), 2.43 (s, 3H), 2.78– 2.83 (m, 2H), 3.2 (s, 3H), 4.52 (t, 2H, J = 6.6), 7.38–7.43 (m, 1H), 7.67–7.74 (m, 1H), 7.74 (d, 1H, J = 8.0), 7.97 (s, 1H), 8.3 (d, 1H, J = 8.0), 8.40 (br s, 1H), 8.5 (d, 1H, J = 8.4), 9.10 (d, 1H, J = 8.4), 11.80 (br s, 1H); HRMS calculated for C28H27N5O5 513.2012 (M + 2H)+, found 513.2017.
7-N-Acetyl-11'-methoxydemethyllavendamycin n-butyl ester (24)
Aldehyde 3 (44 mg, 0.18 mmol) was added to 50 mL of dry anisole and heated with stirring to 65 °C and to the resulting solution, DL-5-methoxytryptophan n-butyl ester (12, 70.8 mg, 0.24 mmol) in 10 mL of dry anisole was added. The reaction mixture was heated for 6 hrs at 70 to 90 °C. The golden yellow solution was evaporated under vacuum and the residue was recrystallized with ethyl acetate, yielding dark red needle crystals (61 mg, 66%): mp 187 °C (dec); Rf = 0.56 (0.05/5 MeOH/CH2Cl2); 1H NMR (CDCl3) Δ 1.06 (t, 3H, J = 7.3), 1.52–1.62 (m, 2H), 1.85–1.95 (m, 2H), 2.36 (s, 3H), 3.98 (s, 3H), 4.51 (t, 2H, J = 6.9), 7.31 (dd, 1H, J = 8.8, 2.2), 7.62 (d, 1H, J = 8.8), 7.66 (d, 1H, J = 2.2), 7.98 (s, 1H), 8.39 (br s, 1H), 8.53 (d, 1H, J = 8.4), 8.91 (s, 1H), 9.17 (d, 1H, J = 8.4), 11.68 (br s, 1H); HRMS calculated for C28H24N4O6 512.1696, found 512.1684.
7-N-Acetyl-11'-fluorodemethyllavendamycin n-butyl ester (25)
Aldehyde 3 (36.6 mg, 0.15 mmol), DL-5-fluorotryptophan n-butyl ester (13, 41.7 mg, 0.15 mmol) in 60 mL dry anisole was stirred and slowly heated to 150 °C over a period of 5 hrs. The orange solution was evaporated under reduced pressure and the residue was recrystallized with ethyl acetate to give 37 mg (51%) of an orange crystalline product: mp > 280 °C; Rf = 0.58 (0.05/5 MeOH/CH2Cl2); 1H NMR (DMSO-d6) Δ 1.01 (t, 3H, J = 7.3), 1.50–1.56 (m, 2H, J = 7.3), 1.78–1.88 (m, 2H, J = 7.3, 7.0), 2.32 (s, 3H), 4.42 (t, 2H, J = 7.0), 7.56–7.65 (m, 1H), 7.70–7.78 (m, 1H), 7.84 (s, 1H), 8.45–8.53 (m, 1H), 8.63 (d, 1H, J = 8.4), 8.95 (d, 1H, J = 8.4), 9.16 (s, 1H), 10.30 (br s, 1H), 12.01 (br s, 1H); HRMS calculated for C27H21FN4O5 500.1496, found 500.1496.
7-N-Chloracetyllavendamycin methyl ester (26)
A mixture of 7-N-Chloroacetyl-2-formylquinoline-5,8-dione (4, 27.8 mg, 0.1 mmol), and 23.2 mg (0.10 mmol) of (2SR,3SR)-β-methyltryptophan methyl ester (14) in 40 mL dried distilled xylene was slowly heated to 135 °C over a 3 hr period and then kept at this temperature for 16 hrs. The mixture was filtered hot to remove the solid impurities and the filtrate was allowed to cool to provide an orange-red solid (22.6 mg, 46%); mp > 280 °C; Rf = 0.49 (1/1 EtOAc/CH2Cl2); 1H NMR (DMSO-d6) Δ 3.08 (s, 3H), 3.97 (s, 3H), 4.64 (s, 2H), 7.42 (dd, 1H, J = 8.0, J = 8.0), 7.69 (dd, 1H, J = 8.4, 8.4), 7.71 (d, 1H, J = 8.4), 7.75 (s, 1H), 8.40 (d, 1H, J = 8.4), 8.50 (d, 1H, J = 8.4), 8.64 (d, 1H, J = 8.4), 10.58 (br s, 1H), 11.95 (br s, 1H); HRMS calculated for C25H17ClN4O5 488.0887, found 488.0871.
7-N-Chloroacetyldemethyllavendamycin n-butyl ester (27)
Aldehyde 4 (37.9 mg, 0.14 mmol) in 83 mL of dry anisole was heated to 80 °C and while the mixture was heated, a solution of L-tryptophan n-butyl ester (15, 35.4 mg, 0.14 mmol) in 30 mL dry anisole was added dropwise over one hour at which time the solution temperature reached to 125 °C. The mixture was heated to 150 °C over a 3 hr period and then maintained at this temperature for 17 hrs. The yellow solution was roto-evaporated to dryness to give a lemon yellow solid (41.5 mg, 57%)): mp 225 °C (dec); Rf = 0.29 (0.2/5 MeOH/CH2Cl2); IR (KBr) 3400-3200 (s., br, H-bonded OH), 3335 (NH), 2959, 1705, 1681, 1655, 1530, 1310, 1257 (phenolic C-O), 744 cm-1; 1H NMR (DMSO-d6) Δ 1.02 (t, 3H, J = 7.3), 1.50–1.58 (m, 2H), 1.75–1..86 (m, 2H), 4.42 (t, 2H, J = 6.6), 4.55 (s, 2H), 7.41 (dd, 1H, J = 8.1, 7.0), 7.71 (dd, 1H, J = 8.1, 7.0), 7.85 (d, 1H, J = 8.1), 7.92 (br s, 1H), 8.53 (d, 1H, J = 8.1), 8.68 (d, 1H, J = 8.8), 8.76 (d, J = 8.8), 9.07 (s, 1H), 9.76 (br s, 1H, OH), 9.93 (br s, 1H, OH), 10.22 (s, 1H), 12.33 (br s, 1H). The last 4 NMR signals disappeared upon deuterium exchange; HRMS calculated for C27H24ClN4O5 (M+H)+ 519.1365, found 519.1317. Oxidation of hydroquinone 27b to the orange colored 27 was accomplished by allowing a solution of 27b in 7 mL acetone to stand in the presence of air overnight at room temperature. The orange solid was filtered, washed with a small amount of acetone, petroleum ether and dried under vacuum (46.3 mg, 64%): mp 247 °C (dec); Rf = 0.58 (MeOH/CH2Cl2 0.2/5); IR (KBr) 1335 (NH), 2958, 1713, 1681, 1515, 1308, 1118, 738 cm-1 (C-Cl); 1H NMR (DMSO-d6) Δ 1.02 (t, 3H, J = 7.3), 1.49–1.58 (m, 2H), 1.79–1.85 (m, 2H), 4.42 (t, 2H, J = 6.6), 4.54 (s, 2H), 7.43 (dd, 1H, J = 7.7, 7.0), 7.68 (dd, 1H, J = 7.7, 7.0), 7.73 (d, 1H, J = 7.7), 7.77 (s, 1H), 8.52 (d, 1H, J = 7.7), 8.57 (d, 1H, J = 8.1), 8.90 (d, 1H, J = 8.1), 9.06 (s, 1H), 10.59 (br s, 1H), 11.89 (br s, 1H); HRMS calculated for C27H21ClN4O5 516.1195, found 516.1173.
7-N-Chloroacetyldemethyllavendamycin n-octyl ester (28)
A mixture of aldehyde 4 (27.9 mg, 0.1 mmol), L-tryptophan n-octyl ester (16, 31.6 mg, 0.1 mmol) in 32 mL dried distilled xylene was slowly heated to 100° C over a period of 6 hrs and maintained at this temperature for 2.5 more hrs. The mixture was hot filtered to remove the solid impurities and the filtrate was evaporated to dryness under reduced pressure to give a dark red solid (30.1 mg, 53%). An analytical sample was obtained by recrystallization with ethyl acetate: mp 215–217 °C; Rf = 0.67 (1/1 EtOAc/CH2Cl2); 1H NMR (CDCl3) Δ 0.88 (t, 3H, J = 6.8), 1.23–1.70 (m, 10H), 1.84–1.95 (m, 2H), 4.29 (s, 2H), 4.48 (t, 2H, J = 6.8), 7.39 (dd, 1H, J = 7.6, 7.6), 7.66 (dd, 1H, J = 7.6, 7.6), 7.97 (s, 1H), 8.24 (d, 1H, J = 7.6), 8.56 (d, 1H, J = 8.4), 8.95 (s, 1H), 9.21 (d, 1H, J = 8.4), 9.58 (br s, 1H), 11.79 (br s, 1H); FAB-HRMS calculated for C35H40ClN4O7S2 [(MH+ + C4H10O2S2 (dithioerythritol; matrix)] 727.2026, found 727.2005.
7-N-Butyryldemethyllavendamycin methyl ester (29)
7-Butyramido-2-formylquinoline-5,8-dione (5, 54.5 mg, 0.2 mmol) was added to a solution of L-tryptophan methyl ester (17, 43.6 mg, 0.2 mmol) in 120 mL dry anisole. The reaction mixture was slowly heated to 130 °C over a 3 hr period and maintained at this temperature for 22 more hrs. The mixture was evaporated under reduced pressure to dryness and to the resulting solid, acetone (2 mL) was added and the solution was allowed to stand at room temperature for two days. The brown precipitate was filtered off (41.7 mg, 59%). The 1H NMR of this product showed it to be a mixture of 29 and its corresponding 5,8-dihydroxy derivative. This product, upon oxidation with DDQ (20.2 mg, 0.09 mmol) in 5 mL of acetone over 24 hrs, gave 30.7 mg of 29 in a 33% overall yield: mp > 276 °C; Rf = 0.39 (0.5/5 MeOH/CH2Cl2); 1H NMR (CDCl3) Δ 1.07 (t, 3H, J = 7.3), 1.78–1.86 (m, 2H), 2.54 (t, 2H, J = 7.3), 4.10 (s, 3H), 7.41 (dd, 1H, J = 7.7, 7.3), 7.66 (m, 1H), 7.77 (d, 1H, J = 7.7), 8.02 (s, 1H), 8.25 (d, 1H, J = 7.7), 8.43 (br s, 1H), 8.59 (d, 1H, J = 8.1), 9.00 (s, 1H), 9.22 (d, 1H, J = 8.1), 11.87 (br s, 1H); HRMS calculated for C26H20N4O5 468.1428, found 468.1425.
7-N-Butyryllavendamycin methyl ester (30)
A mixture of aldehyde 5 (27.2 mg, 0.1 mmol), (2RS,3SR)-β-methyltryptophan methyl ester (14, 23.2 mg, 0.1 mmol), and 30 mL dried and distilled xylene were slowly heated to 130 °C over a 3 hr period and then kept at this temperature for an additional 16 hrs. The mixture was filtered hot to remove the solid impurity and the filtrate was evaporated on a roto-evaporator to the point of crystallization. The mixture was allowed to cool and the orange-red product was filtered and dried (21 mg, 44%): mp 270–273 °C; Rf = 0.74 (1/1 EtOAc/CH2Cl2); 1H NMR (CDCl3) Δ 1.06 (t, 3H, J = 7.6), 1.75-1.90 (m, 2H), 2.52 (t, 2H, J = 7.6), 3.17 (s, 3H), 4.07 (s, 3H), 7.36 (dd, 1H, J = 8.0, 8.0), 7.58–7.72 (m, 2H), 7.92 (s, 1H),8.28–8.35 (m, 2H), 8.41 (d, 1H, J = 8.3), 9.01 (d, 1H, J = 8.3), 11.8 (s, 1H); HRMS calculated for C27H22N4O5 482.1590, found 482.1603.
7-N-butyryldemethyllavendamycin isoamyl ester (31)
Aldehyde 5 (27.9 mg,0.1 mmol), and L-tryptophan isoamyl ester (18, 27.4 mg, 0.1 mmol) in 30 mL of dried xylene were slowly heated to reflux over a 4 hr period and then refluxed for 17 hrs. The mixture was filtered hot to remove the solid impurity and the filtrate was concentrated under reduced pressure to about 2 mL. The solution was allowed to cool and the resulting orange-red product was filtered. The filtrate was evaporated to dryness affording more product (total weight 36.5 mg, 70%): mp 234–235 °C; Rf = 0.80 (1/1 EtOAc/CH2Cl2); 1H NMR (CDCl3) Δ 1.04 (d, 6H, J = 6.8), 1.06 (t, 3H, J = 7.4), 1.75–1.95 (m, 5H), 2.50–2.60 (m, 2H), 4.52 (t, 2H, J = 6.8), 7.39 (dd, 1H, J = 7.8, 7.8), 7.66 (dd, 1H, J = 7.8, 7.8), 7.74 (d, 1H, J = 7.8), 7.79 (s, 1H), 8.24 (d, 1H, J = 8.4), 8.41 (br s, 1H), 8.59(d, 1H, J = 8.4), 9.01 (d, 1H, J = 8.4), 11.9 (br s, 1H); HRMS calculated for C30H28N4O5 524.2059, found 524.2074.
7-N-Butyryldemethyllavendamycin n-octyl ester (32)
In a 50 mL three-necked round-bottomed flask, a mixture of aldehyde 5 (23 mg, 0.085 mmol), L-tryptophan n-octyl ester (16, 27 mg, 0.085 mmol) and 30 mL of dried xylene was slowly heated to 85 °C and maintained at this temperature for an additional 16 hrs. The reaction mixture was filtered hot to remove the impurities and the filtrate was roto-evaporated to dryness affording a dark-red product (38.6 mg, 80%), The crude product was recrystallized with ethyl acetate: mp 166–171 °C; Rf = 0.80 (1/1 EtOAc/CH2Cl2); 1H NMR (CDCl3) Δ 0.86 (t, 3H, J = 6.8), 1.06 (t, 3H, J = 7.3), 1.20–1.60 (m, 10H), 1.65–1.95 (m, 4H), 2.53 (t, 2H, J = 7.5), 4.47 (t, 2H, J = 6.8), 7.36 (dd, 1H, J = 8.0, 8.0), 7.63 (dd, 1H, J = 8.0, 8.0), 7.65 (d, 1H, J = 8.0), 7.93 (s, 1H), 8.19 (d, 1H, J = 8.0), 8.35 (br s, 1H), 8.44 (d, 1H, J = 8.4), 8.87 (s, 1H), 9.08 (d, 1H, J = 8.4), 11.67 (br s, 1H): HRMS calculated for C33H34N4O5 566.2529, found 566.2519.
7-N-Isobutyryldemethyllavendamycin methyl ester (33)
Aldehyde 6 (37.5 mg, 0.137 mmol), L- tryptophan methyl ester (17, 30 mg, 0.137 mmol) and 25 mL dry xylene in a 50 mL three-necked round-bottomed flask were stirred and slowly heated to 130 °C over a 3 hr period and then kept at this temperature for 4 more hrs. The yellow precipitate was filtered and TLC showed it to be nearly pure (40.5 mg, 63%). An analytical sample was obtained by recrystallization with 95% ethanol: mp 255 °C (dec); Rf = 0.50 (0.04/5 MeOH/CH2Cl2); 1H NMR (CDCl3) Δ 1.34 (d, 6H, J = 6.7), 2.70–2.80 (m, 1H), 4.10 (s, 3H), 7.41 (dd, 1H, J = 7.3, 7.3), 7.67–7.81 (m, 2H), 8.02 (s, 1H), 8.25 (d, 1H, J = 8.3), 8.50 (br s, 1H), 8.58 (d, 1H, J = 8.4), 9.00 (s, 1H), 9.20 (d, 1H, J = 8.4), 11.86 (br s, 1H); HRMS calculated for C26H20N4O5 468.1433, found 468.1423.
7-N-Isobutyryldemethyllavendamycin n-butyl ester (34)
A mixture of aldehyde 6 (100 mg, 0.37 mmol), L-tryptophan n- butyl ester (15, 95.7 mg, 0.37 mmol) and 60 mL of dry xylene was stirred and slowly heated to 125 °C over a 5 hr period and kept at this temperature for an additional 3 hrs. The small amount of solid impurity was filtered off and the filtrate was concentrated under reduced pressure to a volume of 4 mL and then allowed to cool to room temperature to give a brown solid (122.5 mg, 65%): mp 220–224 °C (dec); Rf = 0.78 (1/1 hexane/EtOAc); 1H NMR (CDCl3) Δ 1.04 (t, 3H, J = 7.4), 1.33 (d, 6H, J = 6.7), 1.80–2.00 (m, 4H), 2.65–2.80 (m, 1H), 4.49 (t, 2H, J = 6.5), 7.30–7.37 (m, 2H), 7.60–7.80 (m, 2H), 7.73 (dd, 1H, J = 7.3, 7.3), 7.99 (s, 1H), 8.24 (d, 1H, J = 8.0), 8.49 (d, 1H, J = 8.4), 8.55 (s, 1H), 8.94 (s, 1H), 9.18 (d, 1H, J = 8.4), 11.8 (br s, 1H); HRMS calculated for C29H26N4O5 510.1903, found 510.1887.
7-Isobutyramido-2-formylquinoline-5,8-dione (6)
In a 50 mL round-bottomed flask equipped with a magnetic bar, water-cooled condenser and an argon-filled balloon, a mixture of 7-isobutyramido-2-methylquinoline-5,8-dione48 (50, 503 mg, 1.95 mmol), selenium dioxide (217 mg, 1.92 mmol), dried and distilled dioxane (15 mL) and 0.25 mL of water was slowly heated to reflux over a period of 4 hrs. Thin layer chromatography showed the reaction to be completed after 20 more hrs of reflux. Selenium was filtered and the solid was added to 10 mL of dioxane, refluxed for 10 min and then filtered. The solid residue on the filter paper was washed again with 10 mL of dichloromethane. All the filtrates were combined, added to 50 mL of dichloromethane and was washed with a 3% sodium bicarbonate solution (3 × 50 mL). The solution was washed with brine, dried (magnesium sulfate) and evaporated under vacuo to give a bright yellow solid (400 mg, 75%). An analytical sample was obtained by silica gel column chromatography using CH2Cl2/EtOAC (10/1) as the eluent: mp183-184 °C; 1H NMR (CDCl3) Δ 1.29 (d, 1H, J = 7.0), 2.60–2.80 (m, 1H), 8.00 (s, 1H), 8.20 (d, 1H, J = 8.0), 8.35 (br s, 1H), 8.52 (d, 1H, J = 8.0), 10.2 (s, 1H); CIMS, m/z, (relative intensity) 272 (M+ 100), 217 (4.1), 203 (26.4); analysis for C14H12N2O4 calculated C, 61.76; H, 4.44; N, 10.29; found C, 61.83; H, 4.53; N, 10.08.
1-[tert-Butyldimethylsilyl)oxy]-3-methyl-1-aza-1,3-butadiene (65)
A solution of O-(tert-butyldimethylsilyl)hydroxylamine (2.42 g, 16.4 mmol) in dry CH2Cl2 (2.5 mL) was added to a mixture of freshly distilled methacrolein (1.05 g, 15 mmol) and 5.7 g of dry 4-Å molecular sieves in 10 mL of dry CH2Cl2. The mixture was stirred under Ar at room temperature for 48 hrs and then filtered. The filtrate was evaporated under reduced pressure. Vacuum distillation of the crude product (92–94° /3.5 mm-Hg) gave 2.1 g (70%) of 65 in an E/Z ratio of 4/1. Analytical samples of E and Z isomers were obtained by silica gel elution chromatography using CH2Cl2-pet. ether (1/3) as the solvent: (Isomer E); Rf = 0.55 (1/3 CHCl3/pet. ether); 1H NMR (CDCl3) (isomer E) Δ 0.18 (s, 6H), 0.94 (s, 9H), 1.90 (s, 3H), 5.17–5.22 (m, 1H), 5.3–5.35 (m, 1H), 7.85 (s, 1H); (isomer Z): Rf = 0.24 (CHCl3/pet. ether 1/3); 1H NMR (CDCl3) Δ 0.18 (s, 6H), 0.95 (s, 9H), 2.13 (s, 3H), 5.25–5.30 (m, 1H), 5.4–5.45 (m, 1H),7.06 (s, 1H); (Isomer E): MS, m/z, 199 (M+, 20), 184 (23), 142 (100), 128 (39), 115 (16), 102 (53); analysis for C10H21NOSi calculated C, 60.24; H, 10.62; N, 7.03; found C, 60.08, H, 10.70; N, 7.11; HRMS calculated for C20H21NOSi 199.1392 found 199.1372.
7-Acetamido-3-methylquinoline-5,8-dione (51)
Azadiene 6546 (4/1 E/Z mixture,127 mg, 0.64 mmol) and 2-bromo-6-acetamido-1,4-benzoquinone47 (63, 312 mg, 1.28 mmol) were dissolved in 20 mL of dry chlorobenzene and was heated at 120 °C for 44 hrs. The mixture was concentrated under reduced pressure to about 3 mL and then 15 mL of acetonitrile was added and concentrated to about 3 mL. Flash chromatography of this solution on a 0.5 in diameter column using acetonitrile as the solvent yielded 96 mg (66%) of pure 51: mp 278 °C (dec); Rf = 0.11 (EtOAc/CH2Cl2 1/1); 1H NMR (CDCl3) Δ 2.25 (s, 3H), 2.50 (s, 3H), 7.85 (s, 1H), 8.15 (d, 1H, J = 2.2), 8.36 (br s, 1H), 8.76 (d, 1H, J = 2.2); MS, m/z, 230 (M+, 49), 190 (69), 188 (84), 161 (82), 93 (30(, 79 (100); HRMS calculated for C12H10N2O3 230.0691 found 230.0675.
6-Acetamido-2-methylquinoline-5,8-dione (52)
A mixture of 2-bromo-5-acetamidobenzoquinone47 (64, 244 mg, 1 mmol), azadiene 6636 (7/3 E/Z mixture,100 mg, 0.5 mmol) in 22 mL dry chlorobenzene was refluxed under argon for 55 hrs. Chlorobenzene 20 mL was added and the reaction mixture was allowed to cool. This solution was added to a silica gel column (2 × 12.5 cm) and eluted with a 2/1 mixture of EtOAc/pet. ether and then with a 2.5/1 of the same solvent mixture to give 13 mg of pure 52. The column was eluted with 95% ethanol, the resulting solution was evaporated under vacuum to dryness. Benzene (10 mL) was added, the precipitate was filtered off. Evaporation of the filtrate gave another 10 mg of 52 (20% total yield); mp 177 °C (dec); Rf = 0.70 (1/1 MeOH/CHCl3); 1H NMR (CDCl3) Δ 2.27 (s, 3H), 2.78 (s, 3H), 7.50 (d, 1H, J = 8.0), 7.98 (s, 1H), 8.27 (br s, 1H), 8.29 (d, 1H, J = 8.0); HRMS calculated for C12H10N2O3 230.0691, found 230.0691.
7-Acetamidoquinoline-5,8-dione-2-carboxylic acid (53)
In a 50 mL round-bottomed flask equipped with a magnetic bar, 5 mL of glacial acetic acid was heated to 90 °C in an oil bath and to this, aldehyde 3 (488 mg, 2 mmol) was added. The mixture was stirred at this temperature for 15 min and then sodium perborate (1.54 g, 10 mmol) was added to the suspension in small portions over 30 min. An additional 5 mL of acetic acid was added, allowed to stir for an hour and then 10 mL of water was added. The resulting mixture was stirred for a few minutes. The precipitate was filtered to give a yellow solid and the filtrate was reduced to 1/3 of its original volume and then acidified with sulfuric acid to pH = 2, to produce the yellow crystals of 53. The solid was filtered off and added to the first filter cake and dried under vacuum (300 mg, 58%): mp 248 – 250 °C; Rf = 0.23 (95% EtOH); 1H NMR (DMSO-d6) Δ 2.26 (s, 3H), 7.78 (s, 1H), 8.37 (d, 1H, J = 7.2), 8.41 (br s, 1H), 8.47 (d, 1H, J = 7.2), 10.1 (s, 1H); HRMS calculated for C12H8N2O5 260.0428, found 260.0432.
7-Amino-2-formylquinoline-5,8-dione (55)
A mixture of 24.4 mg (0.1 mmol) of 7-acetamido-2-formylquinolie-5,8-dione (3), 5 mL of water and 8 mL of MeOH was flushed with a stream of argon. To this 12.7 mg of sodium bicarbonate was added and was stirred at room temperature. The red mixture was stirred for a total of 20 hrs. Water (10 mL) was added and the solution was extracted with dichloromethane (8 × 20 mL). The combined extracts were concentrated to 5 mL, added on a silica gel column (1.6 × 6 cm) and eluted with EtOAc/pet.ether 2.5/1 to give dione 55 as a red solid (6 mg, 30%): mp 204 °C (dec); Rf = 0.49 (1/10 MeOH/EtOAc) 1H NMR (CDCl3) Δ 5.41 (br s, 2H), 6.14 (s, 1H), 8.26 (d, 1H, J = 8.0), 8.59 (d, 1H, J = 8.0), 10.28 (br s, 1H); HRMS calculated for C10H6N2O3 202.0378, found 202.0373.
7-(4-Hydroxybutylamino)-2-methylquinoline-5,8-dione (57)
To a solution of 7-acetamido-2-methylquinoline-5,8-dione (47, 46 mg, 0.2 mmol) in 14 mL dry THF, concentrated sulfuric acid (1.4 mL) was added dropwise and then refluxed under Ar for 44 hrs. Thin layer chromatography of the reaction mixture showed the immediate generation of the aminodione 54 followed by its conversion to the product 57 as the reaction proceeded. The mixture was neutralized with a concentrated solution of sodium carbonate and then extracted with ethyl acetate (3 × 30 mL). The extracts were washed with brine, dried (MgSO4) and evaporated to give a dark solid. Silica gel flash chromatography of the solid using EtOAc/CH2Cl2 (2/2.5) gave 10 mg (19%) of 57: mp 137 °C (dec); Rf = 0.35 (3/2 EtOAc/CH2Cl2); 1H NMR (CDCl3) Δ 1.59-1.70 (m, 4H), 2.72 (s, 3H), 3.35-3.40 (m, 4H), 4.05 (broad s, 1H), 5.74 (s, 3H), 7.46 (d, 1H, J = 8.1), 8.27 (d, 1H, J = 8.1); HRMS calculated for C14H14N2O2 (M-H2O) 242.1050, found 242.1056
(2RS,3SR)-N-Carbobenzyloxy-β-methyltryptophan (72)
A mixture of β-methyltryptophan49 (4.36 g, 0.02 mol), NaOH (2N, 10 mL), water (20 mL), benzyl chloroformate (3.4 g, 0.02 mol in 2.2 mL of toluene) was stirred in an ice bath. To this, 5 mL of NaOH (4N) was added dropwise over the course of 20 min and then stirred for an additional 10 min. The reaction mixture was acidified to Congo Red with concentrated hydrochloric acid, filtered, washed with cold water, dried (MgSO4) and evaporated to give 6.37 g (93%) of 75: mp 153 °C (dec); Rf = 0.3 (EtOH); 1H NMR (DMSO-d6) Δ 1.32 (d, 3H, J = 7.0), 3.25–3.40 (m, 1H), 3.42–3.51 (m, 1H), 4.30–4.37 (m, 1H), 4.94–5.02 (m, 2H), 6.92–6.98 (m, 1H),7.02–7.14 (m, 2H), 7.18 (s, 1H), 7.23–7.28 (m, 2H), 7.28–7.37 (m, 3H), 7.60 (d, 1H, J = 8.1), 10.85 (br s, 1H); HRMS calculated for C20H20N2O4 352.1418, found 352.1416.
L-N-Carbobenzyloxyltryptophan N,N-dimethylaminoethyl ester (73)
A mixture of L-N- carbobenzyloxytryptophan (commercial 71, 3.384 g, 0.01 mol), 6 mL of a solution of N,N-dimethylaminoethyl chloride (2N in benzene) and 20 mL of EtOAc was stirred and heated. Triethylamine (1.4 mL) was added dropwise and the mixture was refluxed for 5 hrs and then filtered. The filtrate was washed with brine and then with a saturated solution of sodium bicarbonate followed by brine. The solution was dried (MgSO4) and evaporated under reduced pressure to give 3.85 g (92%) of a spongy gel; 1H NMR (CDCl3) Δ 2.15 (s, 6H), 2.42–2.46 (m, 2H), 3.20–3.42 (m, 2H), 4.05–4.15 (m, 2H), 4.60–4.74 (m, 1H), 5.09 (s, 2H), 5.40–5.46 (m, 1H), 6.95 (s, 1H), 7.04–7.08 (m, 1H), 7.13–7.18 (m, 1H), 7.30–7.36 (m, 6H), 7.49–7.55 (m, 1H), 8.22 (br s, 1H); HRMS calculated for C23H27N3O4 409.2001, found 409.2000.
(2RS,3SR)-N-Carbobenzyloxy-β-methyltryptophan N,N-dimethylamioethyl ester(74)
The procedure was the same as that used for the synthesis of 73. The product was obtained as a gel in 84% yield: Rf = 0.64 (EtOH/EtOAc 1/5); 1H NMR (CDCl3), Δ 1.45 (d, 3H, J = 7.3), 2.15 (s, 6H), 2.35–2.45 (m, 2H), 3.75–3.85 (m, 1H), 3.88–4.40 (m, 2H, diastereotopic), 4.65–4.72 (m, 1H), 5.09 (s, 2H), 5.28 (d, 1H, J = 8.8), 7.02 (s, 1H), 7.05 7.11 (m, 1H), 7.13– 7.18 (m, 1H), 7.28–7.38 (m, 6H), 7.60 (d, 1H, J = 8.06), 8.19 (br s, 1H); HRMS calculated for C24H29N3O4 423.2153, found 423.2153.
L-Tryptophan N,N-dimethylaminoethyl ester (10)
A mixture of dry ammonium formate (125 mg), compound 73 (205 mg, 0.5 mmol) and 10% Pd/C (100 mg), in dry DMF was stirred under argon at room temperature for 30 min and then filtered. The filtrate was roto-evaporated to give an oil. Chloroform (10 mL) was added and filtered and the filtrate was roto-evaporated to dryness and then dissolved in 25 ml ethyl acetate. The solution was washed with 3 × 25 mL brine, dried (MgSO4) and then evaporated to give an oily product (101 mg, 74%); Rf = 0.71 (EtOH); 1H NMR (CDCl3) Δ 2.23 (s, 6H), 2.45–2.50 (m, 2H), 3.02–3.07 (m, 1H), 3.20–3.29 (m, 1H), 3.80–3.84 (m, 1H), 4.14–4.18 (m, 2H), 7.05 (s, 1H), 7.07–7.12 (m, 1H), 7.13–7.20 (m, 1H), 7.30–7.38 (m, 1H), 7.54–7.61 (m, 1H), 8.25 (br s, 1H): HRMS calculated for C15H21N3O2 (M+H)+ 276.1713, found 276.1710.
(2RS,3SR)-β-Methyltryptophan N,N-dimethylaminoethyl ester (11)
Using compound 74 as the starting material, the aminoester 11 was prepared by the same procedure as that used for the preparation of tryptophan 10. Compound 11 was obtained as an oil in 67% yield: Rf = 0.12 (1/5 EtOH/ EtOAc); 1H NMR (CDCl3) Δ 1.45 (d, 3H, J = 7.0), 1.67 (br s, 2H), 2.24 (s, 6H), 2.45–2.49 (m, 2H), 3.52–3.58 (m, 1H), 3.68–3.75 (m, 1H), 4.02–4.10 (m, 1H), 4.12–4.19 (m, 1H), 7.04–7.10 (m, 1H), 7.10 (s, 1H), 7.12–7.20 (m, 1H), 7.34 (d, 1H, J = 8.0), 7.61 (d, 1H, J = 8.0), 8.17 (br s, 1H); HRMS calculated for C16H23N3O2 (M + H)+ 290.1869, found 290.1865.
DL-5-Methoxytryptophan n-butyl ester (12)
A solution of DL-5-methoxytryptophan (100 mg, 0.43 mmol), n-butyl alcohol (6 mL) and 1 mL of ether solution of dry HCl (1 M) was stirred and refluxed for 20 hrs. The reaction mixture was roto-evaporated to dryness and the white solid salt was neutralized with a 14% solution of ammonia. Water (5 mL) was added and it was extracted with 2 × 7 mL EtOAc. The combined extracts were washed with brine (2 × 4 mL), dried (Mg SO4) and roto-evaporated to give 85 mg (68%) of a brown oil: Rf = 0.45 (EtOAc); 1H NMR (DMSO-d6) Δ 0.80–0.84 (m, 3H), 1.15–1.24 (m, 2H), 1.38–1.48 (m, 2H), 2.89–3.00 (m, 2H), 3.61–3.65 (m, 1H), 3.75 (s, 3H), 3.93–3.98 (m, 2H), 6.70 (dd,1H, J = 8.8, 2.2), 6.96 (d, 1H, J = 2.2), 7.06 (d, 1H, J = 2.2), 7.21 (d, 1H, J = 8.8), 10.69 (br s, 1H); HRMS calculated for C16H22N2O3 290.1630, found 290.1618.
DL–7-Fluorotryptophan n-butyl ester (13)
This ester was prepared using a similar method to that used for the synthesis of 12 giving white crystals of 13 in 88% yield: mp 83.5–84 °C; Rf = 0.22 (1/100 MeOH/CH2Cl2);); 1H NMR (CDCl3) Δ 0.91 (t, 3H, J = 7.0), 1.25–1.33 (m, 2H), 1.50–1.56 (m, 4H), 2.86–3.20 (m, 1H), 3.15–3.30 (m, 1H), 3.79 (br s, 1H), 4.00–4.20 (m, 2H), 6.85–7.00 (m, 1H), 7.10– 7.20(m, 1H), 7.26 (s, 1H), 7.28 (s, 1H), 8.09 (br s, 1H); HRMS calculated for C15H19N2O2F 278.1430, found 278.1414.
Cell Lines and Culture Conditions
The in vitro cytotoxicity of the N-acyllavendamycin analogues was determined against a panel of five cell types according to the methods described below. Briefly, the stock cultures of normal rat kidney epithelial (NRK-52E) cells and Lewis lung carcinoma cells (3LL) were obtained from the American Type Culture Collection (ATCC, Rockville, MD).70,71 Both cell lines were grown in antibiotic- free culture Dulbecco’s high glucose MEM medium supplemented with 10% fetal bovine serum. The cell number of the 3LL and NRK cell lines was expanded by five passages and the cell lines were stored in liquid nitrogen, all according to standard procedures.72 These cryo-preserved cells were used for all in vitro assays and were grown according to standard cell culture methods as detailed by Freshney72 Any cell culture manipulations were done under gold fluorescent light to prevent damage by photooxidation. Ras oncogene-transformed NRK cells were prepared using rasK, rasN, and rasH oncogenes.
Selection of Transformed Cells and Oncogene Transfections
The plasmids pUCEJ6.6 (no. 41028), containing a transforming human rasH gene, pNRsac (no. 4103) containing a transforming human rasN gene, pKSma (no. 41048), containing a v-rasK gene, and the RSVneo gene were obtained from the ATCC. NRK cells were cotransfected with an oncogene plus the RSVneo gene using standard calcium phosphate coprecipitation and neomycin resistance selection methods.73 Approximately 14 days after transfection and selection with G418 antibiotic ( Sigma Chemical Co., St. Louis, MO) colonies were isolated by the ring cloning procedure.72,74 The clones were stored in liquid nitrogen after expansion of their cell number by four passages in culture medium with G418. Approximately 50 clones were evaluated by several criteria for their use in the in vitro cytotoxicity and in vivo antitumor tests described below. The criteria used to select the clones were the following: (1) the clone had to to have the same growth rate as the parental NRK cells, (2) the growth rate of the clone must be stable with repeated passage ( e.g. up to 100 subcultures), and (3) they must be tumorigenic in immunologically deficient mice. Transformants with this phenotype have a low oncogene copy number and in vitro growth characteristics the same as the nontransformed NRK cells. These criteria were chosen so that compounds were evaluated for antitumor activity, rather than anti-growth activity based on the rate of cell division. Oncogene copy number was determined using standard southern blotting procedures.75 Cell growth characteristics were evaluated using standard methods.72,74 The clones K/1-NRK, transformed by v-rasK gene, H/1.2-NRK, transformed by the human rasH, and N/4.2- NRK transformed by the human rasN gene, were selected for in vitro cytotoxicity and in vivo antitumor testing. These clones have 3 to 4 oncogene copies that are stably integrated. The doubling time of these cells is 24 hrs, which is the same as NRK cells. Adenocarcinomas are produced in female CD1 nu/nu mice approximately 3 days after the sc implantation of 1 × 106 cells. These tumors grow with a doubling time of 24 hrs. In contrast, no tumors are produced in nude mice up to one year after the sc implantation of 1 × 108 parent NRK epithelial cells.
In Vitro Cytotoxicity Assay
Compounds were screened for antitumor activity with an oncogene-based differential cytotoxicity assay. With this assay the cytotoxic action of compounds against oncogene-transformed cells relative to their action against the nontransformed parent epithelial cells was determined. For the evaluation of compounds, the Lewis Lung carcinoma was also used as a murine reference tumor. Briefly, the procedure used for the differential cytotoxicity assay is as follows. Cell suspensions were prepared by trypsin dissociation using standard methods and 50 cells were seeded into each well of a 12-well culture dishes and incubated at 37 °C in 5% CO2.72,74 Groups of triplicate wells were divided into media control, drug vehicle-control and drug treatment groups. One day after seeding, media was replaced with media containing vehicle or drugs at various concentrations ranging from 0.01 to 33 μM. The drugs were dissolved first in dimethyl sulfoxide and were then diluted in warmed culture media before adding to the cells. The cultures were then incubated for an additional 5 days. After exposure to the vehicles or drugs, the cultures were washed, fixed and stained with a modification of Mallory’s stain.76 Colony number were determined with an Artek model 982 image analyzer (Artek System Corp., Farmington, N.Y.). The cytocidal action of the compounds was determined from the colony number as originally described by Puck and Marcus.77 The concentration giving 50% cell kill (LC50) was determined, and a differential index of cytotoxicty was determined by dividing the LC50 value for the normal epithelial cells by the LC50 value for the tumor cells. All data analysis was performed using SAS software.( SAS Institute Inc., Cary, N.C.)
Tumor Bearing Mice
Several N-acyllavendamycin analogs were evaluated against K/1-NRK tumors grown as a xenograft in nude mice. Briefly, female CD1 nu/nu nude and female C57Bl/6 mice were obtained from Charles Rivers, Inc. Animals were housed in plastic shoebox type cages covered with microisolator tops. All animal feed, water, bedding and cages were sterilized before use. The animals were provided water (pH 3.0) and Purina mouse diet ad libitum. All animal manipulations were done with sterile procedures in a 10% exhaust, vertical laminar flow, HEPA filtered hood. To maintain consistent tumor growth, only mice between 4-6 weeks of age were used. Transformed cells obtained by oncogene transfection of NRKE cells as described above were grown in vitro through passage 7 were used to establish xenograft tumors in the female CD 1 nu/nu mice.78 Tumors were initiated by the subcutaneous implantation of 1 × 106 cells approximately 1 cm from the first mammary gland. After implantation, the mice were randomized and divided into treatment groups of 7 mice per group. Starting one day after tumor implantation, the mice were dosed ip with drug or vehicle daily for 8 days as described below. On day 10, tumor mass was determined as described by Tomayko and Reynolds (1989) with the following ellipsoid volume equation: mg tumor = 1/2 (length × width × height).79 Percent inhibition of tumor growth was calculated from the ratio of the tumor mass for the drug treated animals relative to the mass for the vehicle treated animals. All animals were weighed at the beginning and end of tumor growth to determine if inhibition of tumor growth was due to weight loss.
Drug Treatment
Cyclophosphamide was dissolved in isotonic saline and was used as a positive control for treatment of the tumor bearing animals. Lavendamycin analogues, 41 and 42 were dissolved in corn oil and 44 was suspended in 10% emulphor 620 in Dulbecco’s phosphate buffered saline. Starting one day after tumor implantation cyclophosphamide and analogues 41 and 42 were given once daily for 8 days and compound 44 was administered in three doses per day at lower dose levels for 7 days.
Acknowledgments
The financial support given by the National Institutes of Health, American Cancer Society and Eli Lilly and Company is greatly appreciated. We also thank Professor David Williams and staff at the mass spectroscopy laboratory of Indiana University for their kind assistance in obtaining mass spectral data.
Footnotes
mbehforo@bsu.edu
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bar-Sagi D, Hall A. Cell. 2000;103:227. doi: 10.1016/s0092-8674(00)00115-x. [DOI] [PubMed] [Google Scholar]
- 2.Crespo P, Leon J. Cell Mol Life Science. 2000;57:1613. doi: 10.1007/PL00000645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khosravi FR, Der CJ. Cancer and Metastasis Rev. 1994;13:67. doi: 10.1007/BF00690419. [DOI] [PubMed] [Google Scholar]
- 4.Barbacid M. Ann Rev Biochem. 1987;56:779–827. doi: 10.1146/annurev.bi.56.070187.004023. [DOI] [PubMed] [Google Scholar]
- 5.Cox AD, Der CJ. Curr Opin Cell Biol. 1992;4:1008. doi: 10.1016/0955-0674(92)90133-w. [DOI] [PubMed] [Google Scholar]
- 6.Bos JL. Cancer Res. 1989;49:4682. [PubMed] [Google Scholar]
- 7.Barbacid M. Eur J Clin Invest. 1990;20:225. doi: 10.1111/j.1365-2362.1990.tb01848.x. [DOI] [PubMed] [Google Scholar]
- 8.Lowy DR, Williamson BM. Ann Rev Biochem. 1993;62:851. doi: 10.1146/annurev.bi.62.070193.004223. [DOI] [PubMed] [Google Scholar]
- 9.Hancock JF. Nat Rev Mol Cell Biol. 2003;4:373. doi: 10.1038/nrm1105. [DOI] [PubMed] [Google Scholar]
- 10.Rodenhuis S. Semin Cancer Biol. 1992;3:241. [PubMed] [Google Scholar]
- 11.Shields JM, Pruitt K, McFall A, Schaub A, Der CJ. Trends Cell Biol. 2000;10:147. doi: 10.1016/s0962-8924(00)01740-2. [DOI] [PubMed] [Google Scholar]
- 12.Tabin CJ, Bradley SM, Bargman CI, Weinberg RA, Papageorge AG, Scolnick EM, Dhar R, Lowy DR, Chang EH. Nature. 1982;300:143. doi: 10.1038/300143a0. [DOI] [PubMed] [Google Scholar]
- 13.Reddy EP, Reynolds RK, Santos E, Barbacid M. Nature. 1982;300:149. doi: 10.1038/300149a0. [DOI] [PubMed] [Google Scholar]
- 14.Taparowsky E, Suard Y, Fasano O, Shimizu K, Goldfarb M, Wigler M. Nature. 1982;300:762. doi: 10.1038/300762a0. [DOI] [PubMed] [Google Scholar]
- 15.Leon J, Guerrero I, Pellicer A. Mol Cell Biol. 1987;7:1535. doi: 10.1128/mcb.7.4.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fiorucci G, Hall A. Biochem Biophys Acta. 1986;950:81. doi: 10.1016/0167-4781(88)90076-0. [DOI] [PubMed] [Google Scholar]
- 17.Suarez HG, duVillard JA, Severino M, Caillou B, Schlumberger M, Tubiana M, Parmentier C, Monier R. Oncogene. 1990;5:565. [PubMed] [Google Scholar]
- 18.Malumbres M, Pellicer A. Front Biosci. 1998;3:887. doi: 10.2741/a331. [DOI] [PubMed] [Google Scholar]
- 19.Malallanas D, Arozarena I, Berciano M, Aaronson D, Pellicer A, Lafarga M, Crespo P. J Biol Chem. 2003;278:4572. doi: 10.1074/jbc.M209807200. [DOI] [PubMed] [Google Scholar]
- 20.Esteban LM, Vicario-Arbejon C, Fernandez-Salguero P, Fernadez-Melarde A, Swaminathan N, Yienger K, Lopez E, Mckay R, Ward JM, Pellicer A, Santos E. Mol Cell Biol. 2001;21:1444. doi: 10.1128/MCB.21.5.1444-1452.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson L, Greenbaum D, Cichowski K, Mercer K, Murphy E, Schmill E, Bronson MT, Umanoff H, Edelman W, Kuchelapati R, Jacks T. Genes Dev. 1997;11:2468. doi: 10.1101/gad.11.19.2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koera K, Nakamura K, Nakao K, Miyoshi J, Toyoshima K, Hatta T, Otani H, Aiba A, Katsuki M. Oncogene. 1997;15:1151. doi: 10.1038/sj.onc.1201284. [DOI] [PubMed] [Google Scholar]
- 23.Prior IA, Harding A, Yan J, Sluimer J, Parton RG, Hancock JF. Nat Cell Biol. 2001;3:368. doi: 10.1038/35070050. [DOI] [PubMed] [Google Scholar]
- 24.Voice JK, Klemke RL, Le A, Jackson J. J Biol Chem. 1999;274:17164. doi: 10.1074/jbc.274.24.17164. [DOI] [PubMed] [Google Scholar]
- 25.Ehrhardt A, Ehrhardt GR, Guo X, Schrader JW. Exp Hematol. 2002;30:1089. doi: 10.1016/s0301-472x(02)00904-9. [DOI] [PubMed] [Google Scholar]
- 26.Walsh AB, Bar-Sagi D. J Biol Chem. 2001;276:15609. doi: 10.1074/jbc.M010573200. [DOI] [PubMed] [Google Scholar]
- 27.Downward J. Nat Rev Cancer. 2003;3:11. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 28.Doyle TW, Balitz DM, Grulich RE, Nettleton DE, Gould SJ, Tann C-h, Meows AE. Tetrahedron Lett. 1981;22:4595. [Google Scholar]
- 29.Balitz DM, Bush JA, Bradner WT, Doyle TW, O'Herron FA, Nettleton DE. J Antibiotic. 1982;35:259. doi: 10.7164/antibiotics.35.259. [DOI] [PubMed] [Google Scholar]
- 30.Erickson WR, Gould SJ. J Am Chem Soc. 1985;107:5831. [Google Scholar]
- 31.Erickson WR, Gould SJ. J Am Chem Soc. 1987;109:620. [Google Scholar]
- 32.Rao KV, Biemann K, Woodward RBJ. Am Chem Soc. 1963;85:2532. [Google Scholar]
- 33.Hackethal CA, Golbey RB, Tan CTC, Karnofsky DA, Burchenal JH. Antibiot Chemother. 1961;11:178. [PubMed] [Google Scholar]
- 34.Boger DL, Mitscher YM, Drake SD, Kitos PA, Thompson SC. J Med Chem. 1987;30:1918. doi: 10.1021/jm00393a040. [DOI] [PubMed] [Google Scholar]
- 35.Wilson WL, Labra C, Barrist E. Antibiot Chemother. 1961;11:147. [PubMed] [Google Scholar]
- 36.Behforouz M, Gu Z, Cai W, Horn MA, Ahmadian M. J Org Chem. 1993;58:7089. [Google Scholar]
- 37.Behforouz M, Haddad J, Cai W, Arnold MB, Mohammadi F, Sousa AC, Horn MA. J Org Chem. 1996;61:6552. doi: 10.1021/jo960794t. [DOI] [PubMed] [Google Scholar]
- 38.Kende AS, Ebetino FH. Tetrahedron Lett. 1984;25:923. [Google Scholar]
- 39.Boger DL, Duff SR, Panek JS, Yasuda M. J Org Chem. 1985;50:5790. [Google Scholar]
- 40.Behforouz M, Cai W, Stocksdale MG, Lucas JS, Jung JY, Briere D, Wang A, Katen KS, Behforouz NC. J Med Chem. 2003;46:5773. doi: 10.1021/jm0304414. [DOI] [PubMed] [Google Scholar]
- 41.Fang Y, Linardic CM, Richardson DA, Cai W, Behforouz M, Abraham RT. Molecular Cancer Therapeut. 2003;2:517. [PubMed] [Google Scholar]
- 42.Hassani M, Cai W, Holley DC, Lineswala JP, Maharjan BR, Ebrahimian GR, Seradj H, Stocksdale MG, Mohammadi F, Marvin CC, Gerdes JM, Beall HD, Behforouz M. J Med Chem. 2005;48:7733. doi: 10.1021/jm050758z. [DOI] [PubMed] [Google Scholar]
- 43.Seradj H, Cai W, Erasga NO, Chenault DV, Knuckles KA, Ragains JR, Behforouz M. Org Lett. 2004;6:473. doi: 10.1021/ol035381a. [DOI] [PubMed] [Google Scholar]
- 44.Cox ED, Cook JM. Chem Rev. 1995;95:1797. [Google Scholar]
- 45.Behforouz M, West SJ, Chakrabarty C, Rusk DA. 1992;34:483. [Google Scholar]
- 46.Behforouz M, Gu Z, Ahmadian M, Stelzer LS, Haddad J, Scherschel JA. Tetrahedron Lett. 1997;38:2211. [Google Scholar]
- 47.Kelly TR, Echavarren A, Behforouz M. J Org Chem. 1983;48:3849. [Google Scholar]
- 48.Behforouz M, Haddad J, Cai W, Gu Z. J Org Chem. 1998;63:343. [Google Scholar]
- 49.Snyder HR, Matteson DS. J Amer Chem Soc. 1957;79:2217. [Google Scholar]
- 50.Hall LAR, Stephens VC, Burckhalter JH. Org Synth Coll. 1963;4:333. [Google Scholar]
- 51.Kanaoka M, Kimura M. J Pharm Soc Japan. 1975;95:231. doi: 10.1248/yakushi1947.95.2_231. [DOI] [PubMed] [Google Scholar]
- 52.Kanaoka M, Ishida T, Kikuchi T. Chem Pharm Bull. 1978;26:605. [Google Scholar]
- 53.Behforouz M, Zarrinmayeh H, Ogle ME, Riehle TJ, Bell FW. J Heterocycl Chem. 1988;25:1627. [Google Scholar]
- 54.Lown JW. Mol Cell Biochem. 1983;55:17. doi: 10.1007/BF00229240. [DOI] [PubMed] [Google Scholar]
- 55.Bhuyan BK. Antibiotics I. In: Gottlieb ID, Shaw PD, editors. Mechanism of Action. Springer- Verlag; New York: 1967. pp. 173–180. [Google Scholar]
- 56.Cone R, Hasan SK, Lown JW, Morgan AR. Can J Biochem. 1976;54:219. doi: 10.1139/o76-034. [DOI] [PubMed] [Google Scholar]
- 57.Lown JW, Sim SK. Can J Biochem. 1976;54:446. doi: 10.1139/o76-064. [DOI] [PubMed] [Google Scholar]
- 58.Bachur NR, Gordon SL, Gee MV, Kon H. Proc Natl Acad Sci USA. 1979;76:954. doi: 10.1073/pnas.76.2.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hajdu J, Armstrong EC. J Am Chem Soc. 1981;103:232. [Google Scholar]
- 60.Harding MM, Lay PA. J Inorg Biochem. 2004;98:720. doi: 10.1016/j.jinorgbio.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 61.Herlt AJ, Rickards RW, Wu JP. J Antibiot. 1985;38:516. doi: 10.7164/antibiotics.38.516. [DOI] [PubMed] [Google Scholar]
- 62.Rao KV. Cancer Chemother Rep Part 2. 1974;4:11. [PubMed] [Google Scholar]
- 63.Inouye Y, Okada H, Roy SK, Miyasaka T, Hibino S, Tanaka N, Nakamura S. J Antibiot. 1985;38:1429. doi: 10.7164/antibiotics.38.1429. [DOI] [PubMed] [Google Scholar]
- 64.Hodnett EM, Wongwiechintana C, Dunn WJ, III, Marrs P. J Med Chem. 1983;26:570. doi: 10.1021/jm00358a021. [DOI] [PubMed] [Google Scholar]
- 65.Shaikh IA, Johnson F, Grollman AP. J Med Chem. 1986;29:1329. doi: 10.1021/jm00158a002. [DOI] [PubMed] [Google Scholar]
- 66.Reilly HC, Sugiura K. Antibiot Chemother. 1961;11:174. [PubMed] [Google Scholar]
- 67.Oleson JJ, Calderella LA, Mjos KJ, Reith AR, Thie RS, Toplin I. Antibiot Chemother. 1961;11:158. [PubMed] [Google Scholar]
- 68.Sizemore N, Cox AD, Barnard JA, Oldham SM, Reynolds ER, Der CJ, Coffey RJ. Gastroenterology. 1999;117:567. doi: 10.1016/s0016-5085(99)70449-x. [DOI] [PubMed] [Google Scholar]
- 69.Elad-Sfadia G, Haklai R, Balan E, Klog Y. J Biol Chem. 2004;279:34922. doi: 10.1074/jbc.M312697200. [DOI] [PubMed] [Google Scholar]
- 70.De Larco JE, Todaro GJ. J Cell Physiol. 1978;94:335. doi: 10.1002/jcp.1040940311. [DOI] [PubMed] [Google Scholar]
- 71.Suguira K, Stock CC. Cancer (Phil) 1952;5:382. doi: 10.1002/1097-0142(195203)5:2<382::aid-cncr2820050229>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 72.Freshney RI. Culture of Animal Cells: A Manual of Basic Technique. 2. Wiley-Liss; New York: 1987. pp. 6–32. [Google Scholar]
- 73.Davis LG, Kuehl WM, Battey J. In Basic Methods in Molecular Biology. Appleton and Lange; Norwalk, Conn: 1994. pp. 624–629. [Google Scholar]
- 74.Phelps PC, Best CJ, Berezeskky IK, Merriman RL, Tanzer LR, Boder GB, Trump BF. Cancer Lett. 1995;97:7. doi: 10.1016/0304-3835(95)03942-p. [DOI] [PubMed] [Google Scholar]
- 75.Selden RF. In: Southern Blotting, Section IV In Current Protocols in Molecular Biology. Ausubel F, Kingston R, Moore D, Seidman J, Smith J, Struhl K, editors. John Wiley and Sons; New York: 1987–1992. pp. 2.9.1–2.9.10. [Google Scholar]
- 76.Richardson KC, Jarett L, Finke EH. Stain Technology. 1960;35:313. doi: 10.3109/10520296009114754. [DOI] [PubMed] [Google Scholar]
- 77.Puck TT, Marcus PI. JExpMed. 1956;103:653. doi: 10.1084/jem.103.5.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Best CJ, Tanzer LR, Phelps PC, Merriman RL, Boder GG, Trump BF, Elliget KA. In Vitro Cell Dev Biol Anim. 1999;35:205. doi: 10.1007/s11626-999-0028-2. [DOI] [PubMed] [Google Scholar]
- 79.Tomayko M, Reynolds CP. Cancer Chemother Pharmacol. 1989;24:148. doi: 10.1007/BF00300234. [DOI] [PubMed] [Google Scholar]