

Abstract
The APOBEC3 family of mammalian cytidine deaminases, including APOBEC3G (A3G), has been shown to function as innate antiviral factors against retroviruses and can also suppress the replication of the hepatitis B virus (HBV). The mechanism by which A3G inhibits HBV replication remains to be elucidated. In this study, we show that the inhibitory effect of APOBEC3 proteins on HBV replication was mainly at the DNA level, with only a minor effect on viral RNA packaging. The anti-HBV effect of A3G was independent of the DNA-editing function, and the mode of inhibition was not due to HBV DNA degradation. The editing-independent antiviral activity of A3G could target DNA-RNA hybrids as well as single-stranded DNA. Finally, we show that there was a preferential decrease in the accumulation of longer minus-strand DNA by A3G, compared to the shorter minus-strand DNA, and suggest that A3G exerts its inhibitory effect at very early stages during viral reverse transcription.
Hepatitis B virus (HBV) is a major cause of acute and chronic viral hepatitis worldwide (12). An estimated 350 million individuals are chronically infected with HBV; many of these will eventually acquire severe liver diseases, including liver cirrhosis and hepatocellular carcinoma, one of the most common forms of human cancer. HBV belongs to the family Hepadnaviridae, which also includes viruses that infect other mammalian and avian species (e.g., duck HBV [DHBV]). All hepadnaviruses contain a small (ca.-3.2-kb), partially double-stranded (DS), relaxed-circular (RC) DNA genome and replicate this DNA through reverse transcription of an RNA intermediate, the pregenomic RNA (pgRNA) (13, 42, 47), and thus are classified as pararetroviruses. Replication of the DNA genome begins with the encapsidation of the pgRNA and the virally encoded reverse transcriptase (RT) into an immature nucleocapsid formed by the viral core or capsid protein. The immature nucleocapsid then undergoes a process of maturation whereby the RT catalyzes first the synthesis of the single-stranded (SS), minus-strand DNA from the pgRNA template and second the synthesis of the plus-strand DNA from the minus-strand template, resulting in the formation of the characteristic mature DS, RC DNA.
APOBEC3 is a member of the APOBEC family of cytidine deaminases that also includes APOBEC1 and -2 and the activation-induced deaminase (17, 24). In humans, the APOBEC3 subfamily has been expanded to include eight members, APOBEC3A to -H, which lie in tandem on chromosome 22 (17, 24). Recently, it was reported that APOBEC3G (A3G) possesses antiviral activity against a broad range of retroviruses (17). In particular, it decreases the infectivity of the human immunodeficiency virus (HIV) (43), and its effect is counteracted by the virion infectivity factor (Vif) (44). In the presence of Vif, A3G is targeted for degradation (35, 44, 46). However, in the absence of Vif, A3G was found to be packaged into HIV virions during viral assembly (1, 28) and to exert its antiviral effect during reverse transcription. A3G deaminates dC of the viral minus-strand DNA, resulting in extensive C/G-to-T/A hypermutation in the viral genome (16, 31). Other members of the APOBEC3 family, including APOBEC3F (A3F) and, to a somewhat lesser extent, APOBEC3B, display similar antiviral activity (57, 59). Moreover, the DNA-editing function of these deaminases has been shown to block the replication of a broad range of other retroviruses (8, 33, 57).
The known specificity of A3G on SS DNA suggested that it might inhibit any virus with an SS DNA intermediate, such as the pararetrovirus HBV. Indeed, it was recently shown by Turelli et al. that A3G could block HBV DNA replication (52). The in vivo significance of this observation was strengthened by recent reports showing that the normally low expression levels of A3G, as well as several other APOBEC family members, in human hepatoma cell lines and primary human hepatocytes can be strongly enhanced by stimulation with the antiviral cytokine alpha interferon (3, 51). In addition, evidence has been presented recently that suggests that the HBV DNA is edited in vivo during natural infections, albeit at a low level (37, 48). A surprising result initially obtained with studies of HBV was that the inhibition of HBV DNA replication was independent of the catalytic activity of A3G (52). Subsequently, the same editing-independent antiviral function of A3G has been extended to other retroviruses as well (7, 8, 36). At present, the mechanism of this deamination-independent antiviral function of A3G remains an enigma.
We have analyzed the mechanisms of suppression of HBV replication by A3G. We show that the deamination-independent inhibition of HBV DNA by A3G was not a result of enhanced viral DNA degradation. Rather, A3G seemed to inhibit very early steps in viral reverse transcription and to block DNA strand elongation. Furthermore, in contrast to the strict editing specificity for SS DNA by A3G, we show that its deamination-independent antiviral function can also target a DNA-RNA hybrid.
MATERIALS AND METHODS
Cell culture.
The human hepatoblastoma cell line HepG2 was cultured in RPMI. Huh7, a human heptatocelluar carcinoma cell line; 293T, a human embryonic kidney cell line; and LMH, a chicken hepatoma cell line, were cultured in Dulbecco modified Eagle medium-F-12 medium. All cells were maintained at 37°C in complete medium supplemented with 10% fetal bovine serum.
Plasmids.
pCMV-HBV contains the wild-type (WT) HBV 1.1-mer overlength genomic sequence driven by the cytomegalovirus (CMV) promoter (11). pCMV-Y63D, a derivative of pCMV-HBV, harbors a mutation (Y63D) (30) within the terminal protein domain of the RT that abolishes viral DNA synthesis without affecting pgRNA packaging. pCMV-HBV RH− is derived from pCMV-HBV and contains a singe point mutation within the RNase H (RH) domain (D737V) of the RT gene that prevents the degradation of RNA during reverse transcription, leading to the synthesis of DNA-RNA hybrids (39). pCMV-DHBV encodes the entire DHBV pgRNA sequences under the control of the CMV promoter (55). pA3G-Flag is an SRα-driven expression vector carrying the human A3G gene fused to the Flag epitope tag at its amino terminus. The V5-tagged human A3C-, A3F-, A3G-, murine ABOBEC3 (mA3)-, and human APO1-expressing plasmids (59) were obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH. The A3G mutant-expressing plasmids (E259Q, C288S, and C291S) (36) were kindly provided by Ann Sheehy and Michael Malim (King's College London, London, United Kingdom).
Transient transfection.
HepG2 cells were seeded in 60-mm dishes and transfected at 30 to 40% confluency with 4 μg of DNA plasmids using FuGENE6 (Roche). Huh7, LMH, and 293T cells were seeded in 60-mm dishes and transfected at 70% confluency with 10 μg of DNA plasmids using the CalPhos Mammalian Transfection kit (Clontech). Cells were harvested on day 5 posttransfection for DNA analysis and RNA packaging assay.
Southern and Western analyses.
Extraction of HBV DNA replicative intermediates and Southern blotting were performed as previously described (19, 55) with the following modification. Cellular lysates were treated with 60 U of micrococcal nuclease and 5 mM CaCl2 at 37°C for 30 min. Subsequently, an additional 60 U of micrococcal nuclease was added and the mixture was incubated further for another 30 min. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting were conducted using a portion of the cytoplasmic extract that was used for core DNA isolation (19, 55). The rabbit anti-HBV core was from DAKO, rabbit anti-A3G was from the NIH (27, 34, 46), and the rabbit anti-V5 and mouse anti-Flag were from Sigma.
RNA packaging assay by native agarose gel analysis of HBV nucleocapsids.
HBV RNA in cytoplasmic core particles, obtained from the postnuclease lysates (above), was detected by resolving the nucleocapsid particles on agarose gels followed by Southern blot analysis using a 32P-labeled antisense riboprobe (covering nucleotides 1991 to 1803) to detect specifically the packaged pgRNA (18, 55, 56). However, a short DNA probe was used in the packaging assay shown in Fig. 3, as described in its legend. The amount of assembled capsid particles was determined by subsequent reprobing of the same membrane using the anti-HBV core antibody.
FIG. 3.

APOBEC3 proteins did not inhibit packaging of unspliced pgRNA. (A) Structures of the unspliced and spliced HBV pgRNA. ɛ, RNA packaging signal (20, 25); AAA, poly(A) tail. (B to D) The HBV Y63D polymerase mutant was cotransfected into HepG2 (B and C) and Huh7 (D) cells with APOBEC-expressing plasmids. Cells were lysed, and pgRNA packaging into nucleocapsids was measured by native agarose gel analysis as described for Fig. 2, using a probe (as depicted in panel A) that recognizes the region (nucleotides 2426 to 2840) of the pgRNA that is absent from the spliced pgRNAs. In panel C, nucleocapsids in the HepG2 cell lysate were treated with (lanes 3 and 4) or without (lanes 1 and 2) micrococcal nuclease (MNase) before analysis of pgRNA packaging. Levels of RNA packaging were normalized to the corresponding amount of assembled nucleocapsids, and the average normalized RNA packaging levels (RNA/Capsid) are reported.
EPR.
The endogenous polymerase reaction (EPR) was performed using nucleocapsids that were precipitated with polyethylene glycol from transfected cellular lysate (15, 55). Each sample (10 μl of capsid) was treated with a final concentration of 15 U micrococcal nuclease and 5 mM CaCl2 for 30 min at 37°C in a total volume of 21.2 μl. To the nuclease-treated capsids, 8.8 μl reaction mix was added, which contained 3 μl 10× endogenous polymerase buffer (500 mM Tris-HCl, pH 7.5, 100 mM MgCl2, 1% NP-40, 1% β-mercaptoethanol), 1.5 μl 200 mM EGTA, 0.3 μl deoxynucleoside triphosphate-dCTP stock (10 mM each dATP, dGTP, and TTP), 3 μl distilled water, and 1 μl [32P]dCTP (3,000 Ci/mmol; 10 mCi/ml). The reaction mixtures were incubated at 37°C for 30 min followed by the addition of 1 μM cold dCTP. The reaction mixtures were further incubated for the indicated amount of time. At each time point, 5 mM phosphonoformic acid (PFA) was added to arrest further DNA synthesis and the reaction mixtures were incubated at 37°C for an additional 0, 6, and 12 h to allow for DNA degradation. To isolate viral DNA from the core particles, the reactions were treated with 10 mM EDTA, 0.5% SDS, and 0.5 μg/μl proteinase K for 2 h at 50°C. The viral DNA was passed through a Sephadex G-50 QuickSpin column (Roche) to purify the radiolabeled DNA, which was then separated on a 1.5% agarose gel. The gel was washed twice in 2× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) for 15 min and dried at 70°C for 1.5 h, and the labeled DNA products were then detected by autoradiography.
In vivo time course of HBV DNA synthesis and degradation.
HepG2 cells were cotransfected with pCMV-HBV and the A3G-expressing plasmid. Six hours posttransfection, the culture medium was replaced with fresh medium containing a final concentration of 5 mM PFA. The transfected cells were treated with PFA for a total of 3 days with the PFA-containing medium being replaced daily. PFA was then washed off the cells, and fresh medium without PFA was added. The cells were then incubated at 37°C for various times as indicated to allow DNA synthesis. Subsequently, PFA was added back to arrest further DNA synthesis and the cells were cultured for another period of time to allow for the degradation (degradation period) of the presynthesized DNA. The cells were then harvested for core DNA isolation as described above.
RESULTS
Multiple APOBEC3 proteins decreased the levels of HBV core DNA.
To determine which members of the APOBEC3 family can inhibit HBV replication, the human hepatoma cell line HepG2 was cotransfected with HBV- and APOBEC3-expressing plasmids and HBV DNA levels in the cytoplasmic nucleocapsids were analyzed by Southern blotting. Cells expressing the human A3G and A3F and, to a lesser extent, A3C and mA3 had decreased levels of HBV DNA (Fig. 1A). When normalized to the core protein levels, HBV DNA levels in the A3G- and A3F-expressing cells were decreased about sixfold compared to the control cells, while cells expressing A3C and mA3 had somewhat lower effects, inhibiting replication by about threefold (Fig. 1C). In contrast, human APOBEC1, which does not inhibit retrovirus replication (2, 9, 59), also had no effect on HBV replication (Fig. 1 and data not shown) and was used as a negative control.
FIG. 1.

Inhibition of HBV replication by APOBEC3 proteins in HepG2 cells. (A) HepG2 cells were cotransfected with HBV- and APOBEC-expressing plasmids. HBV replicative intermediates were isolated from cytoplasmic nucleocapsids and analyzed by Southern blotting using a radiolabeled HBV DNA (top). The APOBEC1 (APO1)-V5 and Flag plasmids were used as a control for the V5-tagged and Flag-tagged APOBEC3 constructs, respectively. SDS-PAGE and Western blot analysis were used to detect expression of HBV core protein (middle) and APOBEC proteins (bottom). The presence of APOBEC proteins was detected using either anti-V5 epitope (lanes 1 to 5) or anti-A3G antibody (lanes 6 and 7). (B) The HBV polymerase mutant Y63D, which is able to package pgRNA but unable to synthesize DNA, was cotransfected into HepG2 cells along with APOBEC3-expressing plasmids. Cytoplasmic nucleocapsids were extracted from transfected cells, and capsid-associated RNA levels were determined by native agarose gel analysis. pgRNA was detected by using a riboprobe specific for the 5′ end of the plus strand (top). Capsids were detected by probing the same membrane with anti-HBV core protein antibody (bottom). (C) Normalization of HBV DNA (black bars) or RNA (dotted bars) levels to core protein expression or capsid assembly, respectively. Error bars show standard deviations.
The inhibitory effect of A3G on HBV DNA levels was not restricted to HepG2, since it could also be seen in another human hepatoma cell line, Huh7 (Fig. 2A), and the human embryonic kidney cell line 293T (data not shown). The more dramatic reduction (by 18-fold) of HBV DNA by A3G in Huh7 cells (Fig. 2C) compared to HepG2 cells was correlated with a higher expression level of A3G in Huh7 cells (data not shown), consistent with a dose-dependent effect of A3G in both cell lines as also reported recently (41).
FIG. 2.
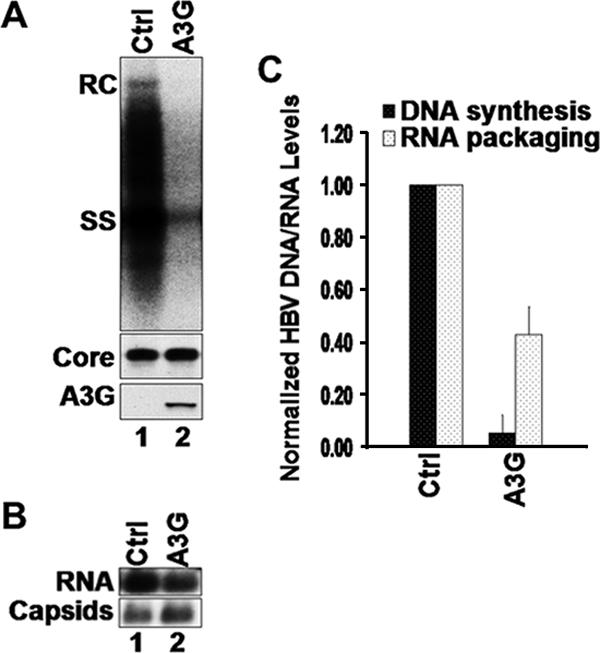
Inhibition of HBV replication by A3G in Huh7 cells. (A) Huh7 cells were cotransfected with HBV- and A3G-expressing plasmids. HBV replicative intermediates were isolated and analyzed by Southern hybridization (top). HBV core protein (middle) and A3G protein (bottom) expression was analyzed by SDS-PAGE and Western blotting with HBV core-specific and A3G-specific antibody, respectively. (B) Huh7 cells were cotransfected with Y63D- and A3G-expressing plasmids. Extraction of cytoplasmic nucleocapsids and analysis of capsid-associated RNA levels were performed as described for Fig. 1. (C) Normalization of HBV DNA (black bars) or RNA (dotted bars) levels to core protein expression or capsid assembly, respectively. Error bars show standard deviations. Ctrl, control.
APOBEC3 proteins had little effect on pgRNA packaging.
To clarify the effect of A3G on RNA packaging without the complication of viral DNA, we used an HBV polymerase mutant, Y63D, which is able to package pgRNA but incapable of viral DNA synthesis (30). We cotransfected HepG2 and Huh7 cells with the Y63D-expressing HBV plasmid along with the APOBEC3-expressing plasmids. Cytoplasmic nucleocapsids were isolated from transfected cells, and core-associated RNA levels were determined by a native agarose gel-based RNA packaging assay, which can measure the levels of assembled nucleocapsids, as well as the amount of pgRNA packaged in the nucleocapsids on the same blot (18, 56). Under conditions where intracellular HBV DNA levels were significantly reduced in HepG2 cells that expressed the APOBEC3 proteins (Fig. 1A and C), pgRNA encapsidation was not or only slightly inhibited (less-than-twofold decrease) (Fig. 1B and C). In Huh7 cells, A3G had a somewhat greater inhibitory effect on RNA packaging but the decrease was still only about 2.5-fold (Fig. 2B and C). This minor effect of A3G on RNA packaging could not account for the dramatic (18-fold) reduction in HBV DNA levels observed in Huh7 cells.
Similar to our findings, a recent report also suggested that A3G does not reduce the levels of pgRNA packaged but rather somehow induces nuclease sensitivity of the full-length, but not a spliced, pgRNA packaged into nucleocapsids (41). To ascertain whether the packaging of the full-length pgRNA was in fact reduced by A3G, we used a probe corresponding to the spliced-out region of the pgRNA, in our packaging assay, to specifically detect the full-length, but not the spliced, pgRNA associated with the nucleocapsids. As shown in Fig. 3, the pgRNA packaging signals detected with this specific probe were also not significantly affected by A3G in either HepG2 (Fig. 3B) or Huh7 (Fig. 3D) cells. Furthermore, the levels of full-length pgRNA packaged, as detected using this probe, were not affected by nuclease digestion of the nucleocapsids, either in the presence or in the absence of A3G. Taken together, these data indicate that the slight inhibition of pgRNA packaging contributed little to the overall decrease in the amount of HBV DNA and that A3G and other APOBEC3 family members mainly act at the viral DNA level, i.e., resulting in either a decrease in viral DNA synthesis or an increase in its degradation.
Inhibition of HBV replication was independent of A3G deaminase activity.
To further examine the mechanism of inhibition by A3G of HBV DNA synthesis, we tested the requirement for its deaminase function. It has been shown that the C-terminal domain of A3G harbors the deamination catalytic center (22). As with the other cytidine deaminases, conserved Zn2+ coordinating Cys residues and a catalytic Glu residue are essential for deaminase activity (22, 32, 38). Three A3G mutants were chosen to test the role of deamination in suppressing HBV replication. The C288S and C291S mutations eliminated the conserved Cys residues required for Zn2+ coordination while the E259Q mutation eliminated the catalytic Glu residue. These mutants have been shown previously to be catalytically inactive and display no cytidine deamination activity in vitro or in vivo but remain competent for nucleic acid binding and viral incorporation (23, 36). In agreement with the previous report (52), HBV DNA replication was still suppressed by all three catalytically inactive A3G mutants to the same extent as the WT A3G, in both HepG2 and Huh7 cells (Fig. 4). Again, the levels of RNA packaged were not significantly affected by the A3G mutants, as was the case with the WT A3G (data not shown). These results thus clearly indicated that the antiviral effect of A3G on HBV replication functioned independently of its deaminase activity.
FIG. 4.
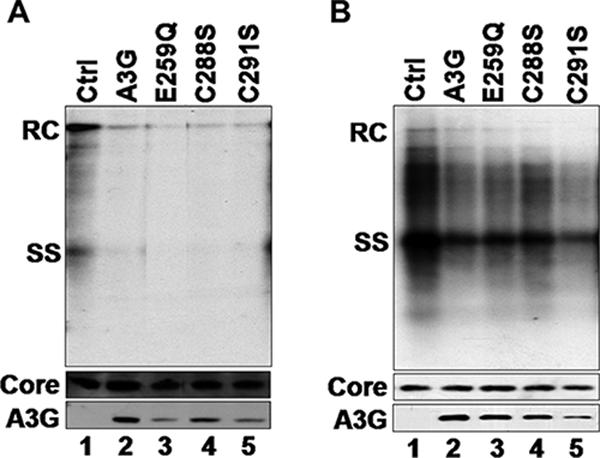
Inhibition of HBV replication by catalytic A3G mutants. HepG2 (A) and Huh7 (B) cells were cotransfected with HBV- and catalytically inactive mutant A3G-expressing plasmids. HBV replicative intermediates were isolated from cytoplasmic nucleocapsids and were analyzed by Southern blotting using a radiolabeled HBV DNA (top). HBV core protein (middle) and A3G protein (bottom) expression was analyzed by SDS-PAGE and Western blotting with HBV core-specific antibody and A3G-specific antibody, respectively. Ctrl, control.
A3G did not enhance HBV DNA degradation or block late stages of DNA synthesis.
To determine if A3G enhanced the degradation of HBV DNA, we first allowed viral DNA synthesis and then looked for evidence of degradation of the different species of DNA that had been already synthesized, in the absence of further DNA synthesis. Thus, transfected HepG2 cells were treated with PFA, a reversible inhibitor of reverse transcription, immediately after transfection. Since PFA blocked reverse transcription without affecting pgRNA packaging, immature, pgRNA-containing nucleocapsids accumulated, with no viral DNA synthesis (21). Three days later, PFA was removed to allow for DNA synthesis. After a period of DNA synthesis for accumulation of increasingly mature viral DNA, PFA was then added back to arrest further DNA synthesis. The cells were cultured for an additional period of time to allow for the putative degradation, if any, of the presynthesized DNA (Fig. 5A). As shown in Fig. 5B, the amounts of viral DNA, either the SS intermediate or the mature RC DNA, were similar at 0, 6, and 12 h after the start of the second PFA treatment (degradation period), in either control (lanes 1 to 6) or A3G-transfected (lanes 7 to 12) cells, indicating that A3G did not enhance HBV DNA degradation.
FIG. 5.

A3G did not enhance HBV DNA degradation or block late DNA synthesis in vivo. (A) In vivo time course. Transfected HepG2 cells were treated with 5 mM PFA for a period of 3 days to allow for the accumulation of pgRNA-containing nucleocapsids. Cells were then washed free of PFA, and DNA synthesis within the nucleocapsids was allowed to proceed for 3 or 24 h. DNA degradation was determined by the readdition of PFA for 0, 6, and 12 h. (B) Southern blot analysis of HBV intracellular nucleocapsid DNA from the in vivo time course. Viral DNA was extracted from nucleocapsids that had synthesized viral DNA for 3 or 24 h (DNA synthesis) or after the second PFA treatment period (DNA degradation) and was detected by Southern blot analysis. Fivefold-more DNA from the A3G samples was used than from the control (Ctrl). Syn, synthesis; Deg, degradation.
It was also apparent from Fig. 5 that the amount of the mature RC DNA relative to that of the SS intermediate was not affected by A3G, suggesting that plus-strand DNA synthesis was not preferentially inhibited by A3G. Instead, A3G may have blocked an earlier step in DNA synthesis (see below). To further analyze the step of HBV DNA synthesis that might be affected by A3G, we isolated immature nucleocapsids, containing mostly SS DNA (viral DNA synthesis in the cell was allowed for 3 h before the nucleocapsids were harvested [Fig. 5]), and subjected them to an EPR, in order to determine if their ability to synthesize viral DNA in vitro was affected by A3G packaged into the capsids (48). In agreement with the in vivo results, A3G did not inhibit the synthesis of mature RC DNA from the SS DNA by these isolated nucleocapsids (Fig. 6, lane 22 versus lane 10). In addition, we also measured any potential degradation of viral DNA in vitro by arresting viral DNA synthesis with PFA, as described above, following the EPR. As observed above in the cells, A3G did not enhance viral DNA degradation in vitro, either during the 6-h period or during the 12-hour period (Fig. 6, particularly lanes 23 and 24 versus lanes 11 and 12).
FIG. 6.
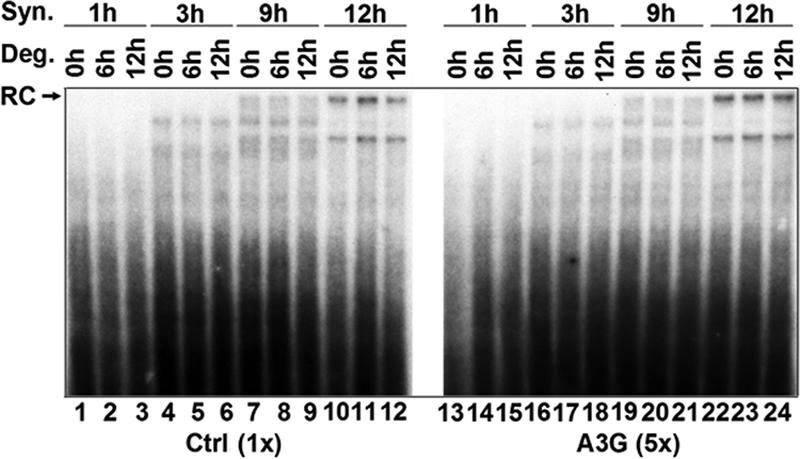
A3G did not enhance DNA degradation or block late HBV DNA synthesis in vitro. Polyethylene glycol-precipitated HBV nucleocapsids were used in an EPR to examine in vitro DNA synthesis in the presence of A3G. Potential DNA degradation of already-synthesized DNA by A3G was determined by the addition of 5 mM PFA to inhibit further HBV DNA synthesis, and nucleocapsids were incubated for an additional 0, 6, and 12 h. Viral DNA was then extracted from the nucleocapsids and separated on an agarose gel, and labeled DNA products were then detected by autoradiography. Fivefold-more DNA from the A3G samples was used than from the control (Ctrl). Syn, synthesis; Deg, degradation.
A3G blocked early steps in viral minus-strand DNA synthesis.
The lack of an effect by A3G on either HBV DNA degradation or the late stages of DNA synthesis suggested that A3G might inhibit viral DNA replication by blocking an early step in viral reverse transcription. Attempts to determine the effect of A3G on the early stage of minus-strand DNA synthesis in vitro using the EPR were inconclusive due to the low efficiency of early viral minus-strand DNA synthesis by the isolated nucleocapsids. As an alternative, we sought to determine if A3G could block an early step in viral minus-strand DNA synthesis in cells. To accumulate nascent viral minus-strand DNA, we elected to use an RH-defective HBV mutant, which contains a single point mutation within the RH domain of the RT gene that prevents RNA degradation during reverse transcription, resulting in a decreased processivity in minus-strand DNA synthesis and accumulation of nascent minus-strand DNA, as well as the absence of any plus-strand DNA (6, 14, 39) (Fig. 7A). Since this mutant synthesized DS DNA-RNA hybrids, it also allowed us to determine if the deaminase-independent antiviral function of A3G would target these hybrids or was strictly dependent on the presence of SS DNA, as is the case with its deaminase activity (4, 23, 49, 58). Intriguingly, A3G could indeed inhibit viral DNA synthesis even in the context of the DNA-RNA hybrids (Fig. 7B). Furthermore, a closer examination of the short nascent minus-strand DNA accumulating in the RH mutant (detected also as minor species in the WT [Fig. 7B]) suggested that A3G might have a smaller effect on these short DNA species, despite the fact that it dramatically reduced the levels of the more mature DNA species in the same cells. To further analyze the nascent minus-strand DNA species, the RNA-DNA hybrids were heat denatured and analyzed by Southern blotting. The results showed that there was indeed a greater reduction by A3G of the levels of longer minus-strand DNA (those longer than approximately 2.0 kb) than of levels of the shorter minus-strand DNA (Fig. 8A and B). Thus, while the longer species V and VI were reduced progressively by A3G (the full-length minus-strand DNA, species VI, was reduced by more than 33-fold), the levels of the short species I to IV were reduced on average by about fivefold. However, the latter results also suggested that there was an even earlier block in viral reverse transcription by A3G, since even the shortest minus strands detected by our assays (ca. 400 bp [Fig. 8, species I]) were already reduced. The same results were obtained with the catalytically inactive A3G mutants (data not shown). In addition, similar results were also obtained with the WT HBV, albeit the levels of the short minus-strand DNA were much lower than those with the RH− mutant. Again, there was an overall reduction in the DNA level, with a preferential inhibition of more mature DNA (data not shown). Thus, these results indicated that (i) A3G could target the DS DNA-RNA hybrids and (ii) A3G may prevent HBV DNA synthesis by inducing early blocks in minus-strand DNA synthesis.
FIG. 7.
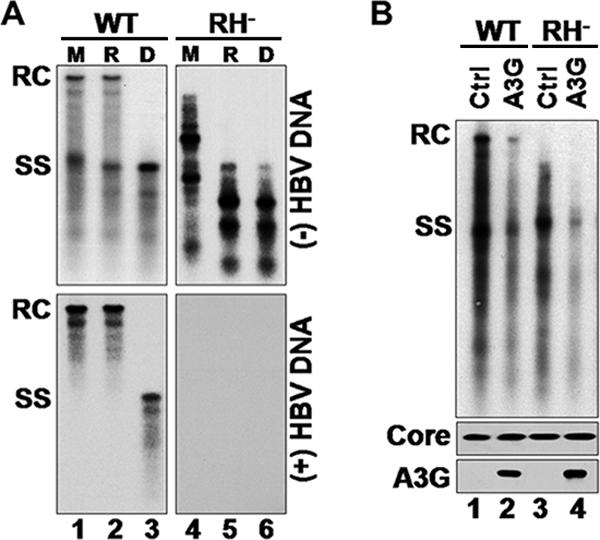
A3G inhibited synthesis of DNA-RNA hybrids. (A) Viral DNA isolated from WT or RH− nucleocapsids, harvested from transfected HepG2 cells, was either mock treated (M), RH treated (R), or heat denatured (D) at 95°C prior to separation on an agarose gel and transferred onto a nylon membrane. The membrane was then probed with a riboprobe specific for minus-strand (top) or plus-strand (bottom) HBV DNA. Note that the RH digestion converted the minus-strand DNA-RNA hybrids (migrating between SS and RC DNA, lane 4) to nascent minus-strand DNA (lane 5) and that there is a complete absence of plus-strand DNA in the RH− mutant. (B) Southern blot analysis of HBV DNA from HepG2 cells transfected with WT or RH-defective (RH−) polymerase mutant together with control (Ctrl) or A3G-expressing plasmid. Viral DNA was extracted from nucleocapsids 5 days posttransfection and analyzed by Southern blotting (top). Shown below are immunoblot assays to detect HBV core protein (middle) and A3G (bottom).
FIG. 8.
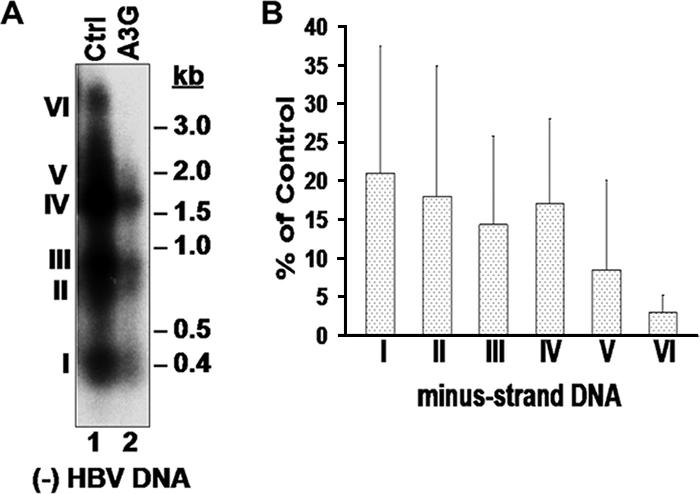
A3G blocked early HBV minus-strand DNA synthesis. (A) HBV DNA isolated from nucleocapsids present in HepG2 cells transfected with RH− HBV and A3G or control (Ctrl). Viral DNA was heat denatured prior to Southern blot analysis and was detected using a minus-strand-specific HBV riboprobe. (B) The relative amounts of minus-strand intermediates, designated I to VI, after normalization to control (percentage of control) are shown. Error bars show standard deviations.
DISCUSSION
The APOBEC3 family of cytidine deaminases has been shown to target a wide range of retroviruses, as well as the pararetrovirus human HBV. In the present study, the effects of the APOBEC3 proteins, in particular A3G, on HBV replication were examined. Our findings showed that A3G, A3F, A3C, and mA3 proteins were able to inhibit HBV DNA replication in two different hepatoma cell lines. Furthermore, the inhibitory effect of APOBEC3 proteins on HBV replication was mainly at the level of viral DNA, with only a minor effect on RNA packaging. The effect of A3G on HBV DNA was clearly independent of its DNA-editing function. Furthermore, the reduction of HBV DNA by A3G was due not to accelerated degradation but rather to very early blocks in viral reverse transcription. Finally, our results showed that the deamination-independent inhibition of HBV DNA synthesis by A3G could also target the DS DNA-RNA hybrid, in contrast to the strict SS DNA specificity of its deaminase activity.
An initial report on the effects of A3G on HBV replication suggested that A3G decreased viral DNA levels by inhibiting pgRNA packaging (52). Subsequently, it was reported that A3G did not block viral RNA packaging per se; rather, the full-length pgRNA packaged in the presence of A3G was somehow rendered sensitive to exogenous nuclease whereas a packaged spliced pgRNA was not affected by A3G (41). In agreement with the latter report, our results showed that A3G, as well as several other APOBEC3 proteins, had only a minor effect or none on pgRNA packaging, which is unlikely to explain its dramatic effect at the DNA level. In an attempt to test the nuclease sensitivity of the packaged viral RNA, we titrated the concentrations of the micrococcal nuclease (over a range of 20-fold) used to digest the cytoplasmic lysate before analysis of pgRNA packaging efficiency (D. H. Nguyen and J. Hu, unpublished results). However, we were unable to confirm the A3G-induced nuclease sensitivity of the packaged full-length pgRNA with any nuclease concentration tested. It remains possible that subtle differences in the exact conditions of nuclease digestion and nucleocapsid isolation might have been responsible for this apparent discrepancy.
Although some evidence has been reported that A3G may induce HBV DNA editing in a small fraction of viral DNA (37, 41, 48), it is clear from our results, as well as those of others (41, 52), that the inhibitory effect of A3G on HBV DNA replication is largely independent of its deaminase activity. Although the DNA deamination function of the APOBEC proteins is well understood, the mechanism of this deamination-independent antiviral function, which also works on other retroviruses and retrotransposons (7, 36, 45), is not yet clear. In principle, the decrease in HBV DNA could result from either an accelerated DNA degradation or a decreased synthesis. It has been suggested that the reduction in retroviral DNA levels by A3G results from the recognition and degradation of the edited (uracil-containing) reverse transcripts by host cellular repair enzymes, such as the uracil DNA glycosylase (UNG), which removes uracil from DNA to generate an abasic site that is subsequently cleaved by the host apurinic/apyrimidinic endonuclease. However, a recent study showed that the loss of UNG did not rescue the levels of viral reverse transcripts that were decreased by A3G (26). We also obtained preliminary results suggesting that UNG activity is not required for the decrease in HBV DNA induced by A3G, since there was no rescue of HBV DNA levels by a bacteriophage UNG inhibitor (Ugi) (10, 26) (Nguyen and Hu, unpublished). Furthermore, attempts to look for enhanced DNA degradation by A3G, either in vivo or in vitro, did not reveal any such effect.
On the other hand, we were able to obtain evidence indicating that A3G may block early stages in viral reverse transcription. While our in vivo and in vitro DNA synthesis assays indicated that A3G did not block the synthesis of late DNA intermediates, as was also reported recently (41), our studies using the RH mutant, which allowed the accumulation of early minus-strand DNA intermediates, demonstrated that A3G showed a stronger inhibitory effect on longer minus-strand DNA than on shorter DNA. This preferential inhibition of longer minus-strand DNA intermediates by A3G was also evident sometimes even with the WT HBV, but it could not be as readily demonstrated because these early intermediates failed to accumulate in the WT virus. An early block in viral DNA synthesis would explain how A3G could reduce HBV DNA without accelerating DNA degradation or its deaminase activity.
The exact mechanism of A3G inhibition of minus-strand DNA synthesis is not yet clear. A3G could block DNA synthesis by inhibiting the polymerase function, or alternatively, it could affect the template function of the pgRNA, e.g., by associating with the viral RNA (4, 50). The reported nuclease sensitivity of the packaged HBV pgRNA induced by A3G (41) would be consistent with the notion that A3G may affect the structure of the pgRNA and, thus, its template function during reverse transcription. The apparent specificity for the synthesis of viral minus-strand, but not plus-strand, DNA by A3G also supports the view that A3G may interfere with the template function of the pgRNA, rather than the polymerase activity per se. The very early block in minus-strand DNA synthesis may suggest that A3G could inhibit the initiation step of minus-strand DNA synthesis, which is primed by the RT protein itself (protein priming) (29, 54), or block minus-strand primer translocation following protein priming (40, 53).
In contrast to the DNA-editing function of A3G, which acts strictly on SS DNA (4, 23, 58), we have shown here that the editing-independent inhibition of viral reverse transcription by A3G could also act on a DS HBV RNA-DNA hybrid. Similar results were obtained with WT and an RH-defective DHBV mutant (S. H. Basagoudanavar and J. Hu, unpublished results). This reinforces the idea that the editing-dependent and -independent antiviral functions of the APOBEC proteins are fundamentally different mechanisms of cellular antiviral defense. Interestingly, A3G did not appear to significantly block HBV second-strand DNA synthesis from the small amounts of minus-strand DNA that escaped A3G inhibition. This suggests that A3G may not be able to function in the context of a DS DNA. In support of this notion, it has recently been shown that certain APOBEC proteins can target SS, but not DS, DNA viruses (5).
In conclusion, our results provide further insights into the antiviral function of A3G. In particular, we have shown that the editing-independent inhibition of HBV replication was not due to viral DNA degradation. Rather, A3G appeared to inhibit very early steps of viral reverse transcription. Additionally, we have shown that A3G clearly inhibited the synthesis of DNA-RNA hybrids as well as SS DNA. The relative ease in detecting and analyzing various reverse transcription intermediates during HBV replication makes it a convenient model system to dissect further the detailed mechanism of A3G-mediated inhibition of reverse transcription.
Acknowledgments
We thank Michael Malim and Ann Sheehy for providing the A3G plasmids. The anti-A3G antibodies were obtained from Warner C. Greene, Jaisri Lingappa, Klaus Strebel, and Sandra Kao; the V5-tagged APOBEC constructs were obtained from B. Matija Peterlin and Yong-Hui Zheng, through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, National Institutes of Health.
This work was supported by a Public Health Service grant, R01 AI43453, from the National Institutes of Health.
Footnotes
Published ahead of print on 21 February 2007.
REFERENCES
- 1.Alce, T. M., and W. Popik. 2004. APOBEC3G is incorporated into virus-like particles by a direct interaction with HIV-1 Gag nucleocapsid protein. J. Biol. Chem. 279:34083-34086. [DOI] [PubMed] [Google Scholar]
- 2.Bishop, K. N., R. K. Holmes, A. M. Sheehy, N. O. Davidson, S. J. Cho, and M. H. Malim. 2004. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr. Biol. 14:1392-1396. [DOI] [PubMed] [Google Scholar]
- 3.Bonvin, M., F. Achermann, I. Greeve, D. Stroka, A. Keogh, D. Inderbitzin, D. Candinas, P. Sommer, S. Wain-Hobson, J. P. Vartanian, and J. Greeve. 2006. Interferon-inducible expression of APOBEC3 editing enzymes in human hepatocytes and inhibition of hepatitis B virus replication. Hepatology 43:1364-1374. [DOI] [PubMed] [Google Scholar]
- 4.Chelico, L., P. Pham, P. Calabrese, and M. F. Goodman. 2006. APOBEC3G DNA deaminase acts processively 3′→5′ on single-stranded DNA. Nat. Struct. Mol. Biol. 13:392-399. [DOI] [PubMed] [Google Scholar]
- 5.Chen, H., C. E. Lilley, Q. Yu, D. V. Lee, J. Chou, I. Narvaiza, N. R. Landau, and M. D. Weitzman. 2006. APOBEC3A is a potent inhibitor of adeno-associated virus and retrotransposons. Curr. Biol. 16:480-485. [DOI] [PubMed] [Google Scholar]
- 6.Chen, Y., and P. L. Marion. 1996. Amino acids essential for RNase H activity of hepadnaviruses are also required for efficient elongation of minus-strand viral DNA. J. Virol. 70:6151-6156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chiu, Y. L., V. B. Soros, J. F. Kreisberg, K. Stopak, W. Yonemoto, and W. C. Greene. 2005. Cellular APOBEC3G restricts HIV-1 infection in resting CD4+ T cells. Nature 435:108-114. [DOI] [PubMed] [Google Scholar]
- 8.Cullen, B. R. 2006. Role and mechanism of action of the APOBEC3 family of antiretroviral resistance factors. J. Virol. 80:1067-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Delebecque, F., R. Suspene, S. Calattini, N. Casartelli, A. Saib, A. Froment, S. Wain-Hobson, A. Gessain, J. P. Vartanian, and O. Schwartz. 2006. Restriction of foamy viruses by APOBEC cytidine deaminases. J. Virol. 80:605-614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Di Noia, J., and M. S. Neuberger. 2002. Altering the pathway of immunoglobulin hypermutation by inhibiting uracil-DNA glycosylase. Nature 419:43-48. [DOI] [PubMed] [Google Scholar]
- 11.Fallows, D. A., and S. P. Goff. 1995. Mutations in the epsilon sequences of human hepatitis B virus affect both RNA encapsidation and reverse transcription. J. Virol. 69:3067-3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ganem, D., and A. M. Prince. 2004. Hepatitis B virus infection—natural history and clinical consequences. N. Engl. J. Med. 350:1118-1129. [DOI] [PubMed] [Google Scholar]
- 13.Ganem, D., and R. J. Schneider. 2001. Hepadnaviridae, p. 2923-2969. In D. M. Knipe and P. M. Howley (ed.), Fields virology. Lippincott Williams & Wilkins, Philadelphia, PA.
- 14.Gerelsaikhan, T., J. Tavis, and V. Bruss. 1996. Hepatitis B virus nucleocapsid envelopment does not occur without genomic DNA synthesis. J. Virol. 70:4269-4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo, J. T., M. Pryce, X. Wang, M. I. Barrasa, J. Hu, and C. Seeger. 2003. Conditional replication of duck hepatitis B virus in hepatoma cells. J. Virol. 77:1885-1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harris, R. S., K. N. Bishop, A. M. Sheehy, H. M. Craig, S. K. Petersen-Mahrt, I. N. Watt, M. S. Neuberger, and M. H. Malim. 2003. DNA deamination mediates innate immunity to retroviral infection. Cell 113:803-809. [DOI] [PubMed] [Google Scholar]
- 17.Harris, R. S., and M. T. Liddament. 2004. Retroviral restriction by APOBEC proteins. Nat. Rev. Immunol. 4:868-877. [DOI] [PubMed] [Google Scholar]
- 18.Hu, J., D. Flores, D. Toft, X. Wang, and D. Nguyen. 2004. Requirement of heat shock protein 90 for human hepatitis B virus reverse transcriptase function. J. Virol. 78:13122-13131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu, J., and C. Seeger. 1996. Hsp90 is required for the activity of a hepatitis B virus reverse transcriptase. Proc. Natl. Acad. Sci. USA 93:1060-1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu, J., and C. Seeger. 1997. RNA signals that control DNA replication in hepadnaviruses. Semin. Virol. 8:205-211. [Google Scholar]
- 21.Hu, J., D. O. Toft, and C. Seeger. 1997. Hepadnavirus assembly and reverse transcription require a multi-component chaperone complex which is incorporated into nucleocapsids. EMBO J. 16:59-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huthoff, H., and M. H. Malim. 2005. Cytidine deamination and resistance to retroviral infection: towards a structural understanding of the APOBEC proteins. Virology 334:147-153. [DOI] [PubMed] [Google Scholar]
- 23.Iwatani, Y., H. Takeuchi, K. Strebel, and J. G. Levin. 2006. Biochemical activities of highly purified, catalytically active human APOBEC3G: correlation with antiviral effect. J. Virol. 80:5992-6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jarmuz, A., A. Chester, J. Bayliss, J. Gisbourne, I. Dunham, J. Scott, and N. Navaratnam. 2002. An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22. Genomics 79:285-296. [DOI] [PubMed] [Google Scholar]
- 25.Junker-Niepmann, M., R. Bartenschlager, and H. Schaller. 1990. A short cis-acting sequence is required for hepatitis B virus pregenome encapsidation and sufficient for packaging of foreign RNA. EMBO J. 9:3389-3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaiser, S. M., and M. Emerman. 2006. Uracil DNA glycosylase is dispensable for human immunodeficiency virus type 1 replication and does not contribute to the antiviral effects of the cytidine deaminase Apobec3G. J. Virol. 80:875-882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kao, S., M. A. Khan, E. Miyagi, R. Plishka, A. Buckler-White, and K. Strebel. 2003. The human immunodeficiency virus type 1 Vif protein reduces intracellular expression and inhibits packaging of APOBEC3G (CEM15), a cellular inhibitor of virus infectivity. J. Virol. 77:11398-11407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khan, M. A., S. Kao, E. Miyagi, H. Takeuchi, R. Goila-Gaur, S. Opi, C. L. Gipson, T. G. Parslow, H. Ly, and K. Strebel. 2005. Viral RNA is required for the association of APOBEC3G with human immunodeficiency virus type 1 nucleoprotein complexes. J. Virol. 79:5870-5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lanford, R. E., L. Notvall, and B. Beames. 1995. Nucleotide priming and reverse transcriptase activity of hepatitis B virus polymerase expressed in insect cells. J. Virol. 69:4431-4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lanford, R. E., L. Notvall, H. Lee, and B. Beames. 1997. Transcomplementation of nucleotide priming and reverse transcription between independently expressed TP and RT domains of the hepatitis B virus reverse transcriptase. J. Virol. 71:2996-3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lecossier, D., F. Bouchonnet, F. Clavel, and A. J. Hance. 2003. Hypermutation of HIV-1 DNA in the absence of the Vif protein. Science 300:1112. [DOI] [PubMed] [Google Scholar]
- 32.MacGinnitie, A. J., S. Anant, and N. O. Davidson. 1995. Mutagenesis of apobec-1, the catalytic subunit of the mammalian apolipoprotein B mRNA editing enzyme, reveals distinct domains that mediate cytosine nucleoside deaminase, RNA binding, and RNA editing activity. J. Biol. Chem. 270:14768-14775. [PubMed] [Google Scholar]
- 33.Mangeat, B., P. Turelli, G. Caron, M. Friedli, L. Perrin, and D. Trono. 2003. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 424:99-103. [DOI] [PubMed] [Google Scholar]
- 34.Mariani, R., D. Chen, B. Schrofelbauer, F. Navarro, R. Konig, B. Bollman, C. Munk, H. Nymark-McMahon, and N. R. Landau. 2003. Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell 114:21-31. [DOI] [PubMed] [Google Scholar]
- 35.Marin, M., K. M. Rose, S. L. Kozak, and D. Kabat. 2003. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat. Med. 9:1398-1403. [DOI] [PubMed] [Google Scholar]
- 36.Newman, E. N., R. K. Holmes, H. M. Craig, K. C. Klein, J. R. Lingappa, M. H. Malim, and A. M. Sheehy. 2005. Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr. Biol. 15:166-170. [DOI] [PubMed] [Google Scholar]
- 37.Noguchi, C., H. Ishino, M. Tsuge, Y. Fujimoto, M. Imamura, S. Takahashi, and K. Chayama. 2005. G to A hypermutation of hepatitis B virus. Hepatology 41:626-633. [DOI] [PubMed] [Google Scholar]
- 38.Prochnow, C., R. Bransteitter, M. G. Klein, M. F. Goodman, and X. S. Chen. 2007. The APOBEC-2 crystal structure and functional implications for the deaminase AID. Nature 445:447-451. [DOI] [PubMed] [Google Scholar]
- 39.Radziwill, G., W. Tucker, and H. Schaller. 1990. Mutational analysis of the hepatitis B virus P gene product: domain structure and RNase H activity. J. Virol. 64:613-620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rieger, A., and M. Nassal. 1996. Specific hepatitis B virus minus-strand DNA synthesis requires only the 5′ encapsidation signal and the 3′-proximal direct repeat DR1. J. Virol. 70:585-589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rosler, C., J. Kock, M. Kann, M. H. Malim, H. E. Blum, T. F. Baumert, and F. von Weizsacker. 2005. APOBEC-mediated interference with hepadnavirus production. Hepatology 42:301-309. [DOI] [PubMed] [Google Scholar]
- 42.Seeger, C., and W. S. Mason. 2000. Hepatitis B virus biology. Microbiol. Mol. Biol. Rev. 64:51-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sheehy, A. M., N. C. Gaddis, J. D. Choi, and M. H. Malim. 2002. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418:646-650. [DOI] [PubMed] [Google Scholar]
- 44.Sheehy, A. M., N. C. Gaddis, and M. H. Malim. 2003. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat. Med. 9:1404-1407. [DOI] [PubMed] [Google Scholar]
- 45.Stenglein, M. D., and R. S. Harris. 2006. APOBEC3B and APOBEC3F inhibit L1 retrotransposition by a DNA deamination-independent mechanism. J. Biol. Chem. 281:16837-16841. [DOI] [PubMed] [Google Scholar]
- 46.Stopak, K., C. de Noronha, W. Yonemoto, and W. C. Greene. 2003. HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol. Cell 12:591-601. [DOI] [PubMed] [Google Scholar]
- 47.Summers, J., and W. S. Mason. 1982. Replication of the genome of a hepatitis B-like virus by reverse transcription of an RNA intermediate. Cell 29:403-415. [DOI] [PubMed] [Google Scholar]
- 48.Suspene, R., D. Guetard, M. Henry, P. Sommer, S. Wain-Hobson, and J. P. Vartanian. 2005. Extensive editing of both hepatitis B virus DNA strands by APOBEC3 cytidine deaminases in vitro and in vivo. Proc. Natl. Acad. Sci. USA 102:8321-8326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Suspene, R., P. Sommer, M. Henry, S. Ferris, D. Guetard, S. Pochet, A. Chester, N. Navaratnam, S. Wain-Hobson, and J. P. Vartanian. 2004. APOBEC3G is a single-stranded DNA cytidine deaminase and functions independently of HIV reverse transcriptase. Nucleic Acids Res. 32:2421-2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Svarovskaia, E. S., H. Xu, J. L. Mbisa, R. Barr, R. J. Gorelick, A. Ono, E. O. Freed, W. S. Hu, and V. K. Pathak. 2004. Human apolipoprotein B mRNA-editing enzyme-catalytic polypeptide-like 3G (APOBEC3G) is incorporated into HIV-1 virions through interactions with viral and nonviral RNAs. J. Biol. Chem. 279:35822-35828. [DOI] [PubMed] [Google Scholar]
- 51.Tanaka, Y., H. Marusawa, H. Seno, Y. Matsumoto, Y. Ueda, Y. Kodama, Y. Endo, J. Yamauchi, T. Matsumoto, A. Takaori-Kondo, I. Ikai, and T. Chiba. 2006. Anti-viral protein APOBEC3G is induced by interferon-alpha stimulation in human hepatocytes. Biochem. Biophys. Res. Commun. 341:314-319. [DOI] [PubMed] [Google Scholar]
- 52.Turelli, P., B. Mangeat, S. Jost, S. Vianin, and D. Trono. 2004. Inhibition of hepatitis B virus replication by APOBEC3G. Science 303:1829. [DOI] [PubMed] [Google Scholar]
- 53.Wang, G. H., and C. Seeger. 1993. Novel mechanism for reverse transcription in hepatitis B viruses. J. Virol. 67:6507-6512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang, G. H., and C. Seeger. 1992. The reverse transcriptase of hepatitis B virus acts as a protein primer for viral DNA synthesis. Cell 71:663-670. [DOI] [PubMed] [Google Scholar]
- 55.Wang, X., N. Grammatikakis, and J. Hu. 2002. Role of p50/CDC37 in hepadnavirus assembly and replication. J. Biol. Chem. 277:24361-24367. [DOI] [PubMed] [Google Scholar]
- 56.Yu, M., and J. Summers. 1994. Multiple functions of capsid protein phosphorylation in duck hepatitis B virus replication. J. Virol. 68:4341-4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu, Q., D. Chen, R. Konig, R. Mariani, D. Unutmaz, and N. R. Landau. 2004. APOBEC3B and APOBEC3C are potent inhibitors of simian immunodeficiency virus replication. J. Biol. Chem. 279:53379-53386. [DOI] [PubMed] [Google Scholar]
- 58.Yu, Q., R. Konig, S. Pillai, K. Chiles, M. Kearney, S. Palmer, D. Richman, J. M. Coffin, and N. R. Landau. 2004. Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nat. Struct. Mol. Biol. 11:435-442. [DOI] [PubMed] [Google Scholar]
- 59.Zheng, Y. H., D. Irwin, T. Kurosu, K. Tokunaga, T. Sata, and B. M. Peterlin. 2004. Human APOBEC3F is another host factor that blocks human immunodeficiency virus type 1 replication. J. Virol. 78:6073-6076. [DOI] [PMC free article] [PubMed] [Google Scholar]