

Abstract
Rotaviruses are characterized by polarized release from the apical side of infected enterocytes, and the rotavirus VP4 spike protein specifically binds to the actin network at the apical pole of differentiated enterocytic cells. To determine the functional consequences of this VP4-actin interaction, fluorescence recovery after photobleaching experiments were carried out to measure the diffusional mobility of VP4 associated with the microfilaments. Results show that VP4 binds to barbed ends of microfilaments by using actin treadmilling. Actin treadmilling inhibition results in the loss of rotavirus apical preferential release, suggesting a major role for actin in polarized rotavirus release.
Rotaviruses belong to the Reoviridae family and are a major cause of infantile viral diarrhea worldwide. The nonenveloped particles contain a segmented double-stranded RNA genome, and rotaviruses are characterized by their enteric tropism. In differentiated enterocytic cells, virions reach the apical pole through an atypical trafficking pathway where they are released without cell lysis (6). Our previous data also show that the spike protein VP4 specifically interacts with and strongly remodels actin bundles of the brush border in both infected and transfected polarized Caco-2 cells (5). Since the structural VP4 protein is located at the most peripheral position of the virus, it could support molecular signals for virus targeting. In the present work, we hypothesized that apical interaction between VP4 and actin may account for the apical release of progeny virions. We found that VP4 interacts with actin through a treadmilling process. Blocking treadmilling with jasplakinolide resulted in random release of rotavirus in polarized intestinal cells.
Analysis of the dynamics of the association of the protein EGFP-VP4 with actin microfilaments.
In order to determine the dynamics of VP4 association with the actin cytoskeleton, fluorescence recovery after photobleaching (FRAP) experiments were performed. Cos-7 cells were transiently transfected with a plasmid encoding enhanced green fluorescent protein (EGFP)-VP4 as previously described (5). Twenty-four hours later, living, EGFP-VP4-expressing Cos-7 cells were observed by confocal microscopy (5). Figure 1 illustrates how a subset of EGFP-VP4-labeled filaments may be photobleached by a focusing high-intensity blue laser light flash. EGFP-VP4 fluorescence recovery was monitored by low-power laser excitation, and the data obtained show that EGFP-VP4 mobility on the actin filament or from the cytoplasm to the filament is rather slow, i.e., 0.4 μm/min as calculated from data recorded from the experiments displayed in Fig. 1 and 2.
FIG. 1.

EGFP-VP4 FRAP experiment on the actin cytoskeleton. Cos-7 cells were transiently transfected with a plasmid encoding EGFP-VP4. At 24 h posttransfection, living Cos-7 cells expressing EGFP-VP4 were observed with a confocal microscope. A part of a EGFP-VP4-labeled filament was photobleached by high-power laser excitation (the photobleached region is indicated by a white arrow). EGPF-VP4 fluorescence recovery was observed for 20 min after photobleaching. Shown are projections of 0.3-μm x-y focal sections taken throughout the height of a VP4-EGFP-expressing cell. EGFP-VP4 fluorescence images were acquired 2 min before photobleaching (Tb − 2 min) and then at 1 min (Tb + 1 min), 6 min (Tb + 6 min), 12 min (Tb + 12 min), 15 min (Tb + 15 min), and 20 min (Tb + 20 min) after photobleaching. Scale bar = 10 μm. This figure is representative of three independent experiments.
FIG. 2.

(A) Quantification of EGFP-VP4 FRAP experiments on the actin cytoskeleton. Quantification was obtained from the measurement of fluorescence intensity from fluorescence image data with METAMORPH software. The analyzed data are from experiments similar to the one presented in Fig. 1. Shown is EGPF-VP4 fluorescence recovery in the photobleached region expressed as a percentage compared of the fluorescence before photobleaching. Fluorescence intensities were standardized by comparison to fluorescence intensities from an unphotobleached part of the cell in order to correct the spontaneous photobleaching which occurs even with low-power laser excitation during image acquisition. Displayed are untreated cells (black line), cells treated for 30 min with 20 μM ML-7 (dotted line), and cells treated for 30 min with 1 μM jasplakinolide (discontinuous dotted line). Data are means ± the standard errors of the means from three independent experiments. (B) To monitor the efficiency of ML-7 treatment, Cos-7 cell lysates were analyzed by Western blotting with anti-diphosphorylated myosin light chain (pp-mlc) from Cell Signaling or anti-myosin light chain (mlc) from Sigma Aldrich. Lane C, control cell lysate; lane DPV, lysate from cells treated for 30 min with 10 μM diperoxovanadate (DPV); lane ML-7 DPV, lysate from cells treated for 30 min with 10 μM DPV and 20 μM ML-7. DPV was prepared as described in reference 1.
Displacement of molecules along actin filaments frequently depends on myosin II motor activity. To establish if the mobility of EGFP-VP4 is dependent on such a motor activity, FRAP experiments were performed with living, EGFP-VP4-expressing Cos-7 cells left untreated or treated for 30 min with 20 μM ML-7, an inhibitor of myosin II phosphorylation. As shown in Fig. 2, ML-7 did not influence EGPF-VP4 mobility on the actin cytoskeleton.
The efficiency of ML-7 treatment was checked by testing the phosphorylation state of myosin II by Western blotting as shown in Fig. 2B. Cells were again treated with ML-7 and with 10 mM of diperoxovanadate, an inducer of myosin II phosphorylation. The efficiency of the inhibition of myosin II phosphorylation with ML-7 confirmed that VP4 does not use myosin II motors to progress along the actin network. These experiments show that VP4 is specifically tethered to actin bundles with high recovery and reduced mobility, a result in agreement with the low observed diffusional mobility compared to the rapid mobility of myosin II (2 to 7 μm/s) (7).
The mobility of EGPF-VP4 on the actin cytoskeleton is sensitive to jasplakinolide.
Another possible mechanism supporting protein mobility on actin results from actin treadmilling, a process in which actin filament length remains approximately constant but actin monomers preferentially join with the barbed ends and dissociate from the pointed ends of filaments. This oriented renewal of actin within microfilaments causes a treadmilling involving both actin monomers and actin-binding proteins. Jasplakinolide is a cyclic peptide isolated from a marine sponge which binds to and stabilizes filamentous actin, inducing a blockade of actin treadmilling. Since the lateral mobility speed of EGFP-VP4 is compatible with an actin treadmilling process, FRAP experiments were performed with cells treated with jasplakinolide. In the meantime, cell treatment with 1 μM jasplakinolide for 30 min did not modify the steady-state appearance of actin bundles (not shown).
As shown in Fig. 2, the recovery of EGFP-VP4 fluorescence was reduced by jasplakinolide treatment of living, EGFP-VP4-expressing Cos-7 cells, suggesting that EGFP-VP4 is an F-actin-binding protein that undergoes actin treadmilling to progress along the actin network.
Note that VP4 mobility is quite compatible with an actin treadmilling velocity of about 0.35 μm/min (4). It could be hypothesized that the inhibition of VP4 mobility would be linked to a competition of VP4 and jasplakinolide for actin microfilament binding rather than to the effect of jasplakinolide on actin treadmilling. However, jasplakinolide has the same actin-binding site as phalloidin, another actin-binding drug (2), and we previously revealed that in Cos-7 cells, VP4 and phalloidin are not competitive for F-actin binding, even in the presence of a large amount of VP4 (5).
Apical release of rotavirus is sensitive to jasplakinolide.
The above-described effect of jasplakinolide on actin-mediated VP4 mobility was studied in the context of rotavirus infection of intestinal Caco-2 cells. It was verified that neither rotavirus infection nor jasplakinolide altered the integrity of tight epithelial Caco-2 cell monolayers grown on membrane filters. In the presence of rotavirus and/or jasplakinolide for 18 h, cell polarity and tight junctions remained unaffected, as shown by the stability of transepithelial electrical resistance (control cells, 210 ± 32 Ω/cm2; rotavirus-infected cells, 206 ± 35 Ω/cm2; jasplakinolide-treated rotavirus infected cells, 200 ± 19 Ω/cm2). Moreover, the lactate dehydrogenase enzymatic activity remained intracellular (96% ± 1.5% of the total activity), indicating an absence of significant cell lysis.
The efficiency of jasplakinolide treatment of Caco-2 cells was monitored by measuring high-density actin polymers. Caco-2 cells left untreated or treated with 1 μM jasplakinolide for 16 h were subjected to ice-cold Triton X-100 detergent extraction, and then cell lysates were analyzed by sucrose density gradient sedimentation as previously described (3). Sucrose density gradients were recovered in 12 fractions from the top to the bottom, the 13th fraction corresponding to the resuspended pellet. The actin content of each fraction was analyzed by Western blotting. As expected, jasplakinolide treatment induced a stabilization of high-density actin polymers, as shown by the high actin content of the pellet (Fig. 3).
FIG. 3.

Jasplakinolide effect on actin polymerization in infected Caco-2 cells. Caco-2 cells were grown until differentiation and infected with RF strain bovine rotavirus at a multiplicity of infection of 10 PFU/cell. From 2 h postinfection, cells were left untreated or treated with 1 μM jasplakinolide. Infection was carried out for 18 h. Caco-2 cells were subjected to 1% ice-cold Triton X-100 extraction and ultracentrifugation in a sucrose density gradient as previously described (3). Actin contents of sucrose density gradient fractions and pellet were analyzed by Western blotting. High-density actin polymers were recovered in the pellet fraction (C).
In order to test if actin-based VP4 mobility is essential for virus production, infectious particles released in the presence of jasplakinolide were quantified. Filter-grown, differentiated Caco-2 cells were infected with RF strain rotavirus (5). Virus release from cells left untreated or treated for 16 h with 1 μM jasplakinolide (drug treatment was started at 2 h postinfection) was quantified at 18 h postinfection. As shown in Fig. 4, jasplakinolide strongly perturbed rotavirus release from Caco-2 cells, since the drug was able to cause a loss of polarized rotavirus release.
FIG. 4.
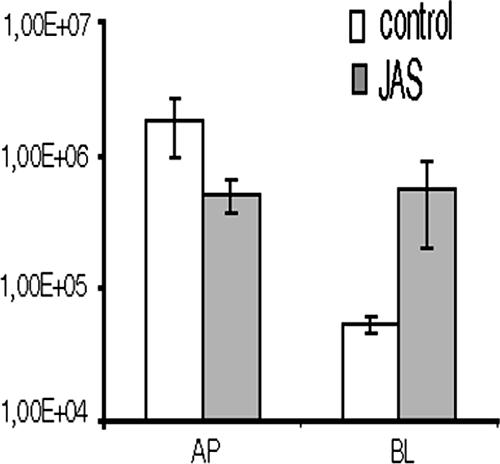
Apical secretion of rotavirus from infected differentiated Caco-2 cells is affected by jasplakinolide (JAS). Caco-2 cells were filter grown, infected, and treated with 1 μM jasplakinolide as described in the legend to Fig. 3. Apical and basolateral (AP, BL) infectious-virus release was quantified. Collected virus was frozen, thawed, and then assayed for virus titration by plaque assay on MA104 cells. The histogram displays the mean values of infectious virus ± the standard deviations from three independent experiments.
The present experiments are the first to explore the functional consequences of VP4 interaction with actin. The results indeed show that association of VP4 with microfilaments is sensitive to the renewal of actin and that VP4 binding to microfilaments is initiated from barbed ends upon renewal of actin monomers. This means that the actin cytoskeleton likely plays an important role in the exocytosis process of progeny virions. Since the total virus production from jasplakinolide-treated or untreated Caco-2 cells remained unchanged, actin-based VP4 mobility does not appear to directly control assembly of VP4 with immature virions. In the meantime, actin treadmilling seems to be essential to ensure polarized release of rotavirus.
Footnotes
Published ahead of print on 14 February 2007.
REFERENCES
- 1.Borbiev, T., S. Nurmukhambetova, F. Liu, A. D. Verin, and J. G. Garcia. 2000. Introduction of C3 exoenzyme into cultured endothelium by Lipofectamine. Anal. Biochem. 285:260-264. [DOI] [PubMed] [Google Scholar]
- 2.Bubb, M. R., A. M. Senderowicz, E. A. Sausville, K. L. Duncan, and E. D. Korn. 1994. Jasplakinolide, a cytotoxic natural product, induces actin polymerization and competitively inhibits the binding of phalloidin to F-actin. J. Biol. Chem. 269:14869-14871. [PubMed] [Google Scholar]
- 3.Delmas, O., A.-M. Durand-Schneider, J. Cohen, O. Colard, and G. Trugnan. 2004. Spike protein VP4 assembly with maturing rotavirus requires a postendoplasmic reticulum event in polarized caco-2 cells. J. Virol. 78:10987-10994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fukui, Y., T. Kitanishi-Yumura, and S. Yumura. 1999. Myosin II-independent F-actin flow contributes to cell locomotion in Dictyostelium. J. Cell Sci. 112(Pt. 6):877-886. [DOI] [PubMed] [Google Scholar]
- 5.Gardet, A., M. Breton, P. Fontanges, G. Trugnan, and S. Chwetzoff. 2006. Rotavirus spike protein VP4 binds to and remodels actin bundles of the epithelial brush border into actin bodies. J. Virol. 80:3947-3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jourdan, N., M. Maurice, D. Delautier, A. M. Quero, A. L. Servin, and G. Trugnan. 1997. Rotavirus is released from the apical surface of cultured human intestinal cells through nonconventional vesicular transport that bypasses the Golgi apparatus. J. Virol. 71:8268-8278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Veigel, C., L. M. Coluccio, J. D. Jontes, J. C. Sparrow, R. A. Milligan, and J. E. Molloy. 1999. The motor protein myosin-I produces its working stroke in two steps. Nature 398:530-533. [DOI] [PubMed] [Google Scholar]