

Abstract
Membrane association of the hepatitis C virus NS5A protein is required for viral replication. This association is dependent on an N-terminal amphipathic helix (AH) within NS5A and is restricted to a subset of host cell intracellular membranes. The mechanism underlying this specificity is not known, but it may suggest a novel strategy for developing specific antiviral therapy. Here we have probed the mechanistic details of NS5A AH-mediated binding to both cell-derived and model membranes by use of biochemical membrane flotation and quartz crystal microbalance (QCM) with dissipation. With both assays, we observed AH-mediated binding to model lipid bilayers. When cell-derived membranes were coated on the quartz nanosensor, however, significantly more binding was detected, and the QCM-derived kinetic measurements suggested the existence of an interacting receptor in the target membranes. Biochemical flotation assays performed with trypsin-treated cell-derived membranes exhibited reduced AH-mediated membrane binding, while membrane binding of control cytochrome b5 remained unaffected. Similarly, trypsin treatment of the nanosensor coated with cellular membranes abolished AH peptide binding to the cellular membranes but did not affect the binding of a control lipid-binding peptide. These results therefore suggest that a protein plays a critical role in mediating and stabilizing the binding of NS5A's AH to its target membrane. These results also demonstrate the successful development of a new nanosensor technology ideal both for studying the interaction between a protein and its target membrane and for developing inhibitors of that interaction.
Hepatitis C virus (HCV) is an important worldwide cause of liver disease and is the leading cause of hepatocellular carcinoma and liver transplantation in the United States (21). Unfortunately, current therapies are inadequate for most of these patients, and it is hoped that a better understanding of HCV molecular virology may lead to the development of improved antiviral strategies. Like other positive-strand RNA viruses, HCV replicates its genome in intimate association with host cell membrane structures (19). That HCV's nonstructural protein 5A (NS5A) might play an important role in this process is suggested by NS5A's tight association with membranes (6, 24). This association is mediated posttranslationally by an amphipathic α-helical segment within the N-terminal segment of NS5A (2, 3). This amphipathic helix (AH) is both necessary and sufficient for conferring membrane association (2, 3). Genetic mutations that introduce charged amino acids into the hydrophobic face of the AH disrupt its amphipathic nature and inhibit membrane association (3). A critical consequence of such mutations is the abrogation of HCV RNA replication (3), highlighting the importance of AH-mediated membrane association for the viral life cycle. Based on nuclear magnetic resonance analyses of the NS5A AH mixed with different detergents that mimic lipids, it has been proposed that the AH associates with membranes in a monotypic fashion wherein the axis of the AH is parallel to the plane of the lipid bilayer with the AH partially inserting into the cytoplasmic leaflet of the membrane (22). Such a model, however, does not readily account for several important experimental observations. Among these is the fact that while NS5A clearly appears to be exclusively associated with host cell membrane structures, this association is restricted to a subset of intracellular membranes. What elements might specify this restricted localization and what might NS5A recognize in its target membranes are important details remaining to be clarified. Further understanding of such details concerning NS5A's membrane association could lead to the development of new therapies for HCV.
In this communication, we hypothesized the existence of a protein receptor. We sought to verify this hypothesis in two independent manners. First, we employed a standard biochemical membrane flotation assay in which AH-mediated membrane association can be conveniently monitored by the use of an in-frame C-terminally fused green fluorescent protein (GFP) tag. Second, we made use of a novel “membrane-on-a-chip” system wherein the binding dynamics of a synthetic peptide corresponding to the NS5A AH to lipid bilayers could be studied in real time using the quartz crystal microbalance (QCM) with dissipation (QCM-D) method (10-12). The latter method allows for measuring precise changes in oscillation frequency (Δf) and energy dissipation (ΔD) that occur when membranes or molecules bind to an oscillating quartz crystal (10). As described and experimentally verified by Sauerbrey, the frequency change (Δf) is a direct function of the mass change, where the frequency decreases with increased bound mass (23). Energy dissipation changes, ΔD, can be measured if the adsorbed film (i.e., bound membrane) is viscous. Energy is dissipated due to the oscillatory motion induced within the bound membrane and reveals the viscoelastic property of the crystal-bound membrane, with complex membranes exhibiting greater energy dissipation than homogenous lipid bilayers (7-9).
MATERIALS AND METHODS
Materials.
Peptides were synthesized by Anaspec Corporation (San Jose, CA). The sequences of AH (corresponding to the amphipathic α-helix of HCV NS5A) and NH (a mutated version of AH containing three point mutations designed to disrupt the amphipathic nature of AH) are Ser-Gly-Ser-Trp-Leu-Arg-Asp-Val-Trp-Asp-Trp-Ile-Cys-Thr-Val-Leu-Thr-Asp-Phe-Lys-Thr-Trp-Leu-Gln-Ser-Lys-Leu-Asp-Tyr-Lys-Asp and Ser-Gly-Ser-Trp-Leu-Arg-Asp-Asp-Trp-Asp-Trp-Glu-Cys-Thr-Val-Leu-Thr-Asp-Asp-Lys-Thr-Trp-Leu-Gln-Ser-Lys-Leu-Asp-Tyr-Lys-Asp, respectively (the charged amino acids introduced into AH to yield NH are underlined). The TAT peptide sequence is Tyr-Gly-Arg-Lys-Lys-Arg-Arg-Glu-Arg-Arg-Arg. Rabbit polyclonal anti-calnexin and horseradish peroxidase-conjugated anti-rabbit secondary antibodies were purchased from Stressgen Corp. and Promega Corp., respectively. Monoclonal antibody against PTP1B (FG6) was purchased from Upstate Biotechnology, Inc.
Membrane preparation.
Small unilamellar vesicles of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) (Avanti Polar Lipids) were prepared by the extrusion method (18). Huh7 membranes were isolated by use of a ball bearing homogenizer from Huh7 cells grown at 37°C with 5% CO2 in a 1:1 mixture of Dulbecco's modified Eagle's medium:RPMI 1640 supplemented with 10% fetal bovine serum, 2 mM l-glutamine, 100 IU/ml penicillin, and 100 μg/ml streptomycin (3).
For the trypsin-treated membranes used in biochemical membrane flotation, Huh7 membranes were incubated with 0.1% trypsin in Dulbecco's phosphate-buffered saline (PBS) for 30 min on ice followed by treatment with 0.1% soybean trypsin inhibitor to stop the protease activity of the trypsin (Sigma).
Western blots.
Aliquots of membranes prepared as described above were solubilized in sample buffer and subjected to Western blot analysis essentially as described previously (3). Briefly, samples were solubilized in sample buffer and analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to nitrocellulose membranes, and probed with anti-calnexin primary antibody (1:1,000) and horseradish peroxidase-conjugated anti-rabbit immunoglobulin G secondary antibody (1:2,500), followed by chemiluminescence (Amersham) development.
Dynamic light scattering.
Dynamic light scattering was performed by a 90Plus particle size analyzer, and the results were analyzed by digital autocorrelator software (Brookhaven Instruments Corporation, New York). All measurements were taken at a scattering angle of 90°, where the reflection effect is minimized.
QCM-D.
A Q-Sense D300 (Q-Sense AB, Gothenburg, Sweden) equipped with a QAFC 301 axial flow chamber was used to conduct QCM-D measurements. AT-cut crystals (Q-Sense) of 14 mm in diameter coated with 50 nm of thermally evaporated silicon dioxide were used for all experiments. Each QCM crystal was treated with oxygen plasma at ∼80 W for ∼3 min prior to measurements (March Plasmod plasma etcher; March Instruments, California). The crystal was driven at its resonance frequency of 5 MHz, and the frequency and dissipation changes for the three overtones at 15, 25, and 35 MHz were also monitored. The temperature of the Q-Sense cell was set at 25.0°C and accurately controlled by a Peltier element in the cell with fluctuation smaller than ±0.1°C. The first 10 min of Δf and that of ΔD were also recorded for each measurement for reference.
Binding dynamics analysis.
To compare the binding dynamics of AH peptide to model and cell-derived membranes, we first used the Sauerbrey linear relationship to calculate the mass of the AH peptide bound to the membrane platform by the change in resonant frequency with the equation 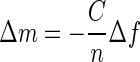 , where C is the mass sensitivity constant with a value of 17.7 ng/cm2Hz for our QCM-D crystal at 5 MHz and n is the overtone number.
, where C is the mass sensitivity constant with a value of 17.7 ng/cm2Hz for our QCM-D crystal at 5 MHz and n is the overtone number.
The rate of AH peptide binding (the association rate constant) was then determined using the following single-phase exponential equation—modeling a nonlinear association between the binding mass, time, and association rate constant—and a nonlinear least-square fitting method using GraphPad software (www.graphpad.com): 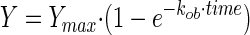 , where Y is the binding mass, Ymax is the maximum bound mass, kob is the observed association rate constant, and time is the time of association. Half-life time (t1/2), indicating the time for the binding mass to increase by 50%, was calculated using the equation t1/2 = ln(2)/kob.
, where Y is the binding mass, Ymax is the maximum bound mass, kob is the observed association rate constant, and time is the time of association. Half-life time (t1/2), indicating the time for the binding mass to increase by 50%, was calculated using the equation t1/2 = ln(2)/kob.
Membrane flotation assay.
Liposomes or isolated biomembranes were mixed with 35S-labeled in vitro translated AH-GFP (GFP fused at its N terminus to the AH of NS5A) or NH-GFP (the corresponding mutant [NH] version) for 30 min at 37°C with gentle agitation in PBS-2 mM EDTA-150 mM NaCl. Samples were mixed with 60% OptiPrep (Sigma) to make a final 40% and loaded in the bottom of tubes. This was then overlaid with step gradients of 30%, 25%, and 5% OptiPrep solutions. After centrifugation at 40,000 rpm in a Beckman SW60 rotor for 4 h, each fraction was collected from the top of the tube, precipitated with methanol-chloroform followed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and transferred to nitrocellulose membranes. Images were obtained and quantified by use of Molecular Imager FX (Bio-Rad) as described previously (3). The percent flotation was then calculated by dividing the amount of AH-GFP (or NH-GFP) in fractions 1 and 2 from the top (containing the low-density membrane fractions of the gradients) by the total amount contained in all fractions. The blots shown in Fig. 4a and b are examples from individual experiments only, while each number graphed in Fig. 4c represents the average of three independent experiments.
FIG. 4.

Biochemical membrane association of AH-GFP and cytochrome b5 proteins to trypsin-treated Huh7-derived membranes. (a and b) Isolated Huh7-derived membranes were either mock treated or treated with 0.1% trypsin on ice for 30 min before the addition of 0.1% soybean trypsin inhibitor. The in vitro-translated AH-GFP and cytochrome b5 proteins were incubated with either the mock-treated or the trypsin-treated Huh7 membranes and were subjected to membrane flotation assays as described for Fig. 1. (c) Membrane association of the proteins determined in three independent experiments was quantified by phosphorimager analysis and plotted. Note that in contrast to that of cytochrome b5, the amount of the membrane-bound AH-GFP was decreased when the trypsin-treated membrane was used.
RESULTS
AH binding to model lipid membranes.
As a first test of the hypothesis that QCM-D can be useful for characterizing AH-mediated membrane association, we sought to compare QCM-D's monitoring of NS5A AH's association with membranes to that obtained with a more classical biochemical technique. We initially used POPC vesicles, which have been previously employed as a simplified model of biological membranes (15). These model lipid membranes were prepared by extruding POPC through a 30-nm track-etched membrane to yield a monodistribution population of vesicles with diameters of 65 ± 1.2 nm. NS5A AH-mediated association with these vesicles was determined by membrane flotation analysis, as described previously (3). Briefly, radioactively labeled AH-GFP (a protein consisting of the NS5A AH with a C-terminally fused GFP tag) was prepared by in vitro transcription/translation, overlaid with a 5 to 40% OptiPrep step density gradient, and subjected to ultracentrifugation. Fractions were collected from the top, and the proteins in each gradient fraction were precipitated and analyzed by Western blotting for GFP. In such assays, non-membrane-associated proteins remain at the bottom of the gradient (high fraction number), whereas membranes and associated proteins “float” towards less dense gradient fractions present at the top (low fraction number).
Figure 1a shows that the AH-GFP fusion protein binds efficiently to the POPC vesicles. Note that a shorter form of the AH-GFP protein lacking the AH domain did not associate with the vesicles and served as an internal control. When the NH-GFP fusion protein (which contains three point mutations introduced into the AH of AH-GFP so as to disrupt its hydrophobic face [3]) was assayed, no association with the POPC vesicles occurred (Fig. 1b). As shown in Fig. 1c and d, comparable results were obtained from QCM-D analysis of AH peptide binding to POPC model lipid bilayers on SiO2 quartz crystals. In particular, for both Fig. 1c and d, an initial frequency change (Δf) of ≈25 Hz was observed upon vesicle addition (arrow 1), corresponding to the mass change associated with the deposition of a lipid bilayer on the SiO2 crystal (8, 11). Coincident with this change in frequency, the dissipation value shifted to close to zero, which indicates the formation of rigid layers on solid substrates. Both of these frequency and dissipation values are characteristic of bilayer formation from POPC vesicles in this system (8, 11) and show complete bilayer formation on the SiO2 quartz crystal surfaces. When a peptide corresponding to the NS5A AH was added at 40 min (Fig. 1c, arrow 3), the frequency change of the resonance showed that AH peptides were bound to the POPC lipid bilayer. In contrast, another peptide, termed NH and differing only in that it contains the three AH-disrupting mutations of NH-GFP, did not bind the POPC bilayers at all (Fig. 1d, arrow 3).
FIG. 1.

NS5A amphipathic alpha-helix-mediated membrane association to model lipid membranes. (a and b) Biochemical membrane flotation analysis of the binding of AH-GFP or NH-GFP proteins to POPC vesicles. POPC vesicles were formed from extrusion through 30-nm track-etched membranes. The effective diameter of the vesicles, as measured by dynamic light scattering, was 65 nm ± 1.2 nm. In vitro-translated AH-GFP (consisting of the NS5A AH with an in-frame C-terminally fused GFP tag) and NH-GFP (same as AH-GFP except that it contains three mutations designed to disrupt the hydrophobic face of the AH [3]) proteins were then mixed with the POPC lipids and subjected to the flotation assay as described previously (3). Note the association of AH-GFP, but not NH-GFP, with the low-density membrane-containing fractions (1 and 2 from the top of the gradient). See text for details. Asterisks indicate a shorter form of the AH-GFP protein. (c) QCM-D analysis of AH peptide binding to quartz crystals coated with POPC model lipid membranes. Bilayers were formed on the SiO2 nanosensors from POPC vesicles prepared as described above. In order to make complete bilayers, we utilized a high NaCl concentration (250 mM) in a PBS buffer. Frequency [Δf(t)] and dissipation [ΔD(t)] changes detected by QCM-D as a function of time were recorded. At 10 min, 0.05 mg/ml of vesicle solution was injected (arrow 1) followed by thorough washing with PBS buffer (arrow 2). After the frequency as well as the dissipation were stabilized at 40 min, 0.1 mg/ml of AH peptide was injected (arrow 3) to investigate the ability of the AH peptide to bind to the model POPC membrane. To ensure that the AH peptide was indeed bound to the membrane, the latter was washed again with PBS buffer (arrow 4) at 50 min. Note the decrease in resonant frequency change associated with the addition of AH peptide, indicating binding. (d) QCM-D analysis of NH peptide binding to POPC model lipid bilayers. Same as above, except that the AH peptide was replaced with NH peptide. The latter differs from AH in that NH contains three mutations designed to disrupt the hydrophobic face of AH (3). Note that the NH peptide does not bind to model POPC membranes, as indicated by lack of both frequency and dissipation changes. Also note that the vertical spikes are due to the injection of materials into the QCM-D sensor chamber and do not affect the changes in frequency and dissipation recorded before and after injections.
QCM-D monitoring of AH binding to cell-derived membranes.
Next, we tested whether the QCM-D crystal could be coated with a more complicated biological membrane. For this, we used membranes prepared from Huh7 cells, a human liver tumor-derived cell line that is widely used for HCV replication studies (1, 16). The formation of the biological membranes and the binding mechanism of target molecules were monitored by QCM-D. Figure 2a and b show the typical QCM-D analysis for forming a biological membrane, in this case, the Huh7 cultured cell membrane, on a SiO2 coated QCM-D crystal. After initial membrane addition (arrow 1) and thorough washing of the membranes with PBS at 70 and 80 min (arrows 2 and 3), we observed that the membranes remained stable on the QCM-D crystal. Again, as with the model POPC membranes, AH peptide added at 90 min exhibited clear binding to the Huh7 membrane, as indicated by the associated frequency change (Δf) (Fig. 2a, arrow 4). However, no such binding was observed upon addition of the NH peptide to the Huh7 membrane (Fig. 2b, arrow 4). This indicates that the helical structure of the AH peptide and/or its amphipathic nature is important to the binding process. In both cases of AH and NH addition, no changes in dissipation were observed. This is to be expected, as such small peptides should not alter the viscoelastic nature of the supported membranes.
FIG. 2.

QCM-D analysis of peptide binding to Huh7-derived membranes. (a) Binding of AH peptide to Huh7-derived membranes formed on SiO2 quartz crystals and detected by Δf(t) and ΔD(t) changes. At 10 min, Huh7 membrane solution (0.25 mg/ml) was injected (arrow 1). After buffer washes at 70 min and again at 80 min (arrows 2 and 3), 0.05 mg/ml of AH peptide was injected at 90 min (arrow 4). In order to confirm the binding of the peptide, an additional wash with the same buffer was performed (arrow 5). The data show that isolated membranes from the cultured Huh7 cells saturated at a higher mass than the model POPC membrane, presumably due to the presence of proteinaceous components. They also show by the dissipation value that those proteinaceous components are associated with higher viscoelastic energy dissipation, as expected. (b) No binding of NH peptide to Huh7-derived membranes formed on a SiO2 surface and detected by Δf(t) and ΔD(t) changes. NH peptide binding was attempted using the same method as in panel a. Membranes derived from cultured Huh7 cells were injected after 10 min (arrow 1). After waiting for saturation binding of membranes to the crystal, the latter was washed with buffer at 70 min and at 80 min (arrows 2 and 3) followed by the application of 0.05 mg/ml of NH peptide at 90 min (arrow 4) and an additional wash with the same buffer (arrow 5). Note that there are no changes in either the frequency or dissipation, which suggests that there is no binding to Huh7-derived membranes.
A key advantage of the QCM technique is that it can provide insight into the kinetics and hence the mechanism of membrane association. Indeed, the binding of AH peptide as a function of time is readily measured, as opposed to just the final steady-state amount associated with the membrane fraction as assessed by the biochemical membrane flotation. By use of the QCM measurements of AH peptide binding to POPC and Huh7-derived membranes, the corresponding kinetic parameters of half-life time, maximum binding mass, and observed association rate constant were determined (Fig. 3).
FIG. 3.

Exponential association binding model fitting for AH-mediated membrane association to model and cell-derived membranes. (a) Normalized frequency changes of AH (3,806.3-Da) peptide binding to POPC model and Huh7 cell-derived membranes. (b) By use of the Sauerbrey linear relationship, the AH binding mass on the two different membrane platforms was calculated and plotted. The exponential association binding equation was then used to fit the specific binding of the AH peptide. (c) By use of the latter curves, the indicated half-life times, maximum binding masses, and observed association rate constants for AH binding to the two types of membranes were calculated.
These data suggest that the mechanism of binding for AH on artificial pure lipid bilayers is different from that on cell-derived membranes. As opposed to a rapid relatively nonspecific interaction of AH with a pure lipid bilayer, the rate of association with the more-complex cell-derived membrane bilayer might reflect the increased time required for the NS5A AH to fully interact with its authentic partner ligand(s). We next sought to investigate the nature of this implied ligand.
AH-mediated membrane binding is sensitive to protease treatment of target membranes.
One important difference between the model POPC membranes and membranes derived from Huh7 cells is that the latter have proteinaceous components on the lipid membranes. As a first test of the hypothesis that proteins present in the membranes could influence AH-mediated membrane association, we performed biochemical membrane flotation assays using target membranes previously treated with the protease trypsin. For this, Huh7 membranes were incubated with, or without, 0.1% trypsin for 30 min on ice followed by soybean trypsin inhibitor, and the amounts of in vitro-translated AH-GFP floating with the respective membrane fractions were determined. As shown in Fig. 4, trypsin treatment of Huh7 membranes was associated with a decrease in binding of AH-GFP to the membrane fraction (fraction 2) (Fig. 4a and c). In contrast, when the membrane association of cytochrome b5, which is known to have a protein-independent mechanism of binding to lipid membranes (4, 14) was assayed in the same manner, unchanged or slightly increased binding of cytochrome b5 was observed (Fig. 4b and c). These results strongly suggest that a cellular membrane protein component contributes to the membrane association of AH-GFP.
To further support this intriguing conclusion, we next sought to perform a similar experiment using the QCM-D technique. After Huh7 membranes were allowed to coat a SiO2 quartz crystal (Fig. 5, arrow 1), the chamber was twice washed with PBS buffer (Fig. 5, black arrows 2 and 3) to confirm the stability of the membranes. Then, 0.1% trypsin was added (Fig. 5, arrow 4), followed by washing thoroughly with PBS buffer four times to remove any residual trypsin (Fig. 5, arrow 5), and AH peptide was added (Fig. 5, arrow 6). In contrast to the binding observed in the absence of prior trypsin treatment (Fig. 2a), the addition of an AH peptide after such treatment no longer changed the resonant frequency of the sensor (Fig. 5a).
FIG. 5.

QCM-D analysis of peptide binding to trypsin-treated Huh7 membranes. (a) AH peptide does not bind to trypsin-treated Huh7 membranes formed on the SiO2 surface, as detected by frequency [Δf(t)] and dissipation [ΔD(t)] changes. At 10 min, Huh7-derived membranes were injected (arrow 1), followed by washing with two sequences of PBS buffer (arrows 2 and 3). Then, 0.1% trypsin was applied to cleave proteinaceous components (arrow 4), followed by thorough washes with buffer. After the washing steps were repeated four times (arrow 5), the AH peptide was injected (arrow 6) to examine its ability to bind to the trypsin-treated Huh7 membrane. Note that there is no associated frequency change upon peptide addition, which would be indicative of binding. (b) Trypsin treatment does not affect the binding of TAT-derived peptide, which binds membranes independently of a protein receptor. AH peptide or TAT-derived peptide was added (arrow 1) to trypsin-treated Huh7 membranes deposited on the SiO2 surface, as in panel a, followed by a buffer washing 20 min later (arrow 2). (c) Mass changes associated with binding of the AH and TAT-derived peptides to the trypsin-treated Huh7 membranes of panel b, as calculated using the Sauerbrey equation (23).
That the trypsin treatment had indeed successfully removed accessible proteins from the surface of the membrane coating the SiO2 crystal is indicated by both the changes in resonant frequency and the energy dissipation observed following trypsin addition. The value of the dissipation, which reflects the viscoelastic character of the functional membrane, declined after trypsin treatment of the membrane surface formed from Huh7 cells on the sensor (Fig. 5a, red tracing). This presumably reflects digestion of proteinaceous components to make a more homogenous membrane with a resulting decreased dissipation value. Similarly, the observed increase in resonant frequency upon trypsin treatment (Fig. 5a, blue tracing) reflects the mass change associated with the removal of surface proteins.
To exclude the possibility that the trypsin treatment had somehow grossly damaged or removed the bilayer from the quartz crystal, we repeated this assay with a TAT-derived peptide that is known to interact with membranes but whose binding is postulated to occur via extracellular glycosaminoglycans (26) rather than protein receptors in the target membrane (Fig. 5b). The positively charged protein transduction domain of the human immunodeficiency virus type 1 TAT (residues 47 to 57 of TAT) is such a peptide. As shown in Fig. 5b and c, the binding levels of TAT-derived and AH peptides to crystals coated with trypsin-treated membranes were compared. Only the addition of TAT-derived peptide was still associated with a significant decrease in the resonance frequency of the membrane sensor (Fig. 5b, red tracing, arrow 1), indicating membrane binding. Thus, the binding mechanisms of the AH and TAT-derived peptides are different, with the binding of the NS5A AH being sensitive to the removal of proteinaceous components from the target membranes.
Because NS5A is localized predominantly in the endoplasmic reticulum (ER) or ER-derived membrane structures, we sought to confirm that the AH-bound membranes contained ER resident markers. For this, aliquots of Huh7-derived membranes used to coat the quartz crystal were analyzed by Western blotting prior to trypsin treatment. As shown in Fig. 6, the AH-bound membranes contained calnexin, an ER resident protein. Finally, we exploited the presence of another ER resident protein, PTP1B (5), to provide an additional control for the above-described trypsin treatment experiments. In particular, the binding of an antibody to PTP1B (FG6 [5]) (Fig. 7a), and the abrogation of this binding by prior trypsin treatment of the target membranes (Fig. 7b), served as a positive control for the detection of receptor-dependent membrane binding by QCM-D.
FIG. 6.
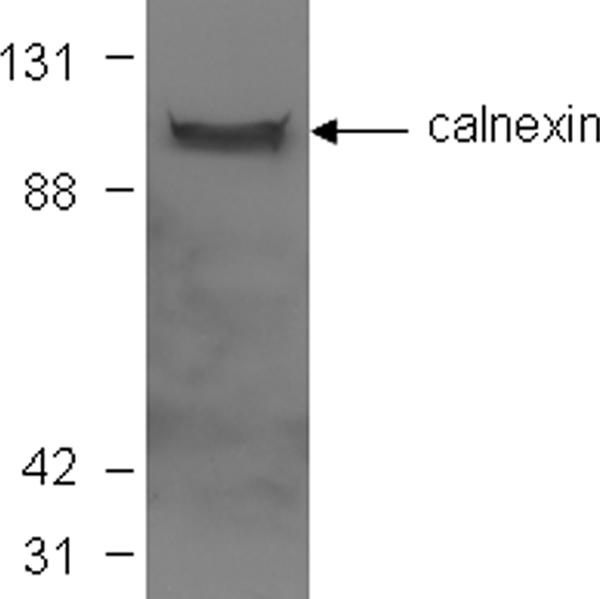
Target membranes in QCM-D analyses contain ER-derived membranes. Aliquots of membranes used to coat the QCM-D sensor crystal were solubilized in sample buffer and subjected to Western blot analysis, in which blots were probed with an antibody to the ER marker calnexin. Molecular mass markers (in kDa) are indicated at the left.
FIG. 7.

QCM-D analysis of monoclonal antibody FG6 binding to its ER membrane receptor PTP1B and sensitivity of binding to prior trypsin treatment of the membranes. (a) Binding of monoclonal antibody FG6 to its ER membrane receptor PTP1B contained in Huh7-derived membranes formed on SiO2 quartz crystals and detected by frequency [Δf(t)] and dissipation [ΔD(t)] changes. At 10 min, Huh7 membrane solution (0.25 mg/ml) was injected (arrow 1). After buffer washes at 70 min and again at 80 min (arrows 2 and 3), 0.05 mg/ml of monoclonal antibody FG6 (5) was injected at 90 min (arrow 4). To ensure that the antibody was indeed bound to its membrane receptor, the membranes were washed again with PBS buffer (arrows 5 and 6). Note the decrease in resonant frequency change associated with the addition of antibody, indicating binding. (b) PTP1B does not bind to trypsin-treated Huh7 membranes formed on the SiO2 surface, as detected by Δf(t). As described for Fig. 5a, at 10 min, Huh7-derived membranes were injected (arrow 1), followed by washing with two sequences of PBS buffer (arrows 2 and 3). Then, 0.1% trypsin was applied to cleave proteinaceous components (arrow 4), followed by thorough washes with buffer. After the washing steps were repeated four times (arrow 5), the anti-human PTP1B (FG6) antibody was injected (arrow 6) to examine its ability to bind to the trypsin-treated Huh7 membrane, followed by additional washing steps (arrows 7 and 8). Note that there is no associated frequency change upon antibody addition, which would be indicative of binding.
DISCUSSION
We hypothesized that the membrane association of HCV NS5A, which is essential for viral replication, is mediated in part by a protein receptor in the host cell target membrane. We sought to test this hypothesis using a novel “membrane-on-a-chip” system wherein the binding dynamics of ligands to lipid bilayers can be studied in real time by use of the QCM-D method (7-10, 12). This method is ideal for measuring mass changes that occur when membranes or molecules bind to an oscillating quartz crystal (10). Our results represent the first application of this sensitive technology to study the binding dynamics of a membrane-associating viral protein. The QCM-D data not only provided the first experimental support for our hypothesis but also were independently validated by more-classical biochemical techniques for monitoring the membrane association of proteins.
QCM-D proved to be a convenient method for monitoring the association of the NS5A AH peptide with a model lipid bilayer. Moreover, we found that significantly more peptide bound when cell-derived membranes were used to coat the quartz crystal nanosensor. This binding was eliminated by prior treatment of the cellular membranes with trypsin. A control peptide, whose interaction with membranes involves trypsin-insensitive glycosaminoglycans, was not altered by the protease treatment. Similar results were obtained using standard biochemical membrane flotation assays of NS5A AH-containing proteins and the control lipid-binding protein cytochrome b5, whose interaction with membranes is independent of a protein receptor.
Together, these results suggest a new model for HCV NS5A binding to host cellular membranes. We propose that in addition to the N-terminal amphipathic helical region's ability to bind model lipid membranes, which would be predicted from the structural analysis alone, the NS5A AH also interacts with a protein receptor in its target membrane. This offers an attractive explanation for how the NS5A proteins are localized to certain types of membranes, such as the ER- or the Golgi-derived membranes, including lipid droplets, but not to the plasma membranes or to the other subcellular membranes (13, 17, 20, 25). This could also explain prior experiments that showed that a peptide mimic of the AH can inhibit NS5A's membrane association in a dose-dependent manner (3). In this context, the high degree of conservation of specific amino acids within the AH across all published HCV isolates may reflect the epitopes involved in mediating interaction with a protein receptor in the target membrane.
Genetic disruption of AH-mediated membrane association of NS5A has been shown to abrogate HCV RNA replication (3). Although this genetically validates AH-mediated membrane association as a potentially potent antiviral target, such a strategy would not be practical for use in patients. If, however, a similar disruption of AH membrane association were achieved pharmacologically, it could be a significant practical clinical therapy. Our results suggest that such pharmacological disruption could be achieved by molecules designed to inhibit AH's interaction with its target membrane receptor.
Finally, our results demonstrate the potential of QCM-D for studying these types of protein-membrane interactions as well as a broad range of problems involving membrane proteins or lipids. The main advantage of the potential biosensor described in this study using QCM-D is that, in addition to providing results comparable to those obtained by traditional biochemical analysis, QCM-D is simple and quick to use and it shows the real-time kinetics of the interactions happening on the lipid bilayer derived from the cells. One major practical application could be to screen and develop precisely the type of inhibitor envisaged above.
Acknowledgments
This work was supported in part by NASA 1025-679-1-REDAL and the Center for Polymer Interfaces and Macromolecular Assemblies, an NSF-MRSEC. This work was also supported by a Burroughs Welcome Fund Career Award (to J.S.G.) and RO1-DK064223.
Footnotes
Published ahead of print on 11 April 2007.
REFERENCES
- 1.Blight, K. J., A. A. Kolykhalov, and C. M. Rice. 2000. Efficient initiation of HCV RNA replication in cell culture. Science 290:1972-1974. [DOI] [PubMed] [Google Scholar]
- 2.Brass, V., E. Bieck, R. Montserret, B. Wolk, J. A. Hellings, H. E. Blum, F. Penin, and D. Moradpour. 2002. An amino-terminal amphipathic alpha-helix mediates membrane association of the hepatitis C virus nonstructural protein 5A. J. Biol. Chem. 277:8130-8139. [DOI] [PubMed] [Google Scholar]
- 3.Elazar, M., K. H. Cheong, P. Liu, H. B. Greenberg, C. M. Rice, and J. S. Glenn. 2003. Amphipathic helix-dependent localization of NS5A mediates hepatitis C virus RNA replication. J. Virol. 77:6055-6061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Enoch, H. G., P. J. Fleming, and P. Strittmatter. 1979. The binding of cytochrome b5 to phospholipid vesicles and biological membranes. Effect of orientation on intermembrane transfer and digestion by carboxypeptidase Y. J. Biol. Chem. 254:6483-6488. [PubMed] [Google Scholar]
- 5.Haj, F. G., J. M. Zabolotny, Y. B. Kim, B. B. Kahn, and B. G. Neel. 2005. Liver-specific protein-tyrosine phosphatase 1B (PTP1B) re-expression alters glucose homeostasis of PTP1B−/−mice. J. Biol. Chem. 280:15038-15046. [DOI] [PubMed] [Google Scholar]
- 6.Hijikata, M., H. Mizushima, Y. Tanji, Y. Komoda, Y. Hirowatari, T. Akagi, N. Kato, K. Kimura, and K. Shimotohno. 1993. Proteolytic processing and membrane association of putative nonstructural proteins of hepatitis C virus. Proc. Natl. Acad. Sci. USA 90:10773-10777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hook, F., B. Kasemo, T. Nylander, C. Fant, K. Sott, and H. Elwing. 2001. Variations in coupled water, viscoelastic properties, and film thickness of a Mefp-1 protein film during adsorption and cross-linking: a quartz crystal microbalance with dissipation monitoring, ellipsometry, and surface plasmon resonance study. Anal. Chem. 73:5796-5804. [DOI] [PubMed] [Google Scholar]
- 8.Hook, F., M. Rodahl, P. Brzezinski, and B. Kasemo. 1998. Measurements using the quartz crystal microbalance technique of ferritin monolayers on methyl-thiolated gold: dependence of energy dissipation and saturation coverage on salt concentration. J. Colloid Interface Sci. 208:63-67. [DOI] [PubMed] [Google Scholar]
- 9.Hook, F., M. Rodahl, B. Kasemo, and P. Brzezinski. 1998. Structural changes in hemoglobin during adsorption to solid surfaces: effects of pH, ionic strength, and ligand binding. Proc. Natl. Acad. Sci. USA 95:12271-12276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kanazawa, K., and J. Gordon. 1985. The oscillation frequency of a quartz resonator in contact with a liquid. Anal. Chim. Acta 175:99-106. [Google Scholar]
- 11.Keller, C. A., K. Glasmastar, V. P. Zhdanov, and B. Kasemo. 2000. Formation of supported membranes from vesicles. Phys. Rev. Lett. 84:5443-5446. [DOI] [PubMed] [Google Scholar]
- 12.Keller, P., and K. Simons. 1998. Cholesterol is required for surface transport of influenza virus hemagglutinin. J. Cell Biol. 140:1357-1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim, J. E., W. K. Song, K. M. Chung, S. H. Back, and S. K. Jang. 1999. Subcellular localization of hepatitis C viral proteins in mammalian cells. Arch. Virol. 144:329-343. [DOI] [PubMed] [Google Scholar]
- 14.Kim, P. K., F. Janiak-Spens, W. S. Trimble, B. Leber, and D. W. Andrews. 1997. Evidence for multiple mechanisms for membrane binding and integration via carboxyl-terminal insertion sequences. Biochemistry 36:8873-8882. [DOI] [PubMed] [Google Scholar]
- 15.Ladokhin, A. S., M. E. Selsted, and S. H. White. 1997. Sizing membrane pores in lipid vesicles by leakage of co-encapsulated markers: pore formation by melittin. Biophys.J. 2:762-1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lohmann, V., F. Korner, J. Koch, U. Herian, L. Theilmann, and R. Bartenschlager.1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110-113. [DOI] [PubMed] [Google Scholar]
- 17.Matto, M., C. M. Rice, B. Aroeti, and J. S. Glenn. 2004. Hepatitis C virus core protein associates with detergent-resistant membranes distinct from classical plasma membrane rafts. J. Virol. 78:12047-12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mayer, L. D., M. J. Hope, and P. R. Cullis. 1986. Vesicles of variable sizes produced by a rapid extrusion procedure. Biochim. Biophys. Acta 858:161-168. [DOI] [PubMed] [Google Scholar]
- 19.Moradpour, D., R. Gosert, D. Egger, F. Penin, H. E. Blum, and K. Bienz. 2003. Membrane association of hepatitis C virus nonstructural proteins and identification of the membrane alteration that harbors the viral replication complex. Antivir. Res. 60:103-109. [DOI] [PubMed] [Google Scholar]
- 20.Moradpour, D., P. Kary, C. M. Rice, and H. E. Blum. 1998. Continuous human cell lines inducibly expressing hepatitis C virus structural and nonstructural proteins. Hepatology 28:192-201. [DOI] [PubMed] [Google Scholar]
- 21.Pearlman, B. L. 2004. Hepatitis C treatment update. Am. J. Med. 117:344-352. [DOI] [PubMed] [Google Scholar]
- 22.Penin, F., V. Brass, N. Appel, S. Ramboarina, R. Montserret, D. Ficheux, H. E. Blum, R. Bartenschlager, and D. Moradpour. 2004. Structure and function of the membrane anchor domain of hepatitis C virus nonstructural protein 5A. J. Biol. Chem. 279:40835-40843. [DOI] [PubMed] [Google Scholar]
- 23.Sauerbrey, G. 1959. Verwendung von Schwingquarzen zur Waegun duenner Schichten und zur Mikrowaegung. Z. Phys. 55:06. [Google Scholar]
- 24.Selby, M. J., Q.-L. Choo, K. Berger, G. Kuo, E. Glazer, M. Eckart, C. Lee, D. Chien, C. Kuo, and M. Houghton. 1993. Expression, identification and subcellular localization of the proteins encoded by the hepatitis C viral genome. J. Gen. Virol. 74:1103-1113. [DOI] [PubMed] [Google Scholar]
- 25.Shi, S. T., S. J. Polyak, H. Tu, D. R. Taylor, D. R. Gretch, and M. M. Lai. 2002. Hepatitis C virus NS5A colocalizes with the core protein on lipid droplets and interacts with apolipoproteins. Virology 292:198-210. [DOI] [PubMed] [Google Scholar]
- 26.Ziegler, A., and J. Seelig. 2004. Interaction of the protein transduction domain of HIV-1 TAT with heparan sulfate: binding mechanism and thermodynamic parameters. Biophys. J. 86:254-263. [DOI] [PMC free article] [PubMed] [Google Scholar]