

Abstract
It has been described that influenza virus polymerase associates with RNA polymerase II (RNAP II). To gain information about the role of this interaction, we explored if changes in RNAP II occur during infection. Here we show that influenza virus causes the specific degradation of the hypophosphorylated form of the largest subunit of RNAP II without affecting the accumulation of its hyperphosphorylated forms. This effect is independent of the viral strain and the origin of the cells used. Analysis of synthesized mRNAs in isolated nuclei of infected cells indicated that transcription decreases concomitantly with RNAP II degradation. Moreover, this degradation correlated with the onset of viral transcription and replication. The ubiquitin-mediated proteasome pathway is not involved in virally induced RNAP II proteolysis. The expression of viral polymerase from its cloned cDNAs was sufficient to cause the degradation. Since the PA polymerase subunit has proteolytic activity, we tested its participation in the process. A recombinant virus that encodes a PA point mutant with decreased proteolytic activity and that has defects in replication delayed the effect, suggesting that PA's contribution to RNAP II degradation occurs during infection.
The genome of influenza virus consists of eight single-stranded RNA molecules of negative polarity. The viral RNA polymerase is composed of three subunits, PB1, PB2, and PA (16, 26, 27), which together with the nucleoprotein perform all the activities required for viral RNA expression (15, 18, 28, 33). The PB2 subunit is able to bind cap 1 structures of host cell hnRNAs (8, 57). The PB1 subunit contains both sequence motifs typical of the viral RNA-dependent RNA polymerases (46), which are essential for RNA synthesis (7), and the endonuclease activity responsible for the cleavage of host mRNA precursors (35). The PA subunit is a phosphoprotein with proteolytic activity (25, 40, 50, 51). The phenotype of viral temperature-sensitive and protease mutants suggests that the PA subunit may be involved in the transition from mRNA transcription to replication (29, 37). The transcription process involves a cap-stealing mechanism by which 5′-capped oligonucleotides derived from newly synthesized RNA polymerase II (RNAP II) transcripts are used as primers and elongated by the viral polymerase (9, 45). In line with this transcription strategy, parental virion RNPs colocalize with active RNAP II in the infected-cell nucleus (I. Salanueva, personal communication). Due to the requirements for cellular capped mRNAs, virus transcription is inhibited by actinomycin D or α-amanitin (38). Viral RNA replication involves the synthesis of cap-independent, full-length positive-stranded RNAs complementary to the genomic viral RNAs (vRNAs), which serve as templates for amplification of the vRNAs and are not sensitive to actinomycin D or α-amanitin (53).
Many viruses induce alterations in host cell gene expression. Among these, changes in the transcriptional machinery of the infected cells are broadly documented. RNAP II, a multiprotein complex composed of 12 subunits, is the enzyme responsible for the transcription of mRNAs. Two major forms of RNAP II are found in cells, hyperphosphorylated and hypophosphorylated on the carboxy-terminal domain (CTD) of its largest subunit. The CTD consists of more than 50 repeats of the Y1S2P3T4S5P6S7 heptapeptide (12), with Ser-2 and Ser-5 (boldface) being the main phosphoacceptor residues (see reference 42 for a review). It has been reported that serine 5 phosphorylation is detected at the promoter region of the transcribing genes, whereas serine 2 phosphorylation is increased as RNAP II leaves the promoter and transcribes the body of the gene. Several cyclin-dependent kinases phosphorylate the CTD. Among them CDK7, a subunit of the transcription factor TFIIH, and CDK9, a subunit of the elongation factor P-TEFb, are found. These kinases associate with one of the several regulatory cyclin T proteins (6, 47, 54). There are many examples of viruses that modify the RNAP II CTD to improve their replication, including human immunodeficiency virus type 1 (13, 31, 59), cytomegalovirus (55), herpes simplex virus type 1 (HSV-1) (14, 17, 21), Epstein-Barr virus (5), and Bunyamwera virus (56).
It has been found that the RNA polymerase complex of influenza virus interacts with the largest subunit of the RNAP II via the CTD. The viral polymerase binds to the hypo- and hyperphosphorylated forms of RNAP II, suggesting that it targets actively transcribing RNAP II (19). We previously identified the cellular protein hCLE/CGI-99 as a factor interacting with the PA subunit of the influenza virus polymerase that associates with active, purified viral RNPs reconstituted in vivo (30). We have also shown that hCLE is a new positive transcription modulator of RNAP II that is present in complexes with both the hypo- and hyperphosphorylated forms of RNAP II (44). All these data prompted us to explore if changes in RNAP II levels or in its phosphorylation pattern occur during influenza virus infection.
MATERIALS AND METHODS
Biological materials.
The cell lines used in this study were HEK293T and HeLa (human), COS-1 (monkey), NIH 3T3 (mouse), and NLB2 (canine). The influenza virus A/Victoria/3/75 (VIC) and A/WSN/33 (WSN) strains were used. Recombinant viruses containing PA, PB1, PB2, and NP genes from the VIC strain and all other genes from the WSN strain, with either wild-type PA or the T157A PA mutant, have been described previously (29). Plasmids pCMVPA, pCMVPB1, and pCMVPB2 have been described previously (20). α-Amanitin and proteasome inhibitor MG132 were from Sigma. The protease inhibitor “Complete” was from Roche.
Virus infection and transfection.
Cells were infected at a multiplicity of infection of 3 PFU/cell. At different hours postinfection (hpi), the cells were collected in phosphate-buffered saline with protease (“Complete”) and phosphatase (500 μM sodium orthovanadate, 500 μM β-glycerophosphate, and 500 μM sodium molybdate) inhibitors, and the cell pellet was resuspended in Laemmli sample buffer (49).
Cell cultures were transfected by the calcium phosphate method (58) with pCMV plasmids expressing PB1, PB2, or PA. After 16 hours of transfection, the cells were collected and used for Western blot analysis.
Western blotting.
Western blotting was done as described previously (30). The following primary antibodies were used: for RNAP II, monoclonal antibodies 8WG16 (1:500), H14 (1:500), and H5 (1:500) from BabCo; for the N-terminal part of the largest RNAP II subunit, polyclonal antibody N-20 from Santa Cruz (1:500); for PA, monoclonal antibodies 2 and 14 (1:20 each) (4); for PB1, a rabbit polyclonal antibody (1:1,000) (24); for PB2, monoclonal antibodies 8 and 28 (1:100 each) (2); for NP protein, a rat polyclonal antibody generated using as antigen a His-NP protein expressed and purified from bacteria (1:2,000); for translation initiation factor 4GI (eIF4GI), a mixture of four rabbit polyclonal antibodies (1:8,000 each) (1); for β-tubulin, a mouse monoclonal antibody (1:15,000) from Sigma; for cyclin T1, a goat polyclonal antibody (1:1,000) from Santa Cruz; for CDK7, a mouse monoclonal antibody (1:1,000) from Santa Cruz; and for ubiquitin, a rabbit polyclonal antibody (1:1,000) from Santa Cruz.
In vitro RNA synthesis.
To analyze total RNA synthesis, cultures of HEK293T cells were mock infected or infected with the VIC or WSN strain and their nuclei were isolated and frozen. These nuclei were used to detect in vitro RNA synthesis by incorporation of [α-32P]GTP (250 μCi/ml) during a 30-min pulse, with or without α-amanitin (5 μg/ml), as described previously (49). After in vitro RNA synthesis, total RNAs were isolated by phenol extraction and ethanol precipitation. The RNAs were quantified by ethidium bromide staining in Bio-Rad Chemi Doc equipment. The radioactivity was quantitated in a phosphorimager after blotting to a nylon membrane.
To analyze the synthesis of hStaufen-1, vimentin, and β-tubulin mRNAs, the in vitro-synthesized RNAs using isolated nuclei from either mock- or influenza virus-infected cells were used as probes for runoff experiments (49). Five hundred nanograms of the corresponding coding sequences was spotted and fixed to nylon membranes and hybridized with the labeled RNAs, and the label was quantitated in a phosphorimager.
Detection of viral RNAs.
Total RNA from infected HEK293T cells was isolated using the Ultraspec RNA isolation reagent from Biotex. Northern blotting was performed using standard conditions (49), and the membranes were hybridized with oligonucleotide probes radiolabeled with [γ-32P]ATP. The probes recognized NP positive sense RNA or vRNA from VIC and WSN strains, and their sequences were 5′-GTCTTCGAGCTCTCGGAC-3′ and 5′-TCTTAGGATCTTTCCCCGC-3′, respectively.
Pulse-chase experiments.
Cultures of HEK293T cells were starved for 2 h in methionine- and cysteine-free Dulbecco's modified Eagle medium (DMEM) and labeled for 1 h with a mixture of [35S]Met/Cys (Promix; Amersham) in 100 μl of the same medium. The chase was carried out by extensive washing with DMEM and incubation in 500 μl of DMEM supplemented with 10% fetal bovine serum. Finally, cells were washed with phosphate-buffered saline buffer, resuspended in 200 μl of DMEM, and used for immunoprecipitation assays as reported previously (50).
Metabolic labeling.
HEK293T cells were mock infected or infected with the VIC strain at a multiplicity of infection of 3 PFU/cell. The cells that were treated with α-amanitin received the treatment at various times postinfection, and the cells tested at 0 hpi were pretreated 1.5 h before the infection. In all cases, the drug was present from its addition until the end of the infection (8 h). At 7 hpi the cells were washed and incubated for 1 hour in the culture medium containing [35S]Met/Cys.
RESULTS
Influenza virus infection induces the specific degradation of hypophosphorylated RNAP II.
It has been found that the influenza virus polymerase associates with the hypo- and Ser-5P-hyperphosphorylated forms of RNAP II, whereas Ser-2P RNAP II does not associate with the viral polymerase (19). To check whether such interactions could produce changes in the accumulation or phosphorylation state of RNAP II during influenza virus infection, HEK293T cells were infected with the VIC strain of influenza virus and total cell extracts used for Western blot analysis with antibodies that recognize different phosphorylated forms of RNAP II. These antibodies have been produced using as epitopes different phosphorylated forms of the Y1S2P3T4S5P5S7 heptapeptide, and their specificities have been extensively studied (42). In Table 1 their recognition pattern for CTD peptides subjected to Biacore analysis is shown (32). As can be observed, 8WG16 antibody mainly detects hypophosphorylated RNAP II; H5 antibody detects phosphorylated RNAP II either at Ser-2 or Ser-5, and H14 antibody detects RNAP II phosphorylated specifically at Ser-5. To assess the progression of the infection, the accumulation levels of nucleoprotein were analyzed and β-tubulin accumulation was also determined as a loading control. Around 4 hpi, the accumulation of the RNAP II forms recognized by the 8WG16 antibody began to decrease, reaching nearly undetectable levels at later times postinfection (10 to 12 h). In contrast, the levels of accumulation of the hyperphosphorylated forms of RNAP II did not show variations as the infection progressed (Fig. 1A). This experiment was repeated more than five times with identical results. The quantitation of the amount of total RNAP II (hypo- plus hyperphosphorylated forms) during influenza virus infection in the representative experiment presented in Fig. 1A is shown at the right of the figure. As observed, a 50% reduction was attained at late times postinfection. Degradation of RNAP II was also observed using an antibody that recognizes the N-terminal part of the largest subunit (Fig. 1, N20). In this case the decline of total RNAP II was lower and mainly affected the RNAP II form with higher electrophoretic mobility, which should correspond to the hypophosphorylated form. On the other hand, no specific cleavage products were found using antibodies that recognize both the N- and C-terminal parts of RNAP II. Thus, these data confirm the above results and indicate that degradation of hypophosphorylated RNAP II does not affect the CTD specifically.
TABLE 1.
Phosphorylation dependence of anti-CTD monoclonal antibodies
| Antibody | Epitope (Y1S2P3T4S5P6S7) | Recognitiona |
|---|---|---|
| 8WG16 | Unphosphorylated | Unphosphorylated repeats (no affinity for repeats containing Ser-2P) |
| H14 | Ser-5P | Ser-5P, Ser-5P + Ser-2P |
| H5 | Ser-2P | Ser-2P, Ser-5P, Ser-2P + Ser-5P |
See reference 32.
FIG. 1.

Influenza virus infection produces degradation of hypophosphorylated RNAP II. (A) HEK293T cells were infected with the VIC strain of influenza virus and at the designated hpi hypophosphorylated RNAP II (8WG16), RNAP II Ser-2P and Ser-5P (H5), RNAP II Ser-5P (H14), total RNAP II (N20), and the indicated proteins were monitored in total cell extracts by Western blotting. Five different experiments were carried out, and a representative experiment is shown. Quantitation of the amount of total RNAP II (hypo- plus hyperphosphoryalated forms) during the virus infection is shown at the right. M, mock-infected cells; V, influenza virus-infected cells. (B) Pulse-chase experiments. Synthesis of hypophosphorylated RNAP II was monitored in HEK293T cells with a mixture of [35S]Met/Cys. The specific 8WG16 antibody was used to immunoprecipitate (Ip) hypophosphorylated RNAP II. Ctrl, unspecific monoclonal antibody. (C) HEK293T cells were infected with the VIC strain of influenza virus and at the designated hpi the presence of the indicated proteins was monitored in total cell extracts by Western blotting.
It is well established that influenza virus infection shuts down host cell protein synthesis (1, 22). Depending on the half-lives of cellular proteins, their amounts may therefore decline during the time of influenza virus infection. Although the different RNAP II forms come from the same mRNA, it could be argued that the specific decline of the hypophosphorylated form may be the consequence of a particularly short half-life. Thus, we studied the half-lives of the different RNAP II forms. With this aim, pulse-chase experiments were carried out with HEK293T cells followed by immunoprecipitation using specific antibodies as described in Materials and Methods. As can be seen (Fig. 1B), immunoprecipitation with the 8WG16 antibody shows that the half-life of the hypophosphorylated form of the RNAP II is around 16 h, indicating that the observed decrease in the accumulation of this RNAP II form is not the consequence of the block in protein synthesis that occurs upon viral infection. Quantitation of the remaining labeled hypophosphorylated RNAP II during the chase time is shown at the bottom of the figure. Similar half-lives were detected for the hyperphosphorylated forms of RNAP II (data not shown). To assess the specificity of the degradation, we probed the membranes with an antibody specific for eIF4GI. This translation factor has a half-life greater than 12 h in human cells, but it is very susceptible to degradation (11, 39). In spite of this susceptibility, no variations were found in eIF4GI levels upon influenza virus infection (Fig. 1C). Finally, we analyzed possible changes in the CTD kinases. Thus, the levels of CDK7 and the regulatory cyclin of CDK9, cyclin T1, were analyzed. No variations were found in their accumulation levels during the infection, in agreement with the unchanged levels of the hyperphosphorylated forms of RNAP II (Fig. 1C).
To check the validity of this observation for other influenza virus strains, we performed similar experiments using the influenza virus WSN strain (a mouse-adapted strain) to infect HEK293T cells. As shown in Fig. 2A, the WSN strain also produced degradation of the hypophosphorylated RNAP II, similarly to the VIC strain. In this case the degradation was delayed starting at around 6 hpi. To explore if this delay could be the consequence of specific requirements of virus-host cell interactions, the same experiment was performed with NIH 3T3 cells of mouse origin. The results using WSN and VIC strains (Fig. 2B) show that degradation of hypophosphorylated RNAP II by WSN infection in NIH 3T3 cells starts at around 4 hpi, whereas the VIC strain produces a somewhat delayed degradation in the NIH 3T3 cell line. This observation was obtained in three different experiments, and here we show a representative experiment. These results indicate that the effect occurs independently of the virus strain, although some modulation depending on specific virus-host cell interactions could take place. Finally, we studied the degradation in additional cell lines such as HeLa, COS-1, and NLB2, of human, monkey, and canine origin, respectively. The cells were infected with the VIC and WSN strains and processed as indicated above at 10 hpi. Results are presented in Fig. 2C and show that, independently of the cell origin, influenza virus infection gives rise to a specific degradation of hypophosphorylated RNAP II. Altogether these results indicate that influenza virus infection produces a specific degradation of the hypophosphorylated form of RNAP II that is independent of the strain and cell origin.
FIG. 2.
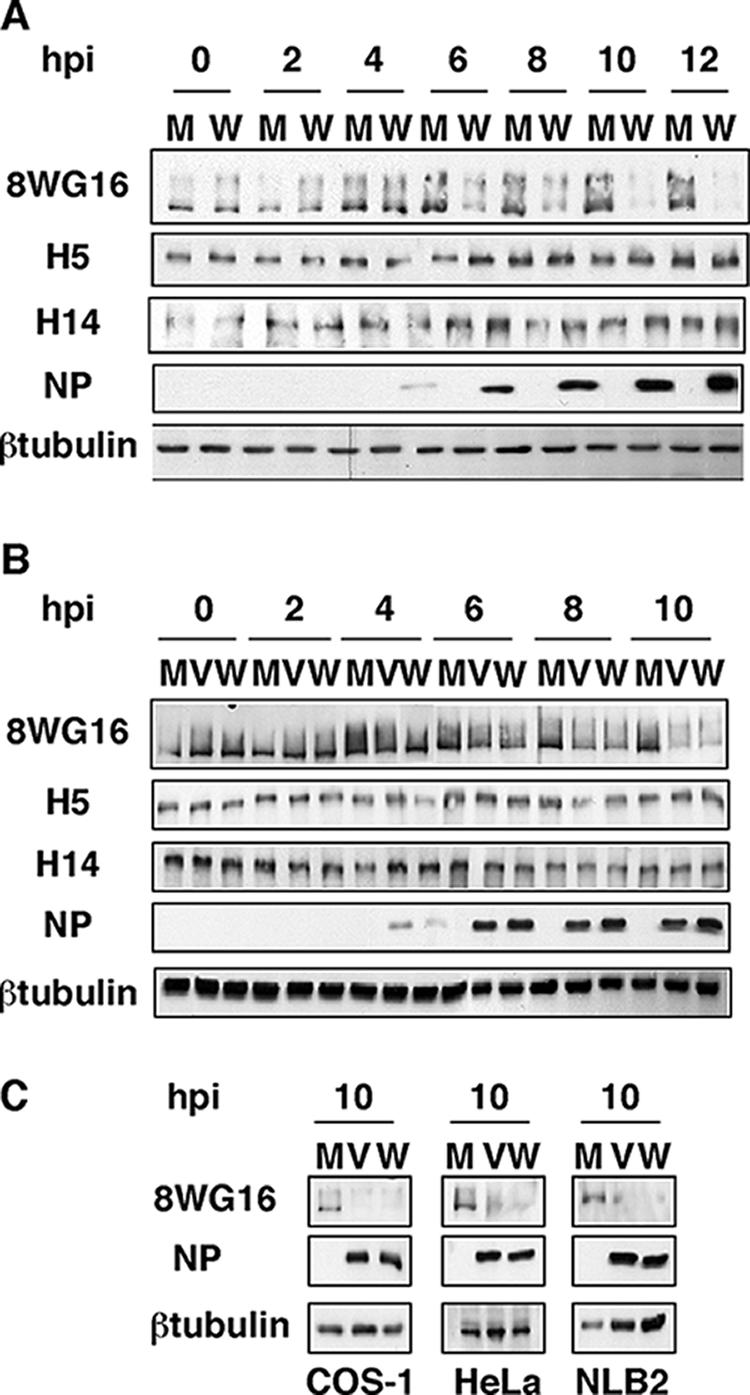
Degradation of hypophosphorylated RNAP II in different virus-host cell systems. (A) HEK293T cells were mock infected (M) or infected with the WSN strain of influenza virus (W) and processed as for Fig. 1. (B) NIH 3T3 cells were mock infected or infected with the VIC (V) or the WSN strain of influenza virus and processed as for Fig. 1. (C) COS-1, HeLa, and NLB2 cells were mock infected or infected with the VIC or WSN strain of influenza virus, and at 10 hpi the presence of RNAP II (8WG16) and the designated proteins was analyzed by Western blotting.
Dependence of cellular mRNA and viral protein synthesis on active RNAP II.
To study the relationship between the virally induced degradation of the hypophosphorylated RNAP II and its activity, we carried out in vitro RNA synthesis. Cultures of HEK293T cells were either mock infected or infected with the VIC or WSN strain, and at the indicated (Fig. 3) hpi the nuclei were isolated. These nuclei were afterward used to perform in vitro RNA synthesis with or without α-amanitin (5 μg/ml) to specifically inhibit RNAP II. The RNA synthesis due to RNAP II activity (plus the RNA produced by viral replication in infected cells that is insensitive to α-amanitin) is calculated as the difference between the values obtained without the drug (total synthesis is synthesis of RNAP I plus RNAP II plus RNAP III) and with the drug (synthesis of RNAP I plus RNAP III). The results are shown in Fig. 3A. The synthesis of cellular mRNAs was unchanged during the 8-h period that was assayed for the mock-infected cells. In contrast, an important inhibition of RNAP II synthesis was observed in the infected cells, which reached its maximum around 4 to 5 hpi and persisted throughout the infection, with an increase at late hpi possibly due to viral replication (see below). To confirm this inhibition, the specific synthesis of several mRNAs corresponding to cellular genes was evaluated. With this aim, in vitro-labeled RNAs from either mock- or influenza virus-infected cells were used as probes in runoff experiments using the cDNAs of hStaufen-1, β-tubulin, and vimentin genes. A progressive reduction in the synthesis of these mRNAs during the infection was observed (Fig. 3B). These results indicate that there is inhibition of cellular mRNA synthesis upon influenza virus infection and that this inhibition correlates with the start of degradation of the hypophosphorylated RNAP II.
FIG. 3.

Dependence of cellular mRNA and viral protein synthesis on active RNAP II. (A) Synthesis of cellular mRNA. HEK293T cells were mock-infected or infected with the VIC or WSN strain of influenza virus, and at the indicated hpi nuclei were isolated and total RNA synthesis was measured by in vitro incorporation of a labeled ribonucleotide with or without α-amanitin (5 μg/ml). The mRNA synthesis due to RNAP II activity is calculated as the difference between the values obtained without the drug (total synthesis is synthesis of RNAP I plus RNAP II plus RNAP III) and with the drug (synthesis of RNAP I plus RNAP III). In the infected cells the contribution of viral replication that is insensitive to α-amanitin should be taken into account. (B) Synthesis of hStaufen-1, β-tubulin, and vimentin mRNAs. Total labeled RNAs from panel A were used as probes for runoff experiments. Five hundred nanograms of the corresponding coding sequences was used, and the label was quantitated in a phosphorimager. Black bars, cells infected with the VIC strain; gray bars, cells infected with the WSN strain. (C) HEK293T cells were mock infected (M) or infected with the VIC strain of influenza virus (V), and α-amanitin (50 μg/ml; α-ama) was added (+) or not (−) to the cell culture at the indicated hpi. At 7 hpi the cells were metabolically labeled with [35S]Met-Cys. Cells were collected at 8 hpi and the synthesized proteins analyzed. At the same times, total cell extracts were used to analyze hypophosphorylated RNAP II by Western blotting (8WG16).
It has been previously shown that active RNAP II is required for primary transcription of the influenza virus genome and that the presence of α-amanitin in the infected cells abolishes viral protein production (34). The addition of α-amanitin during the first 2 h of infection inhibits virus replication, whereas no effect is observed if the drug is added around 4 hpi or later (36). To analyze if there is a correlation between the RNAP II inhibition, its degradation, and its effect on viral progression, we studied the extent of degradation of RNAP II upon α-amanitin treatment in infected cells. With this aim, HEK293T cells were infected with the VIC strain and treated or not with α-amanitin (50 μg/ml) at the indicated (Fig. 3C) times postinfection. In the treated cells, the drug was present from its addition until the end of the infection (8 h). To estimate the synthesis of viral proteins, at 7 hpi the cells were metabolically labeled with [35S]Met-Cys, collected at 8 hpi, and analyzed in denaturing polyacrylamide gels. The results are shown in Fig. 3C. Synthesis of influenza virus proteins was undetectable or very scarce if α-amanitin was present throughout the infection cycle (0 hpi) or added at 0.5 hpi. If the drug was added between 1.5 and 2.5 hpi, a small decrease in viral protein synthesis, compared to untreated cultures, was observed. In contrast, the addition of the drug at later times did not inhibit viral protein synthesis, in agreement with previous results (36). The same samples were used to analyze hypophosphorylated RNAP II levels by Western blot assays. The results are shown in Fig. 3C, bottom panel. The addition of α-amanitin between 0 and 0.5 hpi leads to an almost complete degradation of RNAP II, both in uninfected and infected cells, in agreement with previous data reporting that the degradation rate of RNAP II is α-amanitin dose dependent but is not a consequence of transcriptional arrest (41). From 1.5 to 2.5 hpi the degradation of hypophosphorylated RNAP II increases in the treated and infected cells as a result of the additive effects of both treatments. If the drug is added later, such as at 3.5 hpi, the levels of accumulation of hypophosphorylated RNAP II are similar in treated and uninfected cells and in untreated and infected cells; in this situation viral protein synthesis is normal. Viral protein and hypophosphorylated RNAP II accumulation at 8 hpi in the absence of α-amanitin is presented in Fig. 3C, right, for comparison. The results obtained indicate that a fully active, nondegraded hypophosphorylated RNAP II protein is required during the first 0 to 2.5 hpi to allow appropriate viral protein synthesis; after this time the synthesis becomes independent of the integrity of the cellular transcription machinery. Furthermore, the data also indicate that, if a degradation of hypophosphorylated RNAP II similar to that caused by the infection is produced exogenously by α-amanitin treatment (as described above for 3.5 hpi), it does not impede viral progression if this effect takes place at the same time that it would occur naturally during the infection (compare Fig. 1A, 4 hpi).
The degradation of hypophosphorylated RNAP II correlates with the onset of viral transcription and replication.
To study the relationship between RNAP II degradation and the kinetics of viral transcription and replication, we analyzed the positive and negative sense viral RNA levels during infection. Cultures of HEK293T cells were either mock infected or infected with the VIC or WSN strain, and at the indicated hpi total RNA was isolated and used to detect the positive and negative sense RNA for viral nucleoprotein (Fig. 4, top and bottom panels, respectively). Total positive sense RNA (cRNA plus mRNA) starts accumulating at around 3 hpi and continues increasing until 8 hpi, indicating that its synthesis persists during the infection. As the amount of cRNA is much lower than that of mRNA, the presented data would mainly represent mRNA levels. The NP mRNA levels in the WSN-infected cells were less than those attained in the VIC-infected cells. These results point out that the synthesis of viral messengers takes place even in the context of significantly inactive RNAP II (Fig. 3A), indicating that the availability of 5′-capped cellular premessengers is sufficient to allow viral transcription in these conditions. On the other hand, the accumulation of vRNA starts at around 3 to 4 hpi and attains a maximum at around 6 hpi, with a small decrease at late times postinfection. These data are in agreement with our previous results (29) and different reports (23) using reverse transcription-PCR detection of mRNA, cRNA, and vRNA levels in infected cells. Thus, with different recombinant viruses, an increase of mRNA accumulation until the end of the infection has been shown, whereas cRNA and vRNA accumulation reaches a maximum at around 6 hpi. These results indicate that degradation of hypophosphorylated RNAP II that starts at around 4 hpi (Fig. 1A) correlates with the onset of viral transcription and replication.
FIG. 4.
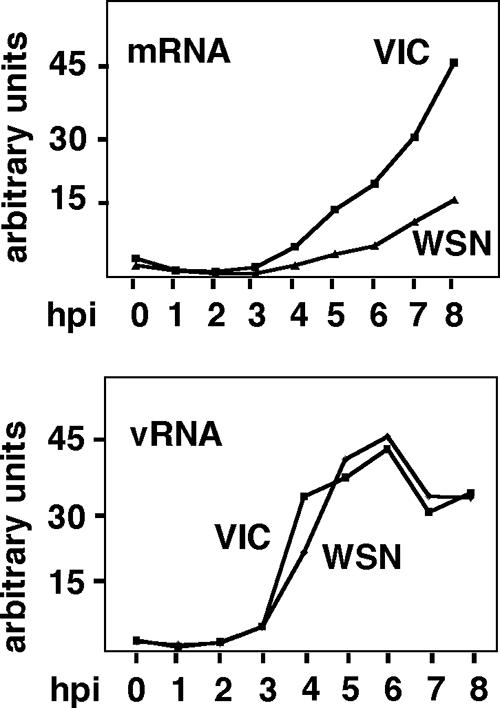
Degradation of hypophosphorylated RNAP II correlates with the onset of viral transcription and replication. HEK293T cells were mock infected or infected with the VIC or WSN strain of influenza virus, and at the indicated hpi total RNAs were isolated and used to detect the positive sense RNA (top) or the vRNA (vRNA; bottom) of viral nucleoproteins by Northern blot analysis.
The viral RNA polymerase complex is sufficient to induce the degradation of RNAP II.
The observed degradation of RNAP II could be the result of either an activity of some viral protein or a cellular proteolytic pathway activated by the infection. Ubiquitin-mediated proteasomal degradation comprises the major proteolytic pathway in eukaryotes. To evaluate its contribution to RNAP II degradation, HEK293T cells were infected or not in the presence or absence of the proteasome inhibitor MG132, which was added 1 h before the infection. At different hpi the levels of accumulation of viral proteins and hypophosphorylated RNAP II were analyzed. The results can be seen in Fig. 5A. The progress of the infection, monitored by observing the appearance of PA and PB1 proteins, was somewhat delayed in the treated cells. In these conditions, almost total degradation of hypophosphorylated RNAP II was observed at 12 hpi, whereas in nontreated cells this effect was observed at 9 hpi. As a control for the effectiveness of the drug, the extracts were probed with antibodies specific for ubiquitin, and an important accumulation of ubiquitinated proteins was observed in the treated cells (Fig. 5B). These results indicate that the proteasome pathway could play some role in influenza virus infection, but it is not responsible for the RNAP II degradation produced by the infection.
FIG. 5.
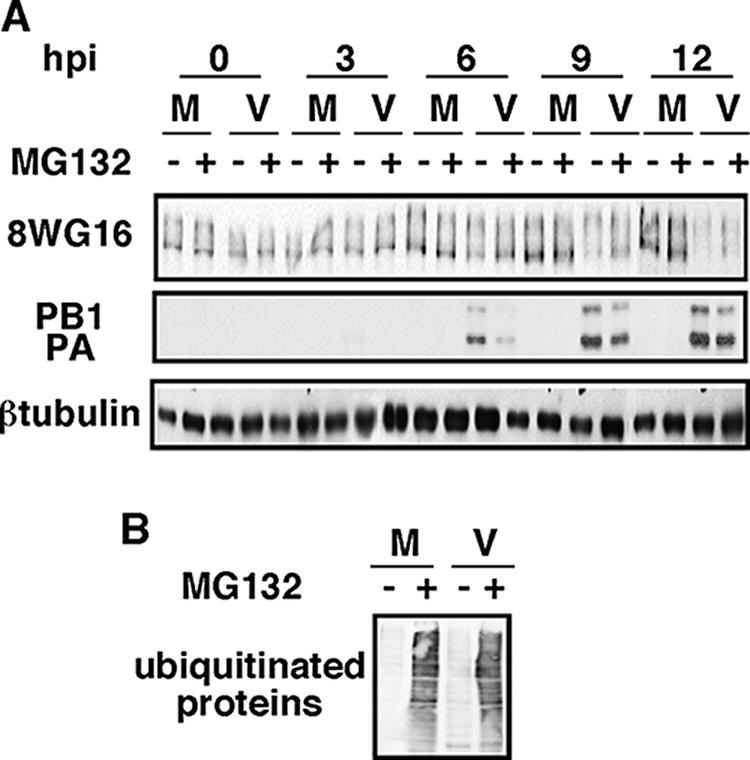
The proteasome pathway is not involved in the degradation of hypophosphorylated RNAP II. (A) HEK293T cells were mock infected (M) or infected with the VIC strain (V), with (+) or without (−) MG132, and at the indicated hpi hypophosphorylated RNAP II (8WG16) and the designated proteins were detected by Western blotting. (B) Western blot using antiubiquitin antibody of mock-infected cells and cells infected at 10 hpi from panel A.
Next, we analyzed the contribution of viral proteins to specific RNAP II degradation. We have previously found that the individual expression of the PA subunit causes its own degradation and that of coexpressed proteins and that the 247 N-terminal amino acids are responsible for this activity (50, 52). The use of protease inhibitors, including those of the proteasome pathway, does not inhibit proteolytic activity (data not shown). Similar to RNAP II degradation upon influenza virus infection, no specific cleavage products of the proteins coexpressed with PA can be observed as a result of its proteolytic activity. The mutation of threonine 157 to alanine in the PA molecule (T157A) diminishes the proteolytic activity (43). Taking these data and the reported association of the viral polymerase with RNAP II into account (19), we explored the possibility that the components of the influenza virus polymerase could produce the degradation of hypophosphorylated RNAP II. HEK293T cells were transfected with plasmids expressing PB1, PB2, or PA or combinations of two plasmids expressing these subunits. After 16 hours of transfection, the amounts of hypophosphorylated and hyperphosphorylated RNAP II were analyzed by Western blotting. The results obtained indicated that neither the individual expression of polymerase subunits nor expression of combinations of two of them induced degradation of hypophosphorylated RNAP II (Fig. 6, left and middle panels). Subsequently, we assayed if the polymerase complex could produce the effect, and hence HEK293T cells were cotransfected with plasmids expressing the three polymerase subunits and processed as above. As can be seen in Fig. 6, in vivo-reconstituted influenza virus polymerase was able to degrade hypophosphorylated RNAP II, whereas hyperphosphorylated RNAP II remained unchanged. These transfection experiments were performed three times with similar results, and the quantitation (mean and standard deviation) of RNAP II degradation upon coexpression of the three subunits is shown at the right of Fig. 6. The decrease of hypophosphorylated RNAP II levels due to the expression of the polymerase was smaller than that observed in influenza virus-infected cells. These results are in agreement with the requirement for coexpression of the three subunits in the same cell. In this context, it should be pointed out that, by performing transfection experiments of HEK293T cells with plasmids expressing PA, PB1, and PB2, we have estimated that the percentage of cells coexpressing the three subunits should be between 30 and 60% (data not shown). Consequently the RNAP II coming from cells that do not express the three polymerase subunits will not be degraded and will mask the degradation that takes place in cotransfected cells. Nevertheless, it is also possible that other unidentified viral or virus-induced proteins could be important for this process.
FIG. 6.

Reconstituted viral polymerase degrades hypophosphorylated RNAP II. HEK293T cells were transfected with plasmids expressing PA, PB1, or PB2 individually (left), with combinations of two of them (middle), or with plasmids expressing PA, PB1, and PB2 together (right), and at 16 h posttransfection the amounts of hypophosphorylated RNAP II (8WG16), hyperphosphorylated RNAP II (H5), and the indicated proteins were detected by Western blotting. Quantitation of RNAP II degradation under conditions of polymerase reconstitution is shown at the right (means and standard deviations).
A recombinant influenza virus encoding a PA mutant defective in proteolysis has a delay in RNAP II degradation.
To analyze if the proteolytic activity of PA could be responsible for the observed degradation, HEK293T cells were infected with recombinant viruses containing a wild-type or a T157A PA mutant gene. The virus with the mutant gene contains a less proteolytic PA protein and does not show changes in virally induced cellular protein translation shutoff (29). At different hpi, samples were taken and the levels of hypophosphorylated RNAP II were analyzed. The results in Fig. 7 show that the recombinant virus containing a wild-type PA produced degradation of the hypophosphorylated RNAP II with the same kinetics as that observed with the VIC strain. In contrast, infection with the recombinant virus that expresses the PA protein with a decreased proteolytic activity led to a delayed degradation of hypophosphorylated RNAP II, although at later times postinfection similar degradation was attained. This experiment has been performed three times, and, although there were small variations in the time of the initiation of degradation, it produced the same pattern on all three occasions. To quantitate this effect, the RNAP II and β-tubulin signals obtained in the Western blots were quantitated by densitometry of autoradiographs, and the obtained RNAP II/β-tubulin ratios from a representative experiment are shown in the bottom part of Fig. 7. Previous results have shown that the recombinant virus containing PA T157A has a delayed transport of PA to the nucleus. While the PA subunit is localized in the nucleus at 4 hpi in the wild-type virus, the PA T157A of the recombinant virus goes to the nucleus at 6 hpi (29). The mutant virus also has a weak reduction in virus growth and a decrease in virus RNA replication (29). Thus, the observed delayed RNAP II degradation could be the consequence of any of these phenotypic alterations or a particular combination of some of them. Nevertheless, Fig. 7 shows that, for example, at 6 hpi, although the levels of PA and PB1 protein accumulation in wild-type and mutant virus-infected cells are similar, there are clearly higher remaining levels of RNAP II in cells infected with the PA T157A virus. These results indicate that the PA protein is directly or indirectly involved in the degradation process and suggest that the proteolytic activity of PA within the viral polymerase complex could contribute to the degradation of the hypophosphorylated RNAP II that occurs during the infection.
FIG. 7.
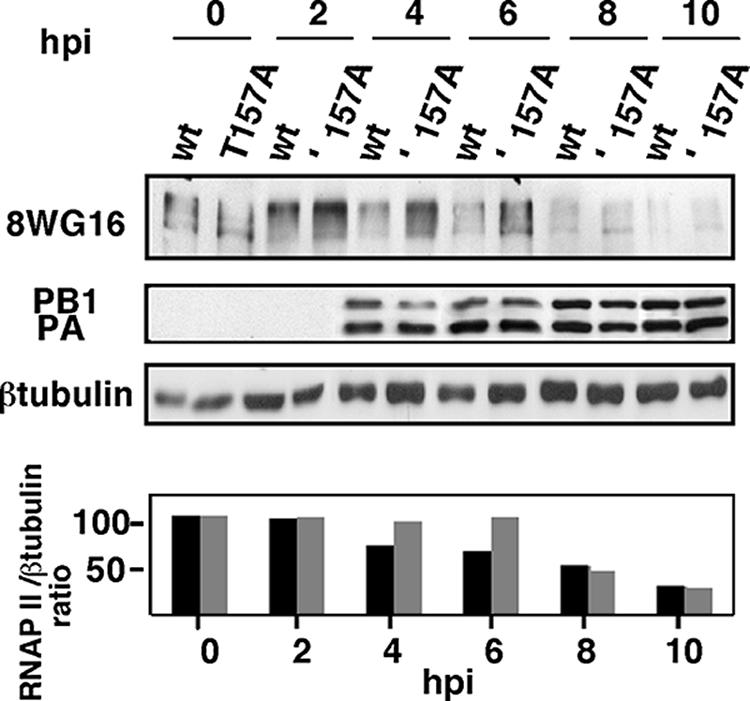
The PA subunit is involved in hypophosphorylated RNAP II degradation. HEK293T cells were infected with rescued recombinant viruses containing wild-type PA (wt) or T157A mutated PA (T157A), and at different hpi hypophosphorylated RNAP II (8WG16) and the indicated proteins were detected by Western blot assays. (Bottom) Quantitation of hypophosphorylated RNAP II levels during the infection. The ratios of RNAP II/β-tubulin accumulation are shown. Black bars, wt PA; gray bars, PA T157A. Three different experiments were carried out, and a representative experiment is shown.
DISCUSSION
Virus-induced modulation of RNAP II.
Some viruses target RNAP II, either increasing or decreasing the degree of phosphorylation of its CTD. HSV-1 is among those whose infection produces loss of serine 2 phosphorylation (14, 21). HSV-1 utilizes the host RNAP II to transcribe its approximately 80 genes in a coordinately regulated cascade. It has been hypothesized that HSV-1 genes, which are short and not configured in standard nucleosomes, may not be subject to transcriptional pausing caused by inherent signals in the gene and/or the action of negative transcription elongation factors. Thus, they may not need Ser-2 phosphorylation of the CTD (21). Bunyamwera virus also prevents CTD phosphorylation at serine 2 (56), suggesting a block in transition from initiation to elongation during infection. Similar to transcription of influenza virus, transcription of bunyaviruses is dependent on host cell mRNAs, since it depends on a cap-snatching mechanism. As the infection should inhibit cellular transcription, it has been postulated that RNAP II activity is dispensable for bunyaviruses and that the pool of cytoplasmic mRNAs would be sufficient to support the viral transcription that takes place in the cytoplasm of the infected cells.
Influenza virus polymerase binds RNAP II both in its hypophosphorylated and serine 5-hyperphosphorylated forms. As the latter is engaged in transcription initiation, this binding could be a manner of placing the viral polymerase close to the sites of generation of the 5′-capped pre-mRNAs. This association might be mediated by the PA-binding protein hCLE, which acts as an RNAP II transcription factor and binds to the hypo- and hyperphosphorylated RNAP II forms (44). Synthesis of mRNAs decreases upon influenza virus infection concomitantly with degradation of hypophosphorylated RNAP II (Fig. 3A and B). The inhibition of RNAP II activity that produces the disappearance of its hypophosphorylated form could be the consequence of a decreased recycling of the hypophosphorylated and hyperphosphorylated forms or the result of an uncharacterized mechanism. Nevertheless, the inhibition should affect specially the transcriptional elongation activity, because in this situation there is active synthesis of viral mRNAs, which depends on 5′-capped oligonucleotides (Fig. 4), indicating that cellular transcription initiation persists. These results are in agreement with recent reports showing that during influenza virus infection there is a decreased association of RNAP II with the coding regions, whereas no variations are found in the RNAP II associated with the promoter regions (10). Both treatment with α-amanitin and influenza virus infection cause degradation of hypophosphorylated RNAP II. Nevertheless, in α-amanitin-treated and infected cells, if the drug is added from 3.5 hpi, the accumulation of viral proteins occurs normally (Fig. 3C). Similarly, viral mRNA levels increase from 3 to 4 hpi despite the degradation of hypophosphorylated RNAP II (Fig. 4). These results suggest that upon infection there are cellular transcription complexes that would be inaccessible to the drug and active for initiation reactions, in spite of the fact that hypophosphorylated RNAP II recycling should be impaired due to its degradation. This effect could represent a strategy to allow the generation of 5′-capped oligonucleotides, avoiding the competition of the RNAP II engaged in mRNA elongation. It should produce cellular transcriptional inhibition and might represent an additional contribution to the general shutoff produced by the infection.
The PA polymerase subunit could be involved in the degradation of cellular RNAP II.
Different viruses use degradation pathways to impair cellular transcription. Thus, HSV-1 ICP27 protein (14) and poliovirus 3Cpro protein (3) are involved in RNAP II Ser-2 and TATA binding protein degradation, respectively. In the case of influenza virus infection the viral polymerase induces the degradation of hypophosphorylated RNAP II (Fig. 6). This is the first case of a viral polymerase that targets cellular RNAP II. Within the polymerase complex, the PA subunit could play a role in this process, as suggested by the delayed effect on RNAP II degradation produced by the mutation of threonine 157 of this subunit (Fig. 7). This residue reduces the proteolytic activity of individually expressed PA and is phosphorylated in vivo (43). Although a cellular proteolytic pathway induced by the expression of the viral polymerase could be involved in hypophosphorylated RNAP II degradation, several facts directly implicate the viral polymerase. (i) Proteasome inhibitors delay but do not impede RNAP II degradation. This holdup could be the consequence of the delayed viral protein accumulation observed in this situation (Fig. 5A). (ii) Individually expressed PA has proteolytic activity modulated by threonine 157 (52), which is not sensitive to protease inhibitors (data not shown). (iii) PA also has a weak serine protease activity in vitro, which resides on the C-terminal part of the molecule (25). Neither the individually expressed polymerase subunits nor the different dimers associate with RNAP II, whereas the trimeric complex possesses this capacity (19). As PA protease activity does not show specificity when individually expressed, it is possible that its association with the polymerase complex could confer the required specificity, rendering hypophosphorylated RNAP II the physiological target for PA proteolytic activity.
Previous reports have shown that PA protease activity appears to be a general characteristic because both human (VIC and PR8) and avian (FPV, MAL, and HK) strains show this phenotype (40). We have previously shown that in vivo-reconstituted RNPs or recombinant influenza viruses containing PA T157A have defects in RNA replication whereas viral transcription remains unaffected (29, 43). The recombinant virus containing PA T157A also has a reduced pathogenicity in mice, indicating that PA proteolytic activity is important for both viral RNA replication and attenuation. Now we provide data suggesting that the PA T157 residue could also be involved in the degradation of RNAP II. Thus, it is possible that degradation of hypophosphorylated RNAP II is important to allow efficient viral RNA replication. Alternatively, the observed RNAP II degradation could be the consequence of viral RNA replication and the impairment shown by PA T157A recombinant virus the result of a defective viral RNA replication process.
Finally, influenza virus and some other viruses whose expression depends on RNAP II inhibit its activity, either by degradation (influenza virus [this report], HSV-1 [14], and La Crosse virus [48]) or by dephosphorylation of its CTD (Bunyamwera virus [56]). These puzzling viral activities could have been selected to avoid the competition of cellular transcription once it is no longer required and would contribute to the overall pathogenesis exerted by the different viruses.
Acknowledgments
We are indebted to J. Ortin, U. Garairgorta, and T. Lutz for critical reviews of the manuscript. The technical assistance of Y. Fernández and N. Zamarreño is gratefully acknowledged.
A. Rodriguez was a fellow from Programa Nacional de Formación de Personal Universitario and A. Pérez-González from the Fondo de Investigaciones Sanitarias. This work was supported by Programa Sectorial de Promoción General del Conocimiento (grant BFU2005-02834).
Footnotes
Published ahead of print on 7 March 2007.
REFERENCES
- 1.Aragón, T., S. de la Luna, I. Novoa, L. Carrasco, J. Ortín, and A. Nieto. 2000. Eukaryotic translation initiation factor 4GI is a cellular target for NS1 protein, a translational activator of influenza virus. Mol. Cell. Biol. 20:6259-6268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arrese, M., and A. Portela. 1996. Serine 3 is critical for phosphorylation at the N-terminal end of the nucleoprotein of influenza virus A/Victoria/3/75. J. Virol. 70:3385-3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Banerjee, R., M. K. Weidman, A. Echeverri, P. Kundu, and A. Dasgupta. 2004. Regulation of poliovirus 3C protease by the 2C polypeptide. J. Virol. 78:9243-9256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bárcena, J., D. l. L. S., M. Ochoa, J. A. Melero, A. Nieto, J. Ortín, and A. Portela. 1994. Monoclonal antibodies against the influenza virus PB2 and NP polypeptides interfere with the initiation step of viral mRNA synthesis in vitro. J. Virol. 68:6900-6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bark-Jones, S. J., H. M. Webb, and M. J. West. 2006. EBV EBNA 2 stimulates CDK9-dependent transcription and RNA polymerase II phosphorylation on serine 5. Oncogene 25:1775-1785. [DOI] [PubMed] [Google Scholar]
- 6.Bensaude, O., F. Bonnet, C. Cassé, M.-F. Dubois, V.-T. Nguyen, and B. Palancade. 1999. Regulated phosphorylation of the RNA polymerase II C-terminal domain. Biochem. Cell Biol. 77:1-7. [PubMed] [Google Scholar]
- 7.Biswas, S. K., and D. P. Nayak. 1994. Mutational analysis of the conserved motifs of influenza A virus polymerase basic protein 1. J. Virol. 68:1819-1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blaas, D., E. Patzelt, and E. Keuchler. 1982. Identification of the cap binding protein of influenza virus. Nucleic Acids Res. 10:4803-4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bouloy, M., S. J. Plotch, and R. M. Krug. 1978. Globin mRNAs are primers for the transcription of influenza viral RNA in vitro. Proc. Natl. Acad. Sci. USA 75:4886-4890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan, A. Y., F. T. Vreede, M. Smith, O. G. Engelhardt, and E. Fodor. 2006. Influenza virus inhibits RNA polymerase II elongation. Virology 351:210-217. [DOI] [PubMed] [Google Scholar]
- 11.Clemens, M. J., M. Bushell, and S. J. Morley. 1998. Degradation of eukaryotic polypeptide chain initiation factor (eIF) 4G in response to induction of apoptosis in human lymphoma cell lines. Oncogene 17:2921-2931. [DOI] [PubMed] [Google Scholar]
- 12.Corden, J. L., J. M. Cadena, J. M. Ahearn, and M. E. Dahmus. 1985. A unique structure at the carboxy terminus of the largest subunit of eukaryotic RNA polymerase II. Proc. Natl. Acad. Sci. USA 82:7934-7938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cujec, T. P., H. Okamoto, K. Fujinaga, J. Meyer, H. Chamberlin, D. O. Morgan, and B. M. Peterlin. 1997. The HIV transactivator TAT binds to the CDK-activating kinase and activates the phosphorylation of the carboxy-terminal domain of RNA polymerase II. Genes Dev. 11:2645-2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dai-Ju, J. Q., L. Li, L. A. Johnson, and R. M. Sandri-Goldin. 2006. ICP27 interacts with the C-terminal domain of RNA polymerase II and facilitates its recruitment to herpes simplex virus 1 transcription sites, where it undergoes proteasomal degradation during infection. J. Virol. 80:3567-3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de la Luna, S., J. Martín, A. Portela, and J. Ortín. 1993. Influenza virus naked RNA can be expressed upon transfection into cells co-expressing the three subunits of the polymerase and the nucleoprotein from SV40 recombinant viruses. J. Gen. Virol. 74:535-539. [DOI] [PubMed] [Google Scholar]
- 16.Detjen, B. M., C. St. Angelo, M. G. Katze, and R. M. Krug. 1987. The three influenza virus polymerase (P) proteins not associated with viral nucleocapsids in the infected cell are in the form of a complex. J. Virol. 61:16-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Durand, L. O., S. J. Advani, A. P. Poon, and B. Roizman. 2005. The carboxyl-terminal domain of RNA polymerase II is phosphorylated by a complex containing cdk9 and infected-cell protein 22 of herpes simplex virus 1. J. Virol. 79:6757-6762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elton, D., P. Digard, L. Tiley, and J. Ortín. 2005. Structure and function of the influenza virus RNP. In Y. Kawaoka (ed.), Contemporary topics in influenza virology. Horizon Scientific Press, Norfolk, VA.
- 19.Engelhardt, O. G., M. Smith, and E. Fodor. 2005. Association of the influenza A virus RNA-dependent RNA polymerase with cellular RNA polymerase II. J. Virol. 79:5812-5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Falcón, A., R. Marión, T. Zürcher, P. Gomez, A. Portela, A. Nieto, and J. Ortín. 2004. Defective RNA replication and late gene expression in temperature-sensitive influenza viruses expressing deleted forms of the NS1 protein. J. Virol. 78:3880-3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fraser, K. A., and S. A. Rice. 2005. Herpes simplex virus type 1 infection leads to loss of serine-2 phosphorylation on the carboxyl-terminal domain of RNA polymerase II. J. Virol. 79:11323-11334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garfinkel, M. S., and M. G. Katze. 1993. Translational control by influenza virus. Selective translation is mediated by sequences within the viral mRNA 5′-untranslated region. J. Biol. Chem. 268:22223-22226. [PubMed] [Google Scholar]
- 23.Gastaminza, P., B. Perales, A. M. Falcón, and J. Ortín. 2003. Mutations in the N-terminal region of influenza virus PB2 protein affect virus RNA replication but not transcription. J. Virol. 77:5098-5108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gonzalez, S., and J. Ortín. 1999. Distinct regions of influenza virus PB1 polymerase subunit recognize vRNA and cRNA templates. EMBO J. 18:3767-3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hara, K., M. Shiota, H. Kido, Y. Ohtsu, T. Kashiwagi, J. Iwahashi, N. Hamada, K. Mizoue, N. Tsumura, H. Kato, and T. Toyoda. 2001. Influenza virus RNA polymerase PA subunit is a novel serine protease with Ser624 at the active site. Genes Cells 6:87-97. [DOI] [PubMed] [Google Scholar]
- 26.Honda, A., J. Mukaigawa, A. Yokoiyama, A. Kato, S. Ueda, K. Nagata, M. Krystal, D. P. Nayak, and A. Ishihama. 1990. Purification and molecular structure of RNA polymerase from influenza virus A/PR8. J. Biochem. (Tokyo) 107:624-628. [DOI] [PubMed] [Google Scholar]
- 27.Horisberger, M. A. 1980. The large P proteins of influenza A viruses are composed of one acidic and two basic polypeptides. Virology 107:302-305. [DOI] [PubMed] [Google Scholar]
- 28.Huang, T. S., P. Palese, and M. Krystal. 1990. Determination of influenza virus proteins required for genome replication. J. Virol. 64:5669-5673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huarte, M., A. Falcón, Y. Nakaya, J. Ortín, A. García-Sastre, and A. Nieto. 2003. Threonine 157 of influenza virus PA polymerase subunit modulates RNA replication in infectious viruses. J. Virol. 77:6007-6013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huarte, M., J. J. Sanz-Ezquerro, F. Roncal, J. Ortín, and A. Nieto. 2001. PA subunit from influenza virus polymerase complex interacts with a cellular protein with homology to a family of transcriptional activators. J. Virol. 75:8597-8604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Isel, C., and J. Karn. 1999. Direct evidence that HIV-1 Tat stimulates RNA polymerase II carboxyl-terminal domain hyperphosphorylation during transcriptional elongation. J. Mol. Biol. 290:929-941. [DOI] [PubMed] [Google Scholar]
- 32.Jones, J. C., H. P. Phatnani, T. A. Haystead, J. A. MacDonald, S. M. Alam, and A. L. Greenleaf. 2004. C-terminal repeat domain kinase I phosphorylates Ser2 and Ser5 of RNA polymerase II C-terminal domain repeats. J. Biol. Chem. 279:24957-24964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kimura, N., M. Mishida, K. Nagata, A. Ishihama, K. Oda, and S. Nakada. 1992. Transcription of a recombinant influenza virus RNA in cells that can express the influenza virus RNA polymerase and nucleoprotein genes. J. Gen. Virol. 73:1321-1328. [DOI] [PubMed] [Google Scholar]
- 34.Lamb, R. A., and P. W. Choppin. 1977. Synthesis of influenza virus polypeptides in cells resistant to alpha-amanitin: evidence for the involvement of cellular RNA polymerase II in virus replication. J. Virol. 23:816-819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li, M. L., P. Rao, and R. M. Krug. 2001. The active sites of the influenza cap-dependent endonuclease are on different polymerase subunits. EMBO J. 20:2078-2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mahy, B. W., N. D. Hastie, and S. J. Armstrong. 1972. Inhibition of influenza virus replication by α-amanitin: mode of action. Proc. Natl. Acad. Sci. USA 69:1421-1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mahy, B. W. J. 1983. Mutants of influenza virus, p. 192-253. In P. Palese and D. W. Kingsbury (ed.), Genetics of influenza viruses. Springer Verlag, Wien, Austria.
- 38.Mark, G. E., J. M. Taylor, B. Broni, and R. M. Krug. 1979. Nuclear accumulation of influenza viral RNA transcripts and the effects of cycloheximide, actinomycin D and α-amanitin. J. Virol. 29:744-752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morley, S. J., P. S. Curtis, and V. M. Pain. 1997. eIF4G: translation's mystery factor begins to yield its secrets. RNA 3:1085-1104. [PMC free article] [PubMed] [Google Scholar]
- 40.Naffakh, N., P. Massin, and S. van der Werf. 2001. The transcription/replication activity of the polymerase of influenza A viruses is not correlated with the level of proteolysis induced by the PA subunit. Virology 285:244-252. [DOI] [PubMed] [Google Scholar]
- 41.Nguyen, V. T., F. Giannoni, M. F. Dubois, S. J. Seo, M. Vigneron, C. Kedinger, and O. Bensaude. 1996. In vivo degradation of RNA polymerase II largest subunit triggered by alpha-amanitin. Nucleic Acids Res. 24:2924-2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Palancade, B., and O. Bensaude. 2003. Investigating RNA polymerase II carboxyl-terminal domain (CTD) phosphorylation. Eur. J. Biochem. 270:3859-3870. [DOI] [PubMed] [Google Scholar]
- 43.Perales, B., J. J. Sanz-Ezquerro, P. Gastaminza, J. Ortega, J. Fernandez-Santarén, J. Ortín, and A. Nieto. 2000. The replication activity of influenza virus polymerase is linked to the capacity of the PA subunit to induce proteolysis. J. Virol. 74:1307-1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pérez-Gonzalez, A., A. Rodriguez, M. Huarte, I. J. Salanueva, and A. Nieto. 2006. hCLE/CGI-99, a human protein that interacts with the influenza virus polymerase, is a mRNA transcription modulator. J. Mol. Biol. 362:887-900. [DOI] [PubMed] [Google Scholar]
- 45.Plotch, S. J., M. Bouloy, I. Ulmanen, and R. M. Krug. 1981. A unique cap(m7GpppXm)-dependent influenza virion endonuclease cleaves capped RNAs to generate the primers that initiate viral RNA transcription. Cell 23:847-858. [DOI] [PubMed] [Google Scholar]
- 46.Poch, O., I. Sauvaget, M. Delarue, and N. Tordo. 1990. Identification of four conserved motifs among the RNA-dependent polymerases encoding elements. EMBO J. 8:3867-3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Price, D. H. 2000. P-TEFb, a cyclin-dependent kinase controlling elongation by RNA polymerase II. Mol. Cell. Biol. 20:2629-2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ruf, M., G. Blakqori, V. Wagner, and F. Weber. 2006. The interferon antagonist NSs of orthobunyaviruses targets the phosphorylated form of RNA polymerase II in a proteasome-dependent manner, p. 150. Abstr. XIII Int. Conf. Negative Strand Viruses.
- 49.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 50.Sanz-Ezquerro, J. J., S. de la Luna, J. Ortín, and A. Nieto. 1995. Individual expression of influenza virus PA protein induces degradation of coexpressed proteins. J. Virol. 69:2420-2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sanz-Ezquerro, J. J., J. Férnandez-Santarén, T. Sierra, T. Aragón, J. Ortega, J. Ortín, G. L. Smith, and A. Nieto. 1998. The PA influenza virus polymerase subunit is a phosphorylated protein. J. Gen. Virol. 79:471-478. [DOI] [PubMed] [Google Scholar]
- 52.Sanz-Ezquerro, J. J., T. Zurcher, S. de la Luna, J. Ortin, and A. Nieto. 1996. The amino-terminal one-third of the influenza virus PA protein is responsible for the induction of proteolysis. J. Virol. 70:1905-1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Scholtissek, C., and R. Rott. 1970. Synthesis in vivo of influenza plus and minus strand RNA and its preferential inhibition by antibiotics. Virology 40:989-996. [DOI] [PubMed] [Google Scholar]
- 54.Svejstrup, J. Q., P. Vichi, and J.-M. Egly. 1996. The multiple roles of transcription/repair factor TFIIH. Trends Biochem. Sci. 21:346-350. [PubMed] [Google Scholar]
- 55.Tamrakar, S., A. J. Kapasi, and D. H. Spector. 2005. Human cytomegalovirus infection induces specific hyperphosphorylation of the carboxyl-terminal domain of the large subunit of RNA polymerase II that is associated with changes in the abundance, activity, and localization of cdk9 and cdk7. J. Virol. 79:15477-15493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thomas, D., G. Blakqori, V. Wagner, M. Banholzer, N. Kessler, R. M. Elliott, O. Haller, and F. Weber. 2004. Inhibition of RNA polymerase II phosphorylation by a viral interferon antagonist. J. Biol. Chem. 279:31471-31477. [DOI] [PubMed] [Google Scholar]
- 57.Ulmanen, I., B. A. Broni, and R. M. Krug. 1981. Role of two of the influenza virus core P proteins in recognizing cap 1 structures (m7GpppNm) on RNAs and in initiating viral RNA transcription. Proc. Natl. Acad. Sci. USA 78:7355-7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wigler, M., A. Pellicer, S. Silverstein, R. Axel, G. Urlaub, and L. Chasin. 1979. DNA-mediated transfer of the adenine phosphoribosyltranferase locus into mammalian cells. Proc. Natl. Acad. Sci. USA 76:1373-1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhou, M., M. A. Halanski, M. F. Radonovich, F. Kashanchi, J. Peng, D. H. Price, and J. N. Brady. 2000. Tat modifies the activity of CDK9 to phosphorylate serine 5 of the RNA polymerase II carboxyl-terminal domain during human immunodeficiency virus type 1 transcription. Mol. Cell. Biol. 20:5077-5086. [DOI] [PMC free article] [PubMed] [Google Scholar]