

Abstract
Rotaviruses are the leading cause of infantile viral gastroenteritis worldwide. Mature enterocytes of the small intestine infected by rotavirus undergo apoptosis, and their replacement by less differentiated dividing cells probably leads to defective absorptive function of the intestinal epithelium, which, in turn, contributes to osmotic diarrhea and rotavirus pathogenesis. Here we show that infection of MA104 cells by the simian rhesus rotavirus strain RRV induced caspase-3 activation, DNA fragmentation, and cleavage of poly(ADP-ribose) polymerase; all three phenomena are features of apoptosis. RRV induced the release of cytochrome c from mitochondria to the cytosol, indicating that the mitochondrial apoptotic pathway was activated. RRV infection of MA104 cells activated Bax, a proapoptotic member of the Bcl-2 family, as revealed by its conformational change. Most importantly, Bax-specific small interfering RNAs partially inhibited cytochrome c release in RRV-infected cells. Thus, mitochondrial dysfunction induced by rotavirus is Bax dependent. Apoptosis presumably leads to impaired intestinal functions, so our findings contribute to improving our understanding of rotavirus pathogenesis at the cellular level.
Rotavirus is a nonenveloped, double-stranded RNA virus belonging to the family Reoviridae. It is the major etiologic agent of severe gastroenteritis in children under the age of 5 years and causes 600,000 deaths per year (40). Rotavirus infection is mainly restricted to the small intestinal villus epithelium, resulting in villus atrophy. Diarrhea associated with rotavirus is a multifactorial process: it involves dysfunctions of both nutrient digestion and enterocyte absorption at the tops of villi, as well as the secretion of water and electrolytes (29, 32, 43). The defective absorptive function and the increased intestinal permeability may be consequences of the replacement of rotavirus-infected mature enterocytes by less differentiated cells, and this could contribute to rotavirus pathogenesis. In a murine model, rotavirus-infected enterocytes undergo apoptosis and are therefore lost (4). It is therefore likely that apoptosis makes a major contribution to diarrhea associated with rotavirus infection.
Cell death by apoptosis is part of the normal development and maintenance of homeostasis (51) but is also involved in pathological situations associated with infections (16, 45) and other causes (3). Apoptosis is characterized by chromatin condensation, cell shrinkage, membrane blebbing, and DNA fragmentation. It can be generally divided into two nonexclusive pathways: the death receptor pathway (extrinsic) and the mitochondrial pathway (intrinsic) (10, 13, 20). In the extrinsic pathway, stimulation of death receptors, such as Fas and tumor necrosis factor receptor 1, leads to the formation of the death-inducing signaling complex (DISC), which allows the activation of caspase-8 and/or caspase-10 and then that of downstream effector caspases, particularly caspase-3 (2, 34). The intrinsic pathway is initiated in response to diverse apoptotic stimuli and leads to the loss of mitochondrial transmembrane potential and the release of several proapoptotic proteins, including cytochrome c and Smac/Diablo, from the mitochondrial intermembrane space to the cytosol. Once released, cytochrome c forms a complex with Apaf-1 and procaspase-9, resulting in activation first of this initiator caspase and then of effector caspases (52). Smac/Diablo promotes caspase activation by directly binding to and inhibiting the caspase inhibitors belonging to the family of inhibitors of apoptosis proteins (50, 52). This mitochondrial apoptotic pathway is tightly controlled by protein members of the Bcl-2 family. Some, including Bcl-2 and Bcl-XL, inhibit apoptosis, whereas others, including Bax and Bid, induce apoptosis (9).
The extrinsic and intrinsic pathways may cross talk through the proapoptotic protein Bid. Indeed, caspase-8 can cleave Bid to generate a truncated form, tBid, that targets mitochondria and activates the proapoptotic protein Bax (26, 27, 30).
There have been very few studies of rotavirus-induced apoptosis in primate cells in vitro. An early study with human carcinoma HT29 cells indicated that rotavirus induced peripheral condensation of the chromatin and fragmentation of the nuclei, suggesting that apoptosis was induced in infected cells (47). A more recent study with fully differentiated Caco-2 cells indicated that rotavirus induced apoptosis in these cells and did so through the mitochondrial pathway (7). However, the precise signaling pathways leading to mitochondrial dysfunction following rotavirus infection have not been investigated.
Here we studied apoptosis induced by the rhesus rotavirus (RRV) strain in the monkey kidney cell line MA104, the cellular model with which the rotavirus cycle has been best characterized. We first confirmed that RRV-induced apoptosis in this model occurs through the mitochondrial pathway as observed in Caco-2 cells. We then investigated the cascade of events related to mitochondrial dysfunction. We report here that the mitochondrial apoptotic pathway in RRV-infected MA104 cells is Bax dependent. It is, to our knowledge, the first demonstration of Bax-dependent apoptosis in rotavirus-infected cells.
MATERIALS AND METHODS
Chemicals.
Protein G (P3296), staurosporine (STS) (S4400), and a mouse anti-tubulin antibody (T5168) were obtained from Sigma-Aldrich. Complete protease inhibitor mixture was obtained from Roche Applied Science. z-VAD-fmk (627610), z-DEVD-fmk (264155), and z-LEHD-fmk (218761) were purchased from Calbiochem. The mouse anti-Cox IV antibody (A21347) was purchased from Molecular Probes. Mouse anti-Bcl-2 (sc-509) and anti-Bax (clone 6A7; sc-23959) antibodies were purchased from Santa Cruz Biotechnology. The rabbit anti-Bax antibody (NT 06-499) and mouse anti-cytochrome c antibody (556433) were obtained from Upstate and BD Pharmingen, respectively. Rabbit anti-Smac/Diablo (2409) was purchased from ProSci Incorporated. Horseradish peroxidase (HRP)-conjugated anti-mouse (NA9310V) and anti-rabbit (NA9340V) secondary antibodies were obtained from Amersham Biosciences. The goat anti-Bid antibody (AF860) and donkey anti-goat (HAF109) HRP-conjugated antibody were purchased from Research & Diagnostic Antibodies. A mouse antibody (clone C-2-10) against poly(ADP-ribose) polymerase (PARP) was obtained from Biomol. Mouse anti-caspase-3 (9668) and anti-caspase-8 (9746) antibodies and Bax (6321) and irrelevant control (6201) small interfering RNAs (siRNAs) were purchased from Cell Signaling.
Cells and virus.
The monkey kidney cell line MA104 was cultured in minimal Eagle's medium supplemented with 10% fetal bovine serum, 2 mM glutamine, and 0.1 mM nonessential amino acids in a 5% CO2 incubator. The rotavirus strain RRV was kindly provided by Didier Poncet (Gif-sur-Yvette, France). Virus stocks were generated in MA104 cells in a serum-free culture medium supplemented with trypsin (0.5 μg/ml). Viruses were activated by treatment with trypsin at 37°C for 30 min, and MA104 cell monolayers were infected at a multiplicity of infection of 0.002 PFU/cell. After 1 h of adsorption at 37°C, the inoculum was removed, and the infected cells were incubated in serum-free medium containing trypsin. When cytopathic effects were complete, the cultures were frozen and then thawed, and the cell debris was removed by centrifugation.
To infect MA104 cells, RRV was activated in serum-free medium with 0.5 μg/ml trypsin. Cells were washed twice in serum-free medium and then infected at a multiplicity of infection of 10. The inoculum was removed 1 h later and replaced with a medium containing 10% fetal bovine serum. This time was defined as 0 h postinfection (p.i.) for all experiments.
Assessment of apoptosis by flow cytometry.
The percentages of cells that were apoptotic were determined by flow cytofluorimetric analysis of aliquots of 2 × 106 cells incubated with acridine orange (AO), a metachromatic nuclear dye (excitation wavelength, 500 nm; emission wavelength, 526 nm), for 15 min at 37°C. AO fluorescence was measured with a FACScan machine (Becton Dickinson). Two populations of cells were separated: one consisting of the living cells, characterized by bright fluorescence labeling, and the second consisting of apoptotic cells, with a characteristic distinct pattern of reduced fluorescence intensity (12). We analyzed at least 10,000 cells for each sample. Data were analyzed with CellQuest software (Becton Dickinson).
Quantification of oligonucleosomal DNA fragmentation.
DNA fragmentation was assayed using a cell death detection enzyme-linked immunosorbent assay (ELISA) kit (Roche Applied Science) with RRV-infected MA104 cells plated in 6-well culture dishes according to the manufacturer's instructions; the test measures the cytosolic histone-associated mono- and oligonucleosomal fragments. The concentration of nucleosomal fragments in cytosolic fractions was determined by a sandwich ELISA using histone-specific antibodies preadsorbed onto microtiter plates and peroxidase-conjugated antibodies against DNA. Peroxidase activity was measured photometrically at 405 nm. The experiments were run in triplicate.
Detection of active caspase-8.
Caspase-8 activation was detected using a carboxyfluorescein (FAM) FLICA apoptosis detection kit (Immunochemistry Technologies, LLC) according to the manufacturer's protocol. This assay is based on a fluorescein-labeled inhibitor (FAM-LETD-fmk) that binds specifically to active caspase-8 (excitation wavelength, 490 nm; emission wavelength, 520 nm). Apoptotic cells containing active caspase-8 were detected with a FACScan (Becton Dickinson) machine. We analyzed at least 10,000 cells for each sample. Data were analyzed with CellQuest software (Becton Dickinson).
Transfection of siRNA.
RNA interference was used to silence Bax gene expression in MA104 cells. Cells were plated in 6-well culture dishes with complete medium and allowed to grow for 24 h so as to reach 50 to 80% confluence. The cells were then transfected with a siRNA (6321; Cell Signaling) targing Bax mRNA. An irrelevant siRNA (6201; Cell Signaling) that does not lead to the specific degradation of any cellular mRNA was used as a negative control. A mixture of OptiMEM medium and LipofectAMINE was incubated for 10 min at room temperature and was then incubated with the siRNA (25 nM) for 20 min at room temperature to allow complex formation. This siRNA mixture was then added to each well according to the manufacturer's suggested protocol. Twenty-four hours after transfection, the medium was changed, and cells were infected with the virus 24 h later. Gene silencing was verified by testing for proteins by immunoblot analysis after transient transfection of MA104 cells with siRNA.
Whole-cell extracts.
Approximately 5·106 cells were collected, washed with phosphate-buffered saline (PBS), and then resuspended in a lysis buffer (20 mM Tris [pH 7.5], 135 mM NaCl, 2 mM EDTA, 1% Triton X-100, 10% glycerol) supplemented with a protease inhibitor mixture (Roche). The cells were homogenized on ice using a Dounce homogenizer and incubated for 10 min at 4°C in the lysis buffer. The lysates were clarified by centrifugation for 10 min at 1,200 × g. The supernatant was collected as the whole-cell extract.
Subcellular fractionation.
A subcellular proteome extraction kit (Calbiochem) was used to isolate the cytosol and heavy-membrane fractions of MA104 cells according to the manufacturer's instructions. Cells (5·106) were harvested, pelleted, washed twice, resuspended in the ice-cold Extraction I buffer containing a protease inhibitor mixture, and incubated for 10 min at 4°C with gentle agitation. The suspension was centrifuged at 1,000 × g and 4°C for 10 min. The supernatant was used as the cytosol fraction. The pellet was resuspended in the ice-cold Extraction II buffer containing a protease inhibitor mixture and was incubated for 30 min at 4°C with gentle agitation. It was then centrifuged at 6,000 × g for 10 min at 4°C, and the supernatant was used as the heavy-membrane fraction.
Detection of Bax conformational change by immunoprecipitation.
Cells were harvested, washed in PBS, and suspended in lysis buffer {10 mM HEPES [pH 7.4], 150 mM NaCl, 1% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate [CHAPS]} containing a mixture of protease inhibitors. The zwitterionic detergent CHAPS was used because it does not affect Bax conformation (18). Cells were homogenized on ice with a Dounce homogenizer, incubated for 2 h in the lysis buffer, and centrifuged for 30 min at 14,000 × g. The resulting supernatant was incubated overnight at 4°C with 20 μl of protein G and 2 μg of the anti-Bax antibody (6A7). The immunoprecipitates were collected by centrifugation at 14,000 × g (4°C, 5 min). The pellets were washed with immunoprecipitation buffer and suspended in 50 μl of Laemmli's buffer containing a reducing agent (Invitrogen).
Western-blot analysis.
Protein concentrations were determined by using a Bio-Rad protein assay kit (Bio-Rad, Richmond, CA). Samples of equal protein content were resuspended in Laemmli's electrophoresis sample buffer containing a reducing agent, denatured by boiling for 5 min, subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (10 to 20% Tricine gels; Novex), and transferred to nitrocellulose membranes (Amersham Biosciences). Nonspecific sites were blocked by incubating the membranes for 1 h at room temperature with 5% nonfat milk and 0.1% Tween 20 in PBS (PBST, pH 7.4), and the membranes were incubated overnight at 4°C or for 2 h at room temperature with the primary antibody. Membranes were then washed in PBST and treated with an appropriate HRP-conjugated secondary antibody for 1 h at room temperature. The immunoblots were washed in PBST and developed using an enhanced chemiluminescence detection kit (Amersham Biosciences). Anti-tubulin and anti-Cox IV antibodies were used to verify equal protein loading.
Statistical analysis.
Data are expressed as means ± standard errors of the means for three independent experiments. Student's t test was used to compare experimental conditions and controls. A P value of <0.05 was considered significant.
RESULTS
RRV infection induces apoptosis in MA104 cells through the mitochondrial pathway.
Apoptosis induced by RRV in MA104 cells was investigated by following the kinetics of DNA fragmentation with the cell death ELISA kit (Roche) (Fig. 1A). At early times p.i. (until 8 h p.i.), DNA fragmentation was not significantly different for infected and uninfected cells. Enrichment of nucleosomal DNA fragments started by 12 h p.i. and increased until 20 h p.i. DNA fragmentation at 20 h p.i. was three- to fourfold that of mock-infected cells and similar to that observed after 14 h of treatment with STS, used as a positive control for DNA fragmentation. These results confirm that rotavirus induced apoptosis in MA104 cells.
FIG. 1.
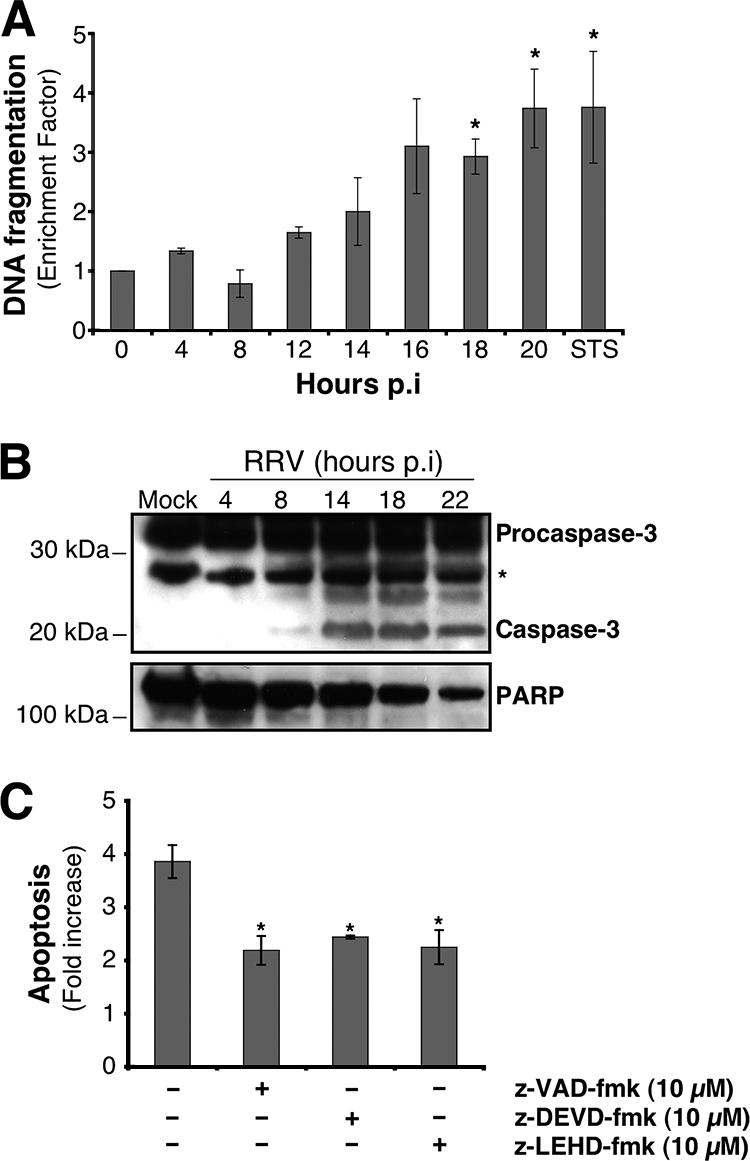
RRV induces apoptosis in MA104 cells. (A) DNA fragmentation in RRV-infected MA104 cells. At the indicated times p.i., DNA fragmentation was analyzed using the cell death detection ELISAPLUS, which detects the appearance of histone-associated low-molecular-weight DNA in the cytoplasm of cells. Mock-infected and STS-treated MA104 cells were used as negative and positive controls, respectively. The enrichment factor was calculated as the absorbance (A405 − A490) for treated cells divided by that for the corresponding untreated cells. Data are means ± standard deviations from three experiments. *, P < 0.05 by a Student t test comparing RRV-infected to mock-infected MA104 cells. (B) RRV infection triggered caspase-3 activation and PARP processing in MA104 cells. At the indicated times p.i., whole-cell extracts were subjected to Western blot analysis with anti-caspase-3 and anti-PARP antibodies. Mock-infected MA104 cells were used as negative controls. The asterisk indicates a nonspecific protein band that was used as a protein loading control. Positions of molecular weight markers are indicated on the left. (C) RRV-induced apoptosis is dependent on caspase activation in MA104 cells. MA104 cells were either left untreated or treated with either the broad caspase inhibitor z-VAD-fmk (10 μM), the specific caspase-9 inhibitor z-LEHD-fmk (10 μM), or the specific caspase-3 inhibitor z-DEVD-fmk (10 μM) 2 h before RRV infection, and inhibitor concentrations were maintained throughout the infection. Mock- and RRV-infected MA104 cells were analyzed 14 h p.i. by flow cytometry after staining with the nuclear dye AO, and the n-fold increase in apoptosis was calculated as the ratio of apoptotic cell percentages for RRV-infected MA104 cells to that for mock-infected MA104 cells. Data are means from three independent experiments. Error bars, standard errors of the mean. *, P < 0.05 by a Student t test comparing untreated to treated MA104 cells.
We then looked for activation of caspase-3, the central executioner in the apoptotic program, in RRV-infected MA104 cells. Cleavage of 32-kDa procaspase-3, revealed by the detection of its 19-kDa cleavage product in a whole-cell lysate, was substantial at about 14 h p.i. (Fig. 1B). To confirm the activation of caspase-3 in RRV-infected MA104 cells, we investigated the processing of one of its characteristic substrates, PARP (Fig. 1B). The amount of full-length PARP in a whole-cell lysate of RRV-infected MA104 cells declined from 14 to 22 h p.i., paralleling the pattern of caspase-3 activation. These results show that rotavirus induced the activation of the caspase executioner in MA104 cells. Furthermore, the peak of caspase-3 activation coincided with that of virus production during a single-cycle infection under the same experimental conditions (data not shown).
To assess the role of caspases in cell death resulting from RRV infection, infected cells were treated with a broad-spectrum caspase inhibitor, z-VAD-fmk (10 μM; Calbiochem). The roles of caspase-9 and caspase-3 were investigated with the specific inhibitors z-LEHD-fmk (10 μM; Calbiochem) and z-DEVD-fmk (10 μM; Calbiochem), respectively. Apoptosis was analyzed at 14 h p.i. by measuring chromatin condensation and fragmentation by flow cytometry after AO staining (Fig. 1C). The three caspase inhibitors significantly reduced the percentage of apoptotic cells following RRV infection relative to that for untreated cells. (The same level of apoptosis inhibition was observed with z-VAD-fmk at a concentration of 100 μM [data not shown].) These results indicate that caspases are at least partly involved in RRV-induced apoptosis. Furthermore, the decrease in apoptosis for cells treated with the caspase-9 inhibitor seems to indicate that the mitochondrial pathway is activated in RRV-infected cells.
To confirm that the mitochondrial pathway is activated during RRV infection in MA104 cells, we investigated the kinetics of cytochrome c and Smac/Diablo efflux from mitochondria into the cytosol. Infected cells were fractionated at various times after infection to separate the cytosolic fraction from the heavy-membrane fraction containing mitochondria, and the fractions were studied by Western blotting (Fig. 2). Cytochrome c and Smac/Diablo were detected in the cytoplasm of infected MA104 cells from 9 h p.i. and 12 h p.i., respectively; their amounts increased in the cytosol thereafter until the last time point investigated (18 h p.i.), whereas their amounts decreased in the heavy-membrane fraction. These findings indicate that rotavirus causes the release of cytochrome c and Smac/Diablo into the cytosol in MA104 cells, consistent with apoptotic mitochondrial dysfunction.
FIG. 2.

The mitochondrial apoptotic pathway is activated in RRV-infected MA104 cells. (Top) Time course of cytochrome c and Smac/Diablo release in RRV-infected MA104 cells. At the indicated times p.i., cells were collected and subjected to subcellular fractionation as described in Materials and Methods. The cytosolic and heavy-membrane fraction proteins were assayed by Western blot analysis for cytochrome c and Smac/Diablo. Mock-infected MA104 cells were used as negative controls. Cox IV and tubulin were used as protein loading controls for the heavy-membrane and cytosolic fractions, respectively. The positions of molecular weight markers are indicated on the left. (Bottom) Cytochrome c and Smac/diablo protein levels of heavy-membrane and cytosolic fractions were determined by densitometry and plotted as ratios relative to the levels of Cox IV and tubulin, respectively.
The RRV-induced mitochondrial apoptotic pathway is independent of caspases and Bid.
We then looked for upstream signals that could trigger the activation of the mitochondrial apoptotic pathway following RRV infection. This pathway can be activated by the BH3-only protein Bid following its cleavage by caspase-8 (26, 30). First, the kinetics of the processing of procaspase-8 in RRV-infected MA104 cells was analyzed by immunoblotting (Fig. 3A). The amounts of procaspase-8 decreased relative to those in mock-infected cells from 14 h p.i., suggesting caspase-8 activation. Caspase-8 activation was confirmed at 14 h p.i. by flow cytometry with a FLICA apoptosis detection kit, based on the specific binding of a fluorescein-labeled inhibitor (FAM-LETD-fmk; Immunochemistry Technologies, LLC) to active caspase (Fig. 3B). The kinetics of Bid processing in RRV-infected MA104 cells was also analyzed by immunoblotting (Fig. 3A). The amounts of Bid and procaspase-8 decreased with respect to those in mock-infected cells, suggesting a cleavage of Bid mediated by activated caspase-8.
FIG. 3.

RRV induces caspase-8 activation and Bid processing in MA104 cells. (A) (Top) Time course of caspase-8 activation and Bid processing in RRV-infected MA104 cells. At the indicated times p.i., whole-cell extracts were subjected to immunoblot analysis with anti-caspase-8 and anti-Bid antibodies. Mock-infected MA104 cells were used as negative controls. Tubulin was used as a control for protein loading. Positions of molecular weight markers are indicated on the left. (Bottom) Bid protein levels were determined by densitometry and plotted as ratios relative to the levels of tubulin. (B) Caspase-8 activation was determined for mock- and RRV-infected MA104 cells (14 h p.i.) by flow cytometry using fluorescein-labeled inhibitor (FAM-LETD-fmk) that binds specifically on active caspase-8, as described in Materials and Methods. A histogram representative of two independent experiments is shown. The percentage of cells positive for activated caspase-8 following RRV infection is given.
To determine whether caspase-8 and Bid were required for engaging the mitochondrial pathway of apoptosis, we assessed cytochrome c release in the presence of the broad-spectrum caspase inhibitor z-VAD-fmk. The release of cytochrome c from the mitochondria into the cytosol in RV-infected MA104 cells (14 h p.i.) was analyzed by Western blotting following subcellular fractionation (Fig. 4). As expected, cleavage of both Bid and PARP was inhibited in the presence of the caspase inhibitor (Fig. 4A). In contrast, z-VAD-fmk was unable to prevent cytochrome c release (Fig. 4B), suggesting that mitochondrial dysfunction in RRV-infected MA104 cells is induced by a caspase- and Bid-independent mechanism. Thus, caspases seem to be involved in RRV-induced apoptosis at a position in the pathway downstream from the mitochondrial dysfunction.
FIG. 4.

Cytochrome c release in RRV-infected MA104 cells is caspase independent. (A) The broad-spectrum caspase inhibitor z-VAD-fmk inhibits Bid and PARP processing in RRV-infected MA104 cells. Mock- and RRV-infected MA104 cells either were not pretreated or were pretreated with 100 μM z-VAD-fmk for 2 h before RRV infection, and treatment was continued during infection. Whole-cell extracts were prepared 14 h p.i., and Bid and PARP levels were assayed by Western blot analysis. Mock-infected MA104 cells were used as negative controls. Tubulin was used as a control for protein loading. Positions of molecular weight markers are indicated on the left. (B) z-VAD-fmk did not inhibit cytochrome c release in RRV-infected MA104 cells. Mock- and RRV-infected MA104 cells either were not pretreated or were pretreated with 100 μM z-VAD-fmk for 2 h before RRV infection, and treatment was continued during infection. Fourteen hours after RRV infection, the cytosolic and heavy-membrane fractions were assayed for cytochrome c by Western blotting. Mock-infected MA104 cells were used as negative controls. Cox IV and tubulin were used as protein loading controls for the heavy-membrane and cytosolic fractions, respectively. Positions of molecular weight markers are indicated on the left.
Bax is activated during RRV infection by a caspase-independent pathway.
The proapoptotic Bcl-2 family protein Bax is a pivotal regulator of cytochrome c release from mitochondria to the cytosol (1, 38, 53). In healthy cells, most Bax is in the cytoplasm, in an inactive form. Bax-mediated cell death occurs through a conformational change and its translocation to the mitochondrial outer membrane, resulting in the loss of mitochondrial membrane potential and the release of proapoptogenic factors including cytochrome c (18, 33, 54). We investigated the kinetics of the conformational change of Bax in RRV-infected cells. Total-cell lysates were prepared at the indicated times p.i. (Fig. 5A) in a lysis buffer (containing CHAPS) that did not affect Bax conformation. Bax was then immunoprecipitated with an anti-Bax antibody (6A7) specific for the active conformation of Bax (18, 19) and was visualized by Western blotting (Fig. 5A). For mock-infected cells, no activated Bax was detected in the immunoprecipitates. From 12 h p.i., Bax was precipitated by antibody 6A7, indicating that RRV infection induced the conformational change of Bax, as observed for STS-treated cells used as positive controls (data not shown). Thus, rotavirus infection triggers a conformational change in Bax in MA104 cells. Furthermore, we showed that Bax activation still occurred in the presence of the broad-spectrum caspase inhibitor z-VAD-fmk (100 μM; Calbiochem) (Fig. 5B), indicating that Bax activation during RRV infection is caspase independent.
FIG. 5.

RRV induces Bax protein activation in MA104 cells. (A) RRV induced a conformational change in Bax in MA104 cells. At the indicated time p.i., MA104 cells were lysed in immunoprecipitation buffer containing the zwitterionic detergent CHAPS (which maintains Bax in its activated conformation). The anti-Bax antibody 6A7 was used to immunoprecipitate conformationally active Bax protein. The immunoprecipitates were analyzed by immunoblotting with an anti-Bax antibody. Asterisk indicates immunoglobulin light chains. Mock-infected MA104 cells were used as negative controls. Positions of molecular weight markers are indicated on the left. (B) z-VAD-fmk did not inhibit Bax conformational change in RRV-infected MA104 cells. RRV-infected MA104 cells either were not pretreated or were pretreated with 100 μM z-VAD-fmk for 2 h before infection, and treatment was continued during infection. Whole-cell lysates were immunoprecipitated with an activated Bax antibody (clone 6A7) and then analyzed by immunoblotting with an anti-Bax antibody. Mock-infected MA104 cells were used as negative controls. Asterisk indicates immunoglobulin light chains. Positions of molecular weight markers are indicated on the left. (C) RRV infection promoted Bcl-2 degradation in MA104 cells. At the indicated times p.i., whole-cell lysates were prepared. Bax and Bcl-2 proteins were detected by Western blotting with anti-Bax and anti-Bcl-2 specific antibodies. Mock-infected MA104 cells were used as negative controls. Tubulin was used as a control for protein loading. Positions of molecular weight markers are indicated on the left.
MA104 cell infection by RRV increases the Bax/Bcl-2 ratio.
The Bcl-2-family, obviously, includes the antiapoptotic protein Bcl-2, which converges on mitochondria and competes with proapoptotic Bax to regulate the release of cytochrome c in response to an apoptotic signal (1, 15, 44). Since the ratio of pro- and antiapoptotic proteins determines, at least in part, the susceptibility of cells to the death signal (38), we studied the time-dependent effects of RRV infection on the abundance of Bax and Bcl-2 in MA104 cells (Fig. 5C). The amount of Bcl-2 decreased as the viral infection proceeded, whereas the total amount of Bax was unaffected. Thus, rotavirus infection increased the Bax/Bcl-2 ratio, leading to an increase in the amount of free Bax. The increase in the amount of free Bax could facilitate its activation and thus contribute to the mitochondrial apoptotic pathway.
The RRV-induced mitochondrial apoptotic pathway is Bax dependent.
These various findings seem to indicate that RRV induces Bax-mediated apoptosis in MA104 cells. To assess the requirement for Bax in RRV-induced mitochondrial dysfunction, MA104 cells were transiently transfected with a specific siRNA to knock down Bax expression. Western blot analysis of MA104 cells transfected with the Bax siRNA (Cell Signaling) and with the irrelevant control siRNA (Cell Signaling) confirmed that the specific siRNA significantly reduced the abundance of Bax (50%) (Fig. 6A). We analyzed cytochrome c release 14 h after infection of MA104 cells previously transfected with siRNA. The Bax siRNA partially inhibited cytochrome c release (52%) in RRV-infected cells, whereas the irrelevant control siRNA had no effect. Furthermore, a much larger proportion of cytochrome c remained in the membrane fraction, containing mitochondria, from RRV-infected cells treated with Bax siRNA than in the membrane fraction from those treated with the irrelevant control (Fig. 6B). We also investigated RRV-induced apoptosis in MA104 cells previously transfected with siRNA. The fraction of apoptotic cells, analyzed at 14 h p.i. by flow cytometry after AO staining, decreased for cells with reduced Bax expression (Fig. 6C). These results demonstrate that Bax plays a key role in the rotavirus-induced mitochondrial pathway of apoptosis.
FIG. 6.

Cytochrome c release induced by RRV infection of MA104 cells is Bax dependent. (A) Knockdown of Bax expression in MA104 cells. (Top) Cells were transfected with a specific Bax siRNA or an irrelevant siRNA. Bax protein was then assayed in whole-cell lysates by immunoblotting. Levels of tubulin were used as protein loading controls. Positions of molecular weight markers are indicated on the left. (Bottom) Bax protein levels were determined by densitometry and plotted as ratios relative to the levels of tubulin. The graph shows the mean percentage of Bax expression inhibition from three independent experiments. Error bars, standard errors of the means. *, P < 0.05 by a Student t test comparing irrelevant-siRNA-transfected to Bax siRNA-transfected MA104 cells. (B) siRNA-induced silencing of the Bax gene results in a reduction of RRV-induced cytochrome c release in MA104 cells. (Top) Cells were infected with RRV 48 h posttransfection, and cytochrome c was analyzed in cytosolic (C) and heavy-membrane (M) fractions by Western blotting 14 h p.i. Mock-infected MA104 cells were used as negative controls. Tubulin and Cox IV were used as controls for the protein loading of the C and M fractions, respectively. Positions of molecular weight markers are indicated on the left. (Bottom) Cytochrome c protein levels of M and C fractions were determined by densitometry and plotted as ratios relative to the levels of Cox IV and tubulin, respectively. (C) siRNA-induced silencing of the Bax gene results in a reduction of RRV-induced apoptosis in MA104 cells. Cells were infected with RRV 48 h posttransfection, and the percentage of apoptosis was determined by flow cytometry after AO staining. MA104 cells transfected with an irrelevant siRNA were used as negative controls.
DISCUSSION
The pathophysiological mechanisms behind rotavirus-induced diarrhea have not been completely described. For mice, apoptosis may be associated with functional changes during rotavirus infection, particularly the modification of digestion and absorption functions. Evidence for apoptosis in intestinal cell lines following rotavirus infection in vitro has been reported (7, 47). However, the precise cellular and molecular mechanisms underlying rotavirus-induced apoptosis have not been defined. Here we investigated the apoptosis induced by the RRV strain in MA104 cells, a monkey kidney epithelial cell line in which the rotavirus cycle has been best characterized.
We showed that several hallmarks of apoptosis, notably DNA fragmentation, caspase-3 activation, and PARP cleavage, were detected following RRV infection of MA104 cells. DNA fragmentation was not observed in a previous study (6) of the same RRV-infected cells, probably because of differences in the sensitivities of the tests used. Moreover, we report the release of cytochrome c and Smac/Diablo from mitochondria into the cytosol, implicating mitochondrial dysfunction in the apoptotic pathway. Cytochrome c release is known to be the proapoptotic signal causing the autocatalytic activation of procaspase-9 that triggers caspase-3 cleavage. We showed that specific inhibition of either caspase-3 or caspase-9 reduced the percentage of apoptotic cells following RRV infection. Thus, RRV-induced apoptosis is at least partly dependent on caspase activation. It is also possible that Smac/Diablo is associated with RRV-induced apoptosis through its action on cellular inhibitors of apoptosis proteins, as observed notably during reovirus-induced apoptosis (22).
We then investigated events upstream from RRV-induced mitochondrial dysfunction. We focused on the Bcl-2 family proteins because they are central regulators of the mitochondrial apoptotic pathway (1, 15, 35, 44) and have been implicated in various models of virus-induced apoptosis (28, 36). Bax, one of the proapoptotic Bcl-2 family proteins, is the farthest downstream activator molecule known of the cytochrome c release machinery (42). We showed that Bax is activated following RRV infection in MA104 cells: we detected its conformational modification, involving the exposure of its N-terminal extremity, required for insertion into mitochondrial membranes (18, 33). Once integrated into the outer mitochondrial membrane, Bax can trigger cytochrome c release, leading to the activation of the effector caspase-3 and apoptotic cell death (11, 14, 23, 53). Our analysis of RRV-infected MA104 cells by using siRNA-mediated gene silencing indicates, for the first time, that the apoptotic mitochondrial pathway is activated through a Bax-dependent mechanism.
The inhibition of the prosurvival function of Bcl-2 is essential for the activation of Bax, because Bcl-2 can compete with Bax to regulate the release of cytochrome c (1, 15, 44). The amount of antiapoptotic Bcl-2 protein decreased in RRV-infected cells without any change in the amount of Bax. This resulted in an increase in the Bax/Bcl-2 ratio in RRV-infected cells, which could favor an increase in the amount of free Bax. Thus, one mechanism by which rotavirus may contribute to Bax activation is decreasing the number of Bax/Bcl-2 complexes at the mitochondrial membrane.
Activated BH3-only proteins such as Bid can suppress the capacity of Bcl-2 to inhibit apoptosis by interacting with it to displace and subsequently activate Bax (24, 25). Bid can be activated by caspase-8, resulting in Bax activation and release of cytochrome c (23). We showed that Bid processing was inhibited by the broad caspase inhibitor z-VAD-fmk in RRV-infected cells; therefore, rotavirus infection causes caspase-8 activation, leading to Bid activation. However, the inhibitor did not inhibit Bax activation and cytochrome c release, suggesting that caspases and Bid do not play a crucial role in the Bax-dependent cytochrome c release pathway during RRV infection. Thus, caspases seem to be involved in RRV-induced apoptosis at a position in the pathway downstream from the mitochondrial dysfunction.
Several mechanisms for promoting Bax activation and mitochondrial dysfunction have been described. These mechanisms include a signal transduction cascade involving mitogen-activated protein kinases such as c-Jun N-terminal protein kinase (JNK) and p38, as described for other models (21, 49). The regulation of mitochondrial dysfunction by JNK following virus infection was first demonstrated with reovirus-infected cells (8). Holloway and Coulson (17) recently showed that JNK and p38 are activated by rotavirus infection in MA104 cells. However, involvement of mitogen-activated protein kinase activation in Bax-dependent mitochondrial dysfunction has not yet been demonstrated for rotavirus-infected cells.
Rotavirus infection of cultured cells induces a progressive rise in the cytosolic Ca2+ concentration (5, 31, 46, 48). This increase may be involved in rotavirus-induced apoptosis, and indeed, the intracellular calcium ion chelator BAPTA-AM partially inhibits apoptosis (7). The rise in the cytosolic Ca2+ concentration in rotavirus-infected cells results from a progressive increase in plasma membrane permeability to Ca2+ and a depletion of endoplasmic reticulum (ER) pools. Xu et al. (55) showed that rotavirus infection leads to ER stress. This stress and ER Ca2+ depletion may lead to Bax activation (39). It would be valuable to determine in future studies whether rotavirus induces apoptosis by inducing both ER stress and ER Ca2+ depletion, leading to mitochondrial dysfunction. Several anti- and proapoptotic members of the Bcl-2 family have been found, in addition to their mitochondrial localization, to be associated with the ER and have been implicated in controlling apoptosis by affecting cellular Ca2+ homeostasis (37, 39, 41). As demonstrated in this study, Bax is involved in mitochondrial dysfunction in RRV infected-cells, but it could also be involved in ER Ca2+ release and thus could amplify the apoptotic signal in RRV-infected cells. It would therefore be interesting to investigate whether Bax plays a role in ER Ca2+ release.
Apoptosis of mature rotavirus-infected enterocytes induces the replacement of these cells by less differentiated dividing cells. This event may well be the cause of the defective absorptive functions of the intestinal epithelium and consequently of the diarrhea associated with rotavirus pathogenesis (4). Thus, the identification of the apoptotic signaling pathways in RRV-infected cells would improve our understanding of the mechanisms by which rotavirus infection causes functional alterations. Our demonstration of the activation of Bax during rotavirus infection and its involvement in the mitochondrial apoptotic pathway provides new insights concerning rotavirus-induced pathogenesis.
Acknowledgments
We thank Didier Poncet (Gif-sur-Yvette, France) for the gift of MA104 cells and the rotavirus RRV strain. We thank Isabelle Pelletier and Aure Saulnier for their help.
This work was supported by the Institut Pasteur, Danone Research Centre Daniel Carasso, and the Ministère de l'Education Nationale, de la Recherche et de la Technologie.
Footnotes
Published ahead of print on 14 February 2007.
REFERENCES
- 1.Adams, J. M., and S. Cory. 2001. Life-or-death decisions by the Bcl-2 protein family. Trends Biochem. Sci. 26:61-66. [DOI] [PubMed] [Google Scholar]
- 2.Ashkenazi, A., and V. M. Dixit. 1998. Death receptors: signaling and modulation. Science 281:1305-1308. [DOI] [PubMed] [Google Scholar]
- 3.Blomgren, K., C. Zhu, X. Wang, J. O. Karlsson, A. L. Leverin, B. A. Bahr, C. Mallard, and H. Hagberg. 2001. Synergistic activation of caspase-3 by m-calpain after neonatal hypoxia-ischemia: a mechanism of “pathological apoptosis”? J. Biol. Chem. 276:10191-10198. [DOI] [PubMed] [Google Scholar]
- 4.Boshuizen, J. A., J. H. Reimerink, A. M. Korteland-van Male, V. J. van Ham, M. P. Koopmans, H. A. Buller, J. Dekker, and A. W. Einerhand. 2003. Changes in small intestinal homeostasis, morphology, and gene expression during rotavirus infection of infant mice. J. Virol. 77:13005-13016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brunet, J. P., J. Cotte-Laffitte, C. Linxe, A. M. Quero, M. Geniteau-Legendre, and A. Servin. 2000. Rotavirus infection induces an increase in intracellular calcium concentration in human intestinal epithelial cells: role in microvillar actin alteration. J. Virol. 74:2323-2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Castilho, J. G., M. V. Botelho, F. Lauretti, N. Taniwaki, R. E. Linhares, and C. Nozawa. 2004. The in vitro cytopathology of a porcine and the simian (SA-11) strains of rotavirus. Mem. Inst. Oswaldo Cruz 99:313-317. [DOI] [PubMed] [Google Scholar]
- 7.Chaibi, C., J. Cotte-Laffitte, C. Sandre, A. Esclatine, A. L. Servin, A. M. Quero, and M. Geniteau-Legendre. 2005. Rotavirus induces apoptosis in fully differentiated human intestinal Caco-2 cells. Virology 332:480-490. [DOI] [PubMed] [Google Scholar]
- 8.Clarke, P., S. M. Meintzer, Y. Wang, L. A. Moffitt, S. M. Richardson-Burns, G. L. Johnson, and K. L. Tyler. 2004. JNK regulates the release of proapoptotic mitochondrial factors in reovirus-infected cells. J. Virol. 78:13132-13138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cory, S., and J. M. Adams. 2002. The Bcl2 family: regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2:647-656. [DOI] [PubMed] [Google Scholar]
- 10.Danial, N. N., and S. J. Korsmeyer. 2004. Cell death: critical control points. Cell 116:205-219. [DOI] [PubMed] [Google Scholar]
- 11.Desagher, S., A. Osen-Sand, A. Nichols, R. Eskes, S. Montessuit, S. Lauper, K. Maundrell, B. Antonsson, and J. C. Martinou. 1999. Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J. Cell Biol. 144:891-901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Estaquier, J., T. Idziorek, F. de Bels, F. Barre-Sinoussi, B. Hurtrel, A. M. Aubertin, A. Venet, M. Mehtali, E. Muchmore, P. Michel, et al. 1994. Programmed cell death and AIDS: significance of T-cell apoptosis in pathogenic and nonpathogenic primate lentiviral infections. Proc. Natl. Acad. Sci. USA 91:9431-9435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Green, D. R., and G. Kroemer. 2004. The pathophysiology of mitochondrial cell death. Science 305:626-629. [DOI] [PubMed] [Google Scholar]
- 14.Gross, A., J. Jockel, M. C. Wei, and S. J. Korsmeyer. 1998. Enforced dimerization of BAX results in its translocation, mitochondrial dysfunction and apoptosis. EMBO J. 17:3878-3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gross, A., J. M. McDonnell, and S. J. Korsmeyer. 1999. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 13:1899-1911. [DOI] [PubMed] [Google Scholar]
- 16.Hofman, P., and P. Auberger. 2000. Roles and mechanisms of apoptosis in infectious diseases. Ann. Pathol. 20:313-322. (In French.) [PubMed] [Google Scholar]
- 17.Holloway, G., and B. S. Coulson. 2006. Rotavirus activates JNK and p38 signaling pathways in intestinal cells, leading to AP-1-driven transcriptional responses and enhanced virus replication. J. Virol. 80:10624-10633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsu, Y. T., and R. J. Youle. 1998. Bax in murine thymus is a soluble monomeric protein that displays differential detergent-induced conformations. J. Biol. Chem. 273:10777-10783. [DOI] [PubMed] [Google Scholar]
- 19.Hsu, Y. T., and R. J. Youle. 1997. Nonionic detergents induce dimerization among members of the Bcl-2 family. J. Biol. Chem. 272:13829-13834. [DOI] [PubMed] [Google Scholar]
- 20.Jin, Z., and W. S. El-Deiry. 2005. Overview of cell death signaling pathways. Cancer Biol. Ther. 4:139-163. [DOI] [PubMed] [Google Scholar]
- 21.Kim, B. J., S. W. Ryu, and B. J. Song. 2006. JNK- and p38 kinase-mediated phosphorylation of Bax leads to its activation and mitochondrial translocation and to apoptosis of human hepatoma HepG2 cells. J. Biol. Chem. 281:21256-21265. [DOI] [PubMed] [Google Scholar]
- 22.Kominsky, D. J., R. J. Bickel, and K. L. Tyler. 2002. Reovirus-induced apoptosis requires mitochondrial release of Smac/DIABLO and involves reduction of cellular inhibitor of apoptosis protein levels. J. Virol. 76:11414-11424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Korsmeyer, S. J., M. C. Wei, M. Saito, S. Weiler, K. J. Oh, and P. H. Schlesinger. 2000. Pro-apoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ. 7:1166-1173. [DOI] [PubMed] [Google Scholar]
- 24.Kuwana, T., L. Bouchier-Hayes, J. E. Chipuk, C. Bonzon, B. A. Sullivan, D. R. Green, and D. D. Newmeyer. 2005. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol. Cell 17:525-535. [DOI] [PubMed] [Google Scholar]
- 25.Letai, A., M. C. Bassik, L. D. Walensky, M. D. Sorcinelli, S. Weiler, and S. J. Korsmeyer. 2002. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2:183-192. [DOI] [PubMed] [Google Scholar]
- 26.Li, H., H. Zhu, C. J. Xu, and J. Yuan. 1998. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94:491-501. [DOI] [PubMed] [Google Scholar]
- 27.Li, S., Y. Zhao, X. He, T. H. Kim, D. K. Kuharsky, H. Rabinowich, J. Chen, C. Du, and X. M. Yin. 2002. Relief of extrinsic pathway inhibition by the Bid-dependent mitochondrial release of Smac in Fas-mediated hepatocyte apoptosis. J. Biol. Chem. 277:26912-26920. [DOI] [PubMed] [Google Scholar]
- 28.Liu, Y., Y. Pu, and X. Zhang. 2006. Role of the mitochondrial signaling pathway in murine coronavirus-induced oligodendrocyte apoptosis. J. Virol. 80:395-403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lundgren, O., and L. Svensson. 2001. Pathogenesis of rotavirus diarrhea. Microbes Infect. 3:1145-1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luo, X., I. Budihardjo, H. Zou, C. Slaughter, and X. Wang. 1998. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 94:481-490. [DOI] [PubMed] [Google Scholar]
- 31.Michelangeli, F., M. C. Ruiz, J. R. del Castillo, J. E. Ludert, and F. Liprandi. 1991. Effect of rotavirus infection on intracellular calcium homeostasis in cultured cells. Virology 181:520-527. [DOI] [PubMed] [Google Scholar]
- 32.Morris, A. P., and M. K. Estes. 2001. Microbes and microbial toxins: paradigms for microbial-mucosal interactions. VIII. Pathological consequences of rotavirus infection and its enterotoxin. Am. J. Physiol. Gastrointest. Liver Physiol. 281:G303-G310. [DOI] [PubMed] [Google Scholar]
- 33.Murphy, K. M., U. N. Streips, and R. B. Lock. 2000. Bcl-2 inhibits a Fas-induced conformational change in the Bax N terminus and Bax mitochondrial translocation. J. Biol. Chem. 275:17225-17228. [DOI] [PubMed] [Google Scholar]
- 34.Muzio, M., A. M. Chinnaiyan, F. C. Kischkel, K. O'Rourke, A. Shevchenko, J. Ni, C. Scaffidi, J. D. Bretz, M. Zhang, R. Gentz, M. Mann, P. H. Krammer, M. E. Peter, and V. M. Dixit. 1996. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell 85:817-827. [DOI] [PubMed] [Google Scholar]
- 35.Narita, M., S. Shimizu, T. Ito, T. Chittenden, R. J. Lutz, H. Matsuda, and Y. Tsujimoto. 1998. Bax interacts with the permeability transition pore to induce permeability transition and cytochrome c release in isolated mitochondria. Proc. Natl. Acad. Sci. USA 95:14681-14686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Natoni, A., G. E. Kass, M. J. Carter, and L. O. Roberts. 2006. The mitochondrial pathway of apoptosis is triggered during feline calicivirus infection. J. Gen. Virol. 87:357-361. [DOI] [PubMed] [Google Scholar]
- 37.Nutt, L. K., A. Pataer, J. Pahler, B. Fang, J. Roth, D. J. McConkey, and S. G. Swisher. 2002. Bax and Bak promote apoptosis by modulating endoplasmic reticular and mitochondrial Ca2+ stores. J. Biol. Chem. 277:9219-9225. [DOI] [PubMed] [Google Scholar]
- 38.Oltvai, Z. N., C. L. Milliman, and S. J. Korsmeyer. 1993. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell 74:609-619. [DOI] [PubMed] [Google Scholar]
- 39.Pan, Z., M. B. Bhat, A. L. Nieminen, and J. Ma. 2001. Synergistic movements of Ca2+ and Bax in cells undergoing apoptosis. J. Biol. Chem. 276:32257-32263. [DOI] [PubMed] [Google Scholar]
- 40.Parashar, U. D., C. J. Gibson, J. S. Bresse, and R. I. Glass. 2006. Rotavirus and severe childhood diarrhea. Emerg. Infect. Dis. 12:304-306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pinton, P., and R. Rizzuto. 2006. Bcl-2 and Ca2+ homeostasis in the endoplasmic reticulum. Cell Death Differ. 13:1409-1418. [DOI] [PubMed] [Google Scholar]
- 42.Puthalakath, H., and A. Strasser. 2002. Keeping killers on a tight leash: transcriptional and post-translational control of the pro-apoptotic activity of BH3-only proteins. Cell Death Differ. 9:505-512. [DOI] [PubMed] [Google Scholar]
- 43.Ramig, R. F. 2004. Pathogenesis of intestinal and systemic rotavirus infection. J. Virol. 78:10213-10220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reed, J. C. 1998. Bcl-2 family proteins. Oncogene 17:3225-3236. [DOI] [PubMed] [Google Scholar]
- 45.Roulston, A., R. C. Marcellus, and P. E. Branton. 1999. Viruses and apoptosis. Annu. Rev. Microbiol. 53:577-628. [DOI] [PubMed] [Google Scholar]
- 46.Ruiz, M. C., J. Cohen, and F. Michelangeli. 2000. Role of Ca2+in the replication and pathogenesis of rotavirus and other viral infections. Cell Calcium 28:137-149. [DOI] [PubMed] [Google Scholar]
- 47.Superti, F., M. G. Ammendolia, A. Tinari, B. Bucci, A. M. Giammarioli, G. Rainaldi, R. Rivabene, and G. Donelli. 1996. Induction of apoptosis in HT-29 cells infected with SA-11 rotavirus. J. Med. Virol. 50:325-334. [DOI] [PubMed] [Google Scholar]
- 48.Tian, P., M. K. Estes, Y. Hu, J. M. Ball, C. Q. Zeng, and W. P. Schilling. 1995. The rotavirus nonstructural glycoprotein NSP4 mobilizes Ca2+ from the endoplasmic reticulum. J. Virol. 69:5763-5772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsuruta, F., J. Sunayama, Y. Mori, S. Hattori, S. Shimizu, Y. Tsujimoto, K. Yoshioka, N. Masuyama, and Y. Gotoh. 2004. JNK promotes Bax translocation to mitochondria through phosphorylation of 14-3-3 proteins. EMBO J. 23:1889-1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van Loo, G., X. Saelens, M. van Gurp, M. MacFarlane, S. J. Martin, and P. Vandenabeele. 2002. The role of mitochondrial factors in apoptosis: a Russian roulette with more than one bullet. Cell Death Differ. 9:1031-1042. [DOI] [PubMed] [Google Scholar]
- 51.Vaux, D. L., and S. J. Korsmeyer. 1999. Cell death in development. Cell 96:245-254. [DOI] [PubMed] [Google Scholar]
- 52.Wang, X. 2001. The expanding role of mitochondria in apoptosis. Genes Dev. 15:2922-2933. [PubMed] [Google Scholar]
- 53.Wei, M. C., W. X. Zong, E. H. Cheng, T. Lindsten, V. Panoutsakopoulou, A. J. Ross, K. A. Roth, G. R. MacGregor, C. B. Thompson, and S. J. Korsmeyer. 2001. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292:727-730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wolter, K. G., Y. T. Hsu, C. L. Smith, A. Nechushtan, X. G. Xi, and R. J. Youle. 1997. Movement of Bax from the cytosol to mitochondria during apoptosis. J. Cell Biol. 139:1281-1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu, A., A. R. Bellamy, and J. A. Taylor. 1998. BiP (GRP78) and endoplasmin (GRP94) are induced following rotavirus infection and bind transiently to an endoplasmic reticulum-localized virion component. J. Virol. 72:9865-9872. [DOI] [PMC free article] [PubMed] [Google Scholar]