

Abstract
The proteins encoded by the A56R and K2L genes of vaccinia virus form a heterodimer (A56/K2) and have a fusion regulatory role as deletion or mutation of either causes infected cells to form large syncytia spontaneously. Here, we showed that syncytia formation is dependent on proteins of the recently described entry fusion complex (EFC), which are also required for virus-cell fusion and low-pH-triggered cell-cell fusion. This finding led us to consider that A56/K2 might prevent fusion by direct or indirect interaction with the EFC. To test this hypothesis, we made a panel of recombinant vaccinia viruses that have a tandem affinity purification tag attached to A56, K2, or the A28 EFC protein. Interaction between A56/K2 and the EFC was demonstrated by their copurification from detergent-treated lysates of infected cells and identification by mass spectrometry or Western blotting. In addition, a purified soluble transmembrane-deleted form of A56/K2 was shown to interact with the EFC. Tagged A56 did not interact with the EFC in the absence of K2, nor did tagged K2 interact with the EFC in the absence of A56. The finding that both A56 and K2 are required for efficient binding to the EFC fits well with prior experiments showing that mutation of either A56 or K2 results in spontaneous fusion of infected cells. Because A56 and K2 are located on the surface of infected cells, they are in position to interact with the EFC of released progeny virions and prevent back-fusion and syncytia formation.
Poxviruses, of which vaccinia virus (VACV) is the prototype, comprise a family of large double-stranded DNA viruses that replicate entirely in the cytoplasm of cells from vertebrate or invertebrate animals (22). The simplest infectious particle, which can be released by cell lysis, is termed a mature virion (MV); it consists of a core structure containing the DNA genome, numerous enzymes, and structural components surrounded by a lipoprotein membrane, likely derived from the endoplasmic reticulum and within which are embedded proteins necessary for fusion with the cell during entry (7, 13, 23). A second type of infectious particle, known as the enveloped virion (EV), is released from the intact cell by exocytosis and is essentially an MV with an additional membrane that is derived from trans-Golgi or endosomal cisternae and discarded before or during the entry process (20, 33).
Entry of the MV can occur by endocytosis (36) as well as by direct fusion with the plasma membrane (6). The latter is greatly accelerated by briefly lowering the pH of the medium below 6, mimicking the usual pH drop following endocytosis (36). In an analogous manner, infected cells with EVs on their surface fuse with each other when the pH is lowered (3, 9, 12). However, syncytia form spontaneously at neutral pH when cells are infected with VACV or the closely related cowpox virus in which the gene A56R, encoding hemagglutinin (HA) (14), or K2L, encoding a serine protease inhibitor (SPI-3) (19, 37, 42), is mutated or deleted. HA is a type I membrane protein that is anchored in the EV and plasma membrane (26, 28), whereas SPI-3 lacks an anchor and is dependent on association with the HA for membrane localization (5, 39). Evidence indicates that membrane association of HA and SPI-3 is required for their antifusion activity, as syncytia form under the following conditions: (i) when cowpox virus-infected cells are incubated with antibodies to HA (28) or SPI-3 (39), (ii) when the membrane anchor is removed from HA (39), or (iii) when the signal sequence is deleted from SPI-3 (5). No putative catalytic motifs are present in HA, and the serine protease inhibitory activity of SPI-3 is not required for fusion inhibition (38).
Recent studies indicate that neutral- and low-pH-induced virus-cell fusion and low-pH-induced fusion of infected cells are each dependent on the same entry fusion complex (EFC), which consists of at least eight viral proteins (A16, A21, A28, G3, G9, H2, J5, and L5) (30, 36). The purpose of the present study was to determine the relationship, if any, between cell-cell fusion at neutral pH, which occurs when either the A56R or K2L gene is deleted, and the EFC. We found that repression of synthesis of a single component of the EFC inhibited formation of syncytia mediated by deletion of either A56R or K2L, suggesting that the fusion mechanism is similar or identical to that occurring during virus entry. Furthermore, a physical association of the HA/SPI-3 heterodimer with the EFC was demonstrated by tandem affinity purification (TAP).
MATERIALS AND METHODS
Cell and virus propagation.
BS-C-1 (ATCC CCL-26) and RK13 (ATCC CCL-37) cells were grown in minimum essential medium with Earle's balanced salt supplement (Quality Biologicals, Gaithersburg, MD) containing l-glutamine and 10% fetal bovine serum. HeLa S3 (ATCC CCL-2.2) suspension cells were cultured in minimum essential medium, Spinner modification (Quality Biologicals), with 5% equine serum and l-glutamine. Unless specified, all recombinant viruses were derived from the Western Reserve (WR) strain (ATCC VR-1354, accession number AY243312). Virus stocks were prepared as described previously (11).
TAP and mass spectrometry.
HeLa S3 cells (1.5 ×109) were infected at a multiplicity of 5 PFU for 24 h. Cells were collected, washed once with ice-cold buffer (150 mM NaCl and 50 mM Tris-HCl, pH 7.4), and lysed by incubating for 1 h at 4°C in streptavidin binding buffer (SBB) (1% Triton X-100, 150 mM NaCl, 50 mM Tris-HCl, pH 7.4, with complete protease inhibitor; Roche, Indianapolis, IN). The lysate was centrifuged for 15 min at 3,000 × g, and the clarified supernatant was collected. The latter, except for 0.3 ml reserved for later analysis, was added to 0.5 ml to 1 ml of streptavidin-Sepharose (GE Healthcare, Piscataway, NJ) that had been washed with SBB, and the mixture was rotated overnight at 4°C. The beads were washed three times with 10 ml of ice cold SBB, and the bound proteins were eluted by three washes with 1 ml of SBB containing 1 mg/ml of d-biotin (USB Corp., Cleveland, OH). The 3 ml of streptavidin eluate was supplemented with Mg acetate, imidazole, and CaCl2 to final concentrations of 1 mM, 1 mM, and 2 mM, respectively. Calmodulin-Sepharose (0.5 ml to 1 ml of packed resin; GE Healthcare) was washed with calmodulin binding buffer ([CBB], which consists of SBB without protease inhibitors and is supplemented with Mg acetate, imidazole, and CaCl2 at final concentrations of 1 mM, 1 mM, and 2 mM, respectively). Calmodulin-Sepharose was added to the supplemented streptavidin eluate along with an additional 2 ml of CBB, and the mixture was rotated overnight at 4°C. The beads were washed three times with 10 ml of CBB and three times with 0.75 ml of CBB containing 25 mM EGTA to elute proteins. The proteins in the calmodulin eluate were concentrated by trichloroacetic acid precipitation, resuspended in lithium-dodecyl sulfate sample buffer (Invitrogen, Carlsbad, CA) containing NuPage sample reducing agent (Invitrogen), and separated on a 4 to 12% NuPage gel (Invitrogen) with 2(N-morpholino)ethanesulfonic acid buffer. Gels were stained with Coomassie blue (GelCode blue stain reagent; Pierce, Rockford, IL), and bands of interest were excised from the polyacrylamide gel and subsequently digested with trypsin. Tandem mass spectrometry and database searching were performed at the National Institute of Allergy and Infectious Diseases (NIAID) core facility.
Recombinant virus construction.
The recombinant viruses constructed and used during this study were the following: vA28iΔA56, vA28iΔK2, vA56tap, vA56TAPΔK2, vK2TAP, vK2TAPΔA56, vA28TAP, vA28TAPΔK2, vK2iΔA56ΔC3, vΔA56ΔK2, and vsA56TAPiΔC3 (where i indicates an inducible gene and Δ indicates a deletion). Viruses vK2TAP, vA28TAP, and vA56TAP were constructed using DNA encoding (i) K2L, A28L, or A56R genes with 300 bp of downstream flanking region, (ii) the TAP tag derived from pCTAP (Stratagene, La Jolla, CA), and (iii) a strong VACV promoter adjacent to the gene for a fluorescent protein, namely, Heteractis crispa red fluorescent protein 1 (HcRed) from Clontech (Mountain View, CA) for K2TAP and enhanced green fluorescent protein (EGFP) from Clonetech for A28TAP. The constructs were prepared by overlapping PCR (Accuprime Pfx; Invitrogen, Carlsbad, CA) so that the TAP tag was appended immediately before the stop codon of the modified gene. The gene encoding the fluorescent reporter was inserted between the stop codon and the 300-bp flanking region. The recombinant PCR product was cloned into pCR-BluntII-TOPO (Invitrogen), and the TOPO plasmids encoding K2TAP and A28TAP were transfected using Lipofectamine 2000 (Invitrogen) into BS-C-1 cells that had been infected 1 h earlier with 1 PFU per cell of VACV. Parental and recombinant viruses were distinguished by fluorescence microscopy, and the latter were clonally purified by three rounds of plaque isolation. vA56TAP was constructed as above except that vΔA56 was used as the parental virus, and the EGFP gene in the A56R locus was replaced with A56TAP; recombinant viruses were distinguished from the parental virus by the absence of green fluorescence.
Deletion of the A56R and K2L genes was achieved by replacing the open reading frames with the DNA encoding EGFP or HcRed, respectively. Briefly, 300 bp of DNA corresponding to the left and right flanks of A56R and K2L were fused by recombinant PCR to the fluorescent protein gene. To construct vA28iΔA56 and vA28iΔK2, BS-C-1 cells were infected in the presence of 100 μM isopropyl-β-d-thiogalactopyranoside (IPTG) with vA28i (31) at 1 PFU per cell and then transfected with the respective A56 or K2 deletion plasmid. Recombinant viruses were distinguished from parental virus by fluorescence microscopy and clonally purified by three rounds of plaque isolation. vΔA56 and vK2TAPΔA56 were constructed by deletion of the A56R gene from VACV strain WR and vK2TAP, respectively, utilizing an approach analogous to that described for vA28iΔA56. The K2L gene was deleted from vΔA56, vA56TAP, and vA28TAP as described for vA28iΔK2, and the resulting viruses were designated vΔA56ΔK2, vA56TAPΔK2, and vA28TAPΔK2, respectively.
vK2i was designed to inducibly overexpress an HA epitope-tagged inducible K2. The corresponding transfer plasmid was assembled by recombinant PCR from (i) 200 bp of DNA upstream of the A56R gene, (ii) bacteriophage T7 promoter and encephalomyocarditis virus leader sequence containing an internal ribosome entry site from pVote 1 (41), (iii) K2L with a C-terminal influenza HA epitope tag, (iv) the HcRed gene regulated by a strong VACV promoter, and (v) 200 bp of DNA downstream of A56R. The final PCR product was cloned into PCR-BluntII-TOPO and sequenced. The K2L expression plasmid was transfected into cells infected with vT7lacOI (2), and the vK2i plaques were detected by fluorescence microscopy. vK2i was clonally purified by three rounds of plaque isolation.
vsA56TAPi, a virus encoding an inducible A56 that is secreted from cells because of deletion of its transmembrane segment and containing the TAP, V5, and ten-histidine tags, was constructed using a modification of a previously described plaque selection system (4). We first constructed vT7lacOIΔF13 by deleting the F13L gene from vT7lacOI. A DNA segment was assembled by overlapping PCR using DNA encoding (i) the T7 promoter, encephalomyocarditis leader sequence, and Escherichia coli lac operator from pVote 1 to provide inducible expression and cap-independent translation; (ii) A56R gene with a V5 tag inserted between amino acids 18 and 19 and replacement of amino acids 280 to 315 with a TAP tag followed by 10 tandem copies of a histidine codon; and (iii) a T7 termination sequences from pVote 1. This DNA was then cloned into pRB21 (4), and the resulting plasmid was used to transfect BS-C-1 cells that had been infected with vT7lacOIΔF13. The new recombinant virus vsA56TAPi formed large plaques and was clonally purified. The C3L gene was deleted from vsA56TAPi and vK2i in a similar fashion as described for deletion of A56 to construct vsA56TAPiΔC3 and vK2iΔA56ΔC3, respectively.
Western blotting.
Affinity-purified protein samples from 2 × 108 to 3 × 108 cells were applied to 10% or 4 to 12% NuPage Bis-Tris gel (Invitrogen). After electrophoresis, the proteins were transferred to nitrocellulose membranes and blocked with Tris-buffered saline supplemented with 5% nonfat dried milk and 0.05% Tween-20 for 1 h at room temperature. The membranes were then incubated with the appropriate primary antibody, washed, incubated with horseradish peroxidase-conjugated secondary antibodies (GE healthcare, Piscataway, NJ), and analyzed with the SuperSignal West Dura or Femto Maximum Sensitivity Substrate chemiluminescence reagents (Pierce, Rockford, IL). Primary and secondary antibodies were removed from the membrane by incubation with Restore Western Blot Stripping Buffer (Pierce) for 30 min at 55°C.
Antibodies.
Rabbit polyclonal antisera used to detect VACV proteins were anti-A21 (35), anti-L5 (34), anti-A16 (25), and anti-p4b/4b (R. Doms and B. Moss, unpublished data). Antibody to A28, prepared by immunizing rabbits with purified recombinant protein, was provided by Gretchen Nelson, NIAID. K2 and A56 rabbit antisera were raised against synthetic peptides PFDITKTRNASFTNKYGTKT, derived from K2 amino acids 176 to 195, and SEKPDYIDNSNCSSVF, derived from A56 amino acids 151 to 166 with the addition of a C-terminal cysteine for conjugation to keyhole limpet hemocyanin (Covance Research Products, Denver, PA). A monoclonal antibody against glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was obtained from Covance.
Protein synthesis and purification.
Ten roller bottles of RK13 cells were incubated with 1 PFU per cell each of vsA56TAPiΔC3 and vK2iΔA56ΔC3 for 2 h at 37°C. After virus adsorption the cells were washed twice with Dulbeccos's phosphate-buffered saline with calcium and magnesium (Quality Biological Inc.). Then, 50 ml of OptiMem (Invitrogen) with 2 mM IPTG was added, and the infection was allowed to proceed for 48 h. The medium was clarified by centrifugation, and any remaining debris was removed by filtering through a 0.45-μm-pore-size membrane. The medium was supplemented with glycerol to a 10% final concentration and NaCl to 400 mM. The protein was purified by binding to wheat germ agglutinin agarose (Vector Laboratories, Burlingame, CA) and eluted with 500 mM N-acetyl-α-d-glucosamine (Calbiochem, La Jolla, CA). The eluate was then bound to streptavidin-Sepharose, washed with phosphate-buffered saline, and eluted with 1 mg/ml of biotin.
RESULTS
The EFC is required for cell-cell fusion at neutral pH.
Our initial question was whether syncytium formation, which occurs spontaneously in cells infected with a VACV that has a mutation in the A56R or K2L gene, is dependent on the same protein complex that mediates virus entry and low pH-induced cell-cell fusion. The function of the protein components of the EFC in virus entry and low-pH-triggered fusion was originally determined by constructing and analyzing conditional lethal mutants in which gene expression was regulated by E. coli lac repressor (15, 24, 25, 29, 30, 32, 34, 35). Repression of any one of the genes encoding an EFC protein resulted in an identical phenotype consisting of a defect in virus entry and loss of ability to mediate low-pH-triggered fusion. Therefore, the present strategy was to determine the effect of deleting the A56R or K2L gene from one of these inducible mutants. If spontaneous fusion of infected cells occurred when EFC gene expression was repressed, it would indicate an alternative pathway of cell-cell fusion.
We chose vA28i as the inducible parental virus because regulation of A28 expression was strongly repressed in the absence of IPTG, and assembly of the EFC was prevented (31). The A56R and K2L genes of vA28i were replaced with EGFP to form vA28iΔA56 and vA28iΔK2, respectively. DNA sequencing confirmed the absence of the A56R or K2L gene. vA28iΔA56 and vA28iΔK2 stocks were prepared in the presence of IPTG so that virions contained A28 and were therefore able to infect cells. After infection in the absence of IPTG, however, the progeny virions would have low levels of A28 and be unable to spread to neighboring cells. As anticipated, both viruses expressed EGFP in the presence or absence of IPTG but formed plaques only under the former conditions. Furthermore, HeLa cells infected with vA28iΔA56 or vA28iΔK2 in the presence of IPTG formed large multinucleated syncytia visualized by fluorescence microscopy (Fig. 1). In the absence of IPTG, however, only a few small syncytia were detected with either vA28iΔA56 or vA28iΔK2 even though the cells were infected, as shown by expression of EGFP (Fig. 1). This result indicated that the A28 protein, in addition to its role in virus entry and low-pH-triggered cell-cell fusion, was required for cell-cell fusion occurring at neutral pH in the absence of the A56R or K2L gene. Extrapolating from this result, we concluded that multicomponent EFC mediates all forms of virus-induced fusion.
FIG. 1.

Fusion of infected cells in the absence of A56 or K2 is dependent on the EFC. HeLa cells were infected with vA28iΔA56 or vA28iΔK2 in the absence (−) or presence (+) of IPTG. After 24 h, the cells were examined under an inverted fluorescence microscope to visualize cells expressing GFP and then photographed. The percentages of nuclei in syncytia containing 3 or more nuclei averaged from two experiments in which the cells were stained with Hoechst dye are indicated.
The A56 and K2 proteins associate with the EFC.
The absence of a recognizable enzymatic motif in the A56 protein and the dispensability of the serine protease inhibitor active site in the K2 protein suggested that their fusion inhibitor activity might depend on protein-protein interactions (from here onward, we refer to the products of the A56R and K2L genes as A56 and K2 rather than as HA and SPI-3, as the latter names are unrelated to their roles in syncytium formation). To identify proteins that associate with the A56 protein, we constructed a recombinant VACV called vA56TAP in which codons for streptavidin and calmodulin binding peptides were appended to the end of the A56R open reading frame. The presence of the TAP tag did not compromise the function of the A56 protein, as syncytia did not form when cells were infected with vA56TAP (data not shown). Infected cells were solubilized with Triton X-100, and the postnuclear supernatant was incubated with Sepharose beads linked to streptavidin. After extensive washing, the bound proteins were eluted with d-biotin and then incubated with Sepharose beads linked to calmodulin. Finally, the calmodulin-Sepharose was washed, and the bound proteins were eluted with EGTA and concentrated by precipitation with trichloroacetic acid. The purified proteins were solubilized with sodium dodecyl sulfate (SDS), resolved by polyacrylamide gel electrophoresis (PAGE), and detected by staining with Coomassie blue. As a negative control, proteins from cells infected with VACV lacking a TAP tag were purified in parallel. Numerous intensely stained bands were evident in the A56TAP sample, whereas they were absent from the control. After bands were cut out and digested with trypsin, the peptides were identified by mass spectrometry. The proteins containing these peptides are indicated next to the bands in Fig. 2. One of the bands corresponded to the K2 protein, confirming an association with A56. The intensity of the K2 band, however, was much less than that of A56, even taking into account the difference in their masses. This suggests that either there is a surplus of A56 relative to K2, not all of the K2 is bound to HA, or the complex partly dissociates during purification. More remarkable was the copurification with A56 of A16, G9, and J5, which are members of the EFC. In other analyses we also detected the additional EFC components A28, H2, and G3. The number of EFC proteins identified in an experiment depended on the amount of protein loaded and the conditions of electrophoresis. The following non-EFC proteins also copurified with A56 and were identified by mass spectrometry (Fig. 2): the VACV products C3, I1, and O2; calmodulin; and mitochondrial ATP synthase and solute carrier family 25 proteins. C3 is a secreted modulator of complement activation (17, 18), I1 is a DNA telomere binding protein (8, 16), and O2 is a nonessential glutaredoxin (1, 27). The biological significance of the interactions of these three VACV proteins with A56 remains to be determined. Calmodulin undoubtedly came from the affinity beads; the presence of two mitochondrial proteins is unexplained.
FIG. 2.
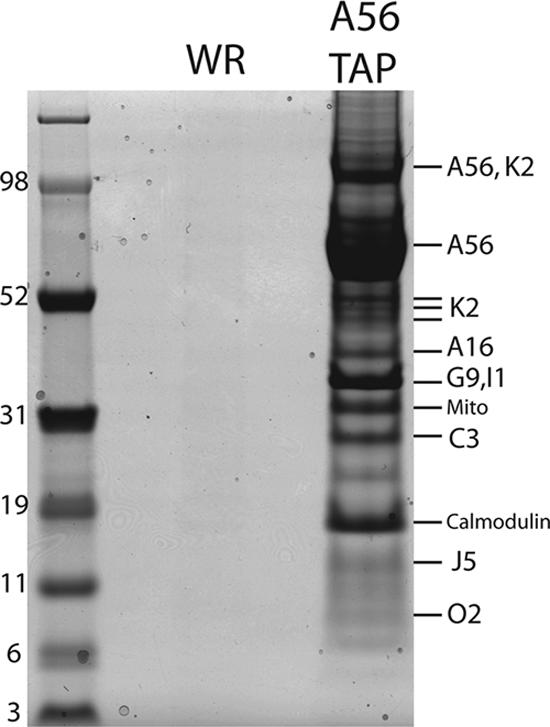
Identification by mass spectrometry of proteins that copurify with A56TAP. HeLa cells were infected with vA56TAP or VACV WR and lysed with Triton X-100, and the postnuclear supernatants were purified successively on streptavidin and calmodulin affinity columns. The bound proteins were eluted, concentrated, resolved by SDS-PAGE, and detected by staining with Coomassie blue. The protein bands were cut from the gel, digested with trypsin, and analyzed by mass spectrometry. Protein identities are indicated to the right of the stained bands. A56 and K2 were resolved as individual polypeptides and in an undissociated heterodimer. Peptides corresponding to two proteins (G9 and I1) were obtained from the same band. The band marked Mito contained peptides corresponding to the mitochondrial protein ATP synthase (gi: 4885079) and solute carrier family 25 member proteins (gi: 32189336). Marker proteins with masses in kilodaltons are indicated at the left.
Western blotting with specific antisera confirmed the specific association of A16, A21, and A28 with TAP-tagged A56 and also revealed the presence of L5, another component of the EFC (Fig. 3). The EFC proteins were not detected when extracts of cells infected with wild-type VACV lacking a TAP tag on A56 were affinity purified in parallel, even though these proteins were present in the starting material (Fig. 3). In addition, antisera to K2 and A56 were used as positive controls, and antisera to D8, an MV membrane protein not associated with the EFC, and the cellular protein GAPDH were used as negative controls. Thus, we could show that all eight EFC proteins associated with A56 either directly or indirectly.
FIG. 3.

Identification by Western blotting of proteins that copurify by tandem affinity chromatography with A56 and dependence on K2. Cells were mock infected (M) or infected with VACV WR, vA56TAP, or vA56TAPΔK2 as indicated and lysed as described in the legend to Fig. 1. Proteins were purified by affinity chromatography, resolved by SDS-PAGE, and detected by Western blotting with antibodies to the proteins indicated. A16, A28, A21, and L5 are protein components of the EFC; D8 is an MV protein that is not part of the EFC; GAPDH is a cellular protein used as a control. PreTAP, proteins analyzed prior to chromatography; TAP, proteins analyzed after chromatography. Viruses are indicated above the lanes.
A56 does not associate with the entry complex in the absence of K2.
Since A56 associates with K2, we were uncertain which protein has the primary role in binding the EFC. To determine if A56 alone could associate with the complex, we replaced the K2L gene of vA56TAP with DNA encoding EGFP. As expected, cells infected with the K2L-deleted virus vA56TAPΔK2 fused to neighboring cells (data not shown). When extracts of cells infected with vA56TAPΔK2 were affinity purified in parallel with extracts of cells infected with vA56TAP, EFC proteins were detected in only the latter (Fig. 3). Nevertheless, expression of A16, A28, L5, and A21 in cells infected with vA56TAPΔK2 was similar to expression in cells infected with either WR or A56TAP (Fig. 3). Although a trace of GAPDH copurified with K2TAP, this contamination was independent of A56. The results suggested that K2 is important for interaction with the entry complex, possibly by direct association.
K2 and A56 are both required for association with the EFC.
Based on the above results, we considered the possibility that the EFC interacted with A56/K2 through K2 and only indirectly with A56. To test this hypothesis, we needed to construct a recombinant VACV in which K2 had a TAP tag and A56 was deleted. The first step was to construct vK2TAP, a recombinant VACV in which the TAP tag was appended to the C terminus of the K2 protein. The presence of the TAP tag slightly affected the function of K2, as there was a small increase in syncytia formation when cells were infected with vK2TAP (data not shown), though this was well below the level of a K2 deletion virus. Western blot analysis of tandem-affinity-purified K2 showed the presence of both A56 and components of the EFC, confirming that the tag did not greatly compromise the function of K2 (shown below). The next step was to delete A56 to determine whether K2 was sufficient for interaction with the EFC. Cells infected with vK2TAPΔA56 formed large multinucleated syncytia, consistent with the phenotype observed for an A56 deletion mutant (data not shown). Initial experiments indicated that EFC proteins did not affinity purify with K2TAP when A56 was not expressed. However, under these conditions, K2 is partially secreted into the medium, as it does not have a membrane anchor. To prevent secretion and potentially allow more opportunity for K2 to interact with the EFC, we employed brefeldin A, an antibiotic that disrupts the Golgi apparatus (10, 21). In cells infected with VACV, the disruption of the Golgi apparatus by brefeldin A has little effect on the formation of MV, while significantly reducing the amount of EV (40).
To determine if A56 is required for binding to the EFC HeLa cells were infected with VACV WR, vK2TAP, or vK2TAPΔA56 in the presence or absence of 10 μg/ml of brefeldin A. Analysis of the cell lysates prior to affinity purification confirmed the absence of A56 synthesis in cells infected with vK2TAPΔA56 (Fig. 4). The mobility of A56 from cells infected with VACV WR or vK2TAP was altered by brefeldin A, as expected from failure to transit the Golgi apparatus (Fig. 4). The amount of A56 also seemed to increase, possibly because shedding of EVs was prevented. Similarly, the mobility of K2 was altered and its expression was increased, particularly in the absence of A56. In contrast, there was no noticeable effect of brefeldin A on the nonglycosylated EFC proteins or on the core protein p4b (Fig. 4). The remainder of each lysate was subjected to TAP and then analyzed by Western blotting. Analysis of proteins purified from cells infected with vK2TAP demonstrated interaction of the TAP-tagged K2 with A56 and entry proteins A16, A28, A21, and L5 in the absence or presence of brefeldin A (Fig. 4). K2 did not interact with p4b, indicating that the EFC interaction was specific. Importantly, K2 was unable to associate with A16, A28, A21, and L5 in the absence of A56. Though brefeldin A increased the amount of K2, this did not result in association with the EFC proteins. Thus, both A56 and K2 are needed for association with the EFC. Although a trace of GAPDH copurified with K2TAP, this contamination was independent of A56.
FIG. 4.

Identification of proteins that copurify with K2TAP and dependence on A56. Cells were mock infected or infected with VACV WR, vK2TAP, or vK2TAPΔA56 as indicated and lysed as described in the legend to Fig. 1; proteins were purified by affinity chromatography, resolved by SDS-PAGE, and detected by Western blotting with antibodies to the proteins A56, K2, A16, A21, L5, and p4b, as indicated. PreTAP, proteins prior to chromatography; TAP, proteins after chromatography. The absence (−) or presence (+) of brefeldin A during the infection is indicated.
Association of A56 and K2 with the TAP-tagged EFC.
Thus far, we have demonstrated association of TAP-tagged A56 and K2 with the EFC. To confirm this interaction, we performed the reciprocal experiment by constructing another recombinant VACV with DNA encoding the TAP tag at the 3′ terminus of the A28L gene. The A28 protein was chosen as it had been shown to exhibit normal function with the addition of the influenza HA epitope (31). The recombinant VACV vA28TAP grew to high titers, indicating that there was no defect in A28 function. We also constructed another recombinant VACV in which the K2L gene of vA28TAP was replaced with the gene encoding EGFP. As expected, vA28TAPΔK2 formed syncytia at neutral pH. Lysates from HeLa cells infected with VACV WR, vA28TAP, or vA28TAPΔK2 were analyzed by Western blotting. We confirmed that the mobility of A28TAP was less than that of A28 due to the increased mass and that K2 was absent from vA28TAPΔK2 (Fig. 5). The remainder of each lysate was subjected to tandem affinity purification followed by Western blotting. Both A56 and K2 copurified with A28TAP from cells infected with vA28TAP (Fig. 5). The specificity of the interaction was demonstrated by the failure of the core protein p4b or the cellular protein GAPDH to copurify with A28TAP and the absence of any bands from affinity-purified extracts of cells infected with VACV WR (Fig. 5). In contrast, the major A56 band was absent when A28TAP was purified from cells infected with vA28TAPΔK2 (Fig. 5). However, a much less intense doublet migrating faster than A56 copurified with A28TAP in the presence or absence of K2 (Fig. 5). Our initial speculation was that the doublet represented cross-reactivity of the anti-A56 peptide antibody, but the same result was obtained when Western blotting was carried out with a monoclonal antibody against A56 (data not shown). If the doublet is really a minor form of A56, then apparently it can bind to A28TAP in the absence of K2. Even so, the absence of the major A56 band confirms the importance of K2 for binding the EFC.
FIG. 5.

Association of A56/K2 with TAP-tagged A28. Cells were infected with VACV WR, vA28TAP, and vA28TAPΔK2 as indicated and lysed as described in the legend to Fig. 1; proteins were purified by affinity chromatography, resolved by SDS-PAGE, and detected by Western blotting with antibodies to the proteins A56, K2, A28, p4b, and GAPDH, as indicated. PreTAP, proteins prior to chromatography; TAP, proteins after chromatography.
Association of soluble A56/K2 with the EFC.
We prepared a soluble form of the A56/K2 heterodimer to further characterize the association between these regulatory proteins with the EFC. Our basic strategy was to prepare two recombinant VACVs that inducibly overexpress K2 or a secreted form of A56 using vT7lacOI (2) as the starting virus. One of the recombinant VACVs had a deleted A56 gene and a K2 gene regulated by the bacteriophage T7 promoter and E. coli lac operator. The second recombinant VACV had an inducible A56 in which the membrane anchor and cytoplasmic tail sequences were replaced by codons for the TAP tag. Initial experiments confirmed secretion of the soluble K2/A56TAP complex from coinfected cells. However, the C3 protein was also associated with the complex. (Note that association of C3 with the full-length K2/A56 complex is shown in Fig. 1). Since C3 is a virus-encoded host defense protein and not required for VACV replication, we replaced the gene with one encoding EGFP in both recombinant VACVs to form vK2iΔA56ΔC3 and vsA56TAPiΔC3 which inducibly express K2 and soluble TAP-tagged A56 in the absence of C3.
A soluble complex of A56 and K2 was isolated by coinfecting RK13 cells with vK2iΔA56ΔC3 and vsA56TAPiΔC3 in medium containing a low concentration of serum and IPTG. IPTG was required to induce expression, while the low serum concentration reduced the level of contaminating proteins. Since both K2 and A56 are glycosylated, we used immobilized wheat germ agglutinin to concentrate the proteins from the medium. This was followed by an affinity step with streptavidin-Sepharose. After the two-step purification, the soluble complex of A56 and K2 was free of major contaminating proteins (Fig. 6A).
FIG. 6.

Interaction of soluble A56/K2 with the EFC. (A) Soluble A56/K2 was purified from the medium of cells coinfected with vK2iΔA56ΔC3 and vsA56TAPiΔC3 in the presence of IPTG by a two-step procedure using immobilized wheat germ agglutinin and streptavidin. After concentration, the purified protein was analyzed by SDS-PAGE and silver staining. (B) A postnuclear lysate from uninfected HeLa cells or HeLa cells infected with vΔA56ΔK2 (ΔΔ) was incubated with purified soluble A56TAP/K2. The proteins were then subjected to TAP and analyzed by SDS-PAGE and Western blotting using antibodies to A56, K2, A16, A28, A21, L5, and p4b, as indicated. Right lane is a control that shows proteins that copurified with A56TAP from cells infected with vA56TAP.
To assess an interaction between purified soluble A56/K2 and the EFC in an infected cell lysate, it was necessary that the latter be free of competing endogenous A56 and K2. Therefore, a recombinant VACV, vΔA56ΔK2, was constructed by sequential replacement of the A56R and K2L genes with DNA encoding EGFP and HcRED, respectively. Western blot analysis confirmed the absence of both proteins (data not shown). Soluble A56/K2 was incubated with the postnuclear lysate of HeLa cells infected with vΔA56ΔK2 or mock infected. Interaction between A56/K2 and the EFC was determined by tandem affinity chromatography, taking advantage of the tag on the recombinant A56 protein. Western blotting confirmed an association between soluble A56/K2 and A16, A28, A21, and L5 but not with the core protein p4b (Fig. 6B). Detection of the EFC was dependent on soluble A56/K2, as none of these proteins was detected in its absence. Note that the secreted form of A56 appears to be a homogenous, fully glycosylated protein, whereas A56 from infected cell lysates (shown on the right of Fig. 6B) is heterogeneous, reflecting incomplete glycosylation.
DISCUSSION
Penetration of the VACV core into the cytoplasm follows fusion of the MV with the plasma membrane at neutral pH (6) or the endosomal membrane at low pH (36). In addition, by lowering the pH of the medium to <6, fusion of the MV with the plasma membrane is greatly enhanced, mimicking endosomal entry (36). VACV also induces cell-cell fusion under several different conditions, e.g., fusion of cells at late times after infection after a brief low-pH shock (called fusion from within) (9, 12), fusion of cells following a low-pH shock immediately after inoculation with large numbers of MVs (called fusion from without) (12), and spontaneous fusion of infected cells at neutral pH to form syncytia in the absence of the regulatory proteins A56 or K2 (14, 19, 37, 42). Fusion from within and fusion due to absence of A56 (and presumably K2) depend on the production of extracellular virus particles (3). Therefore, virus-cell fusion and cell-cell fusion are closely related phenomena. In this context, previous studies had demonstrated that virus-cell fusions as well as low-pH-induced cell-cell fusion depend on the recently discovered EFC (30, 36). The present study completes the picture by showing that neutral-pH cell-cell fusion is also dependent on the EFC. This result fits with the idea that fusion of infected cells is preceded by fusion of surface MVs with the cell, thereby depositing the EFC in the plasma membrane and allowing fusion with neighboring cells (23).
Since the A56 protein has no catalytic motif and mutation of the serpin motif of K2 does not result in syncytia formation, we suspected that A56/K2 might prevent fusion by direct or indirect interaction with the EFC. To test this hypothesis, we made a panel of recombinant VACVs that have a TAP tag attached to A56, K2, or the A28 EFC protein. Interaction between A56/K2 and the EFC was demonstrated by copurification followed by mass spectrometry or Western blotting. Furthermore, TAP-tagged A56 did not interact with the EFC in the absence of K2, nor did TAP-tagged K2 interact with the EFC in the absence of A56. The significance of our detection of an apparently minor form of A56 that copurified with TAP-tagged EFC in the absence of K2 is not yet understood. The finding that both A56 and K2 are required for efficient binding to the EFC fits well with experiments showing that mutation of either A56 or K2 results in spontaneous fusion of infected cells. Whether components of the EFC interact with both A56 and K2 or to only one of these regulatory proteins, which is modified by interaction with the other, remains to be determined.
Although A56 and K2 are present in the EV membrane (26), their synthesis begins early in infection, and the proteins are incorporated into the plasma membrane as well (28). We suspect that the latter location is important in preventing reinfection of cells by progeny virions. However, since the EFC is in the MV membrane, the outer EV membrane must be disrupted before interaction with A56/K2 can occur. A recent study suggests that this disruption may be mediated by glycosaminoglycans on the cell surface (20). Under our experimental conditions, used to characterize the interaction between A56/K2 and the EFC, the cells were lysed with detergent. Therefore, the interaction we observed might have occurred after lysis.
We also characterized a soluble form of A56/K2 that was secreted into the medium by cells coinfected with two recombinant viruses, one of which overexpressed a TAP-tagged A56 protein lacking the cytoplasmic domain and membrane anchor while the other overexpressed K2 but had a deleted A56 gene. The soluble A56/K2 was purified by virtue of the TAP tag on A56 and was shown to bind to the EFC present in infected cell lysates. The soluble A56/K2 should be useful to investigate the binding of individual EF proteins. In addition, a small-molecule mimic of A56/K2 might be developed into a novel antipoxvirus agent.
Acknowledgments
We thank Norman Cooper for providing cell cultures.
The study was supported by NIAID intramural research funds.
Footnotes
Published ahead of print on 4 April 2007.
REFERENCES
- 1.Ahn, B. Y., and B. Moss. 1992. Glutaredoxin homolog encoded by vaccinia virus is a virion-associated enzyme with thioltransferase and dehydroascorbate reductase activities. Proc. Natl. Acad. Sci. USA 89:7060-7064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alexander, W. A., B. Moss, and T. R. Fuerst. 1992. Regulated expression of foreign genes in vaccinia virus under the control of bacteriophage T7 RNA polymerase and the Escherichia coli lac repressor. J. Virol. 66:2934-2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blasco, R., and B. Moss. 1991. Extracellular vaccinia virus formation and cell-to-cell virus transmission are prevented by deletion of the gene encoding the 37,000-Dalton outer envelope protein. J. Virol. 65:5910-5920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blasco, R., and B. Moss. 1995. Selection of recombinant vaccinia viruses on the basis of plaque formation. Gene 158:157-162. [DOI] [PubMed] [Google Scholar]
- 5.Brum, L. M., P. C. Turner, H. Devick, M. T. Baquero, and R. W. Moyer. 2003. Plasma membrane localization and fusion inhibitory activity of the cowpox virus serpin SPI-3 require a functional signal sequence and the virus encoded hemagglutinin. Virology 306:289-302. [DOI] [PubMed] [Google Scholar]
- 6.Carter, G. C., M. Law, M. Hollinshead, and G. L. Smith. 2005. Entry of the vaccinia virus intracellular mature virion and its interactions with glycosaminoglycans. J. Gen. Virol. 86:1279-1290. [DOI] [PubMed] [Google Scholar]
- 7.Condit, R. C., N. Moussatche, and P. Traktman. 2006. In a nutshell: structure and assembly of the vaccinia virion. Adv. Virus Res. 66:31-124. [DOI] [PubMed] [Google Scholar]
- 8.DeMasi, J., S. Du, D. Lennon, and P. Traktman. 2001. Vaccinia virus telomeres: Interaction with the viral I1, I6, and K4 proteins. J. Virol. 75:10090-10105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doms, R. W., R. Blumenthal, and B. Moss. 1990. Fusion of intra- and extracellular forms of vaccinia virus with the cell membrane. J. Virol. 64:4884-4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doms, R. W., G. Russ, and J. W. Yewdell. 1989. Brefeldin A redistributes resident and itinerant Golgi proteins to the enodplasmic reticulum. J. Cell Biol. 109:61-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Earl, P. L., N. Cooper, S. Wyatt, B. Moss, and M. W. Carroll. 1998. Preparation of cell cultures and vaccinia virus stocks, p. 16.16.1-16.16.3. In F. M. Ausubel, R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl (ed.), Current protocols in molecular biology, vol. 2. John Wiley and Sons, New York, NY. [Google Scholar]
- 12.Gong, S. C., C. F. Lai, and M. Esteban. 1990. Vaccinia virus induces cell fusion at acid pH and this activity is mediated by the N terminus of the 14-kDa virus envelope protein. Virology 178:81-91. [DOI] [PubMed] [Google Scholar]
- 13.Husain, M., A. S. Weisberg, and B. Moss. 2006. Existence of an operative pathway from the endoplasmic reticulum to the immature poxvirus membrane. Proc. Natl. Acad. Sci. USA 103:19506-19511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ichihashi, Y., and S. Dales. 1971. Biogenesis of poxviruses: interrelationship between hemagglutinin production and polykaryocytosis. Virology 46:533-543. [DOI] [PubMed] [Google Scholar]
- 15.Izmailyan, R. A., C. Y. Huang, S. Mohammad, S. N. Isaacs, and W. Chang. 2006. The envelope G3L protein is essential for entry of vaccinia virus into host cells. J. Virol. 80:8402-8410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klemperer, N., J. Ward, E. Evans, and P. Traktman. 1997. The vaccinia virus I1 protein is essential for the assembly of mature virions. J. Virol. 71:9285-9294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kotwal, G. J., S. N. Isaacs, R. Mckenzie, M. M. Frank, and B. Moss. 1990. Inhibition of the complement cascade by the major secretory protein of vaccinia virus. Science 250:827-830. [DOI] [PubMed] [Google Scholar]
- 18.Kotwal, G. J., and B. Moss. 1988. Vaccinia virus encodes a secretory polypeptide structurally related to complement control proteins. Nature 335:176-178. [DOI] [PubMed] [Google Scholar]
- 19.Law, K. M., and G. L. Smith. 1992. A vaccinia serine protease inhibitor which prevents virus-induced cell fusion. J. Gen. Virol. 73:549-557. [DOI] [PubMed] [Google Scholar]
- 20.Law, M., G. C. Carter, K. L. Roberts, M. Hollinshead, and G. L. Smith. 2006. Ligand-induced and non-fusogenic dissolution of a viral membrane. Proc. Natl. Acad. Sci. USA 103:5989-5994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lippincott-Schwartz, J., L. C. Yuan, J. S. Bonifacino, and R. D. Klausner. 1989. Rapid redistribution of Golgi proteins into the ER in cells treated with brefeldin A: evidence for membrane cycling from Golgi to ER. Cell 56:801-813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moss, B. 2001. Poxviridae: the viruses and their replication, p. 2849-2883. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 4th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
- 23.Moss, B. 2006. Poxvirus entry and membrane fusion. Virology 344:48-54. [DOI] [PubMed] [Google Scholar]
- 24.Ojeda, S., A. Domi, and B. Moss. 2006. Vaccinia virus G9 protein is an essential component of the poxvirus entry-fusion complex. J. Virol. 80:9822-9830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ojeda, S., T. G. Senkevich, and B. Moss. 2006. Entry of vaccinia virus and cell-cell fusion require a highly conserved cysteine-rich membrane protein encoded by the A16L gene. J. Virol. 80:51-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Payne, L. G. 1979. Identification of the vaccinia hemagglutinin polypeptide from a cell system yielding large amounts of extracellular enveloped virus. J. Virol. 31:147-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rajagopal, I., B. Y. Ahn, B. Moss, and C. K. Mathews. 1995. Roles of vaccinia virus ribonucleotide reductase and glutaredoxin in DNA precursor biosynthesis. J. Biol. Chem. 270:27415-27418. [DOI] [PubMed] [Google Scholar]
- 28.Seki, M., M. Oie, Y. Ichihashi, and H. Shida. 1990. Hemadsorption and fusion inhibition activities of hemagglutinin analyzed by vaccinia virus mutants. Virology 175:372-384. [DOI] [PubMed] [Google Scholar]
- 29.Senkevich, T. G., and B. Moss. 2005. Vaccinia virus H2 protein is an essential component of a complex involved in virus entry and cell-cell fusion. J. Virol. 79:4744-4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Senkevich, T. G., S. Ojeda, A. Townsley, G. E. Nelson, and B. Moss. 2005. Poxvirus multiprotein entry-fusion complex. Proc. Natl. Acad. Sci. USA 102:18572-18577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Senkevich, T. G., B. M. Ward, and B. Moss. 2004. Vaccinia virus A28L gene encodes an essential protein component of the virion membrane with intramolecular disulfide bonds formed by the viral cytoplasmic redox pathway. J. Virol. 78:2348-2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Senkevich, T. G., B. M. Ward, and B. Moss. 2004. Vaccinia virus entry into cells is dependent on a virion surface protein encoded by the A28L gene. J. Virol. 78:2357-2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith, G. L., A. Vanderplasschen, and M. Law. 2002. The formation and function of extracellular enveloped vaccinia virus. J. Gen. Virol. 83:2915-2931. [DOI] [PubMed] [Google Scholar]
- 34.Townsley, A., T. G. Senkevich, and B. Moss. 2005. The product of the vaccinia virus L5R gene is a fourth membrane protein encoded by all poxviruses that is required for cell entry and cell-cell fusion. J. Virol. 79:10988-10998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Townsley, A., T. G. Senkevich, and B. Moss. 2005. Vaccinia virus A21 virion membrane protein is required for cell entry and fusion. J. Virol. 79:9458-9469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Townsley, A. C., A. S. Weisberg, T. R. Wagenaar, and B. Moss. 2006. Vaccinia virus entry into cells via a low pH-dependent-endosomal pathway. J. Virol. 80:8899-8908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Turner, P. C., and R. W. Moyer. 1992. An orthopoxvirus serpin-like gene controls the ability of infected cells to fuse. J. Virol. 66:2076-2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Turner, P. C., and R. W. Moyer. 1995. Orthopoxvirus fusion inhibitor glycoprotein SPI-3 (open reading frame K2L) contains motifs characteristic of serine protease inhibitors that are not required for control of cell fusion. J. Virol. 69:5978-5987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Turner, P. C., and R. W. Moyer. 2006. The cowpox virus fusion regulator proteins SPI-3 and hemagglutinin interact in infected and uninfected cells. Virology 347:88-99. [DOI] [PubMed] [Google Scholar]
- 40.Ulaeto, D., D. Grosenbach, and D. E. Hruby. 1995. Brefeldin A inhibits vaccinia virus envelopment but does not prevent normal processing and localization of the putative envelopment receptor P37. J. Gen. Virol. 76:103-111. [DOI] [PubMed] [Google Scholar]
- 41.Ward, G. A., C. K. Stover, B. Moss, and T. R. Fuerst. 1995. Stringent chemical and thermal regulation of recombinant gene expression by vaccinia virus vectors in mammalian cells. Proc. Natl. Acad. Sci. USA 92:6773-6777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou, J., X. Y. Sun, G. J. P. Fernando, and I. H. Frazer. 1992. The vaccinia virus K2L gene encodes a serine protease inhibitor which inhibits cell-cell fusion. Virology 189:678-686. [DOI] [PubMed] [Google Scholar]