

Abstract
Assembly of retrovirus particles normally entails the selective encapsidation of viral genomic RNA. However, in the absence of packageable viral RNA, assembly is still efficient, and the released virus-like particles (termed “Ψ−” particles) still contain roughly normal amounts of RNA. We have proposed that cellular mRNAs replace the genome in Ψ− particles. We have now analyzed the mRNA content of Ψ− and Ψ+ murine leukemia virus (MLV) particles using both microarray analysis and real-time reverse transcription-PCR. The majority of mRNA species present in the virus-producing cells were also detected in Ψ− particles. Remarkably, nearly all of them were packaged nonselectively; that is, their representation in the particles was simply proportional to their representation in the cells. However, a small number of low-abundance mRNAs were greatly enriched in the particles. In fact, one mRNA species was enriched to the same degree as Ψ+ genomic RNA. Similar results were obtained with particles formed from the human immunodeficiency virus type 1 (HIV-1) Gag protein, and the same mRNAs were enriched in MLV and HIV-1 particles. The levels of individual cellular mRNAs were ∼5- to 10-fold higher in Ψ− than in Ψ+ MLV particles, in agreement with the idea that they are replacing viral RNA in the former. In contrast, signal recognition particle RNA was present at the same level in Ψ− and Ψ+ particles; a minor fraction of this RNA was weakly associated with genomic RNA in Ψ+ MLV particles.
During the assembly of infectious, wild-type retrovirus particles, the viral Gag protein selectively encapsidates the viral genomic RNA. The selection of this RNA for incorporation into the nascent virion is presumably due to the recognition by Gag of a cis-acting “packaging signal,” termed “Ψ,” in the viral RNA. However, the packaging of this RNA is completely dispensable for efficient particle assembly (22, 26). We previously reported that murine leukemia virus (MLV) particles formed in the absence of packageable genomic RNA (“Ψ−” particles) still contain measurable amounts of RNA. Moreover, RNA appears to play a structural role in MLV particles, as RNase digestion of detergent-stripped immature particles solubilizes a substantial fraction of the Gag protein. We observed this effect both with particles containing genomic RNA (“Ψ+” particles) and with Ψ− particles (29).
What is the nature of the RNA present in virions? In addition to two copies of genomic RNA, a wild-type particle is known to contain between 10 and 50 tRNA molecules (two of which are annealed to the genomic RNA and will serve as primers for reverse transcription) and at least one molecule of signal recognition particle (SRP) RNA (initially known as 7SL RNA), the RNA component of the signal recognition particle (4, 5, 10, 25, 39). Several cellular mRNAs have also been detected in purified virus particles (1, 3, 13, 20, 29). It is significant that retroviral genomic RNA is also the mRNA for the Gag and Gag-Pol proteins and that, like most cellular mRNAs, it is capped at its 5′ end and polyadenylated at its 3′ end.
Little is known about the mechanism by which Gag selects Ψ+ RNA for incorporation into the nascent virion. It seems possible that the selection of cellular RNAs for incorporation into retrovirus particles could provide clues regarding this mechanism. We now present a quantitative analysis of the cellular RNAs present in both Ψ− and Ψ+ MLV-derived virus-like particles (VLPs). Our analysis indicates that cellular mRNAs are packaged to replace the genomic RNA in Ψ− particles. There seems to be very little selectivity in this process, as the majority of the thousands of mRNA species present in VLP-producing cells are incorporated into the VLPs simply in proportion to their representation in the cells. However, mitochondrial mRNAs are not packaged in the VLPs. In addition, a small number of cellular mRNAs are very significantly enriched in the VLPs: at least one species is encapsidated at the same efficiency as Ψ+ RNA. Despite this enrichment, these species do not constitute a major fraction of the RNA in the VLPs, because they are at such low levels in the cells. We also expressed the human immunodeficiency virus type 1 (HIV-1) Gag protein in mammalian cells and analyzed the cellular RNAs packaged in the VLPs assembled in these cells; the results were quite similar to those obtained with MLV.
MATERIALS AND METHODS
Production of VLPs.
This study used two Moloney MLV proviral clones. The “Ψ+” clone is the protease-negative (PR−) active-site mutant D32L (12), while the Ψ− clone is a chimera in which the 5′ region of the genome, up to the XhoI site at nucleotide (nt) 1560, is from pPAM3 (27), and the remainder is from the PR− D32L clone. pPAM3 (and thus our Ψ− clone) contains a deletion in the leader RNA sequence from nt 215 to 535 of the MLV genome (27). All MLV genomes are in the plasmid vector pGCcos3neo, a derivative of pSV2neo (36).
The HIV-1 Gag protein was expressed from plasmid pCMV55M1-10 (a kind gift from B. Felber, National Cancer Institute). This plasmid encodes a Rev-independent HIV-1 gag gene, containing a number of silent mutations eliminating the Rev requirement, under the control of the cytomegalovirus major late promoter (35).
VLPs were produced by transient transfection of HEK 293T cells using Transit-293 (Mirus) as recommended by the manufacturer. Twenty-four-hour harvests were collected 48 and 72 h after transfection. Supernatants were filtered through 0.45-μm filters, and VLPs were isolated by pelleting through 20% sucrose in TNE (10 mM Tris-HCl [pH 7.4]-0.1 M NaCl-1 mM EDTA). The pellets were redissolved in TNE before further analysis. In some experiments, the 293T cells had previously been stably transfected with the MLV-derived vector pLXSH (28).
Isolation of RNA from VLPs and Cells.
RNA was isolated from VLPs in PK lysis buffer (50 mM Tris-Cl [pH 7.4], 100 mM NaCl, 10 mM EDTA, 1% sodium dodecyl sulfate, 100 μg/μl proteinase K [Ambion]), followed by phenol-chloroform extraction, exactly as described previously (12), except that GlycoBlue (Ambion), rather than tRNA, was used as a carrier in ethanol precipitations. Extracted RNA was treated with 100 U/ml RQ1 DNase (Promega) for 60 min at 37°C. DNase was then inactivated by the addition of guanidine HCl to 2 M, and the RNA was reprecipitated before further analysis.
Yields of RNA from VLPs were determined by the Ribogreen (Invitrogen) assay, which was performed according to the manufacturer's instructions. Escherichia coli 16S and 23S rRNA provided by the manufacturer were used as a standard.
For the isolation of cellular RNA, transfected cells were first collected in phosphate-buffered saline and washed by centrifugation. The cellular pellet was resuspended in 0.5 ml phosphate-buffered saline, and RNA was then extracted using TRIzol (Invitrogen) according to the manufacturer's protocol. Cell extracts were digested with RQ1 DNase, and the RNA was reprecipitated before further analysis. Cellular RNA was quantitated by measuring the A260. When these RNA concentrations were checked by Ribogreen assay, the results were in excellent agreement.
Isolation of RNA from VLPs for use in Affymetrix microarrays.
Each HIV-1 or MLV (Ψ−) VLP RNA sample was analyzed on a separate Affymetrix Human Genome U133 2.0 Plus array. In each case, the mRNA composition of a VLP sample was compared with that of the transfected cells that produced the VLPs.
Several protocols were used to prepare the VLP RNA for the microarray analyses: in one, the extracted RNA was purified on RNeasy spin columns (QIAGEN) and treated with DNase I; in another, the particles were treated with DNase I prior to RNA extraction; in a third protocol, the VLPs were digested with subtilisin (32) and pelleted through 20% sucrose before RNA extraction. These variations in the experimental procedures appeared to have no effect on the overall results.
Affymetrix Gene Chip and data analysis.
RNAs were first processed using the Invitrogen SuperScript Double-Stranded cDNA synthesis kit and T7-oligo (dT)24 primers to make double-stranded cDNA. The samples were then labeled using the Enzo BioArray High Yield RNA Transcript Labeling kit to generate biotinylated cRNA. After purification by the Affymetrix GeneChip Sample Module Cleanup kit, 12.5 μg of cRNA was fragmented at 94°C for 35 min. The fragmented cRNA was placed in a hybridization cocktail and applied to Affymetrix Human U133 Plus 2.0 arrays (over 47,000 transcripts) for 18 h in the GeneChip Hybridization Oven 640 (Affymetrix). Probe signals were amplified using streptavidin-phycoerythrin and biotinylated antibody stains in the GeneChip Fluidics Station 400. Arrays were then scanned with the GeneChip Scanner 3000. Chip output files were created using GeneChip Operating software v1.4 and normalized to one before comparison as previously described (33, 38). In each case, VLP RNA was compared with RNA from the cells that produced the VLPs. To avoid possible inaccuracies associated with measurements of extremely rare RNAs, we excluded species with signals in the cell RNA preparations that were less than 450 from this analysis. Each probe signal comparison was assigned a call (increase, decrease, or no change) based on the Wilcoxon signed-rank test P values. Comparisons were further analyzed using the Affymetrix Data Mining Tool to calculate signal log ratios. These ratios were converted to “fold change” values with the following formula:
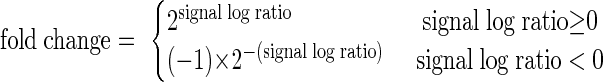 |
Results from this analysis were virtually identical in replicate experiments in which the HIV-1 Gag protein was expressed in 293T cells.
Real-time RT-PCR measurements.
Typically, each real-time reverse transcription-PCR (RT-PCR) reaction mixture contained, in a total of 30 μl, about the equivalent of 100 μl of supernatant or 100 ng of cellular RNA. RNA was reverse transcribed into DNA using random primers (Invitrogen) and Superscript II (Invitrogen) in 1× PCR buffer II supplemented with 2.5 mM MgCl2 (ABI) and 0.5 mM deoxynucleotide triphosphates (Invitrogen). The DNA was then quantitated using real-time PCR. Standard curves were generated by using RNA from in vitro transcription (Promega) of linearized plasmids containing the target sequence of interest. For PGK1 (Mammalian Gene Collection clone 3917985), ASB1 (clone 3842384), and PLEKHB2 (clone 3639839), clones containing the cDNA were obtained from the NIH Mammalian Gene Collection through Invitrogen or the American Type Culture Collection. MLV standards were generated by in vitro transcription of pMXH linearized with HindIII (11) or from a small PCR fragment containing the target sites of both the Mo2421 and SRP (7SL) primer/probe sets. Unincorporated nucleoside triphosphates were removed from RNA standards using G-50 Sephadex spin columns (Roche), and the RNA transcripts were quantitated by measuring the A260. In some experiments, the transcript was further purified by electrophoresis in 6% Tris-borate-EDTA-urea polyacrylamide gels, quantitated by Ribogreen, and stored at −20°C in aliquots. The results were independent of how the standards were prepared and quantitated. Primers/probes were ordered from Applied Biosystems for PGK1 (4310885E), ASB1 (Hs00211548_m1), and PLEKHB2 (Hs00215820_m1) as 20× ready-made primers/probes. Other primer/probe sets were synthesized by Biosource. Primers and probes are detailed in Table 1. Real-time RT-PCR results from multiple experiments were pooled to generate the data presented in this report. Data are presented as means ± standard errors of the means.
TABLE 1.
Primer/probe sets used in real-time RT-PCR analysis
| Primer/probe set | Primera | Target(s) |
|---|---|---|
| PGK1 | 4310885E | Exons 4-5 |
| ASB1 | Hs00211548_m1 | Exons 2-3 |
| PLEKHB2 | Hs00215820_m1 | Exons 1-2 |
| SRP | 5′-GTG CGG ACA CCC GAT CGG CA-3′ | Positions 51-101 of human SRP |
| 5′-TGA GGC TGG AGG ATC GCT TGA G-3′ | ||
| 5′-FAM-AGG AGT TCT GGG CTG TAG TGC-TAMRA-3′ | ||
| MLV-psi | 5′-AGG TCG GGC CAC AAA AAC GGC-3′ | Positions 341 to 404 of MLV |
| 5′-CGG ACC CGT GGT GGA ACT GAC-3′ | ||
| 5′-FAM-ACA CCC GGC CGC AAC CCT-TAMRA-3′ | ||
| MLV genome | 5′-AGG AAA GCG GTA TCG CTG GAC-3′ | Positions 2399 to 2445 of MLV |
| 5′-GAG TGG GTG ACC TTA CCG GT-3′ | ||
| 5′-FAM-ACG GAT CGC AAA GTA CAT CTA GC-TAMRA-3′ |
FAM, 6-carboxyfluorescein; TAMRA, 6-carboxytetramethylrhodamine.
Western blots and quantitation of MLV and HIV-1 Gag.
Proteins were resolved on 4% to 12% sodium dodecyl sulfate-polyacrylamide gels (Invitrogen), and MLV Pr65Gag was detected with rabbit anti-p30CA, while HIV-1 Gag was detected by using goat anti-p24CA antibody (David Ott, AIDS Vaccine Program, SAIC-Frederick), followed by the appropriate horseradish peroxidase-labeled secondary antibody (BioChain Institute, Inc.). The protein of interest was quantitated using Supersignal West Dura extended-duration substrate (Pierce) on an Alpha Innotech Corp. ChemiImager 5500. Highly purified recombinant MLV or HIV-1 Gag proteins (Δp6) (7) (a kind gift of S. A. K. Datta, National Cancer Institute) were used as absolute standards. Multiple dilutions of samples that fell within the standard curve were averaged and reported.
Northern analysis of SRP RNA.
The association of SRP RNA with the dimeric genomic RNA extracted from virions was analyzed by nondenaturing Northern analysis in 1% agarose as described previously (17). In some experiments, the dimeric RNA was extracted from a gel slice using the Zymoclean gel RNA recovery kit (Zymo Research). Specific cleavage of the genomic RNA by RNase H in the presence of an oligodeoxynucleotide complementary to nt 754 to 783 was described previously (18). Viral RNA was detected with a 32P-labeled riboprobe complementary to nt 215 to 739 of the viral RNA, while SRP RNA was detected with a 32P-labeled riboprobe complementary to human SRP RNA. The probes were transcribed from a PCR product that included a T7 polymerase promoter for the transcription of the SRP RNA sequences.
Microarray data accession number.
Microarray results have been deposited in the GEO database (http://www.ncbi.nlm.nih.gov/geo) under the accession number GSE7213.
RESULTS
Presence of SRP RNA in VLPs.
It has long been known that retrovirus particles contain significant amounts of SRP RNA, the ∼300-nt RNA component of the signal recognition particle (5, 10). However, the mechanism of its encapsidation is still not understood. We have quantitated the level of SRP RNA in both MLV and HIV-1 immature VLPs by real-time RT-PCR. In agreement with the findings of Onafuwa-Nuga and colleagues (30, 31), we found approximately 1 copy of SRP RNA per copy of genomic RNA in Ψ+ MLV particles (see below). While this roughly 1:1 stoichiometry might suggest that the association with genomic RNA is responsible for SRP encapsidation, we also found, in further agreement with data reported previously by Onafuwa-Nuga et al. (31), that the level of SRP per ng of Gag protein in Ψ− particles, which have much less genomic RNA than wild-type MLV particles, is indistinguishable from that in the latter. The data supporting these conclusions are included in Fig. 3 to 5. These results show that the incorporation of SRP RNA into VLPs is independent of genomic RNA.
FIG. 3.

Copy numbers of individual RNA species were divided by total amounts of RNA in the respective VLP and cellular RNA preparations.
FIG. 5.

Encapsidation efficiencies of individual RNAs. Encapsidation efficiency is defined as copy numbers per ng RNA in VLPs divided by copy numbers per ng RNA in the virus-producing cells.
We also tested the possibility that SRP RNA might be physically associated with genomic RNA in wild-type particles. We isolated the dimeric genomic RNA from purified wild-type MLV particles (a gift from Julian Bess and Jeff Lifson, AIDS Vaccine Program, SAIC-Frederick) by nondenaturing agarose gel electrophoresis and analyzed it by real-time RT-PCR for the copy numbers of genomic RNA and SRP RNA. (It should be noted that these are PR+, mature particles; all other virions analyzed in this report were immature.) We found that this dimeric RNA fraction contained ∼0.1 copies of SRP RNA per copy of genomic RNA (data not shown). To further investigate the association between the two RNA species, we performed nondenaturing Northern analysis on viral RNA. A faint SRP RNA signal was detected at the position of the dimeric genomic RNA, although as expected, a much more intense signal was near the bottom of the gel, at the position expected for free SRP RNA. Finally, we analyzed the thermostability of the linkage between viral RNA and the minor population of SRP RNA molecules associated with the viral RNA. Total RNA from wild-type MLV particles was progressively heated to a series of temperatures, and aliquots were removed after each heating step for nondenaturing Northern analysis. To “calibrate” the heating, the genomic RNA was cleaved at nt 754 by digestion with RNase H in the presence of an oligodeoxynucleotide complementary to this region of the genome (18), and the dissociation of the fragments of viral RNA was monitored using a probe against nt 215 to 739 of viral RNA. Under these conditions, the genomic RNA is quantitatively cleaved, but weak “tethering” interactions hold all four of the fragments of the cleaved dimeric RNA together until the RNA is heated to ∼50°C. The linkage between the two 5′ fragments, i.e., nt 1 to 754, is significantly more stable than these tethering interactions (Fig. 1A) (18). When the membrane was stripped and reprobed with an SRP RNA probe, we found that the association between SRP RNA and genomic RNA had a thermostability similar to that of the tethering interactions and was almost completely broken by incubation at 50°C (Fig. 1B).
FIG. 1.

Linkage of SRP RNA to MLV dimeric RNA in MLV virions. RNA was extracted from wild-type virions and cleaved with RNase H in the presence of an oligodeoxynucleotide complementary to nt 754 to 783 of the viral genome. The digest was then heated to the temperatures shown above the image and analyzed (A) by nondenaturing Northern blotting using a probe against nt 215 to 739 of viral RNA. The membrane was then stripped and reprobed (B) with a probe complementary to SRP RNA. D, dimeric viral RNA; 5′D, dimer of nt 1 to 754 of viral RNA; 5′F, nt 1 to 754 of viral RNA; S, SRP RNA; Un, unheated.
Quantitation of RNA in Ψ+ and Ψ− VLPs.
We previously reported that Ψ− MLV particles contain approximately the same amount of total RNA as Ψ+ particles (29). We have extended these measurements in the present study. MLV-derived VLPs were produced from 293T cells by transient transfection of a Ψ+ proviral clone or of a proviral clone bearing a deletion in Ψ. In both clones, PR had been inactivated by a point mutation at the active site; thus, the two clones produce immature VLPs. As shown in Table 2, both Ψ− and Ψ+ particles contain ∼37 pg RNA/ng of Gag. Table 2 also shows that a “mock” viral preparation, obtained from a culture transfected with an empty plasmid vector, has far less RNA than the VLP preparations; this result supports the idea that the RNA in our VLP samples is almost entirely associated with VLPs and is not cellular debris.
TABLE 2.
RNA content of retroviral particlesa
| Sample | Cell line | No. of samples | ng RNA/ml supernatant ± SD | pg RNA/ng Gag ± SD | RNA:Gag molecule ratio (bp) |
|---|---|---|---|---|---|
| Ψ− PR− | 293T | 4 | 18 ± 4.0 | 38 ± 7 | 8 |
| PR− | 293T | 3 | 44 ± 2.0 | 36 ± 14 | 7 |
| Mock | 293T | 3 | 1.1 ± 0.1 |
RNA was extracted from particles as described in Materials and Methods and quantitated using Quant-IT Ribogreen. The amount of MLV Pr65Gag or HIV-1 Pr55Gag was estimated by semiquantitative Western analysis as described in Materials and Methods.
We also created a cell line (293/pLXSH) that stably expresses the Ψ+ MLV retroviral vector pLXSH. When VLPs were produced following transient transfection of this cell line with the Ψ− MLV clone, we found that these particles also contained ∼37 pg RNA/ng Gag (data not shown). Taken together, these results demonstrate that under these conditions, there is a constant ratio of RNA to Gag that is independent of the presence of a Ψ+ genome or the context of the Ψ sequence.
Microarray analysis of mRNAs packaged in MLV and HIV-1 VLPs.
The fact that Ψ− particles contain roughly as much RNA as Ψ+ particles indicates that some cellular RNA replaces genomic RNA in Ψ− virions. Previous studies demonstrated the presence of cellular mRNAs in Ψ− retrovirus particles (13, 24, 29). We therefore analyzed the Ψ− RNA preparations for cellular mRNA by using Affymetrix HU-133 Plus 2.0 gene chips. These allow the simultaneous identification and quantitation of over 47,000 cellular transcripts. We compared the mRNA populations in the VLPs with those in the virus-producing cells; thus, we could not only assess the diversity of the encapsidated mRNA species but also estimate the fraction of cellular mRNAs that had been incorporated into the VLPs.
As shown in Table 3, the microarrays detected 16,090 probe sets in the Ψ− MLV particles; this represents about two-thirds of the probe sets found in the RNA from the cells that produced the particles. The relative quantities of the encapsidated mRNAs are discussed below.
TABLE 3.
Enumeration of mRNAs in VLPsa
| Sample | No. of “present” probes | % Present vs cell | No. of “absent” probes | No. of “marginal” probes | Total no. of probes |
|---|---|---|---|---|---|
| MLV VLPs | 16,090 | 70 | 38,021 | 564 | 54,675 |
| MLV cells | 23,073 | 30,661 | 941 | 54,675 | |
| HIV-1 VLP | 19,980 | 86 | 34,292 | 403 | 54,675 |
| HIV-1 cells | 23,112 | 30,605 | 958 | 54,675 |
Affymetrix HU133 Plus 2 gene chips were used to identify cellular mRNAs present in MLV or HIV-1 VLPs and the cells in which the particles were produced. The percentages of the probes in the VLP sample versus the numbers of probes identified in the cellular samples are given.
We also produced HIV-1 VLPs by transient transfection of an HIV-1 Gag expression plasmid in 293T cells (the same cell line that was used in the MLV experiments). Since the plasmid does not encode PR, only immature VLPs are produced. In these experiments, we detected 19,980 probe sets (Table 3); thus, just as with MLV, the HIV-1 VLPs contain the vast majority of the species present in the virus-producing cells.
Relative quantities of mRNAs in VLPs and cells.
The Affymetrix platform is able to quantitatively compare the mRNA levels between two samples. Figure 2 shows, for both MLV and HIV-1, the frequency distribution of the fold change, i.e., the level of each mRNA species in the VLPs relative to its level in the cells. Interestingly, the great majority of the probe sets identified in VLPs cluster near a fold change of 1.0. In other words, these transcripts are evidently represented in the VLPs in proportion to their steady-state expression level in the cells. Thus, these transcripts are simply a representation of the cellular mRNA pool.
FIG. 2.

Frequency distribution of fold change values for individual probe sets.
The most abundant mRNAs (i.e., the probe sets with the highest signals) in the VLPs are identified in Tables 4 and 5. For both MLV and HIV-1, these are almost all mRNAs for ribosomal proteins. They are very abundant in the cells as well as in the VLPs, as is evident from the fact that their fold change values are near one. (A single exception to this generalization is TATA element-modulatory factor 1 mRNA, which, as shown in Table 4, exhibits a high fold change value in the MLV VLPs. Its fold change was also high in the HIV-1 VLPs [data not shown]. In other words, it is enriched during encapsidation. Other mRNAs with this property are investigated in detail below.)
TABLE 4.
Most-abundant mRNAs in MLV VLPs
| Affymetrix Probe ID | MLV VLP signal | Cell signal | Fold change | Gene | Description |
|---|---|---|---|---|---|
| 226131_s_at | 56762.9 | 28547.2 | 1.97 | RPS16 | Ribosomal protein S16 |
| 213414_s_at | 55051.1 | 27659.1 | 2.06 | RPS19 | Ribosomal protein S19 |
| 200095_x_at | 51814.8 | 24602.9 | 1.89 | RPS10 | Ribosomal protein S10 |
| 201429_s_at | 51495.3 | 26922.5 | 1.92 | RPL37A | Ribosomal protein 37A |
| 201254_x_at | 50735.7 | 27560.9 | 1.87 | RPS6 | Ribosomal protein S6 |
| 212391_x_at | 50512.4 | 27271.2 | 1.82 | RPS3A | Ribosomal protein 3A |
| 208692_at | 50365.3 | 22334 | 2.27 | RPS3 | Ribosomal protein S3 |
| 214948_s_at | 49555.6 | 1713.9 | 28.05 | TMF1 | TATA element-modulatory factor 1 |
| 200662_s_at | 49452.8 | 14256.9 | 3.66 | TOMM20 | Translocase of outer mitochondrial membrane 20 |
| 210646_x_at | 49027.3 | 27195.3 | 1.83 | RPL13A | Ribosomal protein L13A |
TABLE 5.
Most-abundant mRNAs in HIV-1 VLPs
| Affymetrix Probe ID | HIV-1 VLP signal | Cell signal | Fold change | Gene | Description |
|---|---|---|---|---|---|
| 201406_at | 31099 | 32890.3 | −1.04 | RPL36A | Ribosomal protein L36A |
| 201254_x_at | 28754.9 | 27682.6 | 1.05 | RPS6 | Ribosomal protein S6 |
| 226131_s_at | 28140.6 | 28181.1 | −1.05 | RPS16 | Ribosomal protein S16 |
| 213414_s_at | 27938.5 | 28583.8 | 1.01 | RPS19 | Ribosomal protein S19 |
| 210646_x_at | 27420.3 | 26431 | 1.01 | RPL13A | Ribosomal protein L13a |
| 200936_at | 27393.7 | 25475.5 | 1.02 | RPL8 | Ribosomal protein L8 |
| 211542_x_at | 26934.6 | 28930.6 | −1.07 | RPS10 | Ribosomal protein S10 |
| 201429_s_at | 26790.4 | 27198.1 | −1.06 | RPL37A | Ribosomal protein L37a |
| 201257_x_at | 26645.3 | 26833.5 | −1.01 | RPS3A | Ribosomal protein S3A |
| 208692_at | 25813.2 | 24262.6 | 1.09 | RPS3 | Ribosomal protein S3 |
A very small peak at the left edge of Fig. 2 indicates that some probe sets exhibit very low fold change values. These are mRNAs that are abundant in cells but, unlike the majority of mRNAs, virtually excluded from the VLPs. The identities of the mRNAs with the lowest fold change values are shown in Table 6. Notably, all of these mRNAs are encoded by mitochondrial rather than nuclear DNA and are translated within the mitochondria (2, 8, 9).
TABLE 6.
mRNAs excluded from VLPsa
| Affymetrix Probe ID | MLV VLP signal | Cell signal | Fold change in MLV VLPs | HIV VLP signal | Cell signal | Fold change in HIV VLPs | Description |
|---|---|---|---|---|---|---|---|
| 238199_x_at | 12.0 | 10630.6 | −484 | 8.9 | 10998.9 | −1,314 | Cytochrome oxidase subunit III |
| 1553570_x_at | 56.1 | 23443.2 | −467 | 7.0 | 26447.7 | −3,373 | Cytochrome c oxidase II |
| 211600_at | 52.4 | 15919.0 | −224 | 15.3 | 16049.2 | −709 | NADH dehydrogenase subunit 5 |
| 224373_s_at | 99.3 | 22482.3 | −207 | 9.8 | 23111.6 | −2,320 | NADH dehydrogenase 4 |
| 1553569_at | 152.2 | 22682.3 | −164 | 13.4 | 24293.6 | −1,722 | Cytochrome c oxidase II |
| 1553538_s_at | 178.1 | 21714.8 | −120 | 60.2 | 22827.4 | −342 | Cytochrome c oxidase I |
| 1553567_s_at | 195.4 | 17332.3 | −97 | 101.1 | 19343.4 | −218 | ATP synthase 6 |
| 1553551_s_at | 242.1 | 19153.7 | −69 | 81.8 | 21195.0 | −191 | NADH dehydrogenase subunit 2 |
| 1555653_at | 170.5 | 15789.5 | −60 | 5.7 | 17050.5 | −1,314 | Homo sapiens NADH dehydrogenase subunit 5 |
| 224372_at | 359.1 | 23390.8 | −49 | 122.5 | 23926.2 | −155 | NADH dehydrogenase subunit 4 |
RNA extracted from MLV and HIV-1 VLPs, and the cells in which the particles were produced, was analyzed using Affymetrix Human Genome U133 Plus 2.0 gene chips. The expression level of individual mRNAs in VLPs was compared to the cellular expression level in cells. The table shows the RNAs with the lowest fold change values in MLV VLPs.
Inspection of Fig. 2 also shows that there is a small class of probe sets with high fold change values representing mRNAs that are significantly enriched in the VLPs. The probe sets with the highest fold change values are identified in Table 7. Interestingly, all of the species enriched in MLV VLPs also show very high fold change values in the HIV-1 VLPs. Table 7 also includes data for PGK-1 mRNA, which is not enriched but was assayed extensively by real-time RT-PCR (see below).
TABLE 7.
mRNAs highly enriched in VLPsa
| Affymetrix Probe ID | MLV VLP signal | Cell signal | Fold change in MLV VLPs | HIV VLP signal | Cell signal | Fold change in HIV VLPs | Gene |
|---|---|---|---|---|---|---|---|
| 220028_at | 32727.1 | 946.6 | 41.07 | 10542.4 | 791.2 | 13.74 | ACVR2B |
| 211383_s_at | 25297.2 | 582.6 | 39.67 | 5192.9 | 503 | 7.73 | WDR37 |
| 212818_s_at | 19777.8 | 666.5 | 39.12 | 6538.2 | 684.2 | 11.24 | ASB1 |
| 226572_at | 17353.4 | 460.8 | 34.78 | 3339.7 | 521.3 | 6.96 | SOCS7 |
| 204181_s_at | 20751.4 | 624.1 | 32.67 | 3156.9 | 521.4 | 6.73 | ZBTB43 |
| 218496_at | 33111.5 | 993.2 | 31.78 | 7356.8 | 1061.3 | 6.73 | RNASEH1 |
| 230085_at | 15230.1 | 451.6 | 31.56 | 2573.1 | 535.1 | 5.90 | PCYT1B |
| 214004_s_at | 18659 | 514.6 | 31.34 | 5298.1 | 471.5 | 9.58 | VGLL4 |
| 224611_s_at | 19328.9 | 917.7 | 29.86 | 2826.1 | 593.1 | 4.32 | DNAJC5 |
| 201411_s_at | 19528.9 | 477.7 | 28.25 | 6034 | 898.8 | 9.19 | PLEKHB2 |
| 200738_s_at | 2642 | 14815 | −5.4 | 4089 | 14252 | −3.58 | PGK1 |
Data obtained for the MLV sample were used to select genes that were enriched compared to their cellular expression level. Very rare mRNAs were eliminated from consideration by removing probe sets with cell signals of <450. The corresponding fold changes in the HIV-1 samples are also shown. ASB1, PLEKHB2, and PGK1 were selected for further analysis by real-time RT-PCR.
Validation and quantification of cellular mRNA in Ψ− MLV and HIV-1 VLPs by real-time RT-PCR.
In order to independently validate the results obtained by the microarray analysis, we used real-time RT-PCR to measure copy numbers of three individual mRNAs that had been identified in the Affymetrix experiments. We chose two species that were significantly enriched in both MLV and HIV-1, i.e., ASB-1 and PLEKHB2, and one species, PGK-1, that is not enriched in either VLP sample.
Figure 3 shows the copy numbers per ng of total RNA of PGK1, ASB1, and PLEKHB2 mRNAs; MLV genomes; and SRP RNA in Ψ− and Ψ+ MLV VLPs and in the corresponding cell cultures as well as in a “mock” cell culture. It is evident that the cellular RNA profiles are unaffected by the transfection of the proviral plasmids. Significantly, the copy numbers of ASB-1 and PLEKHB2 are greater than that of PGK1 in the VLPs, even though PGK1 is considerably more abundant than ASB-1 or PLEKHB2 in the cells. Similar results were also obtained with HIV-1 VLPs (data not shown). These differences in the relative levels of the different mRNA species are completely consistent with the microarray data (Table 7).
Cellular mRNA compensates for missing genome in Ψ− MLV VLPs.
As shown in Table 2, the ratio of total RNA to Gag protein in Ψ− is approximately the same as that in Ψ+ MLV VLPs, despite the deficiency in the packaging of genomic RNA in the Ψ− particles. Thus, some cellular RNA must replace the viral RNA in the latter. We previously suggested (29) that cellular mRNAs are packaged in Ψ− virions to compensate for the lack of viral RNA. To test this possibility, we compared the copy numbers of the three mRNA species, as well as viral RNA and SRP RNA, and normalized the values to the total amount of Gag in the viral pellets. Figure 4 shows that the levels of all three mRNAs are 5- to 10-fold higher in the Ψ− particles than in the Ψ+ particles; in contrast, copies of genomic RNA per ng Gag are ∼60-fold lower in the Ψ− sample than in the Ψ+ sample, and SRP RNA levels are very similar in the two samples. Figure 4 also shows that the ratios between the individual mRNA species are the same in Ψ− and Ψ+ particles.
FIG. 4.

Copy numbers of individual RNA species in MLV VLP preparations were divided by amounts of Gag in the preparation.
Efficiency of cellular mRNA and viral genomic RNA encapsidation into MLV VLPs.
The ratio of the copy number in VLPs to that in cellular RNA is an indication of the selectivity with which a given RNA species is packaged. The fold change reported by the Affymetrix gene chips in Table 4 to Table 7 and Fig. 2 provides this information for many RNAs, but not, of course, for the genomic RNA itself (or SRP RNA). We therefore measured the ratios (copies/ng RNA in VLPs/copies/ng RNA in cells), which we termed the “encapsidation efficiency,” for the three mRNAs as well as viral and SRP RNAs by real-time RT-PCR. Remarkably, as shown in Fig. 5, the efficiency with which the ASB-1 mRNA is packaged into Ψ− particles is somewhat higher than that of the packaging of genomic RNA in Ψ+ particles. Again, the relative efficiencies measured for the three mRNAs are qualitatively consistent with the microarray results.
Encapsidation of nonhomologous retroviral genomes.
As shown above, the great majority of cellular mRNAs are evidently packaged with little if any selectivity (Fig. 2), while Ψ+ genomic RNA is significantly enriched in VLPs (Fig. 5). It seemed possible that Ψ+ RNA is somehow qualitatively different from most mRNAs; for example, perhaps it forms dimers, which might be substrates for efficient, selective packaging, in the cytoplasm. If this were the case, heterologous retroviral genomes might be packaged relatively efficiently. To test this possibility, we generated 293T cells stably transfected with the MLV-derived vector pLXSH (28) and measured the efficiency with which the genomic RNA of this vector was packaged by HIV-1 Gag.
The results of this experiment are shown in Fig. 6. Figure 6A shows that the level of pLXSH RNA per ng of RNA is ∼10-fold higher in MLV Gag-derived VLPs than in HIV-1 Gag VLPs. As shown in Fig. 6B, when normalized to the cellular expression levels, the efficiency of ASB-1 mRNA packaging into MLV VLPs is equal to that of pLXSH RNA packaging. However, in the HIV-1 VLPs, ASB-1 mRNA is packaged with an efficiency that is 10-fold higher than that of pLXSH RNA. These results suggest that the selective packaging of genomic RNAs derives from a specific interaction with the homologous Gag protein, while the encapsidation of ASB-1 mRNA may not involve a specific interaction.
FIG. 6.

Encapsidation of an MLV-derived retroviral vector by MLV and HIV-1 Gag proteins. 293T cells were stably transfected with pLXSH and then transiently transfected with plasmids expressing either Ψ− MLV or HIV-1 Gag. Copies of pLXSH RNA were enumerated using the MLV psi primer/probe set. (A) Copy numbers of individual RNAs per ng of RNA in VLPs or in cells. (B) Encapsidation efficiencies of RNA species.
DISCUSSION
RNA is an essential component of the structure of a retrovirus particle. During assembly, Gag normally selects the Ψ+ genomic RNA for encapsidation into the nascent virion. However, if no genomic RNA is present in the cell, assembly is not detectably impaired, and the resulting “Ψ−” particles still contain RNA. Previous work from this and other laboratories suggested that cellular mRNAs are incorporated into these particles in place of the viral RNA.
The principal focus of this work has been an investigation of the mRNAs present in these Ψ− particles. We also analyzed the state of SRP RNA, which seems to be universally present in retrovirus particles. Our major findings can be summarized as follows: (i) a small fraction of the SRP RNA in a wild-type MLV particle is physically associated with the genomic RNA; (ii) the amount of total RNA per molecule of Gag is not significantly different in Ψ+ or Ψ− MLV VLPs, although the number of viral genomes differs by at least 60-fold; (iii) MLV and HIV-1 retroviral VLPs contain thousands of individual cellular mRNA species, with most of these being encapsidated simply in proportion to their level in the cell; (iv) in contrast, a small proportion of the encapsidated mRNAs are selectively enriched or excluded; (v) mRNAs are also found in Ψ+ VLPs, but at lower levels than in the Ψ− VLPs; (vi) at least one cellular mRNA is enriched as much as an MLV Ψ+ genome or vector; and (vii) an MLV-derived vector is not dramatically enriched in virions assembled from the HIV-1 Gag protein.
The amount of total RNA per Gag was similar in MLV particles produced from a Ψ− genome, a Ψ+ viral genome, or a Ψ− genome in the presence of a Ψ+ MLV vector (data not shown). This amount, ∼35 pg RNA/ng Gag, is very similar to the value obtained in early studies with alpharetrovirus particles (37) and corresponds to ∼7 to 8 bases of RNA per Gag molecule. If we assume that an MLV particle, like an HIV-1 VLP (6), contains 5,000 Gag molecules, then a dimer of genomic RNA would constitute roughly one-half of the total RNA in a virion and would be 2% of the mass of the Gag protein. Despite the relatively small quantity of RNA in the particle, it is evidently crucial in maintaining the structural integrity of the latter (21, 29).
The microarray experiments showed that a Ψ− VLP preparation (either MLV or HIV-1) contains thousands of distinct mRNA species. Somewhat analogous results have previously been reported for the particles produced by the avian cell line SE21Q1b (13). Such a heterogeneous mixture can be very difficult to detect: for example, the mRNAs in Ψ− MLV particles were virtually invisible when viral RNA preparations were end labeled and analyzed by gel electrophoresis and autoradiography (29). In contrast, individual mRNA species have readily been detected in viral preparations (1, 3, 20, 29).
The microarray experiments also showed that the great majority of mRNAs in the MLV and HIV-1 VLPs are neither selectively packaged nor specifically excluded from the VLPs. Rather, they are represented in the viral preparations simply in proportion to their levels in the virus-producing cells (Fig. 2). It is remarkable that the encapsidation mechanism, which is normally highly specific in its selection of Ψ+ RNA, is so indiscriminate in the selection of cellular mRNAs.
One exception to the lack of specificity in mRNA packaging is the exclusion of mitochondrial mRNAs from the VLPs (Table 6): these RNAs are quite abundant in the cells but are scored as “absent” in VLPs in the tabulation of the microarray data. Their absence from the VLPs is not surprising, since these mRNAs are presumably not present in the cytosol, and thus, Gag does not have access to them as it assembles into virus particles in the cytoplasm of the cell. In fact, their absence can be taken as reassuring evidence that the “VLP” preparations are not heavily contaminated with cell debris. This conclusion is also supported by the fact that there is far less RNA in “mock” viral pellets than in VLP preparations (Table 1).
The microarray results also indicated that a limited number of mRNA species are significantly enriched in the VLPs. We verified this finding by the quantitation of two enriched and one nonenriched species using real-time RT-PCR. The extent of this selective packaging is remarkable. For example, as shown in Fig. 5, ASB-1 mRNA is packaged into Ψ+ VLPs about half as efficiently as MLV genomic RNA (and even more efficiently into Ψ− VLPs). However, despite this extraordinary encapsidation efficiency, it is still much less abundant in the virions than genomic RNA. This is because it is such a rare mRNA in the cells: assuming 10 pg total RNA per cell, we estimate that each cell contains only ∼10 copies of ASB-1 mRNA. Figure 3 shows that there are roughly 40 times more copies of genomic RNA than ASB-1 mRNA per ng of cellular RNA. Thus, the level of ASB-1 mRNA in Ψ− particles is only ∼1/20 of that of genomic RNA in Ψ+ particles: even though ASB-1 mRNA is so enriched, it is still present in only a small minority of the virions. It should be noted that steady-state levels of mRNAs in cultured mammalian cells vary over an extremely wide range (15, 16, 19), and our microarray analysis excluded species with very low abundance.
It would obviously be important to understand why specific mRNAs are so highly enriched in retrovirus particles. It is interesting that the same mRNAs are enriched by both MLV Gag and HIV-1 Gag (Table 3). In contrast, HIV-1 Gag does not strongly select the genomic RNA of pLXSH, an MLV-derived vector (Fig. 6). The fact that the same mRNAs are selectively packaged by both Gag proteins, despite this specificity in encapsidation of Ψ+ viral RNAs, suggests that the enriched mRNAs are not packaged because of a resemblance to Ψ+ RNAs, but rather are somehow more available for efficient packaging than the majority of cytoplasmic mRNAs. In addition, analysis by BLAST shows no significant sequence similarity between sequences in ASB-1, or indeed any sequences in the human genome, and nt 215 to 535 of MLV, while the same type of analysis readily identified very similar sequences in the mouse genome (data not shown). It was intriguing that the ASB-1 mRNA detected in this analysis is probably almost 7 kb in length, including almost 6 kb in a long 3′-untranslated region; however, this property is not shared with other dramatically enriched mRNAs (data not shown). We are now trying to identify a packaging signal within ASB-1 RNA.
As noted above, there are many thousands of mRNA species in a VLP preparation, and it is very difficult to quantitate such a mixture. However, we could measure, with some precision, the copy numbers of individual species using real-time RT-PCR. We found that the levels of the three species that we analyzed were ∼5- to 10-fold higher (per ng of Gag) in Ψ− than in Ψ+ preparations. If this difference is representative of the entire population of mRNAs, we could conclude definitively that the increased encapsidation of cellular mRNAs compensates for the absence of genomic RNA in Ψ− particles, as suggested previously (29). However, the fact that the increase in levels of the mRNAs is only 5- to 10-fold also implies that they are present in Ψ+ wild-type viral preparations as well as in Ψ− particles. The data thus suggest that a Ψ+ preparation contains both genomic RNA, comprising about half of the total RNA, and an extremely heterodisperse assortment of cellular mRNAs, constituting in aggregate 10 to 20% of the amount of genomic RNA. Perhaps, under our transient transfection conditions, the level of Gag synthesized is slightly more than enough to encapsidate the packageable genomic RNA in the cells, and the “excess” Gag then packages mRNAs.
We also characterized the SRP RNA in VLPs. It is intriguing (Fig. 3 and 4), as reported previously (31), that the level of this RNA in Ψ− particles is not significantly different from that in Ψ+ particles. Thus, unlike what is observed with cellular mRNAs, there is evidently no competition between SRP RNA and viral RNA for space within the virion. This finding suggests that the mechanism of encapsidation of SRP RNA is different from that of genomic RNA and cellular mRNAs. We also noted that a small fraction of the SRP RNA in Ψ+ particles is physically associated with the dimeric RNA genome; this association was found to be quite thermolabile (Fig. 1). This association might result from the nucleic acid chaperone activity of NC protein within the virion (23, 34). Somewhat analogous observations have previously been made for U6 snRNA (14).
In conclusion, we have found that Ψ− retrovirus particles package cellular mRNAs in place of the viral genome. The bulk of these mRNAs are packaged nonselectively; thus, retroviral Gag proteins can alternatively engage in a highly specific interaction with the genomic RNA or in a largely nonspecific interaction with mRNAs. At the same time, a limited number of low-abundance cellular mRNA species are dramatically enriched during particle assembly. We still do not know whether the preferential packaging of Ψ+ RNA, rather than cellular mRNAs, is due to thermodynamic considerations, such as a higher intrinsic affinity of Gag for Ψ or a greater cooperativity of binding on Ψ+ RNA, or to kinetic considerations, such as the colocalization of Gag with these RNAs. It appears possible that analyses of the selective packaging of cellular mRNAs will shed some light on mechanisms used in the specific encapsidation of genomic RNA.
Acknowledgments
This work was supported in part by the Intramural Research Program of the NIH National Cancer Institute Center for Cancer Research. It was also supported in part by NIH grants R01 HL081205 and P30 ES03819 to Shyam Biswal and R01 GM-21595 and NSF grant MCB-0445841 to Thoru Pederson.
We thank Hannah Lee for technical assistance and Julian Bess, Siddhartha Datta, Barbara Felber, Jeff Lifson, and David Ott for reagents. We also thank Steve Hughes for very helpful discussions.
Footnotes
Published ahead of print on 28 March 2007.
REFERENCES
- 1.Adkins, B., and T. Hunter. 1981. Identification of a packaged cellular mRNA in virions of Rous sarcoma virus. J. Virol. 39:471-480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson, S., A. T. Bankier, B. G. Barrell, M. H. de Bruijn, A. R. Coulson, J. Drouin, I. C. Eperon, D. P. Nierlich, B. A. Roe, F. Sanger, P. H. Schreier, A. J. Smith, R. Staden, and I. G. Young. 1981. Sequence and organization of the human mitochondrial genome. Nature 290:457-465. [DOI] [PubMed] [Google Scholar]
- 3.Aronoff, R., and M. Linial. 1991. Specificity of retroviral RNA packaging. J. Virol. 65:71-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berkowitz, R., J. Fisher, and S. P. Goff. 1996. RNA packaging. Curr. Top. Microbiol. Immunol. 214:177-218. [DOI] [PubMed] [Google Scholar]
- 5.Bishop, J. M., W. E. Levinson, D. Sullivan, L. Fanshier, N. Quintrell, and J. Jackson. 1970. The low molecular weight RNAs of Rous sarcoma virus. II. The 7 S RNA. Virology 42:927-937. [DOI] [PubMed] [Google Scholar]
- 6.Briggs, J. A., M. N. Simon, I. Gross, H. G. Krausslich, S. D. Fuller, V. M. Vogt, and M. C. Johnson. 2004. The stoichiometry of Gag protein in HIV-1. Nat. Struct. Mol. Biol. 11:672-675. [DOI] [PubMed] [Google Scholar]
- 7.Campbell, S., and A. Rein. 1999. In vitro assembly properties of human immunodeficiency virus type 1 Gag protein lacking the p6 domain. J. Virol. 73:2270-2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chomyn, A., M. W. Cleeter, C. I. Ragan, M. Riley, R. F. Doolittle, and G. Attardi. 1986. URF6, last unidentified reading frame of human mtDNA, codes for an NADH dehydrogenase subunit. Science 234:614-618. [DOI] [PubMed] [Google Scholar]
- 9.Chomyn, A., P. Mariottini, M. W. Cleeter, C. I. Ragan, A. Matsuno-Yagi, Y. Hatefi, R. F. Doolittle, and G. Attardi. 1985. Six unidentified reading frames of human mitochondrial DNA encode components of the respiratory-chain NADH dehydrogenase. Nature 314:592-597. [DOI] [PubMed] [Google Scholar]
- 10.Erikson, E., R. L. Erikson, B. Henry, and N. R. Pace. 1973. Comparison of oligonucleotides produced by RNase T1 digestion of 7 S RNA from avian and murine oncornaviruses and from uninfected cells. Virology 53:40-46. [DOI] [PubMed] [Google Scholar]
- 11.Feng, Y. X., D. L. Hatfield, A. Rein, and J. G. Levin. 1989. Translational readthrough of the murine leukemia virus gag gene amber codon does not require virus-induced alteration of tRNA. J. Virol. 63:2405-2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fu, W., and A. Rein. 1993. Maturation of dimeric viral RNA of Moloney murine leukemia virus. J. Virol. 67:5443-5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gallis, B., M. Linial, and R. Eisenman. 1979. An avian oncovirus mutant deficient in genomic RNA: characterization of the packaged RNA as cellular messenger RNA. Virology 94:146-161. [DOI] [PubMed] [Google Scholar]
- 14.Giles, K. E., M. Caputi, and K. L. Beemon. 2004. Packaging and reverse transcription of snRNAs by retroviruses may generate pseudogenes. RNA 10:299-307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hames, B. D., and R. P. Perry. 1977. Homology relationship between the messenger RNA and heterogeneous nuclear RNA of mouse L cells. A DNA excess hybridization study. J. Mol. Biol. 109:437-453. [DOI] [PubMed] [Google Scholar]
- 16.Hastie, N. D., and J. O. Bishop. 1976. The expression of three abundance classes of messenger RNA in mouse tissues. Cell 9:761-774. [DOI] [PubMed] [Google Scholar]
- 17.Hibbert, C. S., J. Mirro, and A. Rein. 2004. mRNA molecules containing murine leukemia virus packaging signals are encapsidated as dimers. J. Virol. 78:10927-10938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hibbert, C. S., and A. Rein. 2005. Preliminary physical mapping of RNA-RNA linkages in the genomic RNA of Moloney murine leukemia virus. J. Virol. 79:8142-8148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holland, C. A., S. Mayrand, and T. Pederson. 1980. Sequence complexity of nuclear and messenger RNA in HeLa cells. J. Mol. Biol. 138:755-778. [DOI] [PubMed] [Google Scholar]
- 20.Ikawa, Y., J. Ross, and P. Leder. 1974. An association between globin messenger RNA and 60S RNA derived from Friend leukemia virus. Proc. Natl. Acad. Sci. USA 71:1154-1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khorchid, A., R. Halwani, M. A. Wainberg, and L. Kleiman. 2002. Role of RNA in facilitating Gag/Gag-Pol interaction. J. Virol. 76:4131-4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Levin, J. G., P. M. Grimley, J. M. Ramseur, and I. K. Berezesky. 1974. Deficiency of 60 to 70S RNA in murine leukemia virus particles assembled in cells treated with actinomycin D. J. Virol. 14:152-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levin, J. G., J. Guo, I. Rouzina, and K. Musier-Forsyth. 2005. Nucleic acid chaperone activity of HIV-1 nucleocapsid protein: critical role in reverse transcription and molecular mechanism. Prog. Nucleic Acid Res. Mol. Biol. 80:217-286. [DOI] [PubMed] [Google Scholar]
- 24.Linial, M., E. Medeiros, and W. S. Hayward. 1978. An avian oncovirus mutant (SE 21Q1b) deficient in genomic RNA: biological and biochemical characterization. Cell 15:1371-1381. [DOI] [PubMed] [Google Scholar]
- 25.Linial, M. L., and A. D. Miller. 1990. Retroviral RNA packaging: sequence requirements and implications. Curr. Top. Microbiol. Immunol. 157:125-152. [DOI] [PubMed] [Google Scholar]
- 26.Mann, R., R. C. Mulligan, and D. Baltimore. 1983. Construction of a retrovirus packaging mutant and its use to produce helper-free defective retrovirus. Cell 33:153-159. [DOI] [PubMed] [Google Scholar]
- 27.Miller, A. D., and C. Buttimore. 1986. Redesign of retrovirus packaging cell lines to avoid recombination leading to helper virus production. Mol. Cell. Biol. 6:2895-2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller, A. D., D. G. Miller, J. V. Garcia, and C. M. Lynch. 1993. Use of retroviral vectors for gene transfer and expression. Methods Enzymol. 217:581-599. [DOI] [PubMed] [Google Scholar]
- 29.Muriaux, D., J. Mirro, D. Harvin, and A. Rein. 2001. RNA is a structural element in retrovirus particles. Proc. Natl. Acad. Sci. USA 98:5246-5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Onafuwa-Nuga, A. A., S. R. King, and A. Telesnitsky. 2005. Nonrandom packaging of host RNAs in Moloney murine leukemia virus. J. Virol. 79:13528-13537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Onafuwa-Nuga, A. A., A. Telesnitsky, and S. R. King. 2006. 7SL RNA, but not the 54-kD signal recognition particle protein, is an abundant component of both infectious HIV-1 and minimal virus-like particles. RNA 12:542-546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ott, D. E., L. V. Coren, D. G. Johnson, R. C. Sowder II, L. O. Arthur, and L. E. Henderson. 1995. Analysis and localization of cyclophilin A found in the virions of human immunodeficiency virus type 1 MN strain. AIDS Res. Hum. Retrovir. 11:1003-1006. [DOI] [PubMed] [Google Scholar]
- 33.Rangasamy, T., C. Y. Cho, R. K. Thimmulappa, L. Zhen, S. S. Srisuma, T. W. Kensler, M. Yamamoto, I. Petrache, R. M. Tuder, and S. Biswal. 2004. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J. Clin. Investig. 114:1248-1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rein, A., L. E. Henderson, and J. G. Levin. 1998. Nucleic acid chaperone activity of retroviral nucleocapsid proteins: significance for viral replication. Trends Biochem. Sci. 23:297-301. [DOI] [PubMed] [Google Scholar]
- 35.Schneider, R., M. Campbell, G. Nasioulas, B. K. Felber, and G. N. Pavlakis. 1997. Inactivation of the human immunodeficiency virus type 1 inhibitory elements allows Rev-independent expression of Gag and Gag/protease and particle formation. J. Virol. 71:4892-4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Southern, P. J., and P. Berg. 1982. Transformation of mammalian cells to antibiotic resistance with a bacterial gene under control of the SV40 early region promoter. J. Mol. Appl. Genet. 1:327-341. [PubMed] [Google Scholar]
- 37.Stromberg, K., N. E. Hurley, N. L. Davis, R. R. Rueckert, and E. Fleissner. 1974. Structural studies of avian myeloblastosis virus: comparison of polypeptides in virion and core component by dodecyl sulfate-polyacrylamide gel electrophoresis. J. Virol. 13:513-528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thimmulappa, R. K., H. Lee, T. Rangasamy, S. P. Reddy, M. Yamamoto, T. W. Kensler, and S. Biswal. 2006. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J. Clin. Investig. 116:984-995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Waters, L. C., and B. C. Mullin. 1977. Transfer RNA in RNA tumor viruses. Prog. Nucleic Acid Res. Mol. Biol. 20:131-160. [DOI] [PubMed] [Google Scholar]