

Abstract
Hepatitis B virus (HBV) contains a small, partially double-stranded, relaxed circular (RC) DNA genome. RC DNA needs to be converted to covalently closed circular (CCC) DNA, which serves as the template for all viral RNA transcription. As a first step toward understanding how CCC DNA is formed, we analyzed the viral and host factors that may be involved in CCC DNA formation, using transient and stable DNA transfections of HBV and the related avian hepadnavirus, duck hepatitis B virus (DHBV). Our results show that HBV CCC DNA formed in hepatoma cells was derived predominantly from RC DNA with a precise junction sequence. In contrast to that of DHBV, HBV CCC DNA formation in cultured cells was accompanied by the accumulation of a RC DNA species from which the covalently attached viral reverse transcriptase (RT) protein was removed (protein-free or PF-RC DNA). Furthermore, whereas envelope deficiency led to increased CCC DNA formation in DHBV, it resulted mainly in increased PF-RC, but not CCC, DNA in HBV, suggesting that the envelope protein(s) may negatively regulate a step in CCC DNA formation that precedes deproteination in both HBV and DHBV. Interestingly, PF-RC DNA, in contrast to RT-linked RC DNA, contained, almost exclusively, mature plus-strand DNA, suggesting that the RT protein was removed preferentially from mature RC DNA.
Hepatitis B virus (HBV) infection is a global public health problem, with over 300 million chronically infected patients worldwide (16). Chronic HBV infection is associated with a high risk of developing severe liver diseases, including cirrhosis and hepatocellular carcinoma, and results in a million deaths annually. HBV is a member of the Hepadnaviridae, a family of small, hepatotropic DNA viruses (37), which also includes related animal viruses, such as duck hepatitis B virus (DHBV) (41) and woodchuck hepatitis virus (WHV) (63). All hepadnaviruses carry a small (ca. 3.2 kb), relaxed circular (RC), partially double-stranded DNA genome (Fig. 1) and replicate through an RNA intermediate, the pregenomic RNA (pgRNA), by reverse transcription (41, 51, 54, 59, 60).
FIG. 1.
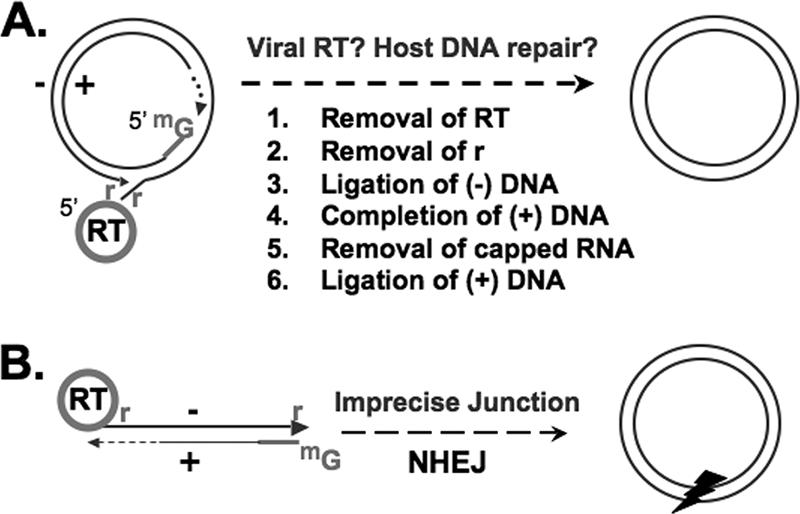
Conversion of RC or DSL DNA to CCC DNA. (A) Structure of genomic RC DNA, which accounts for ca. 90% of reverse transcription products. Shown are the RT protein covalently attached to the 5′ end of the minus-strand DNA [(−) DNA], the r of the minus-strand DNA, and the capped RNA oligomer (denoted by methyl guanosine [mG]) attached to the 5′ end of the incomplete (gapped) plus-strand DNA [(+) DNA], with heterogeneous 3′ ends. Conversion of RC DNA to CCC DNA is a multistep process, as shown. (B) DSL DNA, with ends modified similar to those of RC DNA, except that the 5′ ends of the two strands are not annealed. DSL DNA accounts for ca. 10% of genomic DNA and is thought to be converted to CCC DNA via NHEJ, an imprecise process that results in deletions/insertions at the junction, denoted by an arrowhead.
A critical early step in the HBV infection process is the conversion of the genomic RC DNA, present in the virions, into a covalently closed circular (CCC) DNA, which is found in the host cell nucleus (26, 39, 47, 65). Following entry into the host cells, the virion RC DNA is released into the nucleus for conversion into CCC DNA, which is the first viral product to be made in a newly infected cell. CCC DNA then serves as the viral transcriptional template for the synthesis of all viral RNAs by the host RNA polymerase II. Among the viral RNAs produced is the pgRNA, which is packaged together with the reverse transcriptase (RT) protein into immature nucleocapsids comprised of the viral capsid or core protein (4, 23). The pgRNA is then reverse transcribed within the nucleocapsids to make RC DNA by the multifunctional RT (27), using a reverse transcription pathway that is similar to, yet distinct from, that used by conventional retroviruses (17, 54, 55). A unique feature of hepadnavirus reverse transcription is the RT protein-primed initiation of minus-strand DNA synthesis, which leads to the covalent linkage of the RT protein to the 5′ end of the viral minus-strand DNA (19, 66, 68, 81). The mature, RC DNA-containing nucleocapsids are then enveloped by the viral envelope proteins and secreted extracellularly as virions. Alternatively, RC DNA can be transported directly, without an extracellular (virion) phase, to the nucleus to be converted to more CCC DNA via an intracellular amplification pathway (48, 65, 71). The copy number of CCC DNA per hepatocyte in an infected animal has been estimated to be from 5 to 50 copies per nucleus (35, 71, 77). It is clear that CCC DNA does not undergo semiconservative replication and can be generated only through the product of reverse transcription, RC DNA (65).
Though not integrated into the host chromosomes, the episomal HBV CCC DNA functions equivalently to that of the integrated retroviral provirus and is the molecular basis of HBV persistence. While current antiviral chemotherapy, which inhibits the polymerase activity of the RT, can effectively suppress HBV reverse transcription, it has no direct effect on CCC DNA. In both animal model studies and clinical investigations, it has been found that CCC DNA persists even after years of antiviral therapy and is responsible for the rapid rebound of viral replication upon withdrawal of treatment (33, 34, 38, 43, 80). Although there is no doubt that elimination of CCC DNA is essential to clear a chronic HBV infection, little is currently known about the formation of this critical viral DNA species.
In principle, the conversion of RC DNA to CCC DNA entails the removal of the RT protein from the 5′ end of the minus-strand DNA; the removal of a short RNA oligomer, which is used to prime plus-strand DNA synthesis, from the 5′ end of the plus-strand DNA; the removal of precisely one copy of a short terminal redundancy (r) from the minus-strand DNA; the completion of the plus-strand DNA that is variable in length in RC DNA; and the ligation of both DNA strands (Fig. 1). One reason so little is known about CCC DNA formation is the current lack of detectable intermediates in this complex conversion process.
In addition to RC DNA, all hepadnaviruses synthesize a smaller amount of linear DNA, the so-called double-stranded linear (DSL) form (36, 57). It has been shown that DSL DNA can also be converted to CCC DNA in DHBV and WHV, apparently via intramolecular ligation of the linear DNA ends (probably via nonhomologous end joining [NHEJ]) (Fig. 1) (74, 75). This process is called illegitimate replication, since the junction sequence of the resulting CCC DNA invariably harbors small deletions and/or insertions, mostly within and near the terminal r sequence, thus precluding faithful viral DNA replication. In contrast, the conversion from RC to CCC DNA is a highly accurate and efficient process; a single RC DNA molecule from the infecting virion is able to establish a productive infection (28, 74).
Very little is known about the viral and host factors that may be involved in CCC DNA formation. The only viral protein that is known to affect CCC formation is the viral large surface (LS) protein of DHBV (31, 61, 62), one of two (three in HBV) viral envelope proteins. The DHBV LS protein appears to suppress CCC DNA amplification, using a negative-feedback mechanism. When the CCC level is low, viral transcription and protein expression (including that of LS) are low, and the mature RC DNA is allowed to shuttle to the nucleus to be converted to CCC, increasing the CCC DNA pool size. When the nuclear CCC levels reach 10 to 50 copies, sufficient amounts of LS protein are made to effectively shut off the CCC amplification pathway and redirect the mature nucleocapsids (containing RC DNA) for envelopment and extracellular secretion. Thus, deletion or subtle substitution mutations of the DHBV LS protein increase the CCC copy number per cell more than 10-fold. In contrast, elimination of all HBV surface proteins does not appear to increase HBV CCC DNA in human hepatoma cells (32) or primary human hepatocytes in vitro (56). It remains unclear if this reflects a fundamental difference in the regulation of CCC DNA amplification between HBV and DHBV or a limitation of the experimental systems used, particularly since the levels of HBV CCC DNA detected in the cell cultures were very low.
Similarly, virtually nothing is known about potential host factors involved in CCC formation. No specific host factors that participate directly in CCC formation have been identified. The observation that the livers of HBV-transgenic mice failed to accumulate detectable CCC DNA, despite the presence of high levels of viral DNA (core DNA), including RC DNA, in nucleocapsids, led to the suggestion that CCC formation may require species-specific host factors (20). However, knocking out a liver-enriched transcriptional factor, HNF1α, resulted in detectable, albeit still very low, levels of CCC DNA in transgenic mouse liver (49), suggesting that the physiological state of the host hepatocytes may somehow modulate CCC DNA formation. Also, DHBV CCC DNA formation appears to be more efficient in young ducklings than in older ducks (76), again implicating developmental or adaptive host factors in CCC DNA formation.
In order to analyze the pathway of RC DNA to CCC DNA conversion and the potential viral and host factors involved, we analyzed the accumulation of HBV and DHBV DNA species derived from the RT-linked RC DNA, including CCC DNA and a form of RC DNA that is no longer covalently attached to the RT protein (protein free [PF]). The CCC, as well as the PF-RC, DNA species were examined in the presence or absence of the viral envelope proteins in transiently and stably transfected cells in culture. This study revealed potential pathways of CCC DNA formation and the influence of viral and host factors on the conversion process.
MATERIALS AND METHODS
Cell cultures.
The human hepatoma cell line HepG-2, the chicken hepatoma cell line LMH, and the human embryonic kidney cell line HEK293 were maintained as previously described (25, 67). HepAD38 cells, HepG-2 derivatives that express HBV pgRNA under the control of a tetracycline (Tet)-repressible promoter (30), and Dstet5 cells, LMH derivatives that express an envelope-deficient DHBV pgRNA also under Tet control (22), were maintained in 5 μg/ml of Tet until induction.
Plasmids.
pCMV-HBV directs the expression of wild type (WT) HBV pgRNA under the control of the human cytomegalovirus promoter (12, 25). pCMV-HBV/ENV− was derived from pCMV-HBV by the introduction of translation stop codons into the S open reading frame (21 codons after the pre-S2 start as well as 5 codons after the S start), using mutagenic oligonucleotides and PCR, and is defective in the expression of all HBV surface proteins. pCMV-HBV/POL− was derived from pCMV-HBV by a frameshift mutation introduced into the RT open reading frame after codon 108 and is defective in RT expression. Similarly, pCMV-DHBV directs the expression of WT DHBV pgRNA under the control of the human cytomegalovirus promoter (67, 72). pCMV-DHBV/ENV−, a derivative of pCMV-DHBV that is defective in the expression of all DHBV surface proteins (the 1S mutation) (61), pCMV-DHBV/P−, which harbors two amino acid substitutions in the RT active-site motif that abolishes viral DNA synthesis (YMDD to YMHA) (6, 67), and pCMV-DHBV/P−S− (pCMV-DHBV/ΔXM), which is defective in expression of the viral RT and envelope proteins but functional in core protein expression (67), have been described previously.
Transient transfections.
The HepG-2 cells were transfected by using Fugene 6 (Roche), and the HEK293 and LMH cells were transfected by using a CalPhos Mammalian Transfection kit (BD Biosciences). The transfected cells were harvested 5 days posttransfection.
Isolation of viral DNA.
For isolation of viral DNA from the stable cell lines (HepAD38 and Dstet5), the cells were induced by culturing without Tet for 14 days. Core and PF DNAs were isolated from stably and transiently transfected cells as previously described (22, 25, 46, 67, 73), with minor modifications. Briefly, for isolation of core DNA, cells were lysed in NP-40 lysis buffer (50 mM Tris-HCl [pH 8.0], 1 mM EDTA, 1% NP-40, and 1× protease inhibitor [Roche]). After removal of the nuclear pellet by brief centrifugation, the supernatant (cytoplasmic lysate) was digested with Turbo DNase (Ambion) to remove plasmid DNA input, and proteinase K was then used to digest viral DNA-protein complexes after the nucleocapsids (cores) were precipitated with polyethylene glycol and disrupted by sodium dodecyl sulfate (SDS). Hirt extraction was used for PF DNA isolation (24). Cells in a 60-mm dish were lysed in 1 ml SDS lysis buffer (50 mM Tris-HCl [pH 8.0], 10 mM EDTA, 150 mM NaCl, and 1% SDS). After being incubated for 5 min at room temperature, the cell lysate was transferred to a 1.5-ml microcentrifuge tube, mixed with 0.25 ml of 2.5 M KCl, and incubated at 4°C overnight with gentle rotation. After being centrifugated at 14,000 × g for 20 min, the supernatant was extracted three times with phenol and once with chloroform. The DNA was precipitated with ethanol and washed with 70% ethanol three times, vacuum dried, resuspended in TE (10 mM Tris-HCl-1 mM EDTA [pH 8.0]), and digested with DNase-free RNase before being analyzed. Alternatively, PF DNA was isolated immediately and directly (without the KCl precipitation step) by phenol extraction of the SDS whole-cell lysate. To isolate PF DNA separately from the cytoplasmic and nuclear fractions, the NP-40 cytoplasmic lysate and nuclear pellet (described above for core DNA extraction) were extracted separately, using the Hirt extraction procedure.
Purification of HBV nucleocapsids by isopycnic CsCl density gradient centrifugation.
Cytoplasmic lysate (NP-40 supernatant, described above) from induced HepAD38 cells was digested with Turbo DNase (Ambion) and RNase A (Sigma). The digested lysate was then mixed with an equal volume of 2× CsCl (94.5% wt/vol) in HCB2 buffer (20 mM Tris-HCl [pH 8.0], 50 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% β-mercaptoethanol, and 1× protease inhibitor [Roche]) to a final density of 1.36 g/cm3. The sample was centrifuged at 60,000 rpm for 48 h at room temperature, using a Beckman NVT 65 rotor. The nucleocapsid peak fractions (no. 5 to 8, with densities of 1.35 to 1.39 g/cm3) were washed twice with TE buffer, using Centricon YM-100 microconcentrators (Millipore) to remove CsCl. Equal amounts of each washed fraction were then used for isolation of core DNA (lysis of nucleocapsids with SDS, followed by digestion with proteinase K and phenol extraction) and PF DNA (lysis of nucleocapsids with SDS, followed immediately by phenol extraction without protease digestion).
Southern blot analysis of viral DNA.
The core and PF DNAs isolated from the stable cell lines were either left untreated, heated at the indicated temperatures for 10 min, or digested with the indicated restriction enzymes and then analyzed by Southern blotting. PF DNA samples harvested from transiently transfected cells were digested with DpnI to remove input plasmids. To confirm the nature of CCC DNA, the samples were heat denatured at 95°C for 10 min with or without subsequent EcoRI digestion to linearize the DNA. Agarose gel electrophoresis and Southern blotting were performed as previously described (22, 25, 45, 67).
Two-dimensional agarose gel electrophoresis.
The DNA samples were first resolved by native agarose gel electrophoresis. The gel slice was soaked in a denaturation solution containing 0.5 M NaOH and 1.5 M NaCl for 10 min and then in an alkaline gel running buffer containing 50 mM NaOH and 1 mM EDTA twice for 30 min each. The gel slice was rotated 90° and aligned at the top of an alkaline agarose gel. After second-dimension electrophoresis, the alkaline gel was neutralized and transferred onto a nylon membrane (53). Viral minus- and plus-strand DNAs were detected by using 32P-labeled strand-specific riboprobes.
CCC DNA amplification and sequencing.
Ten femptograms of gel-purified CCC DNA was used as a template for PCR in a reaction mixture (total volume, 25 μl) containing 1 μl reverse primer (10 μM, 5′-CTTCTAGGGGACCTGCCTCGTCGTC-3′, nucleotides [nt] 2376 to 2352), 1 μl forward primer (10 μM, 5′-CGTATCCATGGCTGCTAGGC-3′, nt 1370 to 1389), 1 μl each of 10 mM dATP, dGTP, dCTP, and TTP, 2.5 μl of 10× reaction buffer, and 2.5 U PfuUltra high-fidelity DNA polymerase (Stratagene). The reaction mixture was denatured at 95°C for 2 min, followed by 35 cycles at 95°C for 30 s, 50°C for 30 s, and 68°C for 1.5 min. Under these conditions, at least 10-fold-more RC DNA (i.e., 100 fg) was required to generate a detectable PCR product. Since our gel-purified CCC DNA used in the PCR had less than 10% RC DNA contamination (i.e., less than one fg in the PCR), it is extremely unlikely that any significant amounts of PCR products were derived from RC DNA. The PCR product was gel purified, using a QIAEX II gel extraction kit (QIAGEN) and then digested with BamHI and BglII. The digested product was gel purified again, cloned into pCDNA3, and sequenced.
RESULTS
Hepatoma cells in culture could support HBV and DHBV CCC DNA formation.
As a first step toward understanding how CCC DNA is formed, we analyzed HBV and DHBV CCC DNA formation in transiently and stably transfected hepatoma cells. Two stably transfected and conditional cell lines were employed. The two cell lines, HepAD38 and Dstet5, were derived from the human hepatoma line HepG-2 and the chicken hepatoma line LMH, respectively, and express HBV and DHBV pgRNA in a Tet-repressible manner. As has been previously reported (22), the Dstet5 cell line, which replicates a mutant DHBV genome defective in viral surface proteins, accumulated high levels (estimated to be 200 to 400 copies per cell) of DHBV CCC DNA (Fig. 2B) in an inducible fashion through the aforementioned intracellular CCC DNA amplification pathway. Similarly, HepAD38 cells replicated the WT HBV to high levels (Fig. 2A) (30). Although the HBV CCC DNA level detected in HepAD38 cells was lower than that of DHBV CCC in Dstet5 cells, we could clearly detect it and confirm its supercoiled nature by Southern blot analysis (Fig. 2A). Based on a DNA recovery efficiency of approximately 70% (estimated using an internal plasmid standard added at the beginning of the PF DNA extraction procedure) and the fact that virtually all (more than 95%) induced HepAD38 cells expressed the HBV core protein (as detected by indirect immunofluorescence staining; data not shown), we estimated that approximately 5 to 10 copies of HBV CCC DNA per cell were present in HepAD38 cells, which is similar to results reported in a recent study (78). This number is also close to the average estimate for DHBV-infected duck liver or WHV-infected woodchuck liver (35, 71, 77).
FIG. 2.

Formation of HBV and DHBV CCC DNA in stably transfected hepatoma cell lines. HBV (A) and DHBV (B) DNAs were extracted from the cytoplasmic nucleocapsid (core) with protease digestion or the Hirt supernatant without protease digestion (PF), respectively, from HepAD38 (HBV) and Dstet5 (DHBV) cells cultured in the absence of Tet and analyzed by Southern blotting. The nature of CCC DNA in the HBV PF sample (panel A, lanes 5 and 6) was further confirmed by heating to 95°C, which denatured RC/NC and DSL DNA to SS without denaturing CCC DNA, or at 95°C, followed by EcoRI digestion, which linearized CCC DNA. Note the predominance of CCC DNA and RC/NC DNA in the DHBV and HBV PF DNAs, respectively. El, EcoRI. (C) The kinetics of accumulation of CCC DNA and PF-RC DNA in HepAD38 (HBV) and Dstet5 (DHBV) cells. The PF DNA was extracted after induction for 7, 10, 14, and 17 days for HBV (lanes 1 to 4) and 4, 5, 6, and 7 days for DHBV (lanes 5 to 8) and analyzed by Southern blotting.
HBV PF-RC, not CCC, DNA was the predominant protein-free DNA in the cell lines and accumulated in the cell nucleus.
Curiously, in the HBV CCC DNA sample, which was extracted from HepAD38 cells without protease digestion, the predominant DNA species was not CCC DNA but a species that migrated at the position of RC DNA extracted from cytoplasmic cores with protease digestion (Fig. 2A). Since the replicative, protein-linked core DNA was lost during extraction in the absence of protease digestion (precipitated as potassium dodecyl sulfate complexes or partitioned into the phenol phase; see Material and Methods for details), the DNA species extracted without protease digestion (routinely used to extract CCC DNA) was not linked covalently to protein (thus called protein-free). The relative mobility of the prominent PF DNA suggests that it represented RC DNA with the RT protein removed (deproteinated or protein-free), which approached 30% of the level of core RC DNA and was 5- to 10-fold more abundant than CCC DNA in the same sample (Fig. 2A, lane 3). This pattern was clearly different from that of the DHBV PF DNA sample extracted from the Dstet5 cells in which the predominant DNA species was CCC DNA (Fig. 2B, lane 2). Furthermore, the time course of viral DNA induction showed that the putative HBV PF-RC DNA was detected earlier than the CCC DNA and was much more abundant than CCC DNA at all time points, whereas the DHBV CCC DNA was always the predominant PF DNA species at all time points examined (Fig. 2C).
However, since nicked CCC (NC) DNA would also migrate to the same position as PF RC DNA, we analyzed this presumptive PF-RC DNA further. As RC DNA isolated from the viral cores is held in a circular form only by the cohesive overlap between the 5′ ends of the minus- and plus-strand DNA (Fig. 1), ca. 200 bp in HBV and 50 bp in DHBV, mild heat treatment converted RC DNA to a linear form comigrating with DSL DNA (Fig. 3A and B). In contrast, nicked circular plasmid DNA was resistant to linearization by heating up to 75°C (Fig. 3C). The presumptive PF-RC DNA in the HBV PF DNA preparation indeed behaved like the core RC DNA, being converted almost entirely to the linear form by heating to 75°C (Fig. 3A). There was a small amount of DNA remaining at the RC DNA position after the heat treatment, representing the NC DNA, presumably resulting from nicking of CCC DNA during extraction. In contrast, the DHBV PF DNA sample contained only low levels of the DNA species migrating at the RC position, which were decreased slightly by the heat treatment, indicating that there were only very low levels of PF-RC DNA in the DHBV PF DNA sample (Fig. 3B).
FIG. 3.

Analyses of PF-RC DNA. HBV (A) and DHBV (B) core and PF DNA, extracted as for Fig. 2, or the plasmid pcDNA3 (C) were heated at the indicated temperatures and analyzed by Southern blotting. The diagrams at the bottom depict that only RC, not NC, is linearized at the indicated temperatures. SC, supercoiled plasmid. (D) HBV core or PF DNA was digested with the indicated restriction enzymes and analyzed by Southern blotting. The diagram at the bottom depicts the restriction sites on the RC DNA. E, EcoRI; R, RsrII.
To exclude the possibility that PF-RC DNA was created artificially during the Hirt extraction procedure, we isolated PF DNA from the total cell lysate by direct phenol extraction immediately after lysing the HepAD38 cells with the SDS-containing lysis buffer. The PF DNA isolated using this alternative procedure contained the same three DNA species as those isolated using the Hirt extraction, again with abundant PF-RC DNA and much smaller amounts of CCC (and PF-DSL) DNA (Fig. 4A, lanes 1 and 2). We also extracted PF DNA separately from the cytoplasmic and nuclear fractions and found that virtually all (more than 95%) PF-RC DNA, as well as CCC DNA, was detected in the nuclear fraction (Fig. 4A, lane 3).
FIG. 4.

Detection of HBV PF DNA in the nucleus and nucleocapsid. (A) HBV PF DNA was isolated from the whole-cell lysate, using either Hirt extraction (lane 1) or direct phenol extraction (lane 2), or from the nuclear fraction (N, lane 3) by Hirt extraction. (B) HBV DNA was isolated from the peak fractions of nucleocapsids purified by CsCl gradient centrifugation with protease digestion for core DNA (lanes 1 to 4) or without protease digestion for PF DNA (lanes 5 to 8). HBV core DNA (lane 9) and PF DNA (lane 10) were also extracted from the purified HBV nucleocapsids (fraction 6) after being mixed with the total HepG2 cell lysate. Fr no., fraction number.
However, a very small amount (ca. 1% of the amount of core DNA) of PF-RC DNA was detected in the cytoplasmic fraction (data not shown). Since the cytoplasmic fraction was digested extensively with nuclease, this result suggests that a small amount of PF-RC DNA might be associated with (and protected by) the nucleocapsids. Therefore, we purified HBV nucleocapsids by CsCl density gradient centrifugation and extracted nucleocapsid-associated viral DNA with or without protease digestion. Again, a small amount (ca. 1% of the level of core DNA) of PF-RC DNA seemed to be associated with the purified nucleocapsids (Fig. 4B, lanes 1 to 8). To ascertain whether the presence of cellular components might affect the amount of PF DNA extracted from the nucleocapsids, the purified nucleocapsids were mixed with untransfected HepG-2 cell lysate and core and PF DNAs were again extracted. The presence of the cell lysate did not affect the amount or pattern of PF DNA extracted from the nucleocapsids (Fig. 4B, lanes 9 and 10). As will be discussed later, the apparent association of very small amounts of PF-RC DNA with the nucleocapsids suggests that either residual amounts of core DNA (still linked to the RT protein) were extracted using the PF DNA extraction procedures or that the removal of the RT protein from RC DNA could take place before nucleocapsid disassembly. In any case, our results clearly indicate that the abundant HBV PF-RC DNA (ca. 30% of the level of core RC DNA) detected in the cell nucleus was not created artificially during DNA extraction and could not possibly be accounted for by any residual contamination with cytoplasmic, protein-linked core RC DNA (not more than 1%, if any).
Only mature RC DNA was detected in PF DNA.
In contrast to RC (and a small amount of DSL) DNA, which could be detected in the PF DNA, no single-stranded (SS) DNA species of either HBV or DHBV was ever detected in the PF DNA (Fig. 2 and 3), suggesting that deproteination occurred only on mature RC (DSL) DNA, not on SS DNA. To further analyze the structures of PF RC DNA, we adopted a two-dimensional (native followed by denaturing) agarose gel electrophoresis method to measure the maturity of minus- and plus-strand DNAs in each DNA species. As expected, the minus-strand DNA in the RC, DSL, and CCC species was all full length (data not shown). On the other hand, HBV RC DNA harvested from viral cores contained heterogeneous plus strands, reflecting different maturation stages, as evidenced by the presence of a diagonal signal emanating from the RC spot (Fig. 5, top panel). In contrast, no such diagonal was present in PF-RC DNA. Interestingly, there was a diagonal signal emanating from HBV DSL DNA, though much less in the PF DNA than in the core DNA, suggesting that the plus strands in PF-DSL DNA contained some heterogeneity. A small number of immature plus strands might be contained in PF-RC DNA but did not affect the mobility of PF-RC DNA on the first-dimension gel, accounting for the lack of a diagonal signal and the downward smear below the full-length plus strands, which was also apparent in the core RC DNA (Fig. 3D and below).
FIG. 5.

Two-dimensional agarose gel electrophoresis analyses of core and PF DNAs. The HBV (top panel) and DHBV (bottom panel) core and PF DNAs were extracted as for Fig. 2. The DNA was separated first on a native agarose gel (first dimension), followed by a denaturing alkaline agarose gel (second dimension). After being transferred to a nylon membrane, viral plus-strand DNA was detected by using an antisense riboprobe. Linear DNA markers were run in parallel as size standards (left side). Where indicated, the DNA samples were heated (68°C for DHBV and 75°C for HBV) to convert RC to the DSL form. For clarity, only DNA species migrating above the full-length minus-strand SS DNA (Fig. 2) were analyzed on the second-dimension gel.
We also converted RC DNA to the linear form by mild heating (Fig. 3) and subjected the heated DNA to two-dimensional analysis. Upon this treatment, all the core RC DNA, including its heterogeneous plus strands, migrated to the DSL position and a diagonal emanating from it (Fig. 5, top panel). The same treatment converted PF-RC DNA to the DSL position; the residual DNA remaining at the RC position after heating again likely represented NC DNA. As expected, CCC DNA in the PF DNA sample migrated as a single spot, running ahead of DSL DNA on the first dimension, and being resistant to denaturation, the still-double-stranded circular CCC DNA migrated to approximately the same position as the full-length SS linear DNA on the second-dimension gel. Similar to that of HBV, DHBV core RC DNA contained heterogeneous plus strands, whereas PF-RC DNA exclusively contained full-length plus strands (Fig. 5, bottom panel). However, as is evident in Fig. 3, DHBV PF-RC DNA was much less abundant than CCC or NC DNA. The DHBV DNA signal at the RC/NC position in Fig. 5, which is much stronger than that shown in Fig. 2 and 3, was due to the more-severe nicking of CCC DNA. As described above, the NC DNA was resistant to linearization by mild heating.
On the other hand, restriction digestion of the HBV core and PF-RC DNAs indicated that, although both DNA species were fully double stranded at the EcoRI site, approximately 20 to 30% of the PF-RC DNA, like the core RC DNA, still contained a SS gap at the RsrII site (Fig. 3D). Together, these results suggest that the RT protein was removed preferentially from the nearly mature HBV and DHBV RC DNAs but the plus-strand gap did not have to be filled completely before deproteination could occur.
HBV CCC DNA in HepG-2 cells originated predominantly from RC DNA but also from DSL DNA.
Given the unusual accumulation of PF-RC DNA in HepG-2 cells, we wished to verify that authentic CCC DNA was indeed formed in these cells. As discussed above, CCC DNA derived from RC DNA is expected to have the precise junction sequence at the cohesive-overlap region, whereas CCC DNA derived from DSL DNA by illegitimate replication is known to have short deletions/insertions at the junction. We cloned the HBV CCC DNA sequences spanning the junction by PCR and sequenced 37 individual clones. As depicted schematically in Fig. 6A, the predominant CCC DNA molecules had the precise junction sequence, indicating that they were in fact authentic CCC DNA derived from RC DNA. This indicated that HepG-2 cells could carry out proper conversion of RC DNA to CCC DNA, in addition to accumulating abundant amounts of PF-RC DNA. Two clones harbored deletions near the r sequence, one (no. 28) with a 9-nt deletion (nt 1830 to 1838) immediately downstream from the r sequence (Fig. 6A and B) and the other (no. 15) with a 66-nt deletion (nt 1763 to 1828), including 8 nt from r itself and 58 nt upstream of r (Fig. 6B). The characteristic deletions near r suggest that these CCC molecules originated from the small amounts of DSL DNA via NHEJ, as previously described for DHBV and WHV (73-75). Another clone (no. 7) contained a 35-nt deletion (nt 1449 to 1483) within the SS-gap region of RC (or DSL) DNA (Fig. 6A and B). Still another clone (no. 9) suffered an inversion between nt 1421 and 1429, also within the SS-gap region. These results thus suggest that the SS gap of RC (and perhaps DSL) DNA might be recognized by host DNA repair systems and processed by a cellular nuclease and/or by recombination factors.
FIG. 6.

Sequencing results for CCC DNA at the cohesive-overlap region. The HBV CCC DNA was gel-purified from the PF DNA sample extracted from HepAD38 cells. The sequence spanning the cohesive overlap (Fig. 1) was amplified by PCR, cloned into the plasmid pcDNA3, and sequenced. (A) Schematic diagram summarizing the sequence results. The deletions detected in CCC DNA molecules derived from circularization of DSL DNA are denoted with an arrowhead. The other deletion and inversion events in the CCC DNA were detected in sequences corresponding to the presumed SS gap (arrow) of RC (and DSL, omitted for clarity) DNA. (B) Sequences of four individual clones that suffered deletions or inversion. The sequence on the top line is from the input WT HBV (strain ayw). The deletions and inversion in the sequences shown on the bottom line are indicated by blank spaces (deletion) or a left-pointing arrow (inversion). The nucleotide positions are indicated, as is the 9-nt r sequence. See the text for details.
Elimination of HBV surface proteins increased PF-RC but not CCC DNA.
As discussed above, while DHBV CCC DNA formation is subjected to feedback suppression by the viral LS protein, deletion of the HBV surface proteins did not appear to increase HBV CCC DNA (32, 56). To determine if the HBV surface proteins would have any effect on PF-RC DNA, we transfected an HBV mutant unable to express any surface proteins into HepG-2 cells. Compared with the WT HBV, the mutant only marginally increased (less than 50%) the amounts of CCC DNA (Fig. 7). However, PF-RC DNA and PF-DSL DNA, which accumulated in these transiently transfected HepG-2 cells as in the stably transfected HepAD38 cells (Fig. 2), were increased more significantly (by three- to fivefold) in the absence of the surface proteins (Fig. 7). In sharp contrast, deletion of the DHBV surface proteins dramatically enhanced (more than 10-fold) CCC DNA levels in transfected LMH as well as HEK293 cells, but the low level of PF-RC or PF-DSL DNA was not increased by the deletion (data not shown).
FIG. 7.

HBV core DNA and PF DNA in transiently transfected HepG-2 cells. (A) pCMV-HBV (WT), pCMV-HBV/ENV−, and pCMV-HBV/POL− were transfected into HepG-2 cells. Viral core (lanes 1 to 3) and PF (lanes 4 to 6) DNA samples were harvested 5 days after transfection and analyzed by Southern blotting. The cells were transfected in duplicate, with one of the two dishes used for isolation of core DNA and the other for PF DNA. (B) PF DNA was further analyzed by DpnI digestion (lanes 1 to 3, D) to degrade input plasmids; DpnI digestion, followed by heating to 95°C (lanes 4 to 6, D/H), to denature the plasmid fragments, RC/NC and DSL DNA, but not CCC DNA, to SS DNA; or DpnI digestion, followed by heating and then EcoRI digestion (lanes 7 to 9, D/H/E), to convert CCC DNA to a linear form. The different viral DNA species, as well as the input plasmids, are indicated. This experiment was repeated three times with similar results.
DISCUSSION
In this study, we analyzed the potential pathways and factors involved in the conversion of RC DNA to CCC DNA, an essential event in hepadnavirus replication and persistence. Our results show that conversion of RC DNA to CCC DNA could take place in both HBV- and DHBV-transfected cells in culture. On the other hand, the efficiency of CCC DNA formation differed between HBV and DHBV, suggesting that virus- and/or host-specific factors may affect CCC DNA formation. A deproteinated form of RC DNA, the so-called PF-RC DNA, from which the covalently attached RT protein was removed, accumulated to levels much higher than those of CCC DNA in cultured cells replicating HBV DNA. In contrast, cells replicating DHBV DNA predominantly accumulated CCC DNA, with very little PF-RC DNA. Furthermore, whereas elimination of the viral envelope proteins led to dramatically increased levels of CCC DNA in DHBV, it led mainly to an increase of PF-RC, but not CCC, DNA in HBV. Interestingly, PF-RC DNA contained mostly mature plus-strand DNA, suggesting that the RT protein was preferentially removed from the mature RC DNA. Finally, sequence analyses indicated that HBV CCC DNA was predominantly derived from RC DNA but also from DSL DNA.
It is clear that for RC (or DSL) DNA to be converted to CCC DNA via any pathway, the RT protein that is covalently attached to the 5′ end of the minus strand of RC DNA has to be removed. We have demonstrated here that deproteinated RC DNA, i.e., PF-RC DNA, indeed accumulated to levels much higher than those of CCC DNA in HBV-transfected cells in vitro. A viral DNA species with similar mobility to that of RC DNA, but not covalently attached to the RT protein, was also noted by Summers and colleagues to accumulate in DHBV-infected primary duck hepatocytes when suramin, a viral entry inhibitor, was added after the initial infection (61). A putative PF-RC DNA also appeared to accumulate in the livers of HBV-transgenic mice and in human hepatoma cells transfected or transduced with HBV DNA (1, 7, 10, 11, 49, 78). A small amount of viral DNA that migrated at the RC DNA position was also commonly observed in CCC DNA samples extracted from infected human or animal liver tissues (20, 42, 52, 65, 70, 79). However, since a portion of CCC DNA is inevitably nicked during extraction and the NC DNA also migrates at the same position as RC DNA, it remained unclear if PF-RC DNA was in fact being detected. By carrying out a structural characterization of the presumptive PF-RC DNA, we show here that PF-RC DNA indeed accumulated to high levels in HBV-transfected cell lines.
The following results rigorously eliminated the possibility that the HBV PF-RC DNA we detected might be created artificially during DNA extraction or that it simply represented contaminating core DNA. First, using the same extraction procedure, only HBV PF-RC DNA was detected, whereas little to no DHBV PF-RC DNA was detected. Second, an alternative PF DNA extraction procedure (direct phenol extraction of SDS cell lysate) isolated the same DNA species. Third, PF-RC DNA was detected almost exclusively in the nucleus, whereas RT-linked DNA is within the cytoplasmic nucleocapsids. Fourth, PF-RC DNA contained mostly mature plus strands, and SS DNA was never detected in the PF DNA preparation. Fifth, PF-RC DNA was detected at a level approximately 30% of that of core RC DNA, whereas the PF DNA extraction procedure allowed, at most, 1% of the protein-linked core DNA to be extracted.
In contrast to the accumulation of PF-RC DNA in transfected cell lines in vitro, HBV PF DNA samples extracted from infected human (42) or chimpanzee liver (infected with the same HBV ayw subtype as used here) (2, 70) predominantly contained CCC DNA, with little, if any, PF-RC DNA. This suggests that HBV RC DNA processing and CCC DNA conversion may be different in the cell lines in vitro and in mouse liver, on one hand, and in human and chimpanzee livers on the other. That knocking-out of HNF-1α increased HBV CCC DNA in the livers of HBV-transgenic mice (49) also supports the notion that host cell environment influences CCC DNA formation. One possibility is that host factors required for CCC DNA formation, located downstream of RC DNA deproteination, may be limiting in the cell lines and in mouse liver if PF-RC DNA is in fact a precursor to CCC DNA (Fig. 8). The time courses of PF-RC and CCC DNA accumulations in the HepAD38 cells are consistent with such a precursor-product relationship. Alternatively, in these cells, the RT protein may be removed in a way that precludes RC DNA to CCC DNA conversion, in which case the resulting PF-RC would be a dead-end, off-pathway by-product. In any case, as PF-RC DNA is the only known processing product of RC DNA so far other than CCC DNA itself, further detailed analyses of its formation and structure, which may be different depending on the mechanisms of deproteination, may provide clues about the CCC DNA conversion process. Interestingly, no PF-SS DNA was ever detected, and PF-RC DNA almost exclusively contained nearly full-length plus strands with little to no nascent plus strands, suggesting that the removal of the RT protein, by whatever mechanism, preferentially occurred on mature RC DNA (Fig. 8).
FIG. 8.

Potential viral and host factors influencing RC DNA to CCC DNA conversion. Shown schematically are the immature (containing pgRNA or SS DNA) and mature (containing mature RC DNA) nucleocapsids in the cytoplasm, the hypothetical nuclear RC DNA intermediate still linked to the RT after uncoating (not yet detected), PF-RC DNA (detected in this report), and CCC DNA. Only mature RC DNA, not immature DNA, is shown to be deproteinated, possibly as a result of preferential uncoating of mature nucleocapsids and/or nuclear import of mature RC DNA, which is proposed to be regulated negatively by the viral LS protein. The predominance of HBV and DHBV CCC or RC DNA under different in vivo and in vitro conditions is indicated. RT, reverse transcription. See the text for details.
Two general models of RC DNA deproteination that involve either the RT itself (autorelease) or host DNA repair factors can be envisaged. If the RT protein is removed from RC DNA by a host protease and/or nuclease, which most likely can occur only after nucleocapsid uncoating, these results may reflect the selective uncoating of mature nucleocapsids. On the other hand, if the RT protein could release itself from RC DNA, these results suggest that the timing of this autorelease would be tightly regulated so that the RT was detached from the viral DNA only after DNA synthesis approached completion. The detection of small amounts of PF-RC DNA associated with nucleocapsids, as was also suggested by an earlier report (49), is consistent with the notion that some deproteination may occur prior to nucleocapsid disassembly, thus implicating the RT protein in releasing itself from the RC DNA. However, since the amount of nucleocapsid-associated PF-RC DNA was so low, we cannot yet exclude the possibility that it may simply represent very low-level contamination of protein-linked DNA.
Curiously, in contrast to HBV, which accumulated much more PF-RC DNA than CCC DNA in the HepG-2 cells, DHBV CCC DNA was always the predominant PF DNA species in vivo and in LMH cells. We also transfected both HBV and DHBV DNA into the HEK293 cells and again found that, in these human kidney-derived cells, HBV PF-RC, not CCC, DNA dominated and that DHBV CCC DNA was predominant (W. Gao and J. Hu, unpublished results). These results suggest that viral-specific factors may also play a role in the processing of RC DNA and possibly in the formation of CCC DNA. The nature of the viral factor that accounts for this interesting difference between HBV and DHBV remains to be identified. On the other hand, it is clear from the results presented here and those reported by others (61, 62) that the surface proteins of both HBV and DHBV can regulate the processing of RC DNA and CCC DNA formation. Interestingly, elimination of the HBV surface proteins led to an increase of PF-RC DNA, instead of CCC DNA, in contrast to results for DHBV. Together, these results suggest that the viral surface proteins may suppress a step in the cytoplasm that precedes RC DNA deproteination in the CCC conversion process, e.g., the uncoating of the mature nucleocapsid or nuclear import of RC DNA (Fig. 8). This notion is supported by the fact that the DHBV surface proteins, specifically the LS protein, which is located on cytoplasmic membranes, coordinately regulate the two mutually exclusive fates of the cytoplasmic mature nucleocapsids, i.e., virion formation (occurring in the cytoplasm) and CCC DNA synthesis (requiring nuclear import of the mature nucleocapsids or the RC DNA within them) (5, 31). The failure to increase HBV CCC DNA in the cell lines by elimination of the surface proteins could be explained by postulating a step in CCC DNA formation occurring after RC DNA deproteination that was limiting in these cells, as suggested above by the predominance of PF-RC, instead of CCC, DNA in the WT HBV-transfected cells. We have yet to test if the increase in HBV PF-RC DNA was due to the specific deletion of the HBV LS protein or the other two surface proteins (M and S), which were also eliminated in the mutant HBV used here. However, based on the fact that the pre-S1 domain at the N terminus of the HBV LS protein, like its DHBV counterpart, is able to interact with the mature nucleocapsids for envelopment and virion secretion (31), it is likely that, as in DHBV, the HBV LS protein (and even a similar domain) is also involved in suppressing PF-RC DNA formation.
Sequence analyses of the HBV CCC DNA from human hepatoma cells indicated that it was predominantly converted from RC DNA with precise junctions at the cohesive overlap. However, our results also revealed that HBV CCC DNA could be formed by circularization of DSL DNA via NHEJ, as has been shown previously for DHBV and WHV (73-75). As was shown for DHBV and WHV, the conversion of HBV DSL DNA to CCC DNA was imprecise, resulting in deletions at the circularization junction. The deleted CCC DNA of DHBV was shown to be able to direct new rounds of (imprecise) viral replication through illegitimate replication, which may also occur in HBV-infected cells. In addition, the finding that a fraction of HBV (this report) and WHV (73) CCC DNA suffered deletion/inversion in a region corresponding to the SS gap of RC (and DSL) DNA suggests that the SS gap of RC (and perhaps DSL) DNA could be recognized and processed by host DNA repair systems (e.g., a nuclease and/or recombination factor). Indeed, these aberrant CCC DNA species may participate in further rounds of viral replication, as suggested by the fact that HBV and WHV variants with similar deletions in this region of the viral DNA have been identified previously from infected patients and woodchucks, respectively (13-15, 29).
CCC DNA is the molecular basis of persistent HBV infection. It is particularly refractory to current antiviral therapy, yet its elimination is a prerequisite for any real cure of an HBV infection. Although the half-life of preformed CCC DNA remains a hotly debated issue (21, 33, 40, 43, 44, 58, 70), effective, long-term antiviral treatment to block the synthesis of RC DNA, thus depleting the precursor to CCC DNA, can lead to significant reduction of CCC DNA in vitro and in vivo (1, 3, 8, 11, 18, 22, 49, 64, 69). These results indicate that continuous replenishment of the CCC DNA pool, via either de novo infection or intracellular amplification, is required to maintain persistent infections. The problem with long-term antiviral therapy is the emergence of drug-resistant viruses that render the therapy ineffective (35, 80). Obviously, novel therapies that can directly block RC DNA to CCC DNA conversion or destabilize preformed CCC DNA should greatly accelerate the clearance of CCC DNA and thus terminate persistent infections. Our studies reported here have begun to reveal potential pathways of, and factors involved in, CCC DNA formation. Further studies in this direction should bring much-needed insights into the mechanism of CCC DNA formation, which in turn may facilitate the development of novel antivirals targeted directly to this critical step of viral replication. In addition, these studies also have broad significance for understanding cellular DNA damage repair mechanisms, the malfunction of which underlies a variety of serious human diseases from developmental defects to cancer. In particular, little is currently understood about how covalent protein-DNA abducts are repaired (9, 50). A better understanding of how the RT protein is removed from RC DNA as part of the process of CCC DNA formation may bring important insights into the mechanisms of protein-DNA abduct repair in general.
Acknowledgments
We thank Christoph Seeger for the HepAD38 and Dstet5 cell lines.
This work was supported by Public Health Service grant R01 AI43453 from the National Institutes of Health.
Footnotes
Published ahead of print on 4 April 2007.
REFERENCES
- 1.Abdelhamed, A. M., C. M. Kelley, T. G. Miller, P. A. Furman, E. E. Cable, and H. C. Isom. 2003. Comparison of anti-hepatitis B virus activities of lamivudine and clevudine by a quantitative assay. Antimicrob. Agents Chemother. 47:324-336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Acs, G., M. A. Sells, R. H. Purcell, P. Price, R. Engle, M. Shapiro, and H. Popper. 1987. Hepatitis B virus produced by transfected Hep G2 cells causes hepatitis in chimpanzees. Proc. Natl. Acad. Sci. USA 84:4641-4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson, A. L., K. E. Banks, M. Pontoglio, M. Yaniv, and A. McLachlan. 2005. Alpha/beta interferon differentially modulates the clearance of cytoplasmic encapsidated replication intermediates and nuclear covalently closed circular hepatitis B virus (HBV) DNA from the livers of hepatocyte nuclear factor 1α-null HBV transgenic mice. J. Virol. 79:11045-11052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartenschlager, R., and H. Schaller. 1992. Hepadnaviral assembly is initiated by polymerase binding to the encapsidation signal in the viral RNA genome. EMBO J. 11:3413-3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bruss, V. 1997. A short linear sequence in the pre-S domain of the large hepatitis B virus envelope protein required for virion formation. J. Virol. 71:9350-9357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang, L. J., R. C. Hirsch, D. Ganem, and H. E. Varmus. 1990. Effects of insertional and point mutations on the functions of the duck hepatitis B virus polymerase. J. Virol. 64:5553-5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chou, Y. C., K. S. Jeng, M. L. Chen, H. H. Liu, T. L. Liu, Y. L. Chen, Y. C. Liu, C. P. Hu, and C. Chang. 2005. Evaluation of transcriptional efficiency of hepatitis B virus covalently closed circular DNA by reverse transcription-PCR combined with the restriction enzyme digestion method. J. Virol. 79:1813-1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Colonno, R. J., E. V. Genovesi, I. Medina, L. Lamb, S. K. Durham, M. L. Huang, L. Corey, M. Littlejohn, S. Locarnini, B. C. Tennant, B. Rose, and J. M. Clark. 2001. Long-term entecavir treatment results in sustained antiviral efficacy and prolonged life span in the woodchuck model of chronic hepatitis infection. J. Infect. Dis. 184:1236-1245. [DOI] [PubMed] [Google Scholar]
- 9.Connelly, J. C., and D. R. Leach. 2004. Repair of DNA covalently linked to protein. Mol. Cell 13:307-316. [DOI] [PubMed] [Google Scholar]
- 10.Delaney, W. E., IV, and H. C. Isom. 1998. Hepatitis B virus replication in human HepG2 cells mediated by hepatitis B virus recombinant baculovirus. Hepatology 28:1134-1146. [DOI] [PubMed] [Google Scholar]
- 11.Delaney, W. E., IV, T. G. Miller, and H. C. Isom. 1999. Use of the hepatitis B virus recombinant baculovirus-HepG2 system to study the effects of (−)-β-2′,3′-dideoxy-3′-thiacytidine on replication of hepatitis B virus and accumulation of covalently closed circular DNA. Antimicrob. Agents Chemother. 43:2017-2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fallows, D. A., and S. P. Goff. 1995. Mutations in the epsilon sequences of human hepatitis B virus affect both RNA encapsidation and reverse transcription. J. Virol. 69:3067-3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feitelson, M., L. Lega, J. Guo, M. Resti, M. E. Rossi, C. Azzari, B. S. Blumberg, and A. Vierucci. 1994. Pathogenesis of posttransfusion viral hepatitis in children with beta-thalassemia. Hepatology 19:558-568. [DOI] [PubMed] [Google Scholar]
- 14.Feitelson, M. A., L. X. Duan, J. Guo, and B. S. Blumberg. 1995. X region deletion mutants associated with surface antigen-positive hepatitis B virus infections. Gastroenterology 108:1810-1819. [DOI] [PubMed] [Google Scholar]
- 15.Feitelson, M. A., L. X. Duan, J. Guo, B. Sun, J. Woo, K. Steensma, N. Horiike, and B. S. Blumberg. 1995. X region deletion variants of hepatitis B virus in surface antigen-negative infections and non-A, non-B hepatitis. J. Infect. Dis. 172:713-722. [DOI] [PubMed] [Google Scholar]
- 16.Ganem, D., and A. M. Prince. 2004. Hepatitis B virus infection—natural history and clinical consequences. N. Engl. J. Med. 350:1118-1129. [DOI] [PubMed] [Google Scholar]
- 17.Ganem, D., and R. J. Schneider. 2001. Hepadnaviridae, p. 2923-2969. In B. N. Fields and D. M. Knipe (ed.), Fields virology, 4th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
- 18.Genovesi, E. V., L. Lamb, I. Medina, D. Taylor, M. Seifer, S. Innaimo, R. J. Colonno, D. N. Standring, and J. M. Clark. 1998. Efficacy of the carbocyclic 2′-deoxyguanosine nucleoside BMS-200475 in the woodchuck model of hepatitis B virus infection. Antimicrob. Agents Chemother. 42:3209-3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gerlich, W., and W. S. Robinson. 1980. Hepatitis B virus contains protein covalently attached to the 5′ terminus of its complete DNA strand. Cell 21:801-809. [DOI] [PubMed] [Google Scholar]
- 20.Guidotti, L. G., B. Matzke, H. Schaller, and F. V. Chisari. 1995. High-level hepatitis B virus replication in transgenic mice. J. Virol. 69:6158-6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guidotti, L. G., R. Rochford, J. Chung, M. Shapiro, R. Purcell, and F. V. Chisari. 1999. Viral clearance without destruction of infected cells during acute HBV infection. Science 284:825-829. [DOI] [PubMed] [Google Scholar]
- 22.Guo, J. T., M. Pryce, X. Wang, M. I. Barrasa, J. Hu, and C. Seeger. 2003. Conditional replication of duck hepatitis B virus in hepatoma cells. J. Virol. 77:1885-1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hirsch, R. C., J. E. Lavine, L. J. Chang, H. E. Varmus, and D. Ganem. 1990. Polymerase gene products of hepatitis B viruses are required for genomic RNA packaging as well as for reverse transcription. Nature 344:552-555. [DOI] [PubMed] [Google Scholar]
- 24.Hirt, B. 1967. Selective extraction of polyoma DNA from infected mouse cell cultures. J. Mol. Biol. 26:365-369. [DOI] [PubMed] [Google Scholar]
- 25.Hu, J., D. Flores, D. Toft, X. Wang, and D. Nguyen. 2004. Requirement of heat shock protein 90 for human hepatitis B virus reverse transcriptase function. J. Virol. 78:13122-13131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu, J., and D. Nguyen. 2004. Therapy for chronic hepatitis B: the earlier, the better? Trends Microbiol. 12:431-433. [DOI] [PubMed] [Google Scholar]
- 27.Hu, J., and C. Seeger. 1996. Expression and characterization of hepadnavirus reverse transcriptases. Methods Enzymol. 275:195-208. [DOI] [PubMed] [Google Scholar]
- 28.Jilbert, A. R., D. S. Miller, C. A. Scougall, H. Turnbull, and C. J. Burrell. 1996. Kinetics of duck hepatitis B virus infection following low dose virus inoculation: one virus DNA genome is infectious in neonatal ducks. Virology 226:338-345. [DOI] [PubMed] [Google Scholar]
- 29.Kew, M. C., R. H. Miller, H. S. Chen, B. C. Tennant, and R. H. Purcell. 1993. Mutant woodchuck hepatitis virus genomes from virions resemble rearranged hepadnaviral integrants in hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 90:10211-10215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ladner, S. K., M. J. Otto, C. S. Barker, K. Zaifert, G. H. Wang, J. T. Guo, C. Seeger, and R. W. King. 1997. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: a novel system for screening potential inhibitors of HBV replication. Antimicrob. Agents Chemother. 41:1715-1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lenhoff, R. J., and J. Summers. 1994. Coordinate regulation of replication and virus assembly by the large envelope protein of an avian hepadnavirus. J. Virol. 68:4565-4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ling, R., and T. J. Harrison. 1997. Production of hepatitis B virus covalently closed circular DNA in transfected cells is independent of surface antigen synthesis. J. Gen. Virol. 78:1463-1467. [DOI] [PubMed] [Google Scholar]
- 33.Litwin, S., E. Toll, A. R. Jilbert, and W. S. Mason. 2005. The competing roles of virus replication and hepatocyte death rates in the emergence of drug-resistant mutants: theoretical considerations. J. Clin. Virol. 34(Suppl. 1):S96-S107. [DOI] [PubMed] [Google Scholar]
- 34.Locarnini, S. 2005. Molecular virology and the development of resistant mutants: implications for therapy. Semin. Liver Dis. 25(Suppl. 1):9-19. [DOI] [PubMed] [Google Scholar]
- 35.Locarnini, S., and W. S. Mason. 2006. Cellular and virological mechanisms of HBV drug resistance. J. Hepatol. 44:422-431. [DOI] [PubMed] [Google Scholar]
- 36.Loeb, D. L., R. C. Hirsch, and D. Ganem. 1991. Sequence-independent RNA cleavages generate the primers for plus strand DNA synthesis in hepatitis B viruses: implications for other reverse transcribing elements. EMBO J. 10:3533-3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marion, P. L., and W. S. Robinson. 1983. Hepadna viruses: hepatitis B and related viruses. Curr. Top. Microbiol. Immunol. 105:99-121. [DOI] [PubMed] [Google Scholar]
- 38.Mason, W. S., J. Cullen, G. Moraleda, J. Saputelli, C. E. Aldrich, D. S. Miller, B. Tennant, L. Frick, D. Averett, L. D. Condreay, and A. R. Jilbert. 1998. Lamivudine therapy of WHV-infected woodchucks. Virology 245:18-32. [DOI] [PubMed] [Google Scholar]
- 39.Mason, W. S., M. S. Halpern, J. M. England, G. Seal, J. Egan, L. Coates, C. Aldrich, and J. Summers. 1983. Experimental transmission of duck hepatitis B virus. Virology 131:375-384. [DOI] [PubMed] [Google Scholar]
- 40.Mason, W. S., A. R. Jilbert, and J. Summers. 2005. Clonal expansion of hepatocytes during chronic woodchuck hepatitis virus infection. Proc. Natl. Acad. Sci. USA 102:1139-1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mason, W. S., G. Seal, and J. Summers. 1980. Virus of Pekin ducks with structural and biological relatedness to human hepatitis B virus. J. Virol. 36:829-836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miller, R. H., and W. S. Robinson. 1984. Hepatitis B virus DNA forms in nuclear and cytoplasmic fractions of infected human liver. Virology 137:390-399. [DOI] [PubMed] [Google Scholar]
- 43.Moraleda, G., J. Saputelli, C. E. Aldrich, D. Averett, L. Condreay, and W. S. Mason. 1997. Lack of effect of antiviral therapy in nondividing hepatocyte cultures on the closed circular DNA of woodchuck hepatitis virus. J. Virol. 71:9392-9399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Murray, J. M., S. F. Wieland, R. H. Purcell, and F. V. Chisari. 2005. Dynamics of hepatitis B virus clearance in chimpanzees. Proc. Natl. Acad. Sci. USA 102:17780-17785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perlman, D., and J. Hu. 2003. Duck hepatitis B virus virion secretion requires a double-stranded DNA genome. J. Virol. 77:2287-2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Perlman, D. H., E. A. Berg, P. B. O'Connor, C. E. Costello, and J. Hu. 2005. Reverse transcription-associated dephosphorylation of hepadnavirus nucleocapsids. Proc. Natl. Acad. Sci. USA 102:9020-9025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qiao, M., C. A. Scougall, A. Duszynski, and C. J. Burrell. 1999. Kinetics of early molecular events in duck hepatitis B virus replication in primary duck hepatocytes. J. Gen. Virol. 80:2127-2135. [DOI] [PubMed] [Google Scholar]
- 48.Rabe, B., A. Vlachou, N. Pante, A. Helenius, and M. Kann. 2003. Nuclear import of hepatitis B virus capsids and release of the viral genome. Proc. Natl. Acad. Sci. USA 100:9849-9854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Raney, A. K., C. M. Eggers, E. F. Kline, L. G. Guidotti, M. Pontoglio, M. Yaniv, and A. McLachlan. 2001. Nuclear covalently closed circular viral genomic DNA in the liver of hepatocyte nuclear factor 1α-null hepatitis B virus transgenic mice. J. Virol. 75:2900-2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reardon, J. T., and A. Sancar. 2006. Repair of DNA-polypeptide crosslinks by human excision nuclease. Proc. Natl. Acad. Sci. USA 103:4056-4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Robinson, W. S., D. A. Clayton, and R. L. Greenman. 1974. DNA of a human hepatitis B virus candidate. J. Virol. 14:384-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ruiz-Opazo, N., P. R. Chakraborty, and D. A. Shafritz. 1982. Evidence for supercoiled hepatitis B virus DNA in chimpanzee liver and serum Dane particles: possible implications in persistent HBV infection. Cell 29:129-138. [DOI] [PubMed] [Google Scholar]
- 53.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
- 54.Seeger, C., and J. Hu. 1997. Why are hepadnaviruses DNA and not RNA viruses? Trends Microbiol. 5:447-450. [DOI] [PubMed] [Google Scholar]
- 55.Seeger, C., and W. S. Mason. 2000. Hepatitis B virus biology. Microbiol. Mol. Biol. Rev. 64:51-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sprinzl, M. F., H. Oberwinkler, H. Schaller, and U. Protzer. 2001. Transfer of hepatitis B virus genome by adenovirus vectors into cultured cells and mice: crossing the species barrier. J. Virol. 75:5108-5118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Staprans, S., D. D. Loeb, and D. Ganem. 1991. Mutations affecting hepadnavirus plus-strand DNA synthesis dissociate primer cleavage from translocation and reveal the origin of linear viral DNA. J. Virol. 65:1255-1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Summers, J., A. R. Jilbert, W. Yang, C. E. Aldrich, J. Saputelli, S. Litwin, E. Toll, and W. S. Mason. 2003. Hepatocyte turnover during resolution of a transient hepadnaviral infection. Proc. Natl. Acad. Sci. USA 100:11652-11659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Summers, J., and W. S. Mason. 1982. Replication of the genome of a hepatitis B-like virus by reverse transcription of an RNA intermediate. Cell 29:403-415. [DOI] [PubMed] [Google Scholar]
- 60.Summers, J., A. O'Connell, and I. Millman. 1975. Genome of hepatitis B virus: restriction enzyme cleavage and structure of DNA extracted from Dane particles. Proc. Natl. Acad. Sci. USA 72:4597-4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Summers, J., P. M. Smith, and A. L. Horwich. 1990. Hepadnavirus envelope proteins regulate covalently closed circular DNA amplification. J. Virol. 64:2819-2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Summers, J., P. M. Smith, M. J. Huang, and M. S. Yu. 1991. Morphogenetic and regulatory effects of mutations in the envelope proteins of an avian hepadnavirus. J. Virol. 65:1310-1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Summers, J., J. M. Smolec, and R. Snyder. 1978. A virus similar to human hepatitis B virus associated with hepatitis and hepatoma in woodchucks. Proc. Natl. Acad. Sci. USA 75:4533-4537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sung, J. J., M. L. Wong, S. Bowden, C. T. Liew, A. Y. Hui, V. W. Wong, N. W. Leung, S. Locarnini, and H. L. Chan. 2005. Intrahepatic hepatitis B virus covalently closed circular DNA can be a predictor of sustained response to therapy. Gastroenterology 128:1890-1897. [DOI] [PubMed] [Google Scholar]
- 65.Tuttleman, J. S., C. Pourcel, and J. Summers. 1986. Formation of the pool of covalently closed circular viral DNA in hepadnavirus-infected cells. Cell 47:451-460. [DOI] [PubMed] [Google Scholar]
- 66.Wang, G. H., and C. Seeger. 1992. The reverse transcriptase of hepatitis B virus acts as a protein primer for viral DNA synthesis. Cell 71:663-670. [DOI] [PubMed] [Google Scholar]
- 67.Wang, X., N. Grammatikakis, and J. Hu. 2002. Role of p50/CDC37 in hepadnavirus assembly and replication. J. Biol. Chem. 277:24361-24367. [DOI] [PubMed] [Google Scholar]
- 68.Weber, M., V. Bronsema, H. Bartos, A. Bosserhoff, R. Bartenschlager, and H. Schaller. 1994. Hepadnavirus P protein utilizes a tyrosine residue in the TP domain to prime reverse transcription. J. Virol. 68:2994-2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Werle-Lapostolle, B., S. Bowden, S. Locarnini, K. Wursthorn, J. Petersen, G. Lau, C. Trepo, P. Marcellin, Z. Goodman, W. E. Delaney IV, S. Xiong, C. L. Brosgart, S. S. Chen, C. S. Gibbs, and F. Zoulim. 2004. Persistence of cccDNA during the natural history of chronic hepatitis B and decline during adefovir dipivoxil therapy. Gastroenterology 126:1750-1758. [DOI] [PubMed] [Google Scholar]
- 70.Wieland, S. F., H. C. Spangenberg, R. Thimme, R. H. Purcell, and F. V. Chisari. 2004. Expansion and contraction of the hepatitis B virus transcriptional template in infected chimpanzees. Proc. Natl. Acad. Sci. USA 101:2129-2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wu, T. T., L. Coates, C. E. Aldrich, J. Summers, and W. S. Mason. 1990. In hepatocytes infected with duck hepatitis B virus, the template for viral RNA synthesis is amplified by an intracellular pathway. Virology 175:255-261. [DOI] [PubMed] [Google Scholar]
- 72.Wu, T.-T., L. D. Condreay, L. Coates, C. Aldrich, and W. Mason. 1991. Evidence that less-than-full-length pol gene products are functional in hepadnavirus DNA synthesis. J. Virol. 65:2155-2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yang, W., W. S. Mason, and J. Summers. 1996. Covalently closed circular viral DNA formed from two types of linear DNA in woodchuck hepatitis virus-infected liver. J. Virol. 70:4567-4575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang, W., and J. Summers. 1995. Illegitimate replication of linear hepadnavirus DNA through nonhomologous recombination. J. Virol. 69:4029-4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang, W., and J. Summers. 1998. Infection of ducklings with virus particles containing linear double-stranded duck hepatitis B virus DNA: illegitimate replication and reversion. J. Virol. 72:8710-8717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang, Y. Y., D. P. Theele, and J. Summers. 2005. Age-related differences in amplification of covalently closed circular DNA at early times after duck hepatitis B virus infection of ducks. J. Virol. 79:9896-9903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang, Y. Y., B. H. Zhang, D. Theele, S. Litwin, E. Toll, and J. Summers. 2003. Single-cell analysis of covalently closed circular DNA copy numbers in a hepadnavirus-infected liver. Proc. Natl. Acad. Sci. USA 100:12372-12377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhou, T., H. Guo, J. T. Guo, A. Cuconati, A. Mehta, and T. M. Block. 2006. Hepatitis B virus e antigen production is dependent upon covalently closed circular (ccc) DNA in HepAD38 cell cultures and may serve as a cccDNA surrogate in antiviral screening assays. Antivir. Res. 72:116-124. [DOI] [PubMed] [Google Scholar]
- 79.Zhu, Y., T. Yamamoto, J. Cullen, J. Saputelli, C. E. Aldrich, D. S. Miller, S. Litwin, P. A. Furman, A. R. Jilbert, and W. S. Mason. 2001. Kinetics of hepadnavirus loss from the liver during inhibition of viral DNA synthesis. J. Virol. 75:311-322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zoulim, F. 2005. Combination of nucleoside analogues in the treatment of chronic hepatitis B virus infection: lesson from experimental models. J. Antimicrob. Chemother. 55:608-611. [DOI] [PubMed] [Google Scholar]
- 81.Zoulim, F., and C. Seeger. 1994. Reverse transcription in hepatitis B viruses is primed by a tyrosine residue of the polymerase. J. Virol. 68:6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]