

Abstract
Stable trimeric forms of human immunodeficiency virus recombinant gp140 (rgp140) are important templates for determining the structure of the glycoprotein to assist in our understanding of HIV infection and host immune response. Such information will aid the design of therapeutic drugs and vaccines. Here, we report the production of a highly stable and trimeric rgp140 derived from a HIV type 1 (HIV-1) subtype D isolate that may be suitable for structural studies. The rgp140 is functional in terms of binding to CD4 and three human monoclonal antibodies (17b, b12, and 2G12) that have broad neutralizing activities against a range of HIV-1 isolates from different subtypes. Treatment of rgp140 with protein disulfide isomerase (PDI) severely restricted 17b binding capabilities. The stable nature of the rgp140 was due to the lack of processing at the gp120/41 boundary and the presence of an intermonomer disulfide bond formed by the cysteines of the V3 loop. Further characterization showed the intermonomer disulfide bond to be a target for PDI processing. The relevance of these findings to the roles of the V3 domain and the timing of PDI action during the HIV infection process are discussed.
Human immunodeficiency virus (HIV) belongs to the family Retroviridae, and two major types, HIV type 1 (HIV-1) and HIV-2, have been identified as the pathogenic agents of AIDS (6, 30, 48). HIV-1 can be traced to simian immunodeficiency virus (SIV), which causes AIDS-like symptoms in chimpanzees (3). HIV envelope glycoproteins are synthesized as a gp160 polyprotein precursor that undergoes posttranslational modifications, including glycosylation, trimerization, and cleavage into gp120 and gp41 (by furin-like proteases), which remain weakly associated during final transportation to the cell surface for incorporation into budding virus particles (23, 59).
During the course of HIV infection, the envelope glycoproteins interact with cellular receptors and coreceptors. gp120 initiates infection by binding to CD4 on cells of the host's immune system. Such binding triggers conformational changes within gp120 (34, 70), creating a coreceptor binding site and allowing secondary binding to chemokine receptors, typically CCR5 or CXCR4 (10, 57, 73). During the course of these conformational changes, gp120 may become susceptible to proteolytic cleavage by enzymes such as thrombin (18), which have been shown to increase cell fusion (49), and cell-surface thiol-containing molecules, such as protein disulfide isomerase (PDI) (5, 25, 29, 51) and thioredoxin (61), that are known to break disulfide bonds within gp120. The locations of these broken disulfide bonds and the timing of PDI/thioredoxin action in the HIV infection process are unknown. gp120-chemokine receptor binding triggers conformational changes within gp41, allowing the fusion peptide to insert into the host cell membrane, while further rearrangements of gp41 and the shedding of gp120, possibly assisted by proteolytic events and/or PDI/thioredoxin action, bring the viral and cellular membranes into contact, allowing membrane fusion and hence virus uptake (31, 43, 56, 66).
Expressed recombinant gp140 (rgp140) proteins from which the gp41 transmembrane domain and cytoplasmic tail have been removed show a propensity for oligomerization producing monomers, dimers, trimers, and higher-ordered aggregates (75, 92). Aggregation and the weak association of gp120/41 have hampered biochemical and crystallographic characterization. In attempts to overcome these problems, the use of truncations, gp120/41 cross-linking disulfide bonds (69, 71), and trimeric motifs, such as that from T4 bacteriophage (88), have been used with various degrees of success. rgp140s, produced by constructs based on the env gene sequences of primary isolates of HIV, have been found to display isolate-specific oligomeric patterns, and the extent of oligomerization has been postulated to be clade specific (36).
Despite these problems, and after intense effort, limited structural data for gp120 and gp41 have emerged. Crystallographic structures have been solved for both HIV and SIV gp41 ectodomains (80, 89). These results revealed N- and C-terminal α-helices separated by a glycosylated loop region reported to form contacts with gp120. The N-terminal α-helices of three-gp41 subunits form a trimeric coiled coil, and this provided the basis for gp160 being trimeric. During membrane fusion, the C-terminal helix packs against the N-terminal trimeric coiled coil to create a six-helix bundle, thereby bringing the viral and cellular membranes into contact. Structures for the monomeric “core” of HIV-1 gp120, where N and C termini have been truncated and variable domains 1, 2, and 3 have been removed, have been determined (45, 46). These structures were solved following enzymatic deglycosylation of the cores and binding of the CD4 receptor and the 17b virus neutralizing antibody. Using similar constructs derived from SIVmac32H, structures have been determined for glycosylated cores in the absence of any bound ligands (16, 17). Comparison of the HIV and SIV structures provides an indication of the structural rearrangements that take place upon receptor binding. Recently, a core structure for HIV-1 gp120 carrying the V3 loop, again liganded with CD4 and an antibody, has been reported, showing that the V3 loop would be oriented toward the host cell membrane for coreceptor binding (35). Although important, these gp120 structures represent only approximately 60% of the full-length protein and lack information on functionally important domains. As such, they may not provide the range of information required for effective vaccine and drug design.
In this article, we report the production of a stable, trimeric, proteolytically immature rgp140 (C-terminally truncated gp160), derived from a HIV-1 subtype D strain, that is functional in terms of binding to CD4 and three gp120-specific human monoclonal antibodies with neutralizing activities against a broad range of HIV-1 isolates. Characterization of the rgp140 revealed the presence of an intermonomer disulfide bond formed by cysteines defining the V3 loop. This bond was susceptible to PDI processing, and PDI treatment of rgp140 led to loss of 17b (a coreceptor mimic) binding abilities. The potential relevance of these findings to the HIV-1 infection process is discussed.
MATERIALS AND METHODS
rgp140 expression constructs.
A known expression-competent HIV-1 subtype D gp160 clone, WHO-15_28, was used as a template for the rgp140 construct (20). The required env gene fragment was generated using PCR with native Pfu polymerase (Stratagene) and primers to span the signal peptide-gp120 boundary (SAa/tEKLWV: N120tpa; ATGATCTGATCAGCTRCAGAAAAATTGTGGGT; the BclI site is underlined) and to introduce a premature stop codon 18 residues upstream of the gp41 membrane anchor (NEk/qe/dLLe/aLDK*: Stop664; CACAGAGAATTCTACTTGTCCAATKCCAATAAKTCTTKTTCATT; the EcoRI site is underlined) (21). Such truncation resulted in the disruption and deletion of epitopes for the broadly neutralizing antibodies 2F5 and 4E10, respectively (reviewed in reference 11).
The fragment was digested with BclI/EcoRI and ligated into a BglII/EcoRI-cut vector, pEE14tpagp1203, supplied by P. E. Stephens (37). This resulted in replacement of the WHO-15_28 signal peptide coding sequence with that of tissue plasminogen activator (tpa) and replaced the HIV-1IIIB gp120 coding sequence with the rgp140 gene fragment. The construct was transformed into Escherichia coli DH5α (Invitrogen) and grown on L agar plates/L broth in the presence of 50 μg/ml ampicillin/nafcillin at 30°C. The purified plasmids were sequenced by a gene-walking approach using an ABI 377 sequencer and Big-Dye kits to check for open reading frames prior to the transfection of CHO-K1 cells.
Establishment of constitutively expressing cell lines.
CHO-K1 cells were seeded at 2 × 105 per well into six-well culture plates containing CHO III (A) medium (GIBCO) supplemented with 1× hypoxanthine-thymidine (Invitrogen) and 8 mM l-glutamine. The cells were incubated at 37°C in a 5% CO2 incubator until 80% confluent, at which point the cells were transfected with 1 μg of DNA using Lipofectamine (Invitrogen) according to the manufacturer's protocol. After 72 h, transfected cells were selected for by the addition of 200 μM methylsulfoxamine. Over a period of 2 to 8 weeks, cell colonies were transferred to six-well plates and grown to confluence. Tissue culture supernatants were tested for the secretion of rgp140 by Western blotting, using the murine monoclonal antibody (MAb) 221, which has been mapped to a peptide, GGDMRDNWRSELYKYKVVKI, in the C terminus of HIV-1HXB2 gp120 (9). The highly expressing cell line clone 2.28 was used for subsequent studies.
rgp140 production.
The CHO-K1 cell clone was grown to confluence in 175-cm2 triple flasks using CHO III (A) medium. Confluent monolayers were induced with 2 mM sodium butyrate (Sigma), and protein-containing supernatants were harvested on day 4. To prevent protease cleavage, complete protease inhibitor cocktail tablets (Roche) were added to the postproduction tissue culture supernatants according to the supplier's recommendations. The supernatants were concentrated 20-fold, using a QuixStand cross-flow filtration system (Amersham Biosciences) with 10-kDa cutoff filters prior to purification.
Antibodies used in Western blotting and enzyme-linked immunosorbent assay (ELISA).
In addition to our 221 MAb, murine MAbs directed against a conserved epitope in the gp120-C1 domain (CA13/ARP3119, fine mapped to the sequence EDIISLW; [Centralised Facility for AIDS Reagents [CFAR]) and a glycosylation-dependent epitope in gp41 (T30, mapped to residues 580 to 640, ILAVERY… .NNYTS, and involving the N-linked carbohydrate at N616; [22]) were supplied by N. Almond and B. Moss, respectively. CFAR supplied MAbs 17b (ARP3078), b12 (ARP3065), and 2G12 (EVA3064) and rabbit sera raised against a consensus HIV-1 subtype D V3 peptide, QRTHIGPGQALYTT (EVA437), and the V3 peptide sequence of HIV-1LAI (ARP433). We also synthesized a V3 peptide specific to our rpg140 (INCTRPYHNTRQRTHIGTGQALYTTRIIGDIRKAHCNI) using Fast-moc chemistry on an ABI 431A and cyclized it through cysteine residues using dimethyl sulfoxide oxidation. The oxidized peptide was purified by high-performance liquid chromatography and assayed by mass spectroscopy using electrospray on a Micromass Platform Quadrupole system. Following the inoculation of mice and fusion of spleen cells with Sp2/0-Ag14 myeloma cells, a hybridoma producing the V3-specific MAb 4D8 was generated. For detection of CD4, a sheep antiserum raised against CHO cell-derived recombinant soluble human CD4 was used (ARP404; supplied by CFAR).
Partial purification of rgp140 for antibody production.
Concentrated rgp140-containing medium (100 ml) was partially purified by immunoaffinity chromatography, employing the T30 MAb bound to Affigel-Hz (Bio-Rad). The medium was recirculated over a 10-ml immunoaffinity column for 16 h at 4°C. The column was washed with 20 mM Tris-HCl, 500 mM NaCl, pH 8.0, for 40 column volumes. Bound rgp140 was eluted (50 ml) with 3 M MgCl2, pH 6.8, and dialyzed against 20 mM Tris-HCl, pH 8.0. Eluted rgp140 was subjected to ion-exchange chromatography on a 1-ml Mono Q column (GE Healthcare). A 45-ml linear gradient of 10 to 350 mM NaCl in 20 mM Tris-HCl, pH 8.0 (4 ml/min), was applied to elute bound rgp140, which eluted as a broad peak between 170 and 210 mM NaCl. Pooled fractions were concentrated in a Vivaspin centrifugal concentrator (100,000 molecular weight cutoff [MWCO]; Vivascience) to 0.5 ml. Concentrated rgp140 was applied to a Superdex 200 HR 10/30 gel filtration column (GE Healthcare) equilibrated with 20 mM Tris-HCl, 50 mM NaCl, pH 8.0. Fractions containing rgp140 were pooled and concentrated to a final concentration of 100 μg/ml rgp140, as determined by Galanthus nivalis lectin capture ELISA. Immunization of mice with this material (five intraperitoneal inoculations with 10 μg rgp140 [50 μg total in ribi adjuvant {Sigma M6536}]) and subsequent fusion of spleen cells with Sp2/0-Ag14 myeloma cells yielded a panel of MAbs, one of which, 2E2, gave efficient immunoaffinity purification of rgp140.
rgp140 purification.
An immunoaffinity column was created by cross-linking CNBr-activated Sepharose 4B (Amersham Biosciences) with 2E2 MAb at a concentration of 10 mg antibody per ml beads. The concentrated medium was loaded onto the preequilibrated column (10 mM NaCl, 50 mM Tris, pH 8.0) at a rate of 1 ml/min. After the column was washed with 5 column volumes of 10 mM NaCl, 50 mM phosphate buffer, pH 6.3, rgp140 was eluted using 5 column volumes of 10 mM NaCl, 5 mM glycine, pH 2.5, and immediately neutralized by the addition of 0.05 volume 1 M Tris, pH 9.0. Antibody affinity-purified rgp140 was then subjected to size exclusion chromatography (SEC) using a Superose 6 HR 10/30 column (Amersham Biosciences) equilibrated in 1 M NaCl, 50 mM HEPES, pH 6.5. Fractions containing trimeric rgp140, by native polyacrylamide gel electrophoresis (PAGE), were pooled and concentrated using a 30,000 MWCO concentrator (Vivascience).
Oligomeric structure of rgp140.
Initially, the molecular mass of rgp140 was obtained by comparison to known molecular mass standards (Amersham Biosciences) run on SEC columns. Native PAGE and dynamic light scattering confirmed the molecular mass estimates obtained by SEC.
Sources of rgp120.
Baculovirus-expressed HIV-1IIIB rgp120 (EVA607) was obtained from CFAR. Mammalian-cell-expressed HIV-1IIIB rgp120 was purified from the tissue culture supernatant of a CHO-K1 cell line transfected with pEE14tpagp1203 (37).
CD4-binding activity of rgp140/120.
A two-domain CD4 construct (2DCD4) produced in E. coli (see Acknowledgments) was used in the studies reported here. rgp140/120 was diluted to a concentration of 0.02 μg/μl in sensitizing buffer (7.4 mM NaHCO3, 2.6 mM Na2CO3, pH 9.5), and 50-μl aliquots (1 μg of protein per well; four wells per assay) were dispensed into an ELISA plate, which was incubated at 37°C for 3 h. All subsequent incubations were carried out at 37°C, and the plates were washed (0.3 M NaCl, 0.05% Tween 20) between steps. Next, the wells were blocked with 50 μl 1% skim milk (Marvel) in PBS for 1 h and then incubated overnight (16 h) with either 50 μl/well dilution buffer (0.5% Marvel, 0.05% Tween 20 in PBS) or 50 μl dilution buffer containing 9 mU PDI (Sigma G4251), 1 mM reduced l-glutathione to assess the effect of PDI treatment on CD4 binding. After the plates were washed, 1 μg 2DCD4 (in 50 μl dilution buffer) was added per well, and the plates were incubated for 1 h; 50 μl/well ARP404 (primary antibody) diluted 1/1,000 with dilution buffer was added, and the plates were incubated for 1 h; 50 μl/well horseradish peroxidase-linked secondary donkey anti-sheep immunoglobulin G (IgG) (Sigma; F7634) diluted 1/1,000 in dilution buffer was added, and the plates were incubated for 1 h. The plates were developed using 200 μl substrate (Fast OPD substrate; Sigma) per well and incubated at room temperature in the dark for 30 min. The plates were read on a Multiskan Ascent (Thermo Labsystems) at 450 nm.
Surface plasmon resonance was carried out on a BIAcore 2000 instrument (BIACORE Life Sciences). rgp140 was coupled to the surface of a CM-5 sensor chip using standard amine coupling procedures suggested by the manufacturer. For equilibrium dissociation constant (KD) measurements, the immobilization was 15,000 response units. Binding experiments were carried out at 37°C in HBS-P buffer (BIACORE Life Sciences). Association was measured by passing 2DCD4, in a concentration range of 0.3 to 10 μM, over the rgp140 surface in 5-μl injections at a rate of 2 μl/min. After the 2DCD4 injection, the chip surface was regenerated by injection of 5 μl 1 M NaCl. The KD was calculated using BIAevaluation software, and the data were fitted using a 1:1 binding model.
Antibody-binding activity of rgp140/120.
The antibody-binding activity of rgp140/120 was assayed in an ELISA format similar to that used for CD4 binding. Following binding of rgp140/120 to plates, the plates were washed and 50 ng/well of the required MAb (50 μl of diluted stock, using dilution buffer), which acted as the primary antibody in this assay, was added; the plates were incubated at 37°C for 1 h. After the plates were washed, horseradish peroxidase-conjugated secondary antibody (50 μl/well of either sheep anti-human IgG [Amersham Biosciences; NA933V] or goat anti-mouse IgG [Promega; W4021] diluted 1/1,000 in dilution buffer) was added, and incubation continued for 1 h. The assay was developed by adding substrate, incubating the plates, and reading the results at 450 nm as described above. For rabbit sera, 1/200 dilutions were used and detected with a 1/1,000 dilution of goat anti-rabbit IgG (Sigma; A6154).
Binding of the coreceptor mimic, MAb 17b (76, 79), was monitored after treatment with PDI and in the presence of 2DCD4. Following glycoprotein binding to plates and incubation in the absence/presence of PDI, MAb 17b was added either alone or with 1 μg 2DCD4, and the plates were incubated at 37°C for1 h before the assay was developed as described above.
Enzymatic digestion of rgp140.
Furin (Sigma), thrombin (Roche), and glutamyl endopeptidase-C (Glu-C) (Roche) digestions were carried out using an enzyme/substrate ratio of 1:10 (wt/wt) at pH 8.0 (50 mM Tris-HCl). PDI treatment was performed with 9 mU/μg rgp140 in a similarly buffered solution containing 1 mM reduced l-glutathione. All treatments were performed at 37°C overnight. For dual treatments, where thrombin digestion was followed by PDI treatment, thrombin was first inactivated by the addition of 4-amidinophenylmethanesulfonyl fluoride hydrochloride (Sigma) to a final concentration of 1.7 mg/ml.
N-terminal sequencing of peptide.
N-terminal peptide sequencing was carried out by the Department of Biochemistry at Bristol University. Briefly, rgp140 was digested with Glu-C to release approximately 50 pmol of the V3 loop. The products of digestion were run on sodium dodecyl sulfate (SDS)-PAGE and transferred to polyvinylidene difluoride membranes (Amersham Biosciences). Following transfer, the blot was stained with Coomassie brilliant blue R-250 (0.025% [wt/vol], 40% methanol [vol/vol]). After being destained (50% [vol/vol] methanol), the blot was dried, and the band corresponding to the V3 loop, determined by comparison to a previous Western blot probed with MAb 4D8, was marked prior to submission for sequencing.
RESULTS
Production and purification of HIV rgp140.
Following the concentration of 1 liter of tissue culture supernatant, MAb 2E2 affinity column purification, and Superose 6 SEC, the yield of rgp140 approximated 4.5 mg in fractions 19 to 31, corresponding to 9.5 to 15.5 ml for the elution profile of the Superose 6 column (Fig. 1). Peak fractions from the SEC purification, corresponding to the 12- to 14-ml elution, were pooled, and rgp140 was ascertained to be in a highly pure state by visualization of a single band on Coomassie-stained reducing SDS-PAGE migrating at the correct molecular mass for the protein (Fig. 1, inset B). This pool was used in all subsequent characterization experiments.
FIG. 1.

Gel filtration chromatography of rgp140. MAb 2E2 affinity-purified rgp140 was analyzed on a Superose 6 column, and 0.5-ml fractions were collected. rgp140 eluted over volumes ranging from 9 to 16 ml. The retention volumes of molecular mass standards (thyroglobulin, 669 kDa; ferritin, 440 kDa; catalase, 232 kDa; Amersham Biosciences) are indicated by arrows. (A) Native PAGE (5 to 10%) analysis of 5-μl aliquots from fractions 21 to 28 spanning the major peak (10 to 14 ml, as indicated on the chromatogram), followed by Western blotting with MAb CA13. The locations of aggregated (A) and trimeric (T) rgp140 are indicated. (B) Coomassie-stained reducing SDS-PAGE (4 to 12%) analysis of pooled and concentrated rgp140 peak fractions 25 to 28 (spanning 12 to 14 ml) showing a single highly pure band corresponding to rgp140.
Oligomeric state of rgp140.
Superose 6-SEC produced a single peak that, by comparison to known molecular mass standards, centered on approximately 440 kDa (Fig. 1). To ascertain the oligomeric nature of the protein, native PAGE analysis of the SEC peak fractions (Fig. 1, inset A) was used. This showed rgp140 to exist predominantly in a trimeric state with a small proportion presenting as aggregate, although higher-order aggregates, notably in fractions 21 to 24, may either not have entered the native gel or not have been detected efficiently with MAb CA13. No monomeric or dimeric forms were detectable, as has been reported previously (71). For the four Superose 6 fractions corresponding to 12- to 14-ml elution, dynamic light scattering gave averaged molecular mass estimations of 620, 568, 462, and 443 kDa and polydispersity values of 15.4, 6.8, 12.6, and 12.5%. Values of polydispersity are related to the hydrodynamic radius of the protein in solution, and values of <15% are indicative of a restricted size range that is suitable for crystallization trials (26).
Composition and stability of rgp140.
During maturation of gp160, it undergoes furin digestion at the gp120/41 boundary, producing two moieties of 120 kDa (gp120) and 41 kDa (gp41), which remain weakly associated. Furin maturation of our rgp140 should have resulted in bands migrating to ∼120 kDa (gp120) and ∼30 kDa (truncated gp41). To investigate this, rgp140 was run on SDS-PAGE gels and mapped antigenically via Western blotting. Antibodies directed against the N and C termini of gp120 (MAbs CA13 and 221, respectively) and gp41 (MAb T30) detected a single band migrating to ∼140 kDa, indicating the lack of furin processing (Fig. 2A).
FIG. 2.

Antigenic and proteolytic mapping of rgp140. rgp140 and products of protease digestion were run on SDS-PAGE (4 to 12% gradient) and analyzed by Western blotting using antibodies directed against the N-terminal (MAb CA13) and C-terminal (MAb 221) regions of gp120 and a gp41 domain (MAb T30). (A) For untreated rgp140, detection of a protein by all three MAbs, migrating at the correct molecular mass for full-length rgp140, indicates the lack of proteolytic processing. (B) Full-length rgp140 probed with CA13 (lane 1) compared with the products of furin digestion probed with T30 (lane 2) and CA13 (lane 3). Of the three major bands detected, I represents either undigested rgp140 (lane 2) or a mixture of rgp120/140 (lane 3), while the products running at ∼70 kDa (II) contain either gp120 C termini/gp41 (lane 2) or N termini of gp120, and product III (∼30 kDa; lane 2 only) corresponds to truncated gp41. (C) Full-length rgp140 probed with CA13 (lane 1) compared with the products of thrombin digestion probed with CA13 (lane 2), 221 (lane 3), and T30 (lane 4). As for furin digestion, three major bands were identified corresponding to undigested rgp140 (I), ∼70-kDa products (II) detected by all three MAbs and therefore containing N and C termini of gp120, and gp41, while a weak band (III) corresponding to truncated gp41 was detected with T30 only. (D) Synthetic V3 peptide (a 38-mer with a predicted molecular mass of 4.39 kDa) was incubated at 37°C in the absence (lane 1) and presence (lane 2) of thrombin. The products of digestion were analyzed by SDS-PAGE (15%) and Western blotting using rabbit serum EVA437.
To assess the presence of a gp120/41 processing site, as predicted from gene sequencing, rgp140 was subjected to furin digestion and the products were analyzed by antigenic mapping (Fig. 2B). Furin digestion resulted in products migrating to ∼140 kDa, ∼70 kDa (detected by MAbs T30 and CA13), and ∼30 kDa (detected by MAb T30 only). The presence of a 140-kDa protein detected by MAb T30 after furin digestion suggests incomplete processing. The bands migrating to ∼70 kDa and 30 kDa are therefore the true products of furin digestion. The 30-kDa band detected by MAb T30 corresponds to truncated gp41 and indicates correct processing at the gp120/41 junction. The 70-kDa band was an unexpected product of furin digestion, and antigenic mapping showed it to be heterogeneous, containing the N terminus of gp120 and the gp41 component. This, coupled with the molecular mass, is consistent with cleavage having occurred within gp120 but not at the gp120/41 junction, cutting the protein in half. Previous reports (58, 60, 78) have shown that furin can utilize the thrombin site within the tip of the V3 loop of gp120, which would result in products of 70 kDa, as observed.
Despite our rgp140 V3 loop sequence lacking a consensus thrombin recognition motif (14), thrombin digestion (Fig. 2C) produced the same banding pattern, both in terms of size and antigenic mapping, as seen for furin digestion (Fig. 2B), lending support to the idea that furin utilizes the thrombin site or a site close to it. Probing with MAb 221 (Fig. 2C, lane 3) and MAb CA13 (Fig. 2C, lane 2) showed that both the N and C termini of gp120 were present within the 70-kDa band and indicated the presence of two halves of rgp140. MAb T30 (Fig. 2C, lane 4) detected very little truncated gp41, suggesting that thrombin cannot utilize the gp120/41 processing site effectively. Overall, these results show the production of predominantly trimeric and proteolytically immature rgp140.
Since HIV-1 subtype D gp120s do not have the consensus thrombin cleavage site within the tips of their V3 loops (GPGR↓AF) identified by others (18, 49), our synthetic V3 peptide was subjected to thrombin digestion and SDS-PAGE analysis. While Western blot analysis with a rabbit serum raised against a consensus subtype D V3 peptide showed reactivity with our synthetic V3 peptide, no products were detectable following thrombin digestion (Fig. 2D). This indicates the destruction of antibody recognition sites and confirms the susceptibility of the V3 loop to thrombin. Clements et al. (18) have shown that the V3 loop of HIV-2ROD gp105, despite lacking a consensus cleavage sequence, is susceptible to thrombin, and they suggest that the tertiary structure of the V3 loop, rather than primary amino acid sequence, may be critical in determining thrombin susceptibility.
rgp140 trimeric stability conferred by a disulfide bond and its location.
Previous reports have shown the trimeric stability of rgp140 to be resistant to high temperatures and acidic pH (44). The only conditions capable of breaking the trimeric association of our rgp140 were a combination of high temperature (100°C) and reducing conditions (results not shown). The ability of reducing agents to break such association has been reported by others (47, 69, 88), and these data suggest the presence of at least one intermonomer disulfide bond. To investigate this, the mobilities of rgp140 under reducing SDS-PAGE and nonreducing SDS-PAGE were compared (Fig. 3A). Under reducing conditions, rgp140 migrates as a 140-kDa monomer, while under nonreducing conditions it migrates above the 208-kDa marker, indicative of intermonomer (trimerization) association via disulfide bonding.
FIG. 3.

Presence and location of an intermonomer disulfide bond in trimeric rgp140. (A) rgp140 was run on SDS-PAGE (4 to 12% gradient) under reducing (lane 1) and nonreducing (lane 2) conditions and analyzed by Western blotting using MAb CA13. The presence of an intermonomer disulfide bond is demonstrated by the increase in molecular mass in the absence of reducing agent (5% β-mercaptoethanol). Thrombin digestion of rgp140, followed by running of (B) native PAGE (5 to 10%) and (C) nonreducing SDS-PAGE (4 to 12%), and antigenic mapping by Western blotting were used to study the role of the V3 loop disulfide bond in trimeric rgp140 stability. Lanes 1 contain undigested rgp140, while lanes 2 to 4 show thrombin digestion products detected with the MAbs indicated. Antigenic mapping showed thrombin digestion to have no effect on the native trimeric association of rgp140, while analysis under nonreducing SDS-PAGE indicated intermonomer disulfide bonding. (D) Thirty-five micrograms of rgp140 was digested with Glu-C, and equal amounts were used for SDS-PAGE (15%) analysis under reducing and nonreducing conditions, followed by Western blot detection using MAb 4D8. Comparison of our synthetic V3 peptide (lane 1) and products of rgp140 digestion with Glu-C (lane 2) under reducing conditions (30-s exposure) showed the latter to be considerably larger. This is related to the V3 peptide released by Glu-C spanning 43 amino acid residues (predicted molecular mass, 4.94 kDa) and containing two N-linked carbohydrate moieties in addition to potential O-linked oligosaccharides. The Glu-C-released V3 peptide clearly exists as a higher-ordered structure under nonreducing conditions (lane 3; 20-min exposure). (E) Cartoon of the V3 disulfide-mapping experiments. (i) Native PAGE (panel B). (ii) Nonreducing SDS-PAGE (Panel C). (iii) Reducing SDS-PAGE (Fig. 2C). In all constructs, the gp41 component is represented as a thick line, and for nonreduced constructs, only the V3 disulfide bond is indicated. The locations of CA13 (N), 221 (C), and T30 (g) MAb epitopes are shown, and on the native PAGE-thrombin diagrams, the locations of Glu-C cut sites on either side of the V3 loop are indicated by lines. −, absence; +, presence of thrombin.
A recent report has proposed the possibility of an intermonomer disulfide bond located within the gp41 moiety (90). Current understanding of the disulfide bonding pattern of monomeric gp120 places all the bonds intramonomer (47). Within gp120, the disulfide bonding pattern is complex, with many bonds overlapping each other. However, the disulfide bond formed by cysteines 296 and 331 (the amino acid numbering is based on that used by Kwong et al. [46]) at the base of the V3 loop is one of two that are not overlapped by others. This, coupled with the presence of a thrombin cleavage site within the V3 loop, allowed us to investigate one bond in isolation from the others. Figure 3E shows a cartoon representation of the expected results of thrombin digestion analyzed under native and denaturing nonreducing conditions for either intramonomer or intermonomer V3 loop disulfide bonding patterns.
For this experiment to be meaningful, thrombin digestion should have no effect on the trimeric nature of rgp140. To test this, thrombin-digested rgp140 was run on native PAGE and probed with MAbs to the N and C termini of gp120 and gp41. Only trimeric and higher-order rgp140 proteins were detected, indicating that thrombin treatment alone does not disrupt trimeric association (Fig. 3B). This would be the case whether the V3 loop disulfide bond was intra- or intermonomer (Fig. 3E, i).
Figure 3E, ii, shows the rgp140 species expected to be present on a denaturing nonreducing gel following incubation in the absence and presence of thrombin. The denaturing nonreducing gel showed that thrombin digestion caused the release of monomeric rgp140 that contained the epitopes for all three mapping antibodies (Fig. 3C, lanes 2 to 4). Since thrombin cuts almost exclusively in the V3 loop (Fig. 2C) and results in a reduction in molecular mass corresponding to conversion from trimeric to monomeric forms (Fig. 3C, lanes 1 and 2, respectively), this result indicates the potential for the V3 loop-defining cysteines to form an intermonomer disulfide bond and to be responsible for the stability of our rgp140. However, the retention of trimeric rgp140 in the thrombin-digested samples (Fig. 3C, lanes 2 to 4) suggests either that thrombin digestion was incomplete (as shown in Fig. 2C), despite using thrombin/rgp140 (wt/wt) ratios of 0.1:1 to 320:1, or that more than one intermonomer disulfide bond was present. Incomplete digestion could be related to our rgp140 V3 loop sequence lacking a consensus thrombin recognition motif.
To further investigate the trimeric properties of the V3 loop disulfide bond, proteolytic digestion was used to remove the V3 loop under native conditions for analysis in isolation from the rest of rgp140. Glu-C was predicted to cleave at glutamic acid residues 290 and 335 in our rgp140, on either side of cysteine residues 296 and 331 forming the V3 loop, allowing its removal with local disulfide bonds intact. The major product of Glu-C digestion detected with the 4D8 V3-specific MAb under reducing conditions was considerably larger than the synthetic V3 peptide, presumably due to the attachment of carbohydrates (Fig. 3D, lanes 1 and 2). The same Glu-C digestion product run under nonreducing conditions (Fig. 3D, lane 3) showed an increase in molecular mass compared to reducing SDS-PAGE (Fig. 3D, lane 2), confirming the oligomeric nature of the V3 loop. N-terminal sequencing of the peptide identified in Fig. 3D (lane 2) gave a sequence of SIQINXT, in good agreement with the expected SIQINCT, showing that the V3 loop had been released by Glu-C. This, together with the size of the released peptide (under nonreducing conditions), indicates that cysteines 296 and 331 were solely responsible for the trimeric nature of the excised V3 loop and confirms the intermonomer nature of the V3 loop disulfide bond in our rgp140.
Functional characteristics of rgp140.
Functional characterization of our rgp140 was initiated by assaying for 2DCD4 binding. The ELISA-based method indicated weak binding compared to that seen with baculovirus- and CHO-expressed rgp120s derived from the extensively characterized HIV-1IIIB strain (Fig. 4A). The rgp140 weak binding was confirmed by the 3.34 μM binding affinity (KD) measured by surface plasmon resonance. While the weak binding may be related to strain differences, it falls within the range of previous reports that have shown primary isolates and trimeric rgp140 to have a lower affinity for CD4 than monomeric gp120 (36, 74, 75) and confirms the functionality of our rgp140 in respect to CD4 binding.
FIG. 4.

rgp140/120 binding of antibodies and CD4 and the effects of PDI treatment. The wells of ELISA plates were coated with 1 μg each either rgp140 or baculovirus-produced rgp120 (Bacrgp120) or CHO-K1 cell-produced rgp120 (CHOrgp120), either for 3 h at 37°C (A and C) or for 16 h at 4°C (B). The binding of ligands was then monitored as described in Materials and Methods. To study the effects of PDI on ligand binding, ELISA plate-bound rgp140/120 was treated with PDI (9 mU/well in 50 μl dilution buffer containing 1 mM reduced l-glutathione) overnight at 37°C prior to ligand addition. (A) The abilities of rgp140/120 to bind 2DCD4, and the influence of PDI treatment on binding. (B) The abilities of rgp140 and CHOrgp120 to bind a range of antibody reagents. (C) The abilities of native rgp140/120 and PDI-treated rgp140/120 to bind MAb 17b and 2DCD4/MAb 17b. For all results, the mean and standard errors of duplicate assays (n = 8) were plotted after removal of background readings (range, 0.045 to 0.048).
Additional indications of the correct folding of the gp120 component of the rgp140 were obtained from binding studies related to human MAbs that neutralize a broad range of HIV isolates (Fig. 4B). MAbs 17b, b12, and 2G12 recognize different discontinuous epitopes, with that of 2G12 being generated by certain glycan moieties on gp120 (reviewed in reference 11). While rgp140 and the CHO-expressed rgp120 bound b12 and 17b at comparable levels, binding of 2G12 to rgp140 was twofold less than that observed for CHO-expressed rgp120, though still more efficient than binding of 17b. This probably reflects differences in glycan composition/distribution between the two recombinant glycoproteins. The specificity of the assay was shown with V3-specific reagents, MAb 4D8, and ARP433 and EVA437 sera, with the subtype D rgp140 binding 4D8 and EVA437 while the subtype B CHO-expressed rgp120 bound ARP433 (Fig. 4B).
To further assess rgp140 functionality, we looked at its ability to undergo CD4-induced (CD4i) conformational change to allow the presentation of the coreceptor binding site. Coreceptors for HIV are chemokine receptors, members of a protein superfamily of seven-transmembrane domain receptors coupled to G protein, which are not available as soluble entities. Therefore, we used a coreceptor mimic, MAb 17b, which recognizes a CD4i epitope on gp120. Such epitopes are conserved discontinuous structures that are either formed or better exposed after CD4 binding (76, 79). Antibodies, such as MAb 17b, are able to block the binding of gp120-CD4 complexes to coreceptors (84). The gp120 residues important for MAb 17b and coreceptor binding have been mapped and shown to overlap extensively (64, 65, 76), making MAb 17b a good substitute for the gp120 coreceptor. In an ELISA-based assay, nonliganded rgp140 and the HIVIIIB rgp120s bound MAb 17b relatively weakly at approximately the same levels in independent assays (Fig. 4B/C). When 2DCD4 was present, levels of MAb 17b binding increased twofold for rgp140 and four- to fivefold for rgp120s (Fig. 4C). Overall, these results show that a proteolytically immature and disulfide cross-linked rgp140 is capable of undergoing the CD4i conformational changes necessary for coreceptor binding.
Effect of PDI on CD4 and coreceptor binding.
Using an ELISA-based method, pretreatment of both rgp140 and rgp120 with PDI was found to have negligible effects on CD4 binding (Fig. 4A). Conversely, similar pretreatment resulted in virtual abolishment of endogenous MAb 17b binding, and the enhanced binding in the presence of 2DCD4 was significantly reduced (Fig. 4C). This presumably reflects either disruption or occlusion of the preformed coreceptor binding site in rgp140 by the actions of PDI. Low-level processing of rgp140 by furin/thrombin-like enzymes, as observed in Fig. 2A to C, lanes 1, may contribute to this, as subsequent PDI processing could result in the loss of the N-terminal half of the molecule, which contains the V1/V2 loops, at the base of which are amino acids implicated in the formation of the MAb 17b binding site (64, 86). The low levels of 2DCD4-induced enhancement of MAb 17b binding to PDI-treated rgp140 and rgp120 (Fig. 4C) might reflect incomplete PDI processing, as seen in Fig. 5, lane 3. Overall, these results suggest that PDI processing during the HIV glycoprotein-mediated infection process (66) is likely to occur after coreceptor binding due to its detrimental effects on CD4i coreceptor binding.
FIG. 5.
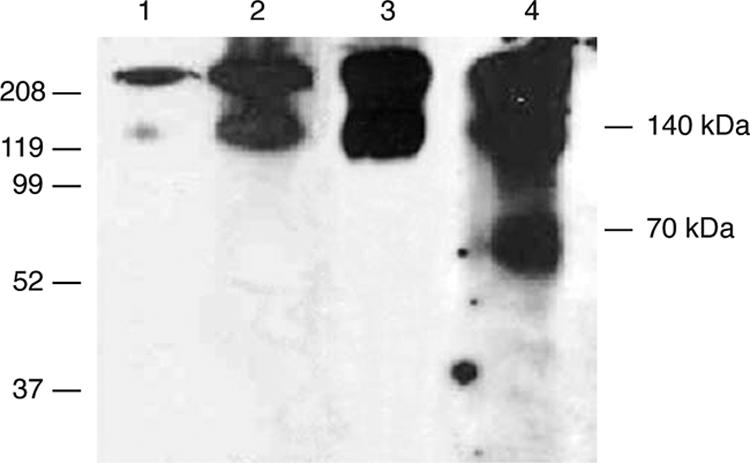
PDI disruption of the intermonomer V3 loop disulfide bond. rgp140 was run on nonreducing SDS-PAGE (4 to 12%) and analyzed by Western blotting using MAb CA13. Lane 1, untreated rgp140 showing the presence of disulfide-bonded trimeric rgp140 (the presence of equivalent bands in lanes 2 to 4 indicates poor cleavage by both thrombin and PDI). Lanes 2 to 4 reveal products of rgp140 digestion with thrombin, PDI, and thrombin followed by PDI, respectively.
The V3 loop disulfide is susceptible to PDI.
At least two disulfide bonds within gp120 are broken by PDI, but their locations are unknown (5). Having established the intermonomer nature of the V3 disulfide bond in our rgp140, we assessed whether it was a candidate for PDI processing. Treatment with PDI destroyed the trimeric nature of our rgp140, yielding monomers (Fig. 5, lane 3). This established the presence of at least one intermonomer disulfide bond processed by PDI. To determine if the V3 loop disulfide was being broken, rgp140 was subjected to thrombin digestion, followed by PDI treatment. When analyzed by nonreducing SDS-PAGE, thrombin cleaved the V3 loop, producing two fragments (the N terminus of gp120 and the C terminus of gp120/gp41) that remained associated via the V3 loop intermonomer disulfide bond as a 140-kDa product (Fig. 3C and Fig. 5, lane 2). PDI caused disruption of this 140-kDa product into its 70-kDa constituents (Fig. 5, lane 4), confirming the susceptibility of the V3 disulfide bond.
DISCUSSION
The expression of rgp140 has resulted in the production of very unstable heterogeneous oligomeric proteins, producing monomers, dimers, trimers, and aggregates (75, 92). In attempts to produce stable trimeric rgp140, the use of trimeric motifs from T4 bacteriophage (88), HIV-SIV chimeras (12), gp120/41-stabilizing disulfide bonds (71), and gp120/41 cleavage site mutants (88) has been tried with various degrees of success. Recent reports have suggested the ability to produce mainly trimeric rgp140 (92) that is highly stable to temperature and pH (44). While a number of the above-mentioned reports are related to transient expression systems, we showed the ability to produce predominantly trimeric and highly stable rgp140 in constitutively expressing CHO cell lines.
During the characterization of our rgp140, it became clear that a lack of processing at the gp120/41 site was a major factor contributing to the high stability of our recombinant protein. Since our rgp140 was furin sensitive (Fig. 2B), the lack of maturation processing was probably “fortuitous,” presumably reflecting a deficiency of furin-like activity in the CHO cell line selected. Such a lack of intracellular processing is consistent with the results of other researchers, who found unprocessed gp160 on the membranes of infected cells (2, 82) and on virus particles (27, 53). Sindbis virus has been shown to bind surface heparan sulfates via intact furin cleavage sites within their glycoproteins (42), and as discussed by Staropoli et al. (75), unprocessed gp160 on virus particles could also bind heparan sulfates, allowing the processing of gp160 by cell surface-expressed furin. This would allow the subsequent shedding of gp120 during membrane fusion.
Although our trimeric rgp140 was resistant to high temperature, acidic pH, and SDS (data not shown), β-mercaptoethanol was able to destroy the trimeric association. β-Mercaptoethanol-induced trimer instability has been shown previously (69, 88), and this, together with our characterization of rgp140 under reducing and nonreducing conditions, led us to postulate the presence of an intermonomer disulfide bond. Characterization of the V3 loop via thrombin digestion indicated that the disulfide bond at its base might be involved in intermonomer contacts. To investigate this further, the V3 loop was excised intact by Glu-C digestion but was shown to exist as a trimer due to the formation of intermonomer disulfide bonds (Fig. 3D). It has been suggested that intermonomer disulfide bonds in gp160 are due to protein misfolding (62); however, we showed that our proteolytically immature, disulfide cross-linked, trimeric rgp140 was functional in terms of weak CD4 binding and its abilities to bind conformation-dependent MAbs and to undergo CD4i conformational changes to bind MAb 17b (Fig. 4). Such a structure, with the associated low-affinity CD4 binding, might represent an early folded form of gp160 destined for transport to the infected cell surface. Partly based on predicted structures for trimeric HIV glycoprotein (55, 85), it has been suggested that such weak binding may be due to steric hindrance within the trimeric structure reducing the access of CD4 (36, 74). Indeed, others have proposed that the trimer might have a flexible structure that oscillates between high- and low-affinity CD4 binding (77), and evidence exists for infectious HIV-1 primary virions expressing at least two forms of gp120 on their surfaces (63). The flexibility of our rgp140 is likely to be restricted, given the intermonomer disulfide bonding and the lack of cleavage at the gp120/41 processing site.
Weak interaction of gp140 with CD4, similar to that observed with our HIV-1 subtype D-derived rgp140, has been reported for a number of primary HIV isolates (36, 74, 75). In extreme cases, strains of HIV and SIV that can infect cells in a CD4-independent manner have been isolated (reviewed in reference 7). Such strains are highly susceptible to neutralizing antibodies directed against CD4i epitopes, suggesting that their coreceptor binding domains are more exposed than in CD4-dependent strains. While the mutations in gp160s leading to CD4 independence are diverse (7), many, as for our rgp140, carry truncations of the gp41 subunit (8, 24, 68). In line with observations of CD4 independence, a study showing that MAb 17b reduced the affinity of gp120 for soluble CD4 (sCD4) led the authors to postulate that coreceptor binding could have a “release effect” on the gp120-CD4 interaction (93). A recent study employing single-molecule force spectroscopy supports this “release effect” hypothesis. Chang et al. (15) showed that, compared to a gp120-CD4 bond, a gp120-coreceptor bond was much longer lived and required significantly greater force to break it. The background levels of MAb 17b binding to all three rgp140/120 constructs were similar, but the level of enhanced MAb 17b binding seen in the presence of 2DCD4 was significantly less for our rgp140 than for either of the rgp120 species (Fig. 4C). This suggests that, compared to the rgp120 species, either a lower proportion of the rgp140 was able to undergo CD4i conformational change or it was in the main already in a CD4i conformation and MAb 17b binding was impeded due to the trimeric nature of the rgp140. If the latter is the case, as might be expected, since truncation of the gp41 cytoplasmic tail has been shown to affect exposure of neutralization epitopes on gp120 (24), it can be hypothesized that CD4 binding is a weak transient interaction whose role is to bring about conformational changes to unmask the coreceptor binding site (7).
A recent report has shown the presence of a potential intermonomer disulfide bond within the gp41 moiety (90). In the latter report, V1/V2 deletion mutants had no effect on disulfide cross-linking while C-terminal gp41 truncation to remove the cysteine residues was found to abrogate trimerization via intermonomer disulfide bonds. The authors further suggested the lack of involvement of gp120, since recombinantly expressed gp120 shows little tendency to self-associate. We hypothesize that the trimerization potential of gp41 is needed to bring the V3 loop cysteines of neighboring gp120 subunits into close enough contact to allow the formation of intermonomer disulfide bonds. Indeed, the C-terminal portion of gp41deleted by Yuan et al. (90) may impose structural constraints on gp120 to enhance gp120 intermonomer bonding.
gp120 binding induces localized and specific conformational changes in CD4 (87), and disulfide exchange has been shown to be important in the HIV entry process, with redox changes in domain 2 of CD4 being required (52). Additionally, PDI is believed to be responsible for processing at least two disulfide bonds within gp120 during the infection process (5). To further characterize the rgp140 V3 intermonomer disulfide bond, we investigated its potential for PDI processing, and although we have not been able to assign an activity to the intersubunit disulfide bond, we have shown that it is potentially one of the disulfide bonds broken by PDI. This agrees with a hypothesis, based on the disulfide bonding pattern of gp120, that three disulfide bonds (spanning V1/V2, V3, and V4) are uncommon cross-strand disulfides (83). Such bonds are under stress and relatively unstable, making them prone to cleavage and release of their stored energy, which might drive conformational changes such as those involved in membrane fusion. With the V3 loop being involved in coreceptor binding, it was suggested that its disulfide bond would be the prime target for PDI processing (83).
The precise point(s) at which cell surface thiol-containing molecules may act in the HIV entry process is unknown, and extrapolation from in vitro experimentation to the in vivo situation should be viewed with caution. However, work by Barbouche et al. (5) suggests that PDI processing takes place post-coreceptor binding, while data presented by others (29, 51) predict that processing occurs after CD4 binding but prior to coreceptor binding. Our results show that pretreatment of rgp140/120 with PDI results in severe inhibition of MAb 17b and CD4i MAb17b binding (Fig. 4C). Therefore, it appears probable that PDI will act post-coreceptor binding in vivo. Molecular-docking studies indicate that PDI probably binds to domain 3 of CD4 (66), while gp120 is known to bind to domain 1 of CD4 (46). It is possible that redox changes in domain 2 of CD4 (52) may allow gp120 and cell surface thiol-containing molecules to come into close proximity. Current models of HIV glycoprotein-induced membrane fusion suggest there is a long-lived “prehairpin intermediate” state in which the gp41 fusion peptide is inserted into the host cell membrane and gp120 is retained in the fusion complex (13, 66). gp41-derived peptide (39, 81) and small-molecule (72) inhibitors target this stage of the fusion process. It is possible that such inhibitors prevent shedding of the gp120 component from the fusion complex, thereby preventing membrane fusion that is induced by the transition of gp41 into a six-helix bundle (50, 54).
The trimeric rgp140 studied here is unlikely to be fusion competent, as it has not undergone gp120/41 processing and it contains intermonomer V3 loop-derived disulfide bonds. Similar introduction of intermonomer disulfide bonding into the HA1 component of influenza hemagglutinin (HA) yielded a molecule that could not undergo pH-induced conformational changes, thereby preventing HA-mediated membrane fusion, but fusion could be restored under conditions that reduced the novel disulfide bonds (32). Similarly, a mutant HIV-1 gp160 containing an intramonomer gp120-gp41 disulfide bond, preventing dissociation of gp120 and gp41, was fusion incompetent, but fusion could be restored by reduction of the disulfide bond (1). There are a growing number of reports implicating PDI processing in HIV infectivity (4, 5, 25, 29, 40, 51, 66, 67). In these studies, inhibition of PDI was found to prevent virus-cell fusion. In the studies presented here, we show that rgp140 contains intermonomer V3-derived disulfide bonds (Fig. 3) that are susceptible to PDI (Fig. 5) and that PDI treatment of rgp120 and rgp140 severely inhibits binding of the coreceptor mimic MAb 17b (Fig. 4C). These observations, together with the V3 domain being a major determinant of chemokine receptor specificity (19, 28, 38), suggest a structural need for V3-derived disulfide bonds. In our current understanding of virus attachment and membrane fusion, there is no known role for gp120 after coreceptor binding (13, 66). However, the conformational changes induced by CD4 and coreceptor binding do not appear adequate to activate gp41 (72), despite the strong interaction with coreceptor (15, 93). Therefore, a role for PDI, possibly aided by thrombin-like processing, can be proposed in the reduction of structure-stabilizing disulfide bonds in gp120, resulting in release from the strong interaction with coreceptor and shedding of gp120 from the fusion complex. This would allow the gp41 component to undergo the further conformational changes required to complete the membrane fusion process. As such, PDI could be considered the trigger for fusion, and its availability and action would be major determinants of the amount of time spent in the “prehairpin intermediate” state during the fusion process (13). Recent work has called into question the specific role of PDI in the HIV entry process and suggests that other cell surface thiol-containing molecules, such as thioredoxin, are more active in reducing disulfide bonds in CD4 and gp120 (61). However, such results do not negate the possibility of the reduction of structure-stabilizing disulfide bonds in gp120 being the trigger for fusion.
Since the V3 domain is a major determinant of HIV-1 tropism and its ability to escape drugs designed to block virus entry, as well as being considered the “principal neutralization determinant” for many years (reviewed in reference 33), our findings are relevant to vaccine design. Indeed, it has been shown for HIV-1ADA that trimeric rgp140 is a better immunogen in guinea pigs than monomeric rgp120 and that the V3-specific neutralizing antibodies induced by it are less susceptible to blocking by ADA-V3 peptide than those raised against rgp120, which are totally blocked (41). This presumably reflects differences between the two immunogens in presentation of epitopes within the V3 domain that may relate to intermonomer disulfide bonding in the trimeric rgp140 resembling more closely the structure present on virus. Recent cryoelectron tomographic studies of SIV glycoprotein on virus particles have resulted in two models for the orientation of the V3 loop, either inward toward the trimer interface with consequent masking of the site or outward, resulting in good exposure to solvent (91). Both forms may exist, depending on the activities of thiol reductases, with the former being less exposed to antibodies. In this context, it is notable that the V3-specific 4D8 MAb detects monomeric V3 more efficiently than its oligomeric (trimeric) form (Fig. 3D).
Current structural data are based upon truncated monomeric versions of HIV-1 (35, 45, 46) and SIV (16, 17) rgp120 with the variable loops deleted. The HIV-1 structures consist of complexes composed of deglycosylated rgp120, 2DCD4, and MAbs to CD4i epitopes. The potential importance of the variable loops is illustrated by the observation that in intact rgp120, the binding of MAb 17b weakens the interactions between gp120 and CD4, an effect that is not seen in constructs with V1/V2 deleted (93). Therefore, the existing rgp120 structures, due to their highly truncated and monomeric nature, lack detail of variable loops, as well as any possible intermonomer disulfide bonds. Given the results presented here, highlighting the importance of the V3 domain, the elucidation of the structure of native trimeric rgp140 is a necessity for better understanding of structure-function relationships in HIV glycoproteins.
Acknowledgments
We thank J. Robinson and the NIBSC Centralised Facility for AIDS Reagents supported by EU Programme EVA (contract QLKZ-CT-1999-00609) and the Medical Research Council for supplying MAb 17b. The 2DCD4 protein used in the work presented here was obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH; sCD4-183 was from Pharmacia.
Footnotes
Published ahead of print on 14 February 2007.
REFERENCES
- 1.Abrahamyan, L. G., R. M. Markosyan, J. P. Moore, F. S. Cohen, and G. B. Melikyan. 2003. Human immunodeficiency virus type 1 Env with an intersubunit disulfide bond engages coreceptors but requires bond reduction after engagement to induce fusion. J. Virol. 77:5829-5836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altmeyer, R., E. Mordelet, M. Girard, and C. Vidal. 1999. Expression and detection of macrophage-tropic HIV-1 gp120 in the brain using conformation-dependent antibodies. Virology 259:314-323. [DOI] [PubMed] [Google Scholar]
- 3.Bailes, E., F. Gao, F. Bibollet-Ruche, V. Courgnaud, M. Peeters, P. A. Marx, B. H. Hahn, and P. M. Sharp. 2003. Hybrid origin of SIV in chimpanzees. Science 300:1713. [DOI] [PubMed] [Google Scholar]
- 4.Barbouche, R., H. Lortat-Jacob, I. M. Jones, and E. Fenouillet. 2005. Glycosaminoglycans and protein disulfide isomerase-mediated reduction of HIV Env. Mol. Pharmacol. 67:1111-1118. [DOI] [PubMed] [Google Scholar]
- 5.Barbouche, R., R. Miquelis, I. M. Jones, and E. Fenouillet. 2003. Protein-disulfide isomerase-mediated reduction of two disulfide bonds of HIV envelope glycoprotein 120 occurs post-CXCR4 binding and is required for fusion. J. Biol. Chem. 278:3131-3136. [DOI] [PubMed] [Google Scholar]
- 6.Barre-Sinoussi, F., J. C. Chermann, F. Rey, M. T. Nugeyre, S. Chamaret, J. Gruest, C. Dauguet, C. Axler-Blin, F. Vezinet-Brun, C. Rouzioux, W. Rozenbaum, and L. Montagnier. 1983. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 220:868-871. [DOI] [PubMed] [Google Scholar]
- 7.Bhattacharya, J., P. J. Peters, and P. R. Clapham. 2003. CD4-independent infection of HIV and SIV: implications for envelope conformation and cell tropism in vivo. AIDS 17(Suppl. 4):S35-S43. [DOI] [PubMed] [Google Scholar]
- 8.Bonavia, A., B. T. Bullock, K. M. Gisselman, B. J. Margulies, and J. E. Clements. 2005. A single amino acid change and truncated TM are sufficient for simian immunodeficiency virus to enter cells using CCR5 in a CD4-independent pathway. Virology 341:12-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bristow, R. G., A. R. Douglas, J. J. Skehel, and R. S. Daniels. 1994. Analysis of murine antibody responses to baculovirus-expressed human immunodeficiency virus type 1 envelope glycoproteins. J. Gen. Virol. 75:2089-2095. [DOI] [PubMed] [Google Scholar]
- 10.Broder, C. C., and R. G. Collman. 1997. Chemokine receptors and HIV. J. Leukoc. Biol. 62:20-29. [DOI] [PubMed] [Google Scholar]
- 11.Burton, D. R., R. L. Stanfield, and I. A. Wilson. 2005. Antibody vs. HIV in a clash of evolutionary titans. Proc. Natl. Acad. Sci. USA 102:14943-14948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Center, R. J., J. Lebowitz, R. D. Leapman, and B. Moss. 2004. Promoting trimerization of soluble human immunodeficiency virus type 1 (HIV-1) Env through the use of HIV-1/simian immunodeficiency virus chimeras. J. Virol. 78:2265-2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chan, D. C., and P. S. Kim. 1998. HIV entry and its inhibition. Cell 93:681-684. [DOI] [PubMed] [Google Scholar]
- 14.Chang, J. Y. 1985. Thrombin specificity. Requirement for apolar amino acids adjacent to the thrombin cleavage site of polypeptide substrate. Eur. J. Biochem. 151:217-224. [DOI] [PubMed] [Google Scholar]
- 15.Chang, M. I., P. Panorchan, T. M. Dobrowsky, Y. Tseng, and D. Wirtz. 2005. Single-molecule analysis of human immunodeficiency virus type 1 gp120-receptor interactions in living cells. J. Virol. 79:14748-14755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen, B., E. M. Vogan, H. Gong, J. J. Skehel, D. C. Wiley, and S. C. Harrison. 2005. Determining the structure of an unliganded and fully glycosylated SIV gp120 envelope glycoprotein. Structure 13:197-211. [DOI] [PubMed] [Google Scholar]
- 17.Chen, B., E. M. Vogan, H. Gong, J. J. Skehel, D. C. Wiley, and S. C. Harrison. 2005. Structure of an unliganded simian immunodeficiency virus gp120 core. Nature 433:834-841. [DOI] [PubMed] [Google Scholar]
- 18.Clements, G. J., M. J. Price-Jones, P. E. Stephens, C. Sutton, T. F. Schulz, P. R. Clapham, J. A. McKeating, M. O. McClure, S. Thomson, M. Marsh, et al. 1991. The V3 loops of the HIV-1 and HIV-2 surface glycoproteins contain proteolytic cleavage sites: a possible function in viral fusion? AIDS Res. Hum. Retrovir. 7:3-16. [DOI] [PubMed] [Google Scholar]
- 19.de Jong, J. J., J. Goudsmit, W. Keulen, B. Klaver, W. Krone, M. Tersmette, and A. de Ronde. 1992. Human immunodeficiency virus type 1 clones chimeric for the envelope V3 domain differ in syncytium formation and replication capacity. J. Virol. 66:757-765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Douglas, N. W., A. I. Knight, A. Hayhurst, W. Y. Barrett, M. J. Kevany, and R. S. Daniels. 1996. An efficient method for the rescue and analysis of functional HIV-1 env genes: evidence for recombination in the vicinity of the tat/rev splice site. AIDS 10:39-46. [DOI] [PubMed] [Google Scholar]
- 21.Douglas, N. W., G. H. Munro, and R. S. Daniels. 1997. HIV/SIV glycoproteins: structure-function relationships. J. Mol. Biol. 273:122-149. [DOI] [PubMed] [Google Scholar]
- 22.Earl, P. L., C. C. Broder, R. W. Doms, and B. Moss. 1997. Epitope map of human immunodeficiency virus type 1 gp41 derived from 47 monoclonal antibodies produced by immunization with oligomeric envelope protein. J. Virol. 71:2674-2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Earl, P. L., B. Moss, and R. W. Doms. 1991. Folding, interaction with GRP78-BiP, assembly, and transport of the human immunodeficiency virus type 1 envelope protein. J. Virol. 65:2047-2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edwards, T. G., S. Wyss, J. D. Reeves, S. Zolla-Pazner, J. A. Hoxie, R. W. Doms, and F. Baribaud. 2002. Truncation of the cytoplasmic domain induces exposure of conserved regions in the ectodomain of human immunodeficiency virus type 1 envelope protein. J. Virol. 76:2683-2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fenouillet, E., R. Barbouche, J. Courageot, and R. Miquelis. 2001. The catalytic activity of protein disulfide isomerase is involved in human immunodeficiency virus envelope-mediated membrane fusion after CD4 cell binding. J. Infect. Dis. 183:744-752. [DOI] [PubMed] [Google Scholar]
- 26.Ferre-D'Amare, A. R., and S. K. Burley. 1994. Use of dynamic light scattering to assess crystallizability of macromolecules and macromolecular assemblies. Structure 2:357-359. [DOI] [PubMed] [Google Scholar]
- 27.Fischer, P. B., G. B. Karlsson, R. A. Dwek, and F. M. Platt. 1996. N-Butyldeoxynojirimycin-mediated inhibition of human immunodeficiency virus entry correlates with impaired gp120 shedding and gp41 exposure. J. Virol. 70:7153-7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fouchier, R. A., M. Groenink, N. A. Kootstra, M. Tersmette, H. G. Huisman, F. Miedema, and H. Schuitemaker. 1992. Phenotype-associated sequence variation in the third variable domain of the human immunodeficiency virus type 1 gp120 molecule. J. Virol. 66:3183-3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gallina, A., T. M. Hanley, R. Mandel, M. Trahey, C. C. Broder, G. A. Viglianti, and H. J. Ryser. 2002. Inhibitors of protein-disulfide isomerase prevent cleavage of disulfide bonds in receptor-bound glycoprotein 120 and prevent HIV-1 entry. J. Biol. Chem. 277:50579-50588. [DOI] [PubMed] [Google Scholar]
- 30.Gallo, R. C., S. Z. Salahuddin, M. Popovic, G. M. Shearer, M. Kaplan, B. F. Haynes, T. J. Palker, R. Redfield, J. Oleske, B. Safai, et al. 1984. Frequent detection and isolation of cytopathic retroviruses (HTLV-III) from patients with AIDS and at risk for AIDS. Science 224:500-503. [DOI] [PubMed] [Google Scholar]
- 31.Gallo, S. A., G. M. Clore, J. M. Louis, C. A. Bewley, and R. Blumenthal. 2004. Temperature-dependent intermediates in HIV-1 envelope glycoprotein-mediated fusion revealed by inhibitors that target N- and C-terminal helical regions of HIV-1 gp41. Biochemistry 43:8230-8233. [DOI] [PubMed] [Google Scholar]
- 32.Godley, L., J. Pfeifer, D. Steinhauer, B. Ely, G. Shaw, R. Kaufmann, E. Suchanek, C. Pabo, J. J. Skehel, D. C. Wiley, and S. Wharton. 1992. Introduction of intersubunit disulfide bonds in the membrane-distal region of the influenza hemagglutinin abolishes membrane fusion activity. Cell 68:635-645. [DOI] [PubMed] [Google Scholar]
- 33.Hartley, O., P. J. Klasse, Q. J. Sattentau, and J. P. Moore. 2005. V3: HIV's switch-hitter. AIDS Res. Hum. Retrovir. 21:171-189. [DOI] [PubMed] [Google Scholar]
- 34.Hsu, S. T., and A. M. Bonvin. 2004. Atomic insight into the CD4 binding-induced conformational changes in HIV-1 gp120. Proteins 55:582-593. [DOI] [PubMed] [Google Scholar]
- 35.Huang, C. C., M. Tang, M. Y. Zhang, S. Majeed, E. Montabana, R. L. Stanfield, D. S. Dimitrov, B. Korber, J. Sodroski, I. A. Wilson, R. Wyatt, and P. D. Kwong. 2005. Structure of a V3-containing HIV-1 gp120 core. Science 310:1025-1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jeffs, S. A., S. Goriup, B. Kebble, D. Crane, B. Bolgiano, Q. Sattentau, S. Jones, and H. Holmes. 2004. Expression and characterisation of recombinant oligomeric envelope glycoproteins derived from primary isolates of HIV-1. Vaccine 22:1032-1046. [DOI] [PubMed] [Google Scholar]
- 37.Jeffs, S. A., J. McKeating, S. Lewis, H. Craft, D. Biram, P. E. Stephens, and R. L. Brady. 1996. Antigenicity of truncated forms of the human immunodeficiency virus type 1 envelope glycoprotein. J. Gen. Virol. 77:1403-1410. [DOI] [PubMed] [Google Scholar]
- 38.Jensen, M. A., F. S. Li, A. B. van 't Wout, D. C. Nickle, D. Shriner, H. X. He, S. McLaughlin, R. Shankarappa, J. B. Margolick, and J. I. Mullins. 2003. Improved coreceptor usage prediction and genotypic monitoring of R5-to-X4 transition by motif analysis of human immunodeficiency virus type 1 env V3 loop sequences. J. Virol. 77:13376-13388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiang, S., K. Lin, N. Strick, and A. R. Neurath. 1993. HIV-1 inhibition by a peptide. Nature 365:113. [DOI] [PubMed] [Google Scholar]
- 40.Jordan, P. A., and J. M. Gibbins. 2006. Extracellular disulfide exchange and the regulation of cellular function. Antioxid. Redox Signal 8:312-324. [DOI] [PubMed] [Google Scholar]
- 41.Kim, M., Z. S. Qiao, D. C. Montefiori, B. F. Haynes, E. L. Reinherz, and H. X. Liao. 2005. Comparison of HIV Type 1 ADA gp120 monomers versus gp140 trimers as immunogens for the induction of neutralizing antibodies. AIDS Res. Hum. Retrovir. 21:58-67. [DOI] [PubMed] [Google Scholar]
- 42.Klimstra, W. B., H. W. Heidner, and R. E. Johnston. 1999. The furin protease cleavage recognition sequence of Sindbis virus PE2 can mediate virion attachment to cell surface heparan sulfate. J. Virol. 73:6299-6306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koshiba, T., and D. C. Chan. 2003. The prefusogenic intermediate of HIV-1 gp41 contains exposed C-peptide regions. J. Biol. Chem. 278:7573-7579. [DOI] [PubMed] [Google Scholar]
- 44.Krell, T., F. Greco, O. Engel, J. Dubayle, A. Kennel, B. Charloteaux, R. Brasseur, M. Chevalier, R. Sodoyer, and R. El Habib. 2004. HIV-1 gp41 and gp160 are hyperthermostable proteins in a mesophilic environment. Characterization of gp41 mutants. Eur. J. Biochem. 271:1566-1579. [DOI] [PubMed] [Google Scholar]
- 45.Kwong, P. D., R. Wyatt, S. Majeed, J. Robinson, R. W. Sweet, J. Sodroski, and W. A. Hendrickson. 2000. Structures of HIV-1 gp120 envelope glycoproteins from laboratory-adapted and primary isolates. Struct. Fold Des. 8:1329-1339. [DOI] [PubMed] [Google Scholar]
- 46.Kwong, P. D., R. Wyatt, J. Robinson, R. W. Sweet, J. Sodroski, and W. A. Hendrickson. 1998. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393:648-659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leonard, C. K., M. W. Spellman, L. Riddle, R. J. Harris, J. N. Thomas, and T. J. Gregory. 1990. Assignment of intrachain disulfide bonds and characterization of potential glycosylation sites of the type 1 recombinant human immunodeficiency virus envelope glycoprotein (gp120) expressed in Chinese hamster ovary cells. J. Biol. Chem. 265:10373-10382. [PubMed] [Google Scholar]
- 48.Levy, J. A., and J. Shimabukuro. 1985. Recovery of AIDS-associated retroviruses from patients with AIDS or AIDS-related conditions and from clinically healthy individuals. J. Infect. Dis. 152:734-738. [DOI] [PubMed] [Google Scholar]
- 49.Ling, H., P. Xiao, O. Usami, and T. Hattori. 2004. Thrombin activates envelope glycoproteins of HIV type 1 and enhances fusion. Microbes Infect. 6:414-420. [DOI] [PubMed] [Google Scholar]
- 50.Markosyan, R. M., F. S. Cohen, and G. B. Melikyan. 2003. HIV-1 envelope proteins complete their folding into six-helix bundles immediately after fusion pore formation. Mol. Biol. Cell 14:926-938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Markovic, I., T. S. Stantchev, K. H. Fields, L. J. Tiffany, M. Tomic, C. D. Weiss, C. C. Broder, K. Strebel, and K. A. Clouse. 2004. Thiol/disulfide exchange is a prerequisite for CXCR4-tropic HIV-1 envelope-mediated T-cell fusion during viral entry. Blood 103:1586-1594. [DOI] [PubMed] [Google Scholar]
- 52.Matthias, L. J., P. T. Yam, X. M. Jiang, N. Vandegraaff, P. Li, P. Poumbourios, N. Donoghue, and P. J. Hogg. 2002. Disulfide exchange in domain 2 of CD4 is required for entry of HIV-1. Nat. Immunol. 3:727-732. [DOI] [PubMed] [Google Scholar]
- 53.McCune, J. M., L. B. Rabin, M. B. Feinberg, M. Lieberman, J. C. Kosek, G. R. Reyes, and I. L. Weissman. 1988. Endoproteolytic cleavage of gp160 is required for the activation of human immunodeficiency virus. Cell 53:55-67. [DOI] [PubMed] [Google Scholar]
- 54.Melikyan, G. B., R. M. Markosyan, H. Hemmati, M. K. Delmedico, D. M. Lambert, and F. S. Cohen. 2000. Evidence that the transition of HIV-1 gp41 into a six-helix bundle, not the bundle configuration, induces membrane fusion. J. Cell Biol. 151:413-423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moore, J. P., Y. Cao, L. Qing, Q. J. Sattentau, J. Pyati, R. Koduri, J. Robinson, C. F. Barbas III, D. R. Burton, and D. D. Ho. 1995. Primary isolates of human immunodeficiency virus type 1 are relatively resistant to neutralization by monoclonal antibodies to gp120, and their neutralization is not predicted by studies with monomeric gp120. J. Virol. 69:101-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moore, J. P., J. A. McKeating, R. A. Weiss, and Q. J. Sattentau. 1990. Dissociation of gp120 from HIV-1 virions induced by soluble CD4. Science 250:1139-1142. [DOI] [PubMed] [Google Scholar]
- 57.Moore, J. P., A. Trkola, and T. Dragic. 1997. Co-receptors for HIV-1 entry. Curr. Opin. Immunol. 9:551-562. [DOI] [PubMed] [Google Scholar]
- 58.Morikawa, Y., E. Barsov, and I. Jones. 1993. Legitimate and illegitimate cleavage of human immunodeficiency virus glycoproteins by furin. J. Virol. 67:3601-3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moulard, M., and E. Decroly. 2000. Maturation of HIV envelope glycoprotein precursors by cellular endoproteases. Biochim. Biophys. Acta 1469:121-132. [DOI] [PubMed] [Google Scholar]
- 60.Moulard, M., S. Hallenberger, W. Garten, and H. D. Klenk. 1999. Processing and routage of HIV glycoproteins by furin to the cell surface. Virus Res. 60:55-65. [DOI] [PubMed] [Google Scholar]
- 61.Ou, W., and J. Silver. 2006. Role of protein disulfide isomerase and other thiol-reactive proteins in HIV-1 envelope protein-mediated fusion. Virology 350:406-417. [DOI] [PubMed] [Google Scholar]
- 62.Owens, R. J., and R. W. Compans. 1990. The human immunodeficiency virus type 1 envelope glycoprotein precursor acquires aberrant intermolecular disulfide bonds that may prevent normal proteolytic processing. Virology 179:827-833. [DOI] [PubMed] [Google Scholar]
- 63.Poignard, P., M. Moulard, E. Golez, V. Vivona, M. Franti, S. Venturini, M. Wang, P. W. Parren, and D. R. Burton. 2003. Heterogeneity of envelope molecules expressed on primary human immunodeficiency virus type 1 particles as probed by the binding of neutralizing and nonneutralizing antibodies. J. Virol. 77:353-365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rizzuto, C., and J. Sodroski. 2000. Fine definition of a conserved CCR5-binding region on the human immunodeficiency virus type 1 glycoprotein 120. AIDS Res. Hum. Retrovir. 16:741-749. [DOI] [PubMed] [Google Scholar]
- 65.Rizzuto, C. D., R. Wyatt, N. Hernandez-Ramos, Y. Sun, P. D. Kwong, W. A. Hendrickson, and J. Sodroski. 1998. A conserved HIV gp120 glycoprotein structure involved in chemokine receptor binding. Science 280:1949-1953. [DOI] [PubMed] [Google Scholar]
- 66.Ryser, H. J., and R. Fluckiger. 2005. Progress in targeting HIV-1 entry. Drug Discov. Today 10:1085-1094. [DOI] [PubMed] [Google Scholar]
- 67.Ryser, H. J., E. M. Levy, R. Mandel, and G. J. DiSciullo. 1994. Inhibition of human immunodeficiency virus infection by agents that interfere with thiol-disulfide interchange upon virus-receptor interaction. Proc. Natl. Acad. Sci. USA 91:4559-4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Saha, K., H. Yan, J. A. Nelson, and B. Zerhouni-Layachi. 2005. Infection of human and non-human cells by a highly fusogenic primary CD4-independent HIV-1 isolate with a truncated envelope cytoplasmic tail. Virology 337:30-44. [DOI] [PubMed] [Google Scholar]
- 69.Sanders, R. W., M. Vesanen, N. Schuelke, A. Master, L. Schiffner, R. Kalyanaraman, M. Paluch, B. Berkhout, P. J. Maddon, W. C. Olson, M. Lu, and J. P. Moore. 2002. Stabilization of the soluble, cleaved, trimeric form of the envelope glycoprotein complex of human immunodeficiency virus type 1. J. Virol. 76:8875-8889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sattentau, Q. J., J. P. Moore, F. Vignaux, F. Traincard, and P. Poignard. 1993. Conformational changes induced in the envelope glycoproteins of the human and simian immunodeficiency viruses by soluble receptor binding. J. Virol. 67:7383-7393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schulke, N., M. S. Vesanen, R. W. Sanders, P. Zhu, M. Lu, D. J. Anselma, A. R. Villa, P. W. Parren, J. M. Binley, K. H. Roux, P. J. Maddon, J. P. Moore, and W. C. Olson. 2002. Oligomeric and conformational properties of a proteolytically mature, disulfide-stabilized human immunodeficiency virus type 1 gp140 envelope glycoprotein. J. Virol. 76:7760-7776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Si, Z., N. Madani, J. M. Cox, J. J. Chruma, J. C. Klein, A. Schon, N. Phan, L. Wang, A. C. Biorn, S. Cocklin, I. Chaiken, E. Freire, A. B. Smith III, and J. G. Sodroski. 2004. Small-molecule inhibitors of HIV-1 entry block receptor-induced conformational changes in the viral envelope glycoproteins. Proc. Natl. Acad. Sci. USA 101:5036-5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Speck, R. F., K. Wehrly, E. J. Platt, R. E. Atchison, I. F. Charo, D. Kabat, B. Chesebro, and M. A. Goldsmith. 1997. Selective employment of chemokine receptors as human immunodeficiency virus type 1 coreceptors determined by individual amino acids within the envelope V3 loop. J. Virol. 71:7136-7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Srivastava, I. K., L. Stamatatos, H. Legg, E. Kan, A. Fong, S. R. Coates, L. Leung, M. Wininger, J. J. Donnelly, J. B. Ulmer, and S. W. Barnett. 2002. Purification and characterization of oligomeric envelope glycoprotein from a primary R5 subtype B human immunodeficiency virus. J. Virol. 76:2835-2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Staropoli, I., C. Chanel, M. Girard, and R. Altmeyer. 2000. Processing, stability, and receptor binding properties of oligomeric envelope glycoprotein from a primary HIV-1 isolate. J. Biol. Chem. 275:35137-35145. [DOI] [PubMed] [Google Scholar]
- 76.Thali, M., J. P. Moore, C. Furman, M. Charles, D. D. Ho, J. Robinson, and J. Sodroski. 1993. Characterization of conserved human immunodeficiency virus type 1 gp120 neutralization epitopes exposed upon gp120-CD4 binding. J. Virol. 67:3978-3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ugolini, S., I. Mondor, and Q. J. Sattentau. 1999. HIV-1 attachment: another look. Trends Microbiol. 7:144-149. [DOI] [PubMed] [Google Scholar]
- 78.Vollenweider, F., S. Benjannet, E. Decroly, D. Savaria, C. Lazure, G. Thomas, M. Chretien, and N. G. Seidah. 1996. Comparative cellular processing of the human immunodeficiency virus (HIV-1) envelope glycoprotein gp160 by the mammalian subtilisin/kexin-like convertases. Biochem. J. 314:521-532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Weinberg, J., H. X. Liao, J. V. Torres, T. J. Matthews, J. Robinson, and B. F. Haynes. 1997. Identification of a synthetic peptide that mimics an HIV glycoprotein 120 envelope conformational determinant exposed following ligation of glycoprotein 120 by CD4. AIDS Res. Hum. Retrovir. 13:657-664. [DOI] [PubMed] [Google Scholar]
- 80.Weissenhorn, W., A. Dessen, S. C. Harrison, J. J. Skehel, and D. C. Wiley. 1997. Atomic structure of the ectodomain from HIV-1 gp41. Nature 387:426-430. [DOI] [PubMed] [Google Scholar]
- 81.Wild, C. T., D. C. Shugars, T. K. Greenwell, C. B. McDanal, and T. J. Matthews. 1994. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc. Natl. Acad. Sci. USA 91:9770-9774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Willey, R. L., J. S. Bonifacino, B. J. Potts, M. A. Martin, and R. D. Klausner. 1988. Biosynthesis, cleavage, and degradation of the human immunodeficiency virus 1 envelope glycoprotein gp160. Proc. Natl. Acad. Sci. USA 85:9580-9584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wouters, M. A., K. K. Lau, and P. J. Hogg. 2004. Cross-strand disulphides in cell entry proteins: poised to act. Bioessays 26:73-79. [DOI] [PubMed] [Google Scholar]
- 84.Wu, H., D. G. Myszka, S. W. Tendian, C. G. Brouillette, R. W. Sweet, I. M. Chaiken, and W. A. Hendrickson. 1996. Kinetic and structural analysis of mutant CD4 receptors that are defective in HIV gp120 binding. Proc. Natl. Acad. Sci. USA 93:15030-15035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wyatt, R., P. D. Kwong, E. Desjardins, R. W. Sweet, J. Robinson, W. A. Hendrickson, and J. G. Sodroski. 1998. The antigenic structure of the HIV gp120 envelope glycoprotein. Nature 393:705-711. [DOI] [PubMed] [Google Scholar]
- 86.Xiang, S. H., N. Doka, R. K. Choudhary, J. Sodroski, and J. E. Robinson. 2002. Characterization of CD4-induced epitopes on the HIV type 1 gp120 envelope glycoprotein recognized by neutralizing human monoclonal antibodies. AIDS Res. Hum. Retrovir. 18:1207-1217. [DOI] [PubMed] [Google Scholar]
- 87.Yachou, A., and R. P. Sekaly. 1999. Binding of soluble recombinant HIV envelope glycoprotein, rgp120, induces conformational changes in the cellular membrane-anchored CD4 molecule. Biochem. Biophys. Res. Commun. 265:428-433. [DOI] [PubMed] [Google Scholar]
- 88.Yang, X., J. Lee, E. M. Mahony, P. D. Kwong, R. Wyatt, and J. Sodroski. 2002. Highly stable trimers formed by human immunodeficiency virus type 1 envelope glycoproteins fused with the trimeric motif of T4 bacteriophage fibritin. J. Virol. 76:4634-4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yang, Z. N., T. C. Mueser, J. Kaufman, S. J. Stahl, P. T. Wingfield, and C. C. Hyde. 1999. The crystal structure of the SIV gp41 ectodomain at 1.47 Å resolution. J. Struct. Biol. 126:131-144. [DOI] [PubMed] [Google Scholar]
- 90.Yuan, W., S. Craig, X. Yang, and J. Sodroski. 2005. Inter-subunit disulfide bonds in soluble HIV-1 envelope glycoprotein trimers. Virology 332:369-383. [DOI] [PubMed] [Google Scholar]
- 91.Zanetti, G., J. A. Briggs, K. Grunewald, Q. J. Sattentau, and S. D. Fuller. 2006. Cryo-electron tomographic structure of an immunodeficiency virus envelope complex in situ. PLoS Pathog. 2:e83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang, C. W., Y. Chishti, R. E. Hussey, and E. L. Reinherz. 2001. Expression, purification, and characterization of recombinant HIV gp140. The gp41 ectodomain of HIV or simian immunodeficiency virus is sufficient to maintain the retroviral envelope glycoprotein as a trimer. J. Biol. Chem. 276:39577-39585. [DOI] [PubMed] [Google Scholar]
- 93.Zhang, W., A. P. Godillot, R. Wyatt, J. Sodroski, and I. Chaiken. 2001. Antibody 17b binding at the coreceptor site weakens the kinetics of the interaction of envelope glycoprotein gp120 with CD4. Biochemistry 40:1662-1670. [DOI] [PubMed] [Google Scholar]