

Abstract
The assembly of most retroviruses occurs at the plasma membrane. Membrane association is directed by MA, the N-terminal domain of the Gag structural protein. For human immunodeficiency virus type 1 (HIV-1), this association is mediated in part by a myristate fatty acid modification. Conflicting evidence has been presented on the relative importance of myristoylation, of ionic interactions between protein and membrane, and of Gag multimerization in membrane association in vivo. We addressed these questions biochemically by determining the affinity of purified myristoylated HIV-1 MA for liposomes of defined composition, both for monomeric and for dimeric forms of the protein. Myristoylation increases the barely detectable intrinsic affinity of the apo-protein for liposomes by only 10-fold, and the resulting affinity is still weak, similar to that of the naturally nonmyristoylated MA of Rous sarcoma virus. Membrane binding of HIV-1 MA is absolutely dependent on the presence of negatively charged lipid and is abrogated at high ionic strength. Forced dimerization of MA increases its membrane affinity by several orders of magnitude. When green fluorescent protein fusions of monomeric or dimeric MA are expressed in cells, the dimeric but not the monomeric protein becomes strongly membrane associated. Computational modeling supports these results and suggests a molecular mechanism for the modest effect of myristoylation on binding, wherein the membrane provides a hydrophobic environment for the myristate that is energetically similar to that provided by the protein. Overall, the results imply that the driving force for membrane association stems largely from ionic interactions between multimerized Gag and negatively charged phospholipids.
In retroviruses, virus assembly is coordinated by a single, multidomain, structural protein called Gag. Several thousand Gag molecules interact with each other, with RNA, and with the plasma membrane to form a budding virus particle, which ultimately pinches off with the aid of cellular protein machinery and thus is released into the extracellular space. Although in the beta retrovirus genus Gag particles are preassembled in the cytoplasm, in most genera assembly takes place on a membrane, and by electron microscopy budding virus particles are observed predominantly or exclusively at the plasma membrane. Thus, Gag has been thought to be targeted directly to the plasma membrane. Several lines of evidence point to the involvement of other membrane compartments in budding. For example, in macrophages infected with human immunodeficiency virus type 1 (HIV-1), budding is observed in what appears to be a late endosomal compartment (46, 55), and similar observations have been made in some other cells (41, 62). However, more recent data suggest alternative interpretations of the nature of the budding site in macrophages (28, 69). Vesicular trafficking and cellular adapter complexes also are implicated in routing monomeric Gag or multimeric Gag complexes to or from the plasma membrane (2, 14). The lipid composition of retroviruses resembles that of membrane microdomains called rafts (1, 49, 52-54), and in some retroviruses budding is inhibited by agents that perturb rafts (37, 42). The data for HIV-1 typically have been interpreted to mean that Gag is targeted to preexisting rafts, although the alternative possibility, that the Gag lattice creates a raft-like microdomain, has not been addressed. Finally, phosphoinositides appear to be involved in proper HIV-1 Gag-Gag interactions (9), in protein-membrane interaction (58, 59), and in membrane targeting and budding in vivo (38).
Membrane binding of Gag is mediated by its N-terminal domain, MA. In most retroviruses MA is myristoylated, and this modification is essential for budding (6, 19). However, in some retroviruses, for example, those in the alpharetrovirus genus (such as Rous sarcoma virus [RSV]), MA bears no fatty acid modification. All retroviral MA proteins have a positively charged surface (36) that also is implicated in membrane interaction (16, 40, 43, 71). The relative contributions of electrostatic interactions with negatively charged lipids and hydrophobic interactions mediated by myristate have been difficult to untangle. In some situations almost the entire MA domain can be replaced with a very short myristoylated sequence without compromising budding and even infectivity (29, 48, 57). At low expression levels in transfected cells, HIV-1 MA is less able to direct Gag to the plasma membrane than short myristoylated N-terminal fragments of MA, in some cases comprising only a few amino acid residues, suggesting that the myristate is not fully exposed in the intact protein (21, 44, 48, 65). Indeed, in the nuclear magnetic resonance (NMR) solution structure of myristoylated MA, the fatty acid moiety is bound in a hydrophobic pocket (67). More recently, HIV-1 MA has been reported to contain a specific binding site for the headgroup of phosphatidylinositol(4,5)bisphosphate [PI(4,5)P2] (58, 59). Another factor in membrane association is Gag multimerization (39, 48, 65), which a priori is expected to increase the overall membrane affinity or avidity of the protein. In addition, multimerization has been shown to favor extrusion of the myristate from its pocket (67).
Numerous studies have attempted to characterize the membrane association of HIV-1 Gag or MA in vivo, but with conflicting results. At steady state, myristoylated Gag was reported to be almost entirely (13, 20, 22, 48, 60, 65, 66, 68, 70, 72) or only ca. 50% or less (30, 39, 40, 42-44) membrane bound. Likewise, nonmyristoylated Gag was reported to be only ca. 5% membrane bound in some studies (22, 40, 43, 44) but significantly more in others (6, 13, 66). For mature MA itself, such estimates ranged from less than 10% (13, 39, 40) up to 50% (60, 65, 66, 72) or to mostly membrane associated (44). The variability in these results likely derives from differences in cell type, membrane-binding assay (fluorescence of individual cells, flotation in density gradients, or centrifugation to form a pellet), and in particular expression level (48). A major limitation of all in vivo systems is that they cannot be manipulated to allow measurements of apparent binding constants and that they cannot distinguish intrinsic properties of the protein from properties imparted by cellular proteins.
The biochemistry of Gag-membrane interaction has been poorly described. Only a few studies have focused on purified MA or Gag proteins (15, 51, 61), but the reported membrane affinities have been questioned (10). Membrane binding of radioactive HIV-1 MA and Gag also has been examined using in vitro-translated proteins in reticulocyte extracts (15, 21, 50, 71, 72). With this method, HIV-1 MA was found to be poorly membrane associated and to contain an electrostatic membrane binding signal near the N terminus (71). However, the reports on the extent of membrane binding of Gag are conflicting, ranging from moderate to nearly complete. Molecular interpretations based on in vitro translation are limited by the vast excess of cold protein in the crude extract.
Using an in vitro liposome flotation assay, we recently described the membrane binding properties of purified RSV MA, a protein that is not naturally myristoylated (10). RSV MA was observed to bind to liposomes weakly and exclusively by ionic interactions, mediated by a basic surface of the protein, and by a biologically relevant concentration of the negatively charged lipid phosphatidylserine (PS). Forced dimerization of MA dramatically increased its affinity. Computational modeling of the interaction of the protein and the membrane surface was fully consonant with the experimental results. The data support a model in which RSV Gag must multimerize (at least to form dimers) to allow stable membrane binding.
We have now extended this analysis to the more complex HIV-1 MA protein. After expression in Escherichia coli, myristoylated MA and nonmyristoylated MA were purified and subjected to liposome flotation at different lipid concentrations and different compositions, both for monomeric and artificially dimerized versions of MA. The results, bolstered by expression of green fluorescent protein (GFP) fusion proteins in transfected cells and by computational modeling, lead to the conclusion that electrostatic interactions and multimerization are essential driving forces for MA-membrane association.
MATERIALS AND METHODS
DNA constructs.
HIV-1 Gag segments were derived from the pNL4-3 strain and cloned into E. coli expression plasmids. The plasmid that coexpresses yeast N-terminal myristoyl transferase (yNMT) and HIV-1 MA (amino acids 1 to 132 HIV-1 Gag) was a gift from Michael Summers. The coexpression vector, originally created by the laboratory of Marilyn Resh, is a modified version of the Invitrogen pET11b vector. The vector contains a pET19a promoter that drives expression of the yNMT gene, and a pET11b promoter that drives expression of the HIV-1 genes. The MA-C-terminal domain (CTD) constructs, in which amino acids 145 to 231 of HIV-1 CA (the CTD) were fused directly to the end of the MA gene, were created by two-step PCR. The CTD Q192A and W184A/M185A mutations also were generated by two-step mutational PCR. The coexpression plasmids were maintained in DH5α E. coli cells and moved into BL21(DE3) codon-plus RIL-competent cells (Stratagene) for protein expression. GFP fusion proteins were cloned by amplifying the genes of interest from the E. coli expression plasmids and inserting them into the Clontech pEGFP N1 vector using the NheI and AgeI restriction sites in the multiple cloning region.
Protein expression.
Overnight cultures of E. coli cells were grown in 2YT medium with appropriate antibiotics. The overnight cultures were diluted 1:100 in 1L fresh medium and grown to an optical density at 600 nm of 0.4, at which time they were induced with 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside). For myristoylated proteins, the cells were supplemented with myristic acid (10 mg/liter, Sigma) 1 h before induction. At 4 h after induction, the cells were harvested by centrifugation and frozen at −20°C.
The frozen cell pellets were resuspended in lysis buffer (20 mM Tris [pH 8], 500 mM NaCl, 10% glycerol, 0.5% Triton X-100, 0.5% deoxycholate, 1 mM phenylmethylsulfonyl fluoride) at 25 ml/liter, the cells were broken by sonication, and the lysates were centrifuged either in a TLA 100.4 rotor at 90,000 rpm for 45 min, or in a Ti 50.2 rotor at 45,000 rpm for 3 h. The supernatants were incubated with 0.05 ml of a 50:50 suspension of Ni2+ resin (QIAGEN) per ml of lysate for 1 h at 4°C and then poured into a column. The resin was washed with 15 ml of binding buffer (20 mM Tris [pH 8], 500 mM NaCl) and then with 10 ml of the same buffer plus 20 mM imidazole. The protein was eluted in 10 ml of binding buffer plus 500 mM imidazole. The myristoylated HIV-1 proteins were further purified by butyl-Sepharose chromatography (Amersham Pharmacia) to remove any nonmyristoylated protein. The nickel eluate was brought to 750 mM (NH4)2SO4 by the addition of 10 ml of 1.5 M (NH4)2SO4 (pH 7.0) while stirring on ice. The protein was loaded onto an AKTA fast protein liquid chromatography column in 50 mM NaPO4 (pH 7)-750 mM (NH4)2SO4 and then eluted with a 20-ml gradient to the same buffer without (NH4)2SO4. Fractions collected at 95 to 100% elution buffer [less than about 35 mM (NH4)2SO4] were dialyzed overnight against elution buffer with 10 mM dithiothreitol and used for liposome binding experiments. Purified proteins were stored in this buffer at 4°C for no more than 1 month. The protein concentration was determined by using advanced protein assay reagent (Cytoskeleton). From a 1-liter preparation typically 10 to 20 mg of protein (at 90 to 95% purity) was recovered after Ni2+ chromatography and approximately 5 mg of myristoylated protein (at >95% purity) was recovered after butyl-Sepharose chromatography.
Liposome preparation and binding assay.
Lipids were purchased from Avanti-Polar Lipids, and liposomes were prepared as described previously (10). In brief, the desired lipids in chloroform were mixed in siliconized glass tubes, and the chloroform was evaporated under a stream of nitrogen. The dried lipids were resuspended in lipid buffer (20 mM Tris [pH 8], 5 mM NaCl, 5 mM EDTA) by using a vortex mixer, typically in 50 μl to yield a final concentration of 50 mg/ml. The resulting multilamellar vesicle suspension was then passed at least 30 times through a 100-nm nitrocellulose filter in an Avanti-Extruder apparatus to yield liposomes of uniform size. Liposomes were prepared fresh before each experiment.
The liposome-binding experiments were performed as previously described. Briefly, 5 μg of purified protein was incubated with liposomes, and each binding reaction was brought to a final volume of 25 μl with lipid buffer (final NaCl concentration of approximately 75 mM), followed by incubation at room temperature for 20 min. A volume of 75 μl of 67% (wt/wt) sucrose in lipid buffer was mixed into each binding reaction by using a vortex mixer, and then 80 μl of this mixture was placed in the bottom of a TLA 100 centrifuge tube and overlaid with 120 μl of 40% (wt/wt) sucrose and then with 40 μl of 4% (wt/wt) sucrose, both in the same buffer. The gradients were centrifuged for 1 h at 87,000 rpm, and 30-μl fractions were collected from the top. The fractions were subjected to electrophoresis on 15% sodium dodecyl sulfate (SDS)-polyacrylamide gels, which were stained with Coomassie brilliant blue R (Sigma) for at least 18 h and then destained for no more than 4 h before being scanned on a UMAX digital scanner. The amount of protein in each lane was quantified by densitometry using ImageQuant (Molecular Dynamics). To confirm that staining was sensitive and linear within the range of protein concentrations recovered in the fractions, serial dilutions of purified serum albumin were subjected to electrophoresis and quantitated as described above. Protein in the top three fractions of the sucrose step gradients, which contained ca. 80% of the recoverable lipid, was counted as liposome bound. This protocol may slightly underestimate the amount of protein actually bound to liposomes. In the figures the error bars represent standard deviations, with the number of independent experiments given in the legends.
To interpret these binding experiments, we applied a simple mass action equation that assumes that the protein binds reversibly to the lipid. Previous studies have shown that although the phenomenon being studied is actually a partitioning event, not a binding event, one can still apply a simple mass action equation to describe the protein-lipid interaction if the concentration of protein is kept much lower than that needed to saturate the surface of the membrane. Since MA does not penetrate the lipid bilayer, the accessible lipid is limited to the outer leaflet of the liposome and therefore is equal to one-half of the total lipid added to the binding reaction. According to the assumptions described previously (10), in all of our binding assays the accessible lipid was always in excess of the area potentially covered by the protein.
Cell culture and subcellular fractionation.
The cell lines DF-1 (chicken fibroblast) and 293T (human embryonic kidney) were maintained in Dulbecco modified Eagle medium supplemented with 5% fetal calf serum, 5% NuSerum (BD Biosciences), 1% heat-inactivated chick serum, standard vitamins, l-glutamine, penicillin, and streptomycin. Transient transfections were performed by using FuGENE (Roche).
For biochemical analysis of membrane binding in vivo, two 100-mm plates of cells were transfected for each construct. At 3 days posttransfection the cells were washed twice with cold hypotonic lysis buffer (1 mM Tris [pH 7.5], 0.1 mM MgCl2, 1 mM phenylmethylsulfonyl fluoride), swollen in 8 ml of the same buffer for 1 h at 4°C, and then broken with 20 strokes in a Teflon pestle homogenizer. Nuclei and cell debris were removed by centrifugation at 1,400 × g for 4 min, and 250 μl of the postnuclear supernatant was mixed with 750 μl of 67% (wt/wt) sucrose (in 20 mM Tris [pH 7.5], 100 mM NaCl, 1 mM EDTA), transferred to an SW60 centrifuge tube, and overlaid with 2.2 ml of 40% (wt/wt) sucrose and then with 800 μl of 4% (wt/wt) sucrose (both in the same buffer). The gradients were centrifuged at 50,000 rpm for 7 h and then fractionated into four 1-ml fractions. Cell membranes banding at the 4%-40% interface were collected in the first fraction, which was then brought to 2 ml with lipid buffer, transferred to a TLS55 centrifuge tube, and centrifuged at 45,000 rpm for 30 min to collect the membranes in a pellet. MA proteins then were analyzed by SDS-polyacrylamide gel electrophoresis (PAGE) and Western blotting using a commercial anti-GFP serum.
Microscopy.
Transfected cells on coverslips were washed with phosphate-buffered saline (PBS) and then fixed with 4% paraformaldehyde in PBS for 20 min. They were then washed twice with PBS and mounted onto microscope slides using Fluoromount-G (Electron Microscopy Sciences). Laser scanning confocal microscopy was performed by using a Leica TCS SP2 microscope. All images were collected with a ×100, 1.40 numerical aperture oil immersion lens. An argon laser with an excitation wavelength of 488 nm was used, and images were analyzed with Leica Confocal Software (Leica Microsystems).
Theoretical model.
In our calculations, MA and the lipid bilayer are represented in atomic detail. The lipid bilayers—PC, 8:1 PC-PS, 4:1 PC-PS, 3:1 PC-PS, 2:1 PC-PS, and 1:1 PC-PS—were constructed as described in previous studies (4). The structure of myristoylated HIV-1 MA (residues 1 to 109) was obtained from the Protein Data Bank coordinate file 1UPH, which comprises a set of 20 NMR models (67). In the analysis we included 10 of these for completeness and as a test of the sensitivity of our approach. The results do not change if any single model is considered alone. Since the structure of nonmyristoylated MA is very similar to the structure of 1UPH with the myristate removed (67), we used the same NMR models for calculations of nonmyristoylated MA. For example, the RMSD between 1UPH and 1HIW (a crystal structure of the nonmyristoylated form) is 1.2 Å over 113 residues with a z-score of 6.6.
For nonmyristoylated MA, the molar partition coefficient, K(nonmyr-MA), is determined according to the following equation:
 |
(1) |
where Kel is the electrostatic component of the binding of MA to the lipid bilayer and is calculated by using equation 3 below. For myristolylated MA, the molar partition coefficient, K(myr-MA), is determined according to:
 |
(2) |
where Kmyr and Kel′ are the nonpolar and electrostatic components of the binding of MA to the lipid bilayer, respectively. Kel′ is calculated by using equation 4 below. The difference between Kel and Kel′ is based on assumptions made about the physical space to which MA is constrained with respect to the membrane surface (Fig. 1). Nonmyristoylated MA is free to adopt any orientation with respect to the membrane (Fig. 1A and C), and each of these orientations is included in the calculation of Kel:
 |
(3) |
where AL is the area per lipid (68 Å2*10−18 dm2), N is Avogadro's number, and ΔGel(R,θ,μ,ϕ) is the electrostatic binding free energy at a distance (R) from the bilayer surface, and a rotation (θ,μ,ϕ) with respect to the bilayer center. The angle brackets denote the average over many randomly sampled orientations of MA with respect to the membrane. The integral represents the Gibbs surface excess of MA at the membrane surface (4).
FIG. 1.

Sampling of myr-seq versus myr-exp HIV-1 MA. The HIV-1 MA domain is depicted by the trace of its C-α atoms, with the N terminus (yellow) and C terminus (dark green) shown in atomistic detail. The three-dimensional electrostatic equipotential profiles for 0.1 M KCl were calculated and visualized with GRASP at +25 mV (blue) and −25 mV (red) (25). The translucent line represents the general position of the membrane envelope. (A and B) MA is depicted in its minimum electrostatic free energy orientation. The N terminus is in a good position to insert the covalently attached myristoyl moiety (not depicted) into the membrane. (C and D) The fundamental differences between the myristate sequestered (myr-seq) and myristate exposed (myr-exp) sampling schemes is depicted (see Materials and Methods). (C) Myr-seq MA is free to sample a full range of protein orientations because it is not anchored to the membrane. (D) Myr-exp MA, however, is anchored to the membrane and is, therefore, more constrained in its sampling.
Myr-MA is assumed to be anchored at the bilayer surface by the myristate anchor (Fig. 1B and D), and only orientations consistent with this constraint are included in the calculation of Kel′. Equation 3 for Kel is reduced to the following as described previously (35):
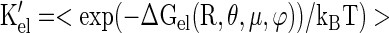 |
(4) |
In equation 2, the Kmyr is determined as:
 |
(5) |
where ΔGmyr is the nonpolar free energy associated with partitioning the myristate into the bilayer either from the aqueous phase, if it is exposed, or from the hydrophobic cleft on MA, if it is sequestered by the protein. Interestingly, ΔGmyr, calculated as the product of (i) a surface tension coefficient, i.e., γ = 25 cal/mol-Å2 for partitioning from an aqueous to a hydrocarbon phase, and (ii) the change in solvent accessible surface area upon either membrane insertion or binding in the MA cleft, is approximately the same, ca. −12 kcal/mol, for both scenarios (26).
Similar calculations were performed previously (4, 34) and, most relevantly, the methodology was applied to the myristoylated and nonmyristoylated peptide mimic of the N terminus of Src (35). The theoretical predictions are in good agreement with the experimental measurements.
Sampling orientations of MA.
In order to approximate the molar binding constant, many different orientations of the protein with respect to the membrane were considered in our calculations. We used the method developed by Murray et al. (35), which applies randomly sampled rigid body rotations to the protein to approximate the spectrum of orientations the protein may experience at the membrane surface. We assume that nonmyristoylated MA is free to absorb and desorb from the bilayer. The minimum free energy orientation was used as the reference orientation for each NMR model, and 100 sampled orientations were produced for each model by applying the appropriate Euler angle rotations (θ,μ,ϕ) about the center of the mass (e.g., Fig. 1A and C). The electrostatic free energies of interaction for each orientation were calculated as a function of distance from the membrane surface, and ΔGel(R,θ,μ,ϕ) was used in equation 3 to determine the Kel.
For myristoylated MA, we assumed that upon membrane association, the myristate was extruded from the protein pocket 100% of the time. Consequently, orientations for myr-exp MA were assumed to be constrained by the myristate at the membrane interface, and rigid body rotations were applied about the nitrogen atom of the N-terminal Gly residue, the atom to which the myristoyl group is covalently bonded (Fig. 1B), rather than the center of mass of MA. We assumed that the myristoyl group confines MA to 2 Å ≤ R ≤ 3 Å and calculated ΔGel(R,θ,μ,ϕ) at R = 2, 2.5, and 3 Å according to previous work.
Electrostatic calculations.
The electrostatic free energy of binding, ΔGel(R,θ,μ,ϕ), was calculated with a modified version of the DelPhi program (17), adapted to solve the nonlinear Poisson-Boltzmann (PB) equation for protein-membrane systems (4, 34). DelPhi produces finite-difference solutions to the PB equation (the FDPB method [26]). The solvent is described in terms of a bulk dielectric constant and a concentration of mobile ions that produce a charge density profile in response to the electrostatic potentials set up by the fixed charges in the system (the mean field approximation). The solutes (here, MA and the membrane) are described in terms of the coordinates of the individual atoms, their atomic radii, and partial charges. Electrostatic free energies are obtained from the calculated potentials, and the electrostatic free energy of interaction is determined as the difference between the electrostatic free energy of MA in a specific orientation with respect to the membrane surface and the electrostatic free energy of MA and the membrane taken separately:
 |
(6) |
The FDPB calculations were performed to a final resolution of three grids/Å with a lattice size of 2893. The precision in the electrostatic free energies of interaction, determined as the difference between the results obtained at the two highest resolution scales (from a series of focusing runs), is 0.3 kcal/mol for all calculations.
RESULTS
To study the membrane-binding properties of purified MA-related proteins (Fig. 2), we coexpressed the HIV-1 proteins together with yeast N-myristoyl transferase and either added myristic acid to the growth medium prior to induction (for myristoylated proteins) or left it out (for nonmyristoylated proteins). Quantitative myristoylation of the relevant purified proteins was verified by mass spectrometry. The purified proteins were incubated with liposomes (diameter, 100 nm) of defined composition as described previously (10) and then subjected to centrifugation and flotation analysis in 0.24-μl sucrose step gradients. The percentage of protein that floated with the liposomes through a 40% sucrose layer was determined by fractionating the gradients, staining the protein after SDS-PAGE, and quantitating the band intensity in the linear range of staining as determined by using a parallel marker protein. Examples of the raw data obtained from flotation experiments are shown in Fig. 3A. In all experiments the liposomes were in excess; i.e., the total available lipid surface exceeded the surface potentially covered by the input protein. One-half of the molar lipid concentration (i.e., that part available for protein binding) at which one-half of the protein bound to the liposomes was taken as a relative measure of membrane affinity. Although the binding of a protein to a membrane is a partition event and therefore is governed by a partition coefficient, not by a true equilibrium constant, for simplicity we use the terms “lipid concentration” and “KD” to describe the relative affinities measured by flotation. This treatment has been described and justified previously (3).
FIG. 2.

Schematic representation of proteins. The top bar represents the HIV-1 Gag protein with its major domains, and the four bars below represent the purified proteins used in the present study. These proteins have a His6 tag at their C termini (data not shown). The bottom two bars represent the two GFP proteins expressed in DF1 and 293T cells. The C-terminal subdomain of CA, CTD, harbors the dimerization interface. In the dimeric proteins MAdim and MAdim-GFP, the CTD contains the Q192 mutation (not shown), which strengthens the dimer interface. In the monomer proteins MAmon and MAmon-GFP, the dark vertical bars represent mutations W184A and M185A, which destroy the dimer interface.
FIG. 3.

Effect of myristoylation and dimerization on liposome interaction. (A) Example of flotation data. A mixture of 8 μg of protein and 400 μg of liposomes was adjusted to 53% sucrose and layered under two sucrose steps, followed by centrifugation. Fractions of 30 μl were collected and the protein in each was analyzed by SDS-PAGE and staining. The two panels show examples of gels in which most of the protein did not bind to liposomes (top panel, 0% PS) or most did bind to the liposomes (bottom panel, 25% PS). Similar gels were used to obtain the data plotted in Fig. 4, 5, and 6. (B) Effect of lipid concentration on MA-lipid interaction. A total of 5 μg of protein was incubated with increasing concentrations of 2:1 PC-PS liposomes. The ratios of bound to total protein are shown on the y axis. Diamonds and dotted line, RSV MA; squares, myristoylated HIV-1 MA; triangles, nonmyristoylated HIV-1 MA. The data represent averages from four to six independent experiments. (C) Effect of dimerization. Proteins were incubated with increasing concentrations of 2:1 PC-PS liposomes and analyzed as described above. The curve for myristoylated MA is reproduced from (B). The data represent averages from two independent experiments. Open circles, myristoylated MA; diamonds, nonmyristoylated MA-CTDdim; squares, myristoylated MA-CTDdim; triangles, myristoylated MA-CTDmon.
Dependence of binding on lipid concentration.
Myristoylated MA bound to liposomes composed of 2:1 PC-PS in a lipid concentration-dependent manner (Fig. 3B). The apparent KD of this binding was 10−3 M, a value that reflects a weak interaction similar to that reported previously for the naturally nonmyristoylated MA protein of RSV (RSV data obtained from Dalton et al. [10] and superimposed for comparison in Fig. 3B). Nonmyristoylated HIV-1 MA also bound detectably to liposomes (Fig. 3B) but too weakly to allow an apparent KD to be determined reliably. In approximate terms, the lack of the fatty acid modification reduced the membrane affinity by 10-fold, to about 10−2 M.
The observation that myristoylated HIV-1 MA interacts with liposomes no more tightly than RSV MA was initially unanticipated. According to both experimental and computational studies on peptides, the myristoyl moiety should contribute approximately 8 kcal/mol in free energy to the protein-lipid interaction, corresponding to a KD of approximately 10−4 M (8, 45). Since the hydrophobic and electrostatic contributions to binding might be expected to be independent and since the electrostatic affinities of RSV MA and HIV-1 MA for acidic membranes should be of roughly similar magnitude based on the clustering of basic residues (36), the binding of HIV-1 MA to membranes might be predicted to have an apparent KD in the range of 10−7 M, several orders of magnitude lower than that observed in the flotation assay. This discrepancy is best explained by the myristoyl moiety being sequestered in a hydrophobic pocket under the conditions used here, as observed more directly in NMR studies (67).
Nonmyristoylated HIV-1 MA crystallizes as a trimer, and a lattice of trimers has been used to model the disposition of the protein inside mature virions (23). The existence of trimers of nonmyristoylated HIV-1 MA in solution has been reported (33) but is controversial. The more biologically relevant myristoylated form of the wild-type HIV-1 MA is reported to exist in a monomer-trimer equilibrium, with 50% of the protein in trimeric form at 70 μM (67). It seemed possible that the presence of membranes could promote myristate extrusion and thus allow trimerization at lower protein concentrations than in solution. We addressed this hypothesis by carrying out flotation experiments at different protein concentrations, in the range 1 to 60 μM (data not shown; the experimental protocols did not permit analysis outside this range). We found that liposome binding was similar at all concentrations tested, implying lack of cooperativity in this concentration range. We speculate that the KD for trimerization, which was determined at pH 5.5 (67), is higher at neutral pH, out of the range in which the flotation experiments were carried out.
Effect of forced dimerization on binding.
The weak affinity of myristoylated HIV-1 MA for acidic liposomes readily explains in vivo observations that MA is predominantly cytosolic in cells. It was recently demonstrated that the myristate moiety becomes exposed at 20-fold lower protein concentrations for the protein MA-CA than for MA itself (67). Since CA forms dimers (KD of ∼10−5 M), myristate exposure apparently is enhanced by protein self-association, in this case mediated by CA-CA interactions. This result leads to a model in which Gag-Gag interactions promote membrane binding. Experimental evidence for this model comes from in vivo studies demonstrating that deletions of the NC domain, which abrogate Gag assembly by blocking Gag extensive multimerization, also block or reduce membrane binding (60). Also, increasing the Gag expression level increases membrane binding (48). However, it is important to note that some studies used sedimentation analysis to quantitate membrane-bound proteins, making it difficult to distinguish membrane binding from protein aggregation.
We designed experiments to demonstrate directly that dimers of HIV-1 MA have a higher affinity for acidic liposomes than do monomers. Since the MA-CA protein is prone to aggregate at the protein concentrations required for the liposome-binding assay, we instead created MA dimers by fusing the HIV-1 CTD of CA (CA residues 145 to 231; Fig. 2), which harbors the dimerization domain, to the C-terminal end of MA. Two nearly identical versions of this protein were constructed, which either abrogate dimerization (W184A, M185A; here called MA-CTDmon) or enhance dimerization by decreasing the KD by 10-fold (Q192A; here called MA-CTDdim, (12). Proteins similar to these, but based on RSV MA instead of HIV-1 MA, previously were verified by velocity sedimentation to be monomeric or dimeric, respectively (10).
To determine the binding properties of the HIV-1 MA-CTD chimeras, myristoylated MA-CTDdim, myristoylated MA-CTDmon, and nonmyristoylated MA-CTDdim were incubated with increasing concentrations of 2:1 PC-PS liposomes. At the protein concentrations used (ca. 5 × 10−6 M) the MA-CTDdim proteins should be dimerized, since the concentration exceeds the reported KD for CA with the Q192A mutation (12) by about fivefold. Myristoylated MA-CTDmon (Fig. 3C) bound liposomes with an affinity indistinguishable from that of myristoylated MA itself (Fig. 3C [the data are reproduced from Fig. 3B for comparison]), ruling out nonspecific effects of the CTD sequence, as was also found for the analogous RSV protein (10). In contrast, myristoylated MA-CTDdim (Fig. 3C) bound to liposomes several orders of magnitude more tightly than the monomeric form. The weak liposome interaction of the nonmyristoylated MA-CTDdim also was increased dramatically upon dimerization (Fig. 3C).
Quantitative interpretation of these results is limited by the restricted range of lipid concentrations over which our experimental protocol could be applied. For the nonmyristoylated MA-CTD, binding of the dimer was about 100-fold tighter than for nonmyristoylated MA itself. This increase in membrane affinity is in the range expected theoretically. In the absence of any effects of geometry in the dimeric complex, the apparent KD for a dimer binding to liposomes (10−4 M) should approximate the product of the apparent KD values of the two monomers, 10−2 M, as was in fact observed. For the more relevant myristoylated proteins, the increase in membrane affinity due to dimerization was too great to be estimated quantitatively but appeared to be several orders of magnitude. Since the product of the observed apparent monomer KD values (10−3 M) is 10−6, the observed tight binding of the dimer might be accounted for quantitatively simply by dimerization. However, since trimerization has been demonstrated explicitly by Tang et al. to lead to increased solvent exposure of the myristate (67), the artificial dimerization in our experiments also might lead to myristate exposure, which then would contribute to the observed tight binding of MA-CTDdim to liposomes. Different techniques would be needed to accurately determine the apparent binding constant of the dimer, in order to learn if the protein binds as tightly as predicted for two fully exposed myristoyl groups that insert into the bilayer plus two positively charged protein surfaces that contact negatively charged lipid.
Overall, our data describing the liposome binding of dimeric proteins strongly suggest that Gag-Gag multimerization (or at least dimerization) is required for membrane binding in vivo. This conclusion is based on an estimate of the concentration of plasma membrane lipid in cells, about 10−3M (32), which would be insufficient to support stable binding of monomeric MA in vitro.
Dependence of binding on lipid composition and ionic strength.
Peptides that are both basic and carry a hydrophobic membrane attachment signal, such as myristate, can bind to uncharged (PC) liposomes; however, this interaction is greatly enhanced by inclusion of anionic lipids such as PS (4, 8, 35, 45). The electrostatic contribution to binding depends on the mol% PS (35). To directly address the role of ionic interactions in membrane binding of HIV-1 MA, we carried out flotation experiments with liposomes of various negative charge densities, prepared with various ratios of PS to PC. The proteins used in these studies were myristoylated MA-CTDmon (shown above to behave like MA, Fig. 3C) and myristoylated MA-CTDdim. The fraction of total protein that floated to the buoyant density of the liposomes at a constant lipid concentration was plotted against the mol% PS.
Binding of monomeric myristoylated MA increased with increasing PS (Fig. 4), reaching a maximum at about 30 mol% PS, which is approximately the concentration of PS present in the inner leaflet of the viral membrane (1). MA-CTDdim also showed increased binding as a function of the mol% PS, but maximum binding occurred at ca. 15% PS (Fig. 4). The lower PS dependence of the dimer might be expected from its higher affinity for membranes. Most important, neither the monomeric nor the dimeric protein showed any binding to 100% PC liposomes. This result is consistent with the poor ability of PC liposomes, compared to PC/PS liposomes, to compete for HIV MA binding to natural membranes (71).
FIG. 4.

Effect of PS on MA-liposome interaction. A total of 5 μg of protein was incubated with 250 μg of liposomes of increasing mol% PS, and analysis was carried out as described in Fig. 3. The ratio of bound protein to total protein for each gradient is plotted on the y axis. The data from one experiment with myristoylated MA and one experiment with myristoylated MA-CTDmon were averaged to generate the curve for monomeric MA (diamonds). The data from two independent experiments with MA-CTDdim were used to generate the curve for dimeric MA (squares).
To further address the relationship between the electrostatic and hydrophobic contributions of the bipartite membrane-binding signal of HIV-1 MA, we measured the effect of ionic strength upon the MA-lipid interaction. At high ionic strength, the electrostatic component of membrane binding should be abrogated, leaving only the hydrophobic contribution from the myristate moiety. The monomeric or dimeric myristoylated proteins MA-CTDmon or MA-CTDdim were incubated with liposomes at a fixed lipid concentration at different NaCl concentrations, and then flotation analysis was carried out at the same ionic strength. For both proteins, binding decreased with increasing ionic strength. At 300 mM NaCl binding was greatly reduced, while at 500 mM NaCl it was almost undetectable (Fig. 5). These results imply that the electrostatic contribution to binding is essential, a finding consistent with the absence of detectable interaction with 100% PC liposomes.
FIG. 5.

Effect of ionic strength on MA-liposome interaction. A total of 5 μg of protein was incubated with 250 μg of 2:1 PC-PS liposomes in buffer containing increasing [NaCl] from 20 mM to 200 mM. In a separate experiment, 5 μg of protein was incubated with 250 μg of 2:1 PC-PS liposomes in buffer containing either 300 or 500 mM NaCl. The bars show the ratio of bound protein to total protein for each gradient. The data are averages of two independent experiments. Error bars represent standard deviations from the means.
Effects of other lipids or lipid metabolites on binding of MA to liposomes.
Relative to the plasma membrane of host cells, the lipid envelope of HIV-1, like that of other retroviruses, is enriched in sphingomyelin (sph) and cholesterol (chol) (1), hallmarks of liquid-ordered or so-called raft microdomains. Also, HIV-1 Gag appears to be raft-associated in cells (20, 24, 37, 42), although not all published data are in agreement (13). To address the question of whetehr myristoylated MA has a specific affinity for lipid rafts, we incubated the protein with increasing amounts of liposomes of composition 1:1:1 PC-PS-sph or 1:1:1:3 PC-PS-sph-chol, corresponding approximately to the ratios of these lipids in the viral envelope. Binding of myristoylated MA to these raft-like liposomes was indistinguishable from binding to 2:1 PC-PS liposomes (Fig. 6), suggesting that the distinctive lipid composition of virions is not a consequence of preferential interaction of the MA domain with lipid rafts during assembly. However, it is important to note that neither of these lipid compositions accurately reflects the composition on the inner leaflet in rafts, which is poorly defined but presumably is devoid of sphingolipids, since these are outer leaflet markers. Perhaps the raft-like character of HIV-1 virions is an indirect consequence of Gag-Gag interactions, since multimerization has been suggested to promote raft localization (20, 30, 42).
FIG. 6.

Effect of other lipids on MA-liposome interaction. (A) A total of 5 μg of myristoylated MA was incubated with increasing amounts of liposomes with compositions 2:1 PC-PS, 1:1:1 PC-sph-PS, or 1:1:1:3 PC-Sph-PS-chol. The data are from one experiment, with the curve and error bars for 2:1 PC-PS reproduced from Fig. 2B. An independent experiment at a single concentration of liposomes near the midpoint of these curves yielded similar results.
Inositol phosphates (IPs) and/or their glycerol lipid counterparts (PIPs) appear to be involved in HIV-1 assembly and budding. First, the in vitro assembly of purified HIV-1 Gag protein into virus-like-particles of correct size and shape requires either inositol pentakisphosphate (IP5) or inositol hexakisphosphate (IP6), or the analogous short-chain PIPs (9). This requirement is manifested only when the MA domain is present. Less highly phosphorylated IP species do not function in a similar manner. Second, perturbing the pools of PIPs in vivo affects HIV-1 assembly (38), in that depletion of the pool of PI(4,5)P2 reduces Gag association with the plasma membrane and reduces budding. In contrast, elevating the cellular pool of PI(4,5)P2 redirects budding to a subset of endosomal membranes. Third, HIV-1 MA contains a specific binding pocket for the head group of PI(4,5)P2, and binding of short-chain PI(4,5)2 analogues promotes myristate extrusion from the pocket (58, 59).
To examine the impact of IPs on membrane association of HIV-1 MA, we incubated myristoylated MA with 2:1 PC-PS liposomes in the presence or absence of 50 μM IP6. This concentration is in the range measured in Jurkat T-lymphocytes and is similar to levels of IP6 required for proper in vitro assembly of VLPs. Addition of IP6 had no impact upon membrane binding at two different lipid concentrations (data not shown). Since the lipid concentrations used in these experiments approximate that of the plasma membrane in most cell types, these results suggest that IP6 does not directly affect membrane binding of HIV-1 MA. Higher concentrations of IP6 (200 μM or 2 mM) did inhibit the liposome binding of dimeric myristoylated and nonmyristoylated MA (data not shown). This inhibition may be nonspecific due to the increase in ionic strength of the solution. For example, given the high negative charge density on the molecule, a solution of 2 mM IP6 would be equivalent in ionic strength to about 300 mM NaCl. A complication in interpreting the effects of IP6 on MA stems from the observation that tight binding of IP6 to Gag requires not only the MA domain but also the NC domain (11).
In preliminary experiments we also sought to establish if PI(4,5)P2 enhances membrane binding of HIV-1 MA. Myristoylated MA was incubated with increasing concentrations of 2:1 PC-PS liposomes, with or without added 1% PI(4,5)P2 to approximate the concentration of this lipid in the plasma membrane. A modest effect upon the membrane binding was observed, corresponding approximately to a 1.5-fold increase in affinity at three different lipid concentrations (data not shown). This enhancement is in the range expected for simple electrostatic effects. According to our theoretical calculations, 1% PI(4,5)P2 in the context of 2:1 PC:PS would increase the membrane affinity by fivefold (not shown). Thus, under these conditions the in vitro binding assay does not provide support for an essential role for PI(4,5)P2 in membrane binding. More careful experiments will be required to quantitatively tease apart electrostatic effects and the predicted specific binding effects in the interaction of HIV-1 MA with liposomes carrying physiological levels of PS and PI(4,5)P2.
Computational modeling of liposome binding of monomeric and dimeric proteins.
To help interpret the experimental results, we used computational modeling (see Materials and Methods). The electrostatic interaction was calculated for the adsorption of the positively charged surface of MA, based on its known structure, to the negatively charged membrane, based on a defined mol% PS. A pictorial example of this interaction is shown in Fig. 1A, with the blue and red meshes corresponding to the +1 kT/e (+25 mV) and −1 kT/e (+25 mV) equipotential contours, respectively. The hydrophobic interaction of the myristate with the groove in MA and with the lipid also was calculated based on burial of solvent exposed surface. These calculations showed that the membrane and the MA groove provide an equivalent nonpolar environment for the myristate. In other words, the nonpolar energy lost upon extrusion of the myristate from the groove on MA is compensated for by the nonpolar energy gained upon insertion of the myristate into the membrane phase. Therefore, there is no change in nonpolar energy as the myristate is extruded from MA and inserted completely into the membrane.
Given this conclusion, how can the 10-fold experimental difference in the KD of myristoylated MA-CTDmon and nonmyristoylated MA-CTDmon be explained? The answer in principle is that the myristate anchors MA at the membrane surface in such a manner that the basic patch is restricted to electrostatically favorable binding orientations (Fig. 1B and D). In contrast, nonmyr-MA is free to interact with the membrane surface in any orientation (Fig. 1A and C). Thus, the modest increase in binding due to myristoylation is essentially an electrostatic phenomenon. In quantitative terms, we calculate that for 2:1 PC-PS liposomes the predicted KD values for monomeric nonmyristoylated and myristoylated MA are approximately 10−2 and 10−3 M, respectively, a finding consistent with the experimentally measured values. Previous work has shown that theoretical calculations are more reliable in predicting relative than absolute binding, and therefore the fact that both theory and experiment yield a 10-fold effect of the myristate suggests that this modeling captures the essential features of the membrane-binding reaction.
The computational model further suggests that membrane association of MA alone could facilitate the exposure and subsequent membrane insertion of the myristoyl group. Thus, the interaction of MA with phospholipids may serve the same “myristate exposing function” that MA-MA interactions have been proposed to perform (67). Once MA is adsorbed to the membrane by electrostatic interactions, the membrane phase, whose “effective concentration” is much greater than that of the hydrophobic groove, can easily outcompete the binding pocket for the myristate moiety.
The modeling also predicts that the electrostatic free energy of a dimer should be twice that of a monomer, i.e., that the KD value for the dimer is the product of those for two monomers. One possible example of a dimeric MA-CTD interacting electrostatically with a membrane containing 20% PS is shown in Fig. 7A. The actual orientations and positions of the two juxtaposed MA domains are difficult to predict since MA is not part of the CTD dimerization domain itself but is only tethered to it. The prediction of additivity is born out experimentally for nonmyristoylated MA-CTDmon (KD of ∼10−2 M) and MA-CTDdim (KD = 10−4 M). However, the technical limitations of the liposome flotation assay did not permit this prediction to be tested for the myristoylated monomeric and dimeric species, because of their stronger liposome binding.
FIG. 7.

Myr monomeric and non-myr dimeric forms of HIV-1 MA. Proteins are represented as molecular surfaces (white and light yellow), which are obtained by rolling a water molecule over the surface of the protein, with the N terminus (yellow) and C terminus (green) mapped to the molecular surface, and the myristate (pink) depicted atomistically. The three-dimensional electrostatic equipotential profiles for 0.1 M KCl were calculated and visualized with GRASP at +25 mV (blue) and −25 mV (red) (25). The translucent line represents the general position of the membrane envelope. The dimer model was produced with combinatorial extension (63) using the EIAV dimer structure (PDB code, 1hek).
Finally, computational modeling also agrees with the experimental measurements with respect to increasing the mol% PS in the membrane and increasing the ionic strength of the solution. For example, modeling predicts that the myristoylated MA dimer should bind to liposomes with 20% PS as tightly as the monomer should bind to liposomes with 33% PS, close to the results determined experimentally (Fig. 4). Similarly, according to the modeling, the binding constant for myristylated MA dimers and monomers at 0.1 M KCl should decrease by 25,000- and 160-fold, respectively, when the KCl concentration is increased to 0.5 M. This is in qualitative agreement with the salt effects observed experimentally (Fig. 5). The congruence between experimental and theoretical results reinforces the conclusion that MA-membrane binding is driven mainly by ionic interactions.
Membrane binding in vivo.
To assess the relevance of the in vitro liposome binding experiments, we expressed GFP fusion proteins of MA-CTDmon and MA-CTDdim by transient transfection of human 293T cells or of chicken DF1 cells and then analyzed the cells by fluorescence microscopy and subcellular fractionation. Both types of cells gave similar results. Fluorescence of MA-CTDmon-GFP appeared primarily cytoplasmic but with some staining of the plasma membrane (Fig. 8A). In contrast, the fluorescence of MA-CTDdim-GFP appeared to be concentrated at the plasma membrane, especially at membrane protrusions such as microvilli, ruffles, and retraction fibers. These interpretations of the fluorescence images were bolstered by biochemical analysis of postnuclear supernatants of lysed cells. In membrane flotation experiments, the majority of the monomeric protein remained with the cytoplasmic fraction at the bottom of the sucrose step gradient (Fig. 8B), implying only weak affinity for membranes, similar to results reported by others (13, 39, 40, 65). In contrast, the majority of the dimeric protein floated with cellular membranes (Fig. 8B). We conclude from these observations that the properties of purified monomeric and dimeric HIV-1 MA proteins assessed in vitro in the liposome binding experiments predict the behavior of the proteins in living cells, at the expression levels realized in these experiments. The results reaffirm that membrane association in vivo requires protein-protein interactions.
FIG. 8.

Membrane binding of monomeric and dimeric MA proteins in cells. (A) Fluorescence microscopy. DNAs encoding myristoylated MA-CTDmon-GFP or MA-CTDdim-GFP were transfected into 293T cells, and representative fluorescence images were captured 3 days later by confocal microscopy. The images are of the plane closest to the glass coverslip. (B) Membrane fractionation. Transfected cells were swollen in hypotonic buffer and then lysed by homogenization. The postnuclear supernatant was adjusted to 55% sucrose and layered under a 4-ml step sucrose gradient. After centrifugation, the floating membrane fractions and the fractions at the bottom of the gradient were collected and subjected to SDS-PAGE and Western blotting with an anti-CA antiserum. 10%, one-tenth of the postnuclear supernatant; M, membrane fraction; C, cytosol fraction.
DISCUSSION
We have studied the binding of purified, myristoylated HIV-1 MA protein to liposomes of defined composition. This represents the first use of a direct, quantitative assay to measure the interaction of HIV-1 MA with lipid vesicles in the absence of cellular factors. MA-membrane interaction was found to be driven predominantly by electrostatic interactions, since binding was absolutely dependent on the acidic lipid PS and was abrogated at high ionic strength. The weak apparent dissociation constant, about 1 mM, would not be expected to allow stable membrane association in vivo. An artificially dimerized version of MA bound to acidic membranes much more tightly than monomeric MA itself, directly demonstrating the importance of protein-protein interactions in membrane interaction. Analysis of membrane binding of monomeric or dimeric MA-GFP in transfected cells supported this conclusion. Computational modeling provided further qualitative and quantitative support for the role of electrostatics and protein multimerization in membrane binding.
Prior to our work, affinities of purified HIV-1 proteins for liposomes were estimated by quenching of intrinsic protein fluorescence, in some cases for proteins derived by proteolytic cleavage of purified full-length Gag (15, 61). Nonmyristoylated MA was reported to bind to PS-containing liposomes several orders of magnitude more tightly than we found and to have the same membrane-binding affinity as myristoylated HIV-1 Gag. The HIV-1 CA protein, used as a control in some of these studies, was reported to interact with membranes as strongly as MA itself, which is difficult to reconcile with the known role of CA in assembly. In our hands, negligible amounts of HIV-1 CA bound to PS-containing liposomes under any conditions (data not shown). In the most recent study using fluorescence quenching, the KD values of myristoylated and nonmyristoylated HIV-1 Gag for binding to PS-containing liposomes were reported to be similar, about 10 μM (51). These measurements were made at 0.5 M NaCl, at which concentration we find almost no interaction of MA with liposomes. The origin of these large discrepancies is unclear. They could derive from the sensitive nature of the fluorescence quenching assay, which might detect small amounts of denatured or otherwise abnormal protein.
Other in vitro studies of the binding of myristoylated HIV-1 proteins to membranes used proteins derived from a rabbit reticulocyte in vitro transcription and translation system (15, 21, 50, 71, 72), which yields a radiochemically pure protein that is easily monitored. The data from this system were the first to imply that the HIV-1 MA domain harbors an electrostatic signal that acts synergistically with the myristate in binding of the protein to membranes containing acidic lipids (71, 72). However, in these early studies no attempts were made to underpin the conclusions by quantitative assessment of binding constants. In addition, a complicating factor in interpreting data from in vitro translation is that the radioactive proteins always are present in a vast excess of unlabeled proteins from the reticulocyte crude lysate.
It is well established that budding of an intact HIV-1 Gag requires both myristoylation and an adjoining positively charged surface on MA. However, a mechanistic understanding of the role for myristate has remained elusive. For example, in transfected cells virtually the entire MA domain of Gag, including the basic surface, can be functionally replaced by a short myristoylated peptide with only one or a few basic residues (29, 57). Compared to such a peptide, MA apparently has an attenuated ability to promote binding of Gag to membranes in vivo (48, 65). In the in vitro translation studies, C-terminally extended forms of MA bound liposomes more tightly than MA itself. This observation led to the hypothesis of a myristoyl switch in HIV-1 Gag, in which the myristate is sequestered in MA but exposed in Gag (21, 65, 71). In the original form of this hypothesis it was assumed that a conformational change in MA brings about exposure of the myristate. From the more recent structural and biophysical studies of myristoylated MA (67), it is clear that the myristoylated MA polypeptide actually adopts a structure almost identical to that of the nonmyristoylated polypeptide. These seemingly contradictory results can be explained in part by the observed sequestration of the myristoyl moiety in a hydrophobic pocket in MA. In the model proposed by these authors, trimerization (or in principle some other form of multimerization) of the MA domain leads to extrusion of the myristate, making it available for insertion into the membrane. In addition, binding of short-chain analogues of PI(4,5)P2 also promotes extrusion of the myristate from its pocket (58, 59), a reaction that is presumed to increase membrane affinity of the protein.
In our experiments, myristoylation of MA raised its membrane affinity by only 10-fold. A free myristoyl group should increase the binding constant by about 103- to 104-fold, as found, for example, for the peptide corresponding to the N terminus of the Src oncoprotein in liposome-binding studies similar to ours (8). Using computational modeling, we explain these differences by proposing that the nonpolar interactions due to myristate insertion into the membrane are energetically similar to those due to sequestration in the MA hydrophobic pocket. The modest increase in affinity due to myristoylation then can be accounted for by the difference in the way the protein sees the membrane in the presence or absence of the fatty acid modification. The nonmyristoylated protein randomly samples all orientations, only some of which provide favorable positioning of the basic surface next to the acidic lipid bilayer. In contrast, even transient insertion of the myristate moiety into the membrane constrains the protein to sample only the electrostatically favorable orientations, since the fatty acid is located in the positive surface of the protein. This model quantitatively accounts for the 10-fold difference in membrane affinity and can be viewed as an alternative to the model in which MA trimerization promotes myristate extrusion (59). Nevertheless, it is difficult to reconcile the modest 10-fold effect of the fatty acid in vitro with the observation that myristoylation is absolutely required in vivo for budding, even at high expression levels that might be expected to overcome the lack myristoylation. This apparent discrepancy might reflect the extensive multimerization of Gag that underlies virus assembly and budding in vivo. The myristoyl groups may be involved in the lateral targeting at the plasma membrane surface, i.e., in formation of the Gag lattice. In addition, since PI(4,5)2 binding to MA promotes extrusion of the myristate, a subtle synergistic effect of this membrane constituent and the myristate might be revealed in vitro only under special conditions such as, for example, at low PS concentrations.
A vexing challenge in HIV-1 membrane biology is elucidating the cellular location and the role of Gag multimerization in membrane binding. In the broad sense used here, this term is meant to refer to any form of Gag in which protein-protein interactions bring together two or more molecules. This nomenclature also has been used to refer to ensembles of Gag molecules that are larger than dimers. There is a lack of consensus as to what extent multimerization is required for the membrane association of HIV-1 Gag. The C-terminal domain of CA mediates Gag dimerization, with dissociation constant of about 10−5 M (12, 18), which is similar for CA and for Gag (11). The cytoplasmic concentration of Gag in infected cells is arguably of this same order of magnitude, suggesting that the expression level may determine whether Gag is a monomer or a dimer as a cytoplasmic protein. The HIV-1 MA domain is reported to trimerize in HIV-1 (67), and Gag itself appears to trimerize in some conditions (11). In alpharetroviruses (47) and in human T-cell leukemia virus type 1 (56) MA appears to dimerize, and full-length RSV MA crystallizes as a dimer (R. Kingston, unpublished data). In all retroviruses massive multimerization of Gag is mediated by the NC domain, apparently indirectly via its binding to RNA (7, 27, 31, 60). To the degree that this large-scale multimerization is irreversible, membrane binding probably also is irreversible.
Understanding the mechanism by which multimerization enhances membrane binding is of fundamental importance. Based on the model of Tang et al. (67), trimerization should increase membrane affinity for two reasons. First, as we have shown directly for dimers, a complex of more than one MA will have increased avidity for acidic lipids because of electrostatic effects. Second, the trimerization-induced exposure of the three myristates and their insertion into the bilayer should further augment binding. Our experimental protocol did not allow binding constants to be assigned for dimerized, myristoylated MA. A more sensitive binding assay with a wider dynamic range will be needed to quantitatively and separately discern the two predicted effects of dimerization.
The published results on the role IPs or PIPs in HIV-1 assembly and budding are difficult to interpret in a unitary fashion. Interfering with PI(4,5)P2 metabolism strongly perturbs budding (38), MA has a specific binding site for short-chain fatty acid versions of PI(4,5)P2 (59), and this binding leads to release of the myristate from its pocket. Although the reported dissociation constant for PI(4,5)P2 is relatively weak, about 150 μM at physiological ionic strength and pH 5.5 (59), together the in vitro and in vivo results clearly argue for a role for this lipid in HIV-1 budding. Therefore, it may be unexpected that our preliminary experiments showed only a modest effect of PI(4,5)P2 on MA affinity for 2:1 PC-PS liposomes, suggesting that PI(4,5)P2 does not play a paramount role in determining membrane affinity in vitro under these conditions. Possibly, the electrostatic interactions between the basic surface of MA and the liposome surface have a strong enough myristate-exposing function in vitro to largely mask any additional effect of PI(4,5)P2. However, we cannot exclude that technical complications due to the solubility properties of long-chain PI(4,5)P2 interfered with the in vitro binding assays used.
The bridge between biochemical measurements and observations on living cells may not be direct. For example, although liposome flotation presumably reflects equilibrium binding, membrane targeting in vivo may be controlled by kinetic factors. In assessing the role PI(4,5)P2 in directly promoting HIV-1 Gag-membrane interactions, it is important to note that the MA domains of retroviruses other than HIV-1 are not known to interact with PI(4,5)P2, and yet these Gag proteins typically bind to and bud from the plasma membrane with an efficiency similar to that of HIV-1 Gag. Finally, if every HIV-1 Gag molecule brought one PIP2 molecule into the virion, and assuming 5,000 Gag molecules in a complete immature spherical HIV-1 particle (5), the resulting amount of PIP2 is likely to be incompatible with the lipid composition of HIV-1. The published composition shows that PI is selectively excluded from virions (1). However, that analysis did not include an explicit quantitation of phosphorylated PI species, which therefore can be estimated only by inference about the identity of unassigned radioactive spots on the thin layer chromatogram. Several assumptions render such estimates uncertain. Perhaps, as suggested by Saad et al. (59), PI(4,5)P2 is not used stoichiometrically but only to seed budding of the virus.
IP6 and IP5, but not the PIP2 headgroup IP3, are reported to support proper in vitro assembly of HIV-1 Gag as a purified protein (9). The fact that IP4 and dibutyryl PIP3 do promote Gag assembly, although to a lesser degree than IP6 or IP5, suggests that the relevant molecular species in vivo might be PIP3 and not PIP2. A complication in interpreting the in vitro assembly experiments is that IP6 does not bind tightly to MA itself but only to the larger Gag molecule that includes not only MA both also the CA and NC domains (11). Furthermore, binding of this small molecule to Gag leads to chemical protection of two lysine residues in MA (64), which in the NMR studies are not visibly affected by binding of PIP2 to the hydrophobic pocket (59). Therefore, it seems possible that IPs and PIPs bind to Gag in different ways. Our results do not clarify these several apparent discrepancies.
In summary, our results quantitatively establish the importance both of electrostatic interactions with acid lipids and of multimerization in membrane binding of HIV-1 MA. The present study should serve as a platform for more detailed analyses that address the mechanisms by which lipids like PI(4,5)P2 and the myristate modification modulate membrane interaction.
Acknowledgments
We thank Michael Summers for the expression plasmid for myristoylated MA, Wesley Sundquist for the gift of purified HIV-1 CA protein, and Gerald Feigenson for technical advice.
This study was supported by USPHS grants CA20087 to V.M.V. and AI54167 and GM66147 to D.M., by a grant for supercomputing MCB030023 from National Computational Science Alliance to D.M., and by a seed grant to support collaborative research between the Weill Cornell Medical College and the Cornell main campus. D.A.-A. was supported by minority supplement CA20087-S1.
Footnotes
Published ahead of print on 28 March 2007.
REFERENCES
- 1.Aloia, R. C., H. Tian, and F. C. Jensen. 1993. Lipid composition and fluidity of the human immunodeficiency virus envelope and host cell plasma membranes. Proc. Natl. Acad. Sci. USA 90:5181-5185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Batonick, M., M. Favre, M. Boge, P. Spearman, S. Honing, and M. Thali. 2005. Interaction of HIV-1 Gag with the clathrin-associated adaptor AP-2. Virology 342:190-200. [DOI] [PubMed]
- 3.Ben-Tal, N., B. Honig, C. Miller, and S. McLaughlin. 1997. Electrostatic binding of proteins to membranes: theoretical prediction and experimental results with charybdotoxin and phospholipid vesicles. Biophys. J. 73:1717-1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ben-Tal, N., B. Honig, R. M. Peitzsch, G. Denisov, and S. McLaughlin. 1996. Binding of small basic peptides to membranes containing acidic lipids: theoretical models and experimental results. Biophys. J. 71:561-575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Briggs, J. A., M. N. Simon, I. Gross, H. G. Krausslich, S. D. Fuller, V. M. Vogt, and M. C. Johnson. 2004. The stoichiometry of Gag protein in HIV-1. Nat. Struct. Mol. Biol. 11:672-675. [DOI] [PubMed] [Google Scholar]
- 6.Bryant, M., and L. Ratner. 1990. Myristoylation-dependent replication and assembly of human immunodeficiency virus 1. Proc. Natl. Acad. Sci. USA 87:523-527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burniston, M. T., A. Cimarelli, J. Colgan, S. P. Curtis, and J. Luban. 1999. Human immunodeficiency virus type 1 Gag polyprotein multimerization requires the nucleocapsid domain and RNA and is promoted by the capsid-dimer interface and the basic region of matrix protein. J. Virol. 73:8527-8540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buser, C. A., C. T. Sigal, M. D. Resh, and S. McLaughlin. 1994. Membrane binding of myristylated peptides corresponding to the NH2 terminus of Src. Biochemistry 33:13093-13101. [DOI] [PubMed] [Google Scholar]
- 9.Campbell, S., R. J. Fisher, E. M. Towler, S. Fox, H. J. Issaq, T. Wolfe, L. R. Phillips, and A. Rein. 2001. Modulation of HIV-like particle assembly in vitro by inositol phosphates. Proc. Natl. Acad. Sci. USA 98:10875-10879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dalton, A. K., P. S. Murray, D. Murray, and V. M. Vogt. 2005. Biochemical characterization of Rous sarcoma virus MA protein interaction with membranes. J. Virol. 79:6227-6238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Datta, S. A., Z. Zhao, P. K. Clark, S. Tarasov, J. N. Alexandratos, S. J. Campbell, M. Kvaratskhelia, J. Lebowitz, and A. Rein. 2007. Interactions between HIV-1 Gag molecules in solution: an inositol phosphate-mediated switch. J. Mol. Biol. 365:799-811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.del Alamo, M., J. L. Neira, and M. G. Mateu. 2003. Thermodynamic dissection of a low affinity protein-protein interface involved in human immunodeficiency virus assembly. J. Biol. Chem. 278:27923-27929. [DOI] [PubMed] [Google Scholar]
- 13.Ding, L., A. Derdowski, J. J. Wang, and P. Spearman. 2003. Independent segregation of human immunodeficiency virus type 1 Gag protein complexes and lipid rafts. J. Virol. 77:1916-1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dong, X., H. Li, A. Derdowski, L. Ding, A. Burnett, X. Chen, T. R. Peters, T. S. Dermody, E. Woodruff, J. J. Wang, and P. Spearman. 2005. AP-3 directs the intracellular trafficking of HIV-1 Gag and plays a key role in particle assembly. Cell 120:663-674. [DOI] [PubMed] [Google Scholar]
- 15.Ehrlich, L. S., S. Fong, S. Scarlata, G. Zybarth, and C. Carter. 1996. Partitioning of HIV-1 Gag and Gag-related proteins to membranes. Biochemistry 35:3933-3943. [DOI] [PubMed] [Google Scholar]
- 16.Freed, E. O., J. M. Orenstein, A. J. Buckler-White, and M. A. Martin. 1994. Single amino acid changes in the human immunodeficiency virus type 1 matrix protein block virus particle production. J. Virol. 68:5311-5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gallagher, K., and K. A. Sharp. 1998. Electrostatic contributions to heat capacity changes of DNA-ligand binding. Biophys. J. 75:769-776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gamble, T. R., S. Yoo, F. F. Vajdos, U. K. von Schwedler, D. K. Worthylake, H. Wang, J. P. McCutcheon, W. I. Sundquist, and C. P. Hill. 1997. Structure of the carboxyl-terminal dimerization domain of the HIV-1 capsid protein. Science 278:849-853. [DOI] [PubMed] [Google Scholar]
- 19.Gottlinger, H. G., J. G. Sodroski, and W. A. Haseltine. 1989. Role of capsid precursor processing and myristoylation in morphogenesis and infectivity of human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. USA 86:5781-5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Halwani, R., A. Khorchid, S. Cen, and L. Kleiman. 2003. Rapid localization of Gag/GagPol complexes to detergent-resistant membrane during the assembly of human immunodeficiency virus type 1. J. Virol. 77:3973-3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hermida-Matsumoto, L., and M. D. Resh. 1999. Human immunodeficiency virus type 1 protease triggers a myristoyl switch that modulates membrane binding of Pr55(Gag) and p17MA. J. Virol. 73:1902-1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hermida-Matsumoto, L., and M. D. Resh. 2000. Localization of human immunodeficiency virus type 1 Gag and Env at the plasma membrane by confocal imaging. J. Virol. 74:8670-8679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hill, C. P., D. Worthylake, D. P. Bancroft, A. M. Christensen, and W. I. Sundquist. 1996. Crystal structures of the trimeric human immunodeficiency virus type 1 matrix protein: implications for membrane association and assembly. Proc. Natl. Acad. Sci. USA 93:3099-3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holm, K., K. Weclewicz, R. Hewson, and M. Suomalainen. 2003. Human immunodeficiency virus type 1 assembly and lipid rafts: Pr55(Gag) associates with membrane domains that are largely resistant to Brij98 but sensitive to Triton X-100. J. Virol. 77:4805-4817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Honig, B., and A. Nicholls. 1995. Classical electrostatics in biology and chemistry. Science 268:1144-1149. [DOI] [PubMed] [Google Scholar]
- 26.Honig, B. H., and A. Nicholls. 1995. Classical electrostatics in biology and chemistry. Science 268:1144-1149. [DOI] [PubMed] [Google Scholar]
- 27.Johnson, M. C., H. M. Scobie, Y. M. Ma, and V. M. Vogt. 2002. Nucleic acid-independent retrovirus assembly can be driven by dimerization. J. Virol. 76:11177-11185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jouvenet, N., S. J. Neil, C. Bess, M. C. Johnson, C. A. Virgen, S. M. Simon, and P. D. Bieniasz. 2006. Plasma membrane is the site of productive HIV-1 particle assembly. PLoS Biol. 4:e435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee, P. P., and M. L. Linial. 1994. Efficient particle formation can occur if the matrix domain of human immunodeficiency virus type 1 Gag is substituted by a myristylation signal. J. Virol. 68:6644-6654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lindwasser, O. W., and M. D. Resh. 2001. Multimerization of human immunodeficiency virus type 1 Gag promotes its localization to barges, raft-like membrane microdomains. J. Virol. 75:7913-7924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma, Y. M., and V. M. Vogt. 2004. Nucleic acid binding-induced Gag dimerization in the assembly of Rous sarcoma virus particles in vitro. J. Virol. 78:52-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McLaughlin, S., and A. Aderem. 1995. The myristoyl-electrostatic switch: a modulator of reversible protein-membrane interactions. Trends Biochem. Sci. 20:272-276. [DOI] [PubMed] [Google Scholar]
- 33.Morikawa, Y., W. H. Zhang, D. J. Hockley, M. V. Nermut, and I. M. Jones. 1998. Detection of a trimeric human immunodeficiency virus type 1 Gag intermediate is dependent on sequences in the matrix protein, p17. J. Virol. 72:7659-7663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mulgrew-Nesbitt, A., K. Diraviyam, J. Wang, S. Singh, P. Murray, Z. Li, L. Rogers, N. Mirkovic, and D. Murray. 2006. The role of electrostatics in protein-membrane interactions. Biochim. Biophys. Acta 1761:812-826. [DOI] [PubMed] [Google Scholar]
- 35.Murray, D., L. Hermida-Matsumoto, C. A. Buser, J. Tsang, C. T. Sigal, N. Ben-Tal, B. Honig, M. D. Resh, and S. McLaughlin. 1998. Electrostatics and the membrane association of Src: theory and experiment. Biochemistry 37:2145-2159. [DOI] [PubMed] [Google Scholar]
- 36.Murray, P. S., Z. Li, J. Wang, C. L. Tang, B. Honig, and D. Murray. 2005. Retroviral matrix domains share electrostatic homology: models for membrane binding function throughout the viral life cycle. Structure 13:1521-1531. [DOI] [PubMed] [Google Scholar]
- 37.Nguyen, D. H., and J. E. Hildreth. 2000. Evidence for budding of human immunodeficiency virus type 1 selectively from glycolipid-enriched membrane lipid rafts. J. Virol. 74:3264-3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ono, A., S. D. Ablan, S. J. Lockett, K. Nagashima, and E. O. Freed. 2004. Phosphatidylinositol(4,5)bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc. Natl. Acad. Sci. USA 101:14889-14894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ono, A., D. Demirov, and E. O. Freed. 2000. Relationship between human immunodeficiency virus type 1 Gag multimerization and membrane binding. J. Virol. 74:5142-5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ono, A., and E. O. Freed. 1999. Binding of human immunodeficiency virus type 1 Gag to membrane: role of the matrix amino terminus. J. Virol. 73:4136-4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ono, A., and E. O. Freed. 2004. Cell-type-dependent targeting of human immunodeficiency virus type 1 assembly to the plasma membrane and the multivesicular body. J. Virol. 78:1552-1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ono, A., and E. O. Freed. 2001. Plasma membrane rafts play a critical role in HIV-1 assembly and release. Proc. Natl. Acad. Sci. USA 98:13925-13930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ono, A., J. M. Orenstein, and E. O. Freed. 2000. Role of the Gag matrix domain in targeting human immunodeficiency virus type 1 assembly. J. Virol. 74:2855-2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Paillart, J. C., and H. G. Gottlinger. 1999. Opposing effects of human immunodeficiency virus type 1 matrix mutations support a myristyl switch model of Gag membrane targeting. J. Virol. 73:2604-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peitzsch, R. M., and S. McLaughlin. 1993. Binding of acylated peptides and fatty acids to phospholipid vesicles: pertinence to myristoylated proteins. Biochemistry 32:10436-10443. [DOI] [PubMed] [Google Scholar]
- 46.Pelchen-Matthews, A., B. Kramer, and M. Marsh. 2003. Infectious HIV-1 assembles in late endosomes in primary macrophages. J. Cell Biol. 162:443-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pepinsky, R. B., D. Cappiello, C. Wilkowski, and V. M. Vogt. 1980. Chemical crosslinking of proteins in avian sarcoma and leukemia viruses. Virology 102:205-210. [DOI] [PubMed] [Google Scholar]
- 48.Perez-Caballero, D., T. Hatziioannou, J. Martin-Serrano, and P. D. Bieniasz. 2004. Human immunodeficiency virus type 1 matrix inhibits and confers cooperativity on Gag precursor-membrane interactions. J. Virol. 78:9560-9563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pessin, J. E., and M. Glaser. 1980. Budding of Rous sarcoma virus and vesicular stomatitis virus from localized lipid regions in the plasma membrane of chicken embryo fibroblasts. J. Biol. Chem. 255:9044-9050. [PubMed] [Google Scholar]
- 50.Platt, E. J., and O. K. Haffar. 1994. Characterization of human immunodeficiency virus type 1 Pr55gag membrane association in a cell-free system: requirement for a C-terminal domain. Proc. Natl. Acad. Sci. USA 91:4594-4598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Provitera, P., R. El-Maghrabi, and S. Scarlata. 2005. The effect of HIV-1 Gag myristoylation on membrane binding. Biophys. Chem. 119:23-32. [DOI] [PubMed]
- 52.Quigley, J. P., D. B. Rifkin, and M. H. Einhorn. 1972. Determination of phospholipid composition of RNA tumor viruses by 32 P labeling of infected cell cultures. Anal. Biochem. 47:614-619. [DOI] [PubMed] [Google Scholar]
- 53.Quigley, J. P., D. B. Rifkin, and E. Reich. 1972. Lipid studies of Rous sarcoma virus and host cell membranes. Virology 50:550-557. [DOI] [PubMed] [Google Scholar]
- 54.Quigley, J. P., D. B. Rifkin, and E. Reich. 1971. Phospholipid composition of Rous sarcoma virus, host cell membranes and other enveloped RNA viruses. Virology 46:106-116. [DOI] [PubMed] [Google Scholar]
- 55.Raposo, G., M. Moore, D. Innes, R. Leijendekker, A. Leigh-Brown, P. Benaroch, and H. Geuze. 2002. Human macrophages accumulate HIV-1 particles in MHC II compartments. Traffic 3:718-729. [DOI] [PubMed] [Google Scholar]
- 56.Rayne, F., A. V. Kajava, J. Lalanne, and R. Z. Mamoun. 2004. In vivo homodimerisation of HTLV-1 Gag and MA gives clues to the retroviral capsid and TM envelope protein arrangement. J. Mol. Biol. 343:903-916. [DOI] [PubMed] [Google Scholar]
- 57.Reil, H., A. A. Bukovsky, H. R. Gelderblom, and H. G. Gottlinger. 1998. Efficient HIV-1 replication can occur in the absence of the viral matrix protein. EMBO J. 17:2699-2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Saad, J. S., E. Loeliger, P. Luncsford, M. Liriano, J. Tai, A. Kim, J. Miller, A. Joshi, E. O. Freed, and M. F. Summers. 2007. Point mutations in the HIV-1 matrix protein turn off the myristyl switch. J. Mol. Biol. 366:574-585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Saad, J. S., J. Miller, J. Tai, A. Kim, R. H. Ghanam, and M. F. Summers. 2006. Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc. Natl. Acad. Sci. USA 103:11364-11369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sandefur, S., V. Varthakavi, and P. Spearman. 1998. The I domain is required for efficient plasma membrane binding of human immunodeficiency virus type 1 Pr55Gag. J. Virol. 72:2723-2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Scarlata, S., L. S. Ehrlich, and C. A. Carter. 1998. Membrane-induced alterations in HIV-1 Gag and matrix protein-protein interactions. J. Mol. Biol. 277:161-169. [DOI] [PubMed] [Google Scholar]
- 62.Sherer, N. M., M. J. Lehmann, L. F. Jimenez-Soto, A. Ingmundson, S. M. Horner, G. Cicchetti, P. G. Allen, M. Pypaert, J. M. Cunningham, and W. Mothes. 2003. Visualization of retroviral replication in living cells reveals budding into multivesicular bodies. Traffic 4:785-801. [DOI] [PubMed] [Google Scholar]
- 63.Shindyalov, I. N., and P. E. Bourne. 1998. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 11:739-747. [DOI] [PubMed] [Google Scholar]
- 64.Shkriabai, N., S. A. Datta, Z. Zhao, S. Hess, A. Rein, and M. Kvaratskhelia. 2006. Interactions of HIV-1 Gag with assembly cofactors. Biochemistry 45:4077-4083. [DOI] [PubMed] [Google Scholar]
- 65.Spearman, P., R. Horton, L. Ratner, and I. Kuli-Zade. 1997. Membrane binding of human immunodeficiency virus type 1 matrix protein in vivo supports a conformational myristyl switch mechanism. J. Virol. 71:6582-6592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Spearman, P., J. J. Wang, N. Vander Heyden, and L. Ratner. 1994. Identification of human immunodeficiency virus type 1 Gag protein domains essential to membrane binding and particle assembly. J. Virol. 68:3232-3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tang, C., E. Loeliger, P. Luncsford, I. Kinde, D. Beckett, and M. F. Summers. 2004. Entropic switch regulates myristate exposure in the HIV-1 matrix protein. Proc. Natl. Acad. Sci. USA 101:517-522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tritel, M., and M. D. Resh. 2000. Kinetic analysis of human immunodeficiency virus type 1 assembly reveals the presence of sequential intermediates. J. Virol. 74:5845-5855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Welsch, S. O. T. Keppler, A. Habermann, I. Allespach, J. Krijnse-Locker, and H.-G. Kräusslich. 2007. HIV-1, buds predominantly at the plasma membrane of primary human macrophages. PLoS Pathology 3:e36. [DOI] [PMC free article] [PubMed]
- 70.Yuan, X., X. Yu, T. H. Lee, and M. Essex. 1993. Mutations in the N-terminal region of human immunodeficiency virus type 1 matrix protein block intracellular transport of the Gag precursor. J. Virol. 67:6387-6394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhou, W., L. J. Parent, J. W. Wills, and M. D. Resh. 1994. Identification of a membrane-binding domain within the amino-terminal region of human immunodeficiency virus type 1 Gag protein which interacts with acidic phospholipids. J. Virol. 68:2556-2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhou, W., and M. D. Resh. 1996. Differential membrane binding of the human immunodeficiency virus type 1 matrix protein. J. Virol. 70:8540-8548. [DOI] [PMC free article] [PubMed] [Google Scholar]