

Abstract
During eukaryotic mRNA transcription, the synthetic activity and mRNA processing factor interactions of RNA polymerase II (RNAP II) are regulated by phosphorylation of its carboxyl-terminal domain (CTD), with modification occurring primarily on serines 2 and 5 of the CTD. We previously showed that herpes simplex virus type 1 (HSV-1) infection rapidly triggers the loss of RNAP II forms bearing serine 2 phosphorylation (Ser-2P RNAP II). Here we show that the HSV-1 immediate-early (IE) protein ICP22 is responsible for this effect during the IE phase of infection. This activity does not require the viral UL13 protein kinase, which is required for several other regulatory functions of ICP22. Additionally, we show that transient expression of ICP22 can trigger the loss of Ser-2P RNAP II in transfected cells. Thus, the ability of ICP22 to cause the loss of Ser-2 RNAP II does not require other viral factors or the context of the infected cell. Expression of the HSV-1 ICP22-related protein US1.5, which corresponds to residues 147 to 420 of ICP22, also triggers a loss of Ser-2P RNAP II in transfected cells, whereas expression of the varicella-zoster virus ICP22 homolog, ORF63, does not. Our study also provides evidence for a second, viral late gene-dependent pathway that triggers loss of Ser-2P RNAP II in infected cells, consistent with the recent work of Dai-Ju et al. (J. Q. Dai-Ju, L. Li, L. A. Johnson, and R. M. Sandri-Goldin, J. Virol. 80:3567-3581, 2006). Therefore, it appears that HSV-1 has evolved redundant mechanisms for triggering the loss of a specific phosphorylated form of RNAP II.
Herpes simplex virus type 1 (HSV-1) is a common human pathogen that can replicate in epithelial cells and other cells of the host or alternatively can remain latent in peripheral neurons. During productive infections, HSV-1 genes are transcribed in the nucleus by the host cell RNA polymerase II (RNAP II). Expression of the ∼80 viral genes occurs in a coordinately activated cascade that consists of the sequential expression of immediate-early (IE), delayed-early (DE), and late (L) genes.
Although transcription of viral genes is mediated by RNAP II, viral regulatory proteins modulate the specificity of RNAP II during infection (reviewed in reference 47). IE promoters are the only class utilized initially by RNAP II, with the assistance of the viral tegument protein VP16. This leads to the expression of the five IE proteins. Subsequent expression of DE/L genes requires the action of IE proteins. Four of these, infected cell protein 0 (ICP0), ICP4, ICP22, and ICP27, have been implicated in gene expression. ICP4 is the most critical for further viral gene expression, being required for the transcription of nearly all DE and L genes (for a recent discussion of ICP4, see reference 60). ICP4 binds to DNA, interacts with basal cell transcription factors, and helps to promote the formation of RNAP II transcription initiation complexes on DE/L promoters. The molecular functions of ICP0, ICP22, and ICP27 are less well understood, but each has been implicated in inducing expression of viral DE/L genes (9, 20, 39, 44, 49, 52), and recent evidence shows that ICP27 physically interacts with RNAP II (10, 62). At the same time that RNAP II transcribes viral DE/L genes, its transcriptional activity on many cellular genes decreases dramatically (21, 25, 54-56). The mechanisms by which RNAP II is redirected from host to viral genes are not well understood but may involve physical changes to RNAP II itself, as discussed below.
RNAP II is a multiprotein enzyme consisting of 12 subunits (for a review, see reference 59). The C-terminal domain (CTD) of the large subunit (LS) of RNAP II consists of numerous repeats (52 in humans) of the heptapeptide consensus sequence YSPTSPS and is the site of abundant phosphorylation by cellular CTD kinases. These modifications regulate many aspects of RNAP II transcription, including initiation, elongation, and the recruitment of mRNA processing factors to the transcription complex (22, 27, 33, 35, 36, 50). As a result of its extensive CTD phosphorylation, RNAP II exists in either of two distinct states in eukaryotic cells: a hypophosphorylated form, termed RNAP IIA, or a hyperphosphorylated form, termed RNAP IIO. The LS forms in RNAP IIA and RNAP IIO are designated IIa and IIo, respectively, and migrate on a sodium dodecyl sulfate-polyacrylamide gel with distinct apparent molecular masses of 210 and 240 kDa. The two different forms of RNAP II have distinct roles in transcription: RNAP IIA is recruited to promoters to initiate transcription, whereas RNAP IIO is actively engaged in mRNA elongation. Multiple cellular CTD kinases have been identified, and these appear to mainly target serine 2 or 5 (Ser-2 and Ser-5) of the heptapeptide repeat. Two of the most important of these are cyclin-dependent kinase 7 (cdk7) and cdk9. cdk7 is a component of the general transcription factor TFIIH and targets Ser-5 during the promoter clearance phase of transcription. Phosphorylation on Ser-5 is also crucial for recruitment of mRNA capping factors. cdk9 is a component of positive transcriptional elongation factor b (P-TEFb) and targets Ser-2 at a postinitiation stage of transcription. Modification on Ser-2 is associated with enhanced elongation efficiency and the recruitment of polyadenylation factors. In addition to CTD kinases, several cellular CTD phosphatases have been identified, although these are not as well characterized as the CTD kinases.
We and others have shown that HSV-1 infection has major effects on RNAP II and its CTD phosphorylation. Early in infection, the hyperphosphorylated RNAP IIO form disappears and is replaced by a novel form in which the LS has an intermediate electrophoretic mobility, likely reflecting an intermediate level of CTD phosphorylation (46). We designated this form RNAP III (for intermediate) and the corresponding form of LS IIi. Whereas RNAP IIO bears phosphorylation on both Ser-2 and Ser-5, RNAP III is phosphorylated almost exclusively on Ser-5 (16). Consistent with this, RNAP II forms bearing Ser-2 phosphorylation (hereafter referred to as Ser-2P RNAP II) are rapidly lost following infection with HSV-1 (10, 16) in a process that involves proteasome-mediated degradation of RNAP II (10). However, the induction of RNAP III appears to be mechanistically separate from the loss of Ser-2P RNAP II, since the former but not the latter requires the HSV-1 proteins ICP22 and UL13 (16, 24).
In this study, we sought to identity the viral factor (or factors) which trigger the loss of Ser-2P RNAP II. Dai-Ju et al. recently provided evidence that viral L gene transcription can trigger the loss of Ser-2P RNAP II (10). However, viral L gene expression cannot be the sole trigger, since we previously found that Ser-2P RNAP is lost in cells infected with an ICP4 mutant which is unable to express DE/L genes (16). This suggests the existence of another pathway for triggering Ser-2P RNAP II loss. In this study, we show that the IE protein ICP22 is responsible for mediating the loss of Ser-2P RNAP II early in infection. Thus, it appears that HSV-1 has evolved two distinct mechanisms for triggering the loss of a specific phospho-species of host RNAP II.
MATERIALS AND METHODS
Cells, viruses, and infections.
Vero cells (African green monkey kidney cells) were obtained from the American Type Culture Collection. E5 (13) and V22 (31) cells are Vero cells that contain stably transfected copies of the HSV-1 ICP4 and ICP22 genes, respectively. Vero cells were grown in Dulbecco modified Eagle medium supplemented to contain 5% heat-inactivated fetal bovine serum, 50 U of penicillin/ml, and 50 μg of streptomycin/ml. The medium for the E5 and V22 cells was the same except that it also contained 300 μg/ml G418.
The wild-type (WT) strain of HSV-1 used in these studies was KOS1.1 (19). Viral ICP0 (n212) (7), ICP4 (d120) (12), ICP22 (d22lacZ) (24), ICP27 (d27-1) (44), and UL13 (d13lacZ) (24) mutants have been described, as well as an ICP22-UL13 double mutant (d22/13) (16). The construction of the ICP4-ICP22 double mutant d120/22 is described below. F22 is a derivative of KOS1.1 that expresses N-terminally FLAG epitope-tagged ICP22 (C. Spencer and S. Rice, unpublished). Infections were carried out at a multiplicity of infection of 10 in phosphate-buffered saline containing 0.1% glucose and 0.1% heat-inactivated newborn calf serum. Viral absorption was for 1 h at 37°C, at which time the viral inoculum was replaced with 199 medium containing 2% heat-inactivated newborn calf serum, 50 U of penicillin/ml, and 50 μg of streptomycin/ml. All infections were incubated thereafter at 37°C. The inoculum and/or medium for some infections contained 50 μg/ml cycloheximide (CH), 400 μg/ml phosphonoacetic acid (PAA), or 10 μg/ml of actinomycin D (ActD).
The ICP4-ICP22 double mutant d120/22 was constructed by a marker transfer procedure (44). Briefly, infectious viral DNA isolated from d120 was cotransfected into E5 cells with AgeI-digested pBglOZ DNA, which contains an Escherichia coli lacZ gene-disrupted ICP22 gene (24). Viral recombinants that expressed beta-galactosidase were identified by plaquing the cell lysates on E5 cells in the presence of 300 μg/ml 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside. A blue isolate was plaque purified three times in E5 cells and designated d120/22. As expected, d120/22 is unable to replicate in Vero cells but can replicate efficiently in E5 cells. Immunoblot analysis confirmed that d120/22-infected Vero cells do not express either ICP4 or ICP22.
Plasmids and transfections.
An ICP22 expression plasmid, pcDNA22, was constructed in the following manner. The ICP22 coding region of strain KOS1.1 was PCR amplified from plasmid pBamN-β (24) using specific primers (5′, GATCGGATCCATGGCCGACATTTCCCCAGG; 3′, GATCGAATTCTCGGGGCGACCGGACTCACG). The PCR product was digested with BamHI and EcoRI and cloned into pCMV-Tag2B (Stratagene). This construct, designated pCMVTag22, encodes an ICP22 molecule having an N-terminal FLAG epitope tag. DNA sequencing confirmed that the ICP22 sequence in pcDNA22 was identical to that in pBamN-β. However, transfection experiments indicated that ICP22 expression from pCMVTag22 was low, similar to what has been observed by some others using this expression vector (58). Therefore, the FLAG-ICP22 coding region was subcloned into an alternate expression vector, pcDNA3.1(−) (Invitrogen), using the NotI and EcoRI sites, giving rise to pcDNA22. Plasmid pcDNAUS1.5 was constructed in a manner analogous to that of pcDNA22, except that the initial PCR was carried out using the 5′ primer GATCGGATCCATGGGGCGGGTCCGGTCTAC, which corresponds to codon 147 of the ICP22 gene (the start codon for translation of US1.5) (29).
An expression vector for the varicella-zoster virus (VZV) ORF63 gene, pcDNAorf63, was made by the following steps. Plasmid pSstf15 (11), which has a genomic insert of VZV DNA and was a kind gift of Andrew Davison, was digested with PciI. This cuts at the initiation codon of ORF70 (which is identical to ORF63). The 5′ overhanging ends were made blunt by a fill-in reaction using the Klenow fragment, and the DNA was cleaved with BamHI, which cuts 3′ to the ORF70 gene. The ORF70-containing fragment was then cloned into SrfI/BamHI-digested pCMV-Tag2B, giving rise to pCMVTagorf63. This plasmid encodes FLAG-tagged ORF63. To increase expression of ORF63, the FLAG-ORF63 gene was moved into the pcDNA3.1(−) vector using the NotI and EcoRI sites, as described above. This plasmid was designated pcDNAorf63.
In some experiments, the plasmids pCMVβ-c (34) and pEGFP-C2 (Clontech), which express beta-galactosidase and enhanced green fluorescent protein, respectively, from the cytomegalovirus immediate-early (CMV IE) promoter, were used as controls.
Transfections and immunoblots.
Transfection of Vero cells was done using Lipofectamine 2000 (Invitrogen) according to the manufacturer's recommended protocol. For immunoblotting, slightly subconfluent cells in 25-cm2 tissue culture flasks were transfected with 8 μg of plasmid DNA. For immunofluorescence, subconfluent cells plated on coverslips in 4-cm2 wells were transfected with 1.6 μg of plasmid DNA per well. When less than these amounts of plasmid were used (e.g., in Fig. 6D), salmon sperm DNA was added to bring the total amount of DNA transfected to either 8 or 1.6 μg.
FIG. 6.

ICP22 expression in transfected cells triggers loss of Ser-2P RNAP II. (A) Immunoblot analysis of transfected plasmids. Vero cells were transfected with pCMVβgal (lane 1), pcDNAUS1.5 (lane 3), pcDNA22 (lane 4), or pcDNAORF63 (lane 5), and protein extracts were harvested 2 days posttransfection. The proteins were analyzed by immunoblotting using an anti-FLAG antiserum. As a positive control, a protein extract from F22, a recombinant virus expressing an amino-terminally FLAG-tagged ICP22, was also analyzed (lane 2). (B) ICP22 expression triggers loss of Ser-2P RNAP II. Vero cells transfected with pcDNA22 were fixed 2 days posttransfection and stained for ICP22 (panel a) or Ser-2P LS (panel b). Nuclei positive for ICP22 expression are marked by white arrowheads. (C) Specificity of ICP22-induced loss of Ser-2P RNAP II. Cells were transfected with pcDNA22 (panels a to i) or pCMV-βgal (panels j to l) and analyzed by immunofluorescence at 2 days posttransfection for the antigens indicated. (D) Effect of US1.5 or VZV ORF63 expression on Ser-2P RNAP II. Cells were transfected with 0.1 μg pcDNAUS1.5 (panels a to c) or 1.6 μg pcDNAorf63 (panels d to f) per well. Control immunoblotting experiments indicated that these amounts of plasmid express comparable total amounts of US1.5 and ORF63 proteins based on anti-FLAG reactivity. At 2 days posttransfection, the cells were analyzed by immunofluorescence for the FLAG and Ser-2P LS antigens. The Ser-2P LS patterns shown are typical of the large majority (>80%) of FLAG-positive cells.
Analysis of protein expression by immunoblotting was carried out as described previously (16, 24, 34). To detect Ser-2P RNAP II, the mouse monoclonal antibody H5 (Covance, Denver PA) (5, 33) was used at a dilution of 1:500. The cellular early endosomal antigen (EEA1) was detected using a mouse monoclonal antibody (BD Biosciences, San Jose, CA) diluted 1:2,500. The mouse monoclonal antibodies for ICP4 (H1114), ICP0 (H1112), ICP27 (H1119), and ICP8 (H1115) were purchased from the Rumbaugh-Goodwin Institute for Cancer Research (Plantation, FL) and were used at dilutions of 1:800, 1:1,000, 1:5,400, and 1:300, respectively. Rabbit polyclonal antiserum specific for ICP22 was a generous gift from John Blaho (Mt. Sinai School of Medicine) and was used at a dilution of 1:500. Rabbit anti-FLAG polyclonal antiserum (Immunology Consultants, Newberg, OR) was used at a 1:1,000 dilution. The secondary antibodies used for immunoblot detection were horseradish peroxidase-conjugated goat anti-mouse and anti-rabbit immunoglobulin G, purchased from Jackson ImmunoResearch (West Grove, PA), and were both diluted 1:7,500. Secondary antibodies were detected with enhanced chemiluminescence (ECL) Western blotting detection reagents (Amersham).
Quantitation of LS levels by immunoblotting was done by two methods. In the first (see Fig. 2 and 3), LS and EEA1 autoradiography signals were quantitated using a Bio-Rad GS-700 imaging densitometer. To control for variation in protein recovery, the LS signal for each sample was normalized to the EEA1 signal for that sample. In the second method (Fig. 4), detection of the secondary antibody was done directly from the immunoblot by chemifluorescence imaging using ECL-Plus reagents (catalog no. RPN2132; Amersham) and a Molecular Dynamics Storm 840 imager. Again, LS signals for each sample were normalized to the corresponding EEA1 signals.
FIG. 2.

Loss of Ser-2P RNAP II is ICP22 dependent under IE conditions. Replicate cultures of Vero cells were mock infected or infected with KOS1.1 or d22lacZ. In one set of infections, the cells were left untreated (lanes 1, 4, and 7). In the others, the cells were treated with 100 μg/ml CH for 5 h, followed by 2 h without drugs (labeled CHR; lanes 2, 5, and 8) or 2 h in the presence of 10 μg/ml ActD (labeled CHR/A; lanes 3, 6, and 9). Proteins were harvested from all infections at 7 hpi. (A) Immunoblotting analysis of Ser-2P LS, EEA-1, and ICP8. (B) Quantitation of Ser-2P RNAP II levels. The Ser-2P LS signals from the immunoblot shown in panel A were quantitated by densitometry and normalized to EEA1 levels to correct for protein recovery. The level of Ser-2P RNAP II found in untreated, mock-infected cells (lane 1) was set at 100%.
FIG. 3.

Evidence that ICP22 and an L-gene-dependent factor combine to trigger loss of Ser-2P RNAP II. (A) Effect of PAA on Ser-2P RNAP II loss. Replicate cultures of Vero cells were mock infected or infected with WT HSV-1 or the mutants indicated. In one set of infections, the overlay medium contained 400 μg/ml PAA. Protein extracts were prepared at 8 hpi and analyzed for Ser-2P LS and EEA1 by immunoblotting. (B) Effect of ICP22 and PAA on Ser-2P RNAP II levels in a time-course assay. Replicate cultures of Vero cells were mock infected or infected with WT HSV-1 or d22lacZ in the presence or absence of 400 μg/ml PAA added at 1 hpi. Protein extracts were harvested at the times indicated and analyzed for Ser-2P RNAP II and EEA1 by immunoblotting and densitometry. Ser-2P RNAP II levels were normalized to those for EEA1 to correct for protein recovery. The level of Ser-2P LS found in mock-infected cells was set at 100%.
FIG. 4.

Ser-2 RNAP II is retained in cells infected with an ICP4-ICP22 double mutant. (A) Immunoblot analysis. Replicate cultures of Vero, V22, or E5 cells were mock infected or infected with WT HSV-1, d22lacZ, d120, or d120/22. Protein extracts were prepared at 8 hpi and analyzed by immunoblotting as described in the legend to Fig. 1. (B and C) Quantitation of Ser-2P LS loss in cells infected with an ICP4 ICP22 double mutant. Cells were infected as indicated and analyzed by immunoblotting for Ser-2P LS and EEA1 (B). For quantitation (C), Ser-2P LS and EEA1 signals were obtained directly from the blot by chemifluorescence imaging, and Ser-2P LS levels were normalized to those for EEA1 to correct for protein recovery. The level in mock-infected cells was set to 100%.
Indirect immunofluorescence.
For infection experiments, fixation and permeabilization were done as described by Zeng et al. (61). Briefly, the cells were permeabilized for 2 min with 0.5% Triton X-100 in CSK buffer (10 mM PIPES, pH 7, 1 mM EGTA, 3 mM MgCl2, 20% sucrose) and then fixed in 3.7% formaldehyde in the same buffer for 20 min. For transfection experiments, fixation was done immediately in formaldehyde, followed by acetone permeabilization (43). For fluorescent staining, coverslips were incubated at 37°C for 1 h with various primary antibodies. The primary antibodies were H5, diluted 1:100; ARNA3 (Fitzgerald, Concord MA), which recognizes the body of the RNAP II LS, diluted 1:100; H14 (Covance), which recognizes the CTD when it is phosphorylated on Ser-5 (33), diluted 1:300; rabbit immunoglobulin G specific for beta-galactosidase (Rockland Immunochemicals, Gilbertsville PA), diluted 1:200; rabbit anti-ICP22 antisera, diluted 1:250; and anti FLAG antisera, diluted 1:300. After primary incubation, cells underwent secondary staining for 1 h at 37°C. When a combination of mouse and rabbit primary antibodies was used, secondary staining was done with a 1:2,000 dilution of Cy3-conjugated goat anti-mouse immunoglobulin G and a 1:500 dilution of Cy2-conjugated goat anti-rabbit immunoglobulin G. When cells were costained for Ser-2P RNAP II and ICP27, secondary staining was done with a 1:1,000 dilution of Cy3-conjugated goat anti-mouse immunoglobulin M μ chain antibody and a 1:200 dilution of Cy2-conjugated antimouse immunoglobulin G Fc fragment antibody. All secondary antibodies were purchased from Jackson ImmunoResearch.
RESULTS
ICP22 triggers loss of Ser-2P RNAP II in infected cells.
We previously showed that HSV-1 infection induces the loss of Ser-2P RNAP II and that this effect is dependent upon viral gene expression, since Ser-2P RNAP II is not lost when cells are infected in the presence of the protein synthesis inhibitor CH (16). Dai-Ju et al. confirmed these results and provided evidence that a factor associated with HSV-1 L gene expression is responsible (10). However, our previous results (16) indicated that Ser-2P RNAP II loss can be triggered by infection with an ICP4 null mutant, which is unable to express DE or L genes. This suggests that an IE factor (or factors) can also trigger Ser-2P RNAP II loss. However, using viral IE mutants, we previously were unable to implicate any of the four known IE regulatory proteins (ICP0, ICP4, ICP22, and ICP27) (16).
We reasoned that in our past experiments, the effect of IE proteins on Ser-2P RNAP II loss could have been obscured by the L-gene-dependent pathway described by Dai-Ju et al. (10). To further explore loss of Ser-2P under IE conditions, we decided to enhance the IE phase of infection through the use of a CH reversal protocol (18). In this procedure, cells are infected in the presence of the protein synthesis inhibitor CH, which leads to the accumulation of high levels of IE mRNAs. Upon removal of CH (i.e., reversal), IE proteins are synthesized at enhanced levels for a period of time before gene expression transitions to the DE/L genes. To carry out the analysis, Vero cells were infected in the presence of 100 μg/ml CH for 5 h, followed by a 2-h reversal without CH, at which time the proteins were harvested. For comparison, we also carried out a 7-h infection without CH treatment. The protein samples were analyzed by immunoblotting using the phospho-specific anti-CTD monoclonal antibody H5, which reacts with LS only when it bears Ser-2 phosphorylation (33). The results (Fig. 1A) showed that under CH reversal conditions, cells infected with the WT strain KOS1.1 exhibited a near-complete loss of Ser-2P RNAP II (lane 4) compared to results for mock-infected cells (lane 2). Similarly, HSV-1 strains defective in ICP4 (d120), ICP0 (n212), and ICP27 (d27-1) also showed a dramatic loss of this form of RNAP II (lanes 8, 10, and 12, respectively). In contrast, cells infected with the ICP22 deletion mutant d22lacZ exhibited relatively high levels of Ser-2P RNAP II after the CH reversal (lane 6). In fact, even without CH treatment, there was somewhat more Ser-2P RNAP II in the d22lacZ infection (lane 5) than in any of the other nontreated infections (although there was still a substantial loss). As a control, a separate region of the same immunoblot was probed for the cellular protein EEA1, which does not change in abundance after HSV-1 infection (16). EEA1 levels were similar in all samples, indicating that comparable levels of proteins were loaded in each lane. Additional immunoblotting analysis confirmed that all of the viral strains expressed the expected set of IE proteins (Fig. 1A, bottom panels). Thus, these results indicate that at least under CH reversal conditions, ICP22 plays a key role in triggering the loss of Ser-2P RNAP II.
FIG. 1.
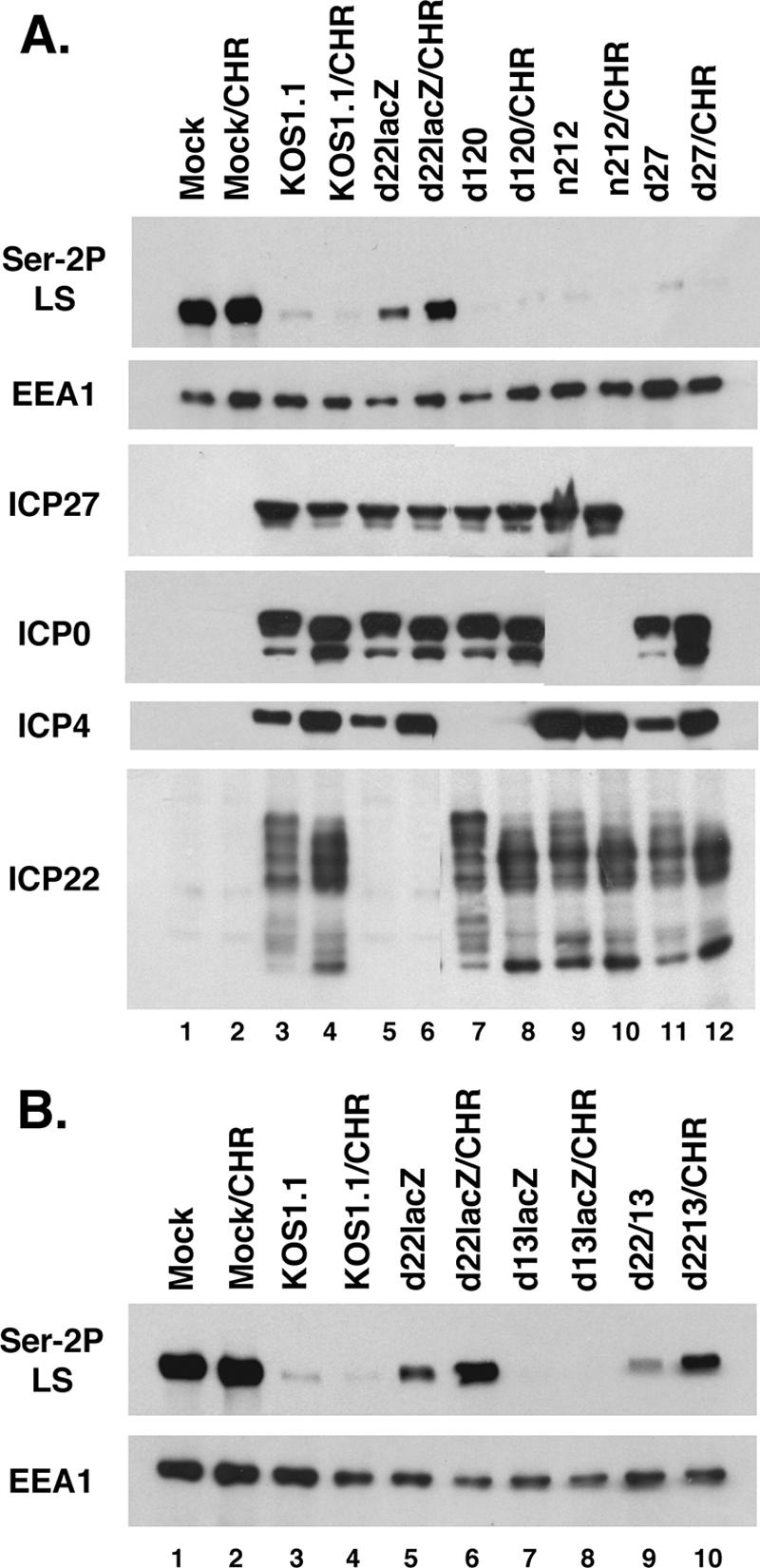
ICP22 promotes loss of Ser-2P RNAP II in infected cells. (A) Immunoblot analysis of viral IE mutants after CH reversal. Duplicate cultures of Vero cells were mock infected or infected with WT HSV-1 strain KOS1.1 or the mutants indicated. In one set of infections (lanes 1, 3, 5, 7, 9, and 11), the cells were not treated with drugs, and total proteins were harvested at 7 hpi. In the other set (labeled CHR; lanes 2, 4, 6, 8, 10, and 12), cells were infected and maintained in the presence of 100 μg/ml CH for 5 h. The CH was then removed, and the cells were incubated for two more hours prior to harvesting. Protein extracts were analyzed by immunoblotting for Ser-2P RNAP II, EEA1, ICP27, ICP0, ICP4, and ICP22. (B) Analysis of ICP22 and UL13 mutants after CH reversal. The experiment was performed as described for panel A.
Many of ICP22's regulatory functions, including its ability to induce the intermediately phosphorylated RNAP III form (24), require the HSV-1 protein kinase UL13, which has been implicated in phosphorylating ICP22 (42). To see whether UL13 is involved in loss of Ser-2P RNAP II, we carried out the CH reversal analysis with the HSV-1 UL13 mutant d13lacZ. As can be seen in Fig. 1B, the UL13 mutant (lane 8) resembled KOS1.1 (lane 4) in its ability to trigger Ser-2P RNAP II loss. In contrast, both the ICP22 mutant d22lacZ and an ICP22-UL13 double mutant d22/13 (lanes 6 and 10, respectively) were deficient in this activity. Thus, the ability of ICP22 to trigger loss of Ser-2P RNAP II does not require UL13.
The conditions used in the above CH reversal experiments enhance IE protein synthesis, but they do not completely prevent the ensuing expression of DE/L proteins. However, if the transcriptional inhibitor ActD is added during the reversal, then DE and L genes cannot be transcribed and protein synthesis is tightly restricted to IE proteins (18). We used this protocol modification to ask whether Ser-2P RNAP II loss is triggered under strict IE conditions and, if so, whether ICP22 is responsible. Vero cells were infected with KOS1.1 or d22lacZ in the presence of CH for 5 h, followed by a 2-h reversal, in either the presence or absence of ActD. Another set of infections was not treated with any drugs. All infections were terminated at 7 h postinfection (hpi), and the proteins were analyzed by immunoblotting for Ser-2 RNAP II, EEA1, and the DE protein ICP8 (Fig. 2A). In addition, the results of the Ser-2P LS blot were quantified by densitometry; in order to control for protein recovery, the LS signals were normalized to those for EEA1 (Fig. 2B). Inspection of the ICP8 blot revealed that this DE protein was not detectably expressed when ActD was present during the CH reversal (lanes 6 and 9), confirming that ActD restricted expression to IE proteins. Several conclusions can be made from the data. First, in the KOS1.1 infection reversed in the presence of ActD, there was significant loss of Ser-2P RNAP II, to ∼50% of the mock value (lane 6). Thus, IE gene expression is sufficient to trigger loss of Ser-2P RNAP II. Second, no loss occurred in the d22lacZ infection reversed in the presence of ActD (lane 9). This indicates that ICP22 is the IE factor which is responsible for loss of Ser-2P RNAP II. Third, there was significantly more loss of Ser-2P RNAP II in the KOS1.1 and d22lacZ infections when ActD was absent in the reversal than when it was present (compare lane 5 to lane 6 and lane 8 to lane 9). This suggests that a factor dependent upon DE/L gene expression can also trigger loss of Ser-2P RNAP II, consistent with the work of Dai-Ju et al. (10).
Evidence that a viral L factor also triggers loss of Ser-2P RNAP II.
The results above suggest that HSV-1 may trigger dual pathways, one involving ICP22 and one involving DE/L gene expression, that can each induce the loss of Ser-2P RNAP II. We wished to further explore the role of DE/L gene expression and if possible to differentiate between the effects of DE and L genes. One way that DE and L genes can be distinguished is by the effects of viral DNA replication inhibitors, such as PAA, upon their expression (17). PAA potentiates the expression of DE genes but inhibits or prevents the expression of L genes. To see how PAA affects the loss of Ser-2P RNAP II, we mock infected Vero cells or infected them with KOS1.1 or various IE mutants in the presence or absence of 400 μg/ml PAA. Total proteins were harvested at 8 hpi and analyzed for Ser-2P RNAP II levels (Fig. 3A). PAA had little if any effect on mock- or virus-infected cells, with one striking exception. When it was added to the d22lacZ infection, significantly more Ser-2P RNAP II was retained (lane 6). Thus, in the absence of ICP22, PAA significantly inhibits the loss of Ser-2P RNAP II. This suggests that ICP22 and an L-phase factor combine to fully trigger the loss.
We next carried out a time course experiment to observe the individual and combined effects of ICP22 and PAA addition on the loss of Ser-2P RNAP II. Vero cells were infected with KOS1.1 or d22lacZ in the presence or absence of PAA, and protein samples were prepared every 1.5 h through 9 h of infection. Ser-2P RNAP II and EEA1 levels were analyzed by quantitative immunoblotting. The results, shown in Fig. 3B, indicate that neither the deletion of ICP22 nor the addition of PAA to WT HSV-1 infection was alone sufficient to prevent the overall depletion (to <25%) of Ser-2P RNAP II by 9 hpi. However, when d22lacZ-infected cells were treated with PAA, loss of Ser-2P RNAP II was substantially suppressed, with ∼60% still remaining at 9 hpi. Thus, both ICP22 and a viral late factor appear to be needed to fully trigger loss of Ser-2P RNAP II. We also noted that PAA treatment of the KOS1.1 infection led to an acceleration of Ser-2P RNAP II loss, compared to the untreated infection, beginning at about 4.5 hpi. We considered the possibility that this effect was due to enhanced ICP22 expression in the presence of PAA. However, immunoblot analysis of these samples indicated that PAA treatment did not increase ICP22 expression in KOS1.1-infected cells (data not shown). Thus, the reason for the PAA-induced acceleration of Ser-2P RNAP II loss in the WT infection is currently unknown.
An ICP22-ICP4 double mutant fails to induce loss of Ser-2P RNAP II.
The above experiments, coupled with the work of Dai-Ju et al. (10), suggest that ICP22 and viral late gene expression represent dual pathways by which HSV-1 triggers loss of Ser-2P RNAP II. To gain independent evidence of this, we employed a genetic approach by constructing an ICP4-ICP22 double mutant, which we designated d120/22 (see Materials and Methods for details). Since DE/L gene expression is highly dependent upon ICP4 (12, 57), this mutant cannot express either ICP22 or L genes. Thus, according to our model, it would be predicted to be unable to trigger any loss of Ser-2P RNAP II. To test this, Vero cells were infected with KOS1.1, d22lacZ, d120, or d120/22. As controls, infections were also carried out in V22 (31) or E5 (13) cells, which are Vero derivatives that express ICP22 and ICP4, respectively, upon infection. Proteins were harvested at 8 hpi and analyzed by immunoblotting. As expected, by 8 hpi, KOS1.1 had triggered a dramatic decrease in Ser-2P RNAP II in Vero cells (Fig. 4A, lane 2), as had d22lacZ (lane 5) and d120 (lane 8). However, d120/22-infected Vero cells retained most if not all Ser-2P RNAP II (lane 11). Significantly, d120/22 triggered Ser-2P RNAP II loss in both V22 or E5 cells (lanes 12 and 13, respectively), indicating that expression of either ICP22 or ICP4 can restore the ability of this mutant to trigger the loss. Immunoblotting analyses confirmed that all viruses and cells used in the experiment expressed the expected set of IE proteins (Fig. 4A, bottom panels). It was noted that the electrophoretic mobility of ICP22 detected in V22 cells varied depending upon the mutant used for the infection. That is, the mobility of ICP22 was relatively slow in the KOS1.1 and d22lacZ infections (Fig. 4A, lanes 3 and 6) but faster in the d120 and d120/22 infections (lanes 9 and 12). This most likely reflects UL13-dependent posttranslational modification of ICP22. Purves et al. have shown that it is newly expressed UL13, not virion UL13, that is responsible for the modification of ICP22 (41). Since UL13 is almost certainly not expressed in the d120 and d120/22 infections due to the absence of ICP4, the ICP22 made under these conditions is likely incompletely modified, thus explaining its enhanced electrophoretic mobility.
To obtain quantitative data concerning the loss of Ser-2P RNAP II, we repeated the above infection, and in this case, Ser-2 RNAP II signal intensities were obtained directly from the immunoblots by chemifluorescence imaging, again normalizing the signals to those for EEA1 to control for protein recovery (Fig. 4B and C). The results indicated that as expected, Ser-2P RNAP II levels were reduced significantly in cells infected with KOS1.1, d22lacZ, or d120. However, no reduction compared to results for mock-infected cells was seen in the d120/22 infection. Thus, deletion of both ICP22 and ICP4 completely abrogates HSV-1's ability to trigger the loss of Ser-2P RNAP II. This is consistent with a model in which both ICP22 and L gene expression independently trigger the loss.
In our previous study, we used immunofluorescence to confirm that there is a significant loss of Ser-2P RNAP after HSV-1-infection (16). In the course of those experiments, we observed that a residual fraction of Ser-2P RNAP II is not lost after HSV-1 infection but is instead relocalized into nuclear speckle structures. Similar results were reported by Dai-Ju et al. (10). Nuclear speckles are normal components of mammalian cell nuclei and contain many cellular splicing proteins and some other factors involved in mRNA production (23). To see what effect d120/22 infection has on Ser-2P RNAP II subcellular localization, we carried out an immunofluorescence analysis of mock- or HSV-1-infected Vero cells that had been fixed at 8 hpi (Fig. 5). As expected, KOS1.1 infection induced a striking loss of Ser-2P RNAP II (panel d). In addition, some of the residual antigen relocalized to nuclear speckles, many of which costained for ICP27, as previously reported (panels e and f) (37). Infections with d22lacZ and d120 gave similar results (panels g to i and j to l). In contrast, there was little if any diminution of the Ser-2P RNAP II signal in d120/22-infected cells (panel m), consistent with our immunoblotting analyses. Interestingly, infection with d120/22 still induced the relocalization of a fraction of Ser-2P RNAP II into nuclear speckles that partially costained for ICP27 (panels n and o). This indicates that the HSV-1-induced movement of Ser-2P RNAP II into nuclear speckles is independent of the loss of Ser-2P RNAP II and does not require either ICP22 or ICP4.
FIG. 5.

Immunofluorescence analysis of Ser-2P RNAP II in cells infected with an ICP4-ICP22 double mutant. Vero cells were mock infected or infected with WT HSV-1 or the mutants indicated. At 8 hpi, the cells were fixed and processed for immunofluorescence using antibodies specific for Ser-2P LS or ICP27. The cells shown are representative of the majority of cells (>80%) in the sample.
Expression of ICP22 by transfection triggers loss of Ser-2P RNAP II.
Since our results showed that ICP22 plays a key role in the loss of Ser-2P RNAP II in infected cells, we investigated the possibility that ICP22 might affect Ser-2P RNAP II in uninfected cells. To test this, we constructed an expression plasmid, pcDNA22, that encodes an N-terminally FLAG-tagged ICP22 molecule (Fig. 6A, lane 4). Vero cells were transfected with pcDNA22 and analyzed by immunofluorescence at 2 days posttransfection. Intriguingly, we consistently observed that cells which were positive for ICP22 almost invariably exhibited minimal to no staining for Ser-2 RNAP II (Fig. 6B, compare panels a and b).
Additional experiments were carried out to address the specificity of ICP22's effects on RNAP II. As seen in Fig. 6C, although ICP22 expression consistently triggered loss of Ser-2P RNAP II staining (panels a to c), staining for Ser-5P RNAP II, detected by monoclonal antibody H14, was unaffected (panels d to f). Similar analysis with monoclonal antibody ARNA3, which binds to the body of the RNAP II LS and thus detects all forms of the protein, indicated that ICP22 did not appreciably affect the levels of total LS (panels g to i). Since ICP22 expression from pcDNA22 is driven by the robust human CMV IE promoter, we considered the possibility that high-level transcription of any transfected gene might trigger loss of Ser-2P RNAP II. However, this was not the case, since CMV IE promoter-driven expression of β-galactosidase (Fig. 6C, panels j to l) or enhanced green fluorescent protein (data not shown) did not alter Ser-2P RNAP II staining. Furthermore, ICP22's ability to induce loss of Ser-2P RNAP II did not require the CMV IE promoter, since similar results were observed when ICP22 was expressed from its endogenous promoter on plasmid pBamN (24) (data not shown). Finally, ICP22's ability to trigger loss of Ser-2P RNAP II was not specific to Vero cells, since we observed similar results in transfected HeLa and ARPE-19 (pigmented retinal epithelial) cells (data not shown). Based on these results, we conclude that ICP22 expressed in transfected cells can specifically trigger the loss of the Ser2-phosphorylated form of RNAP II.
Carter and Roizman have shown that the C-terminal region of the ICP22 gene harbors a related gene, designated US1.5, that encodes a protein corresponding to a C-terminal segment of ICP22 (residues 147 to 420) (8). Thus, it was of interest to ask whether the US1.5 protein is capable of causing the loss of Ser-2P RNAP II in transfected cells. Additionally, we were curious as to whether the VZV homolog of ICP22, ORF63, might also carry out this activity. To answer these questions, we constructed plasmids which express either FLAG-tagged US1.5 or ORF63 and transfected them into Vero cells. Immunoblot analysis confirmed that these constructs expressed FLAG-reactive proteins of the expected sizes, although expression of ORF63 was consistently lower than that of ICP22 and US1.5 (Fig. 6A, lanes 3 and 5). To see whether US1.5 or ORF63 can affect RNAP II, we transfected the plasmids into Vero cells and performed immunofluorescence at 2 days posttransfection. The results showed that the US1.5 protein triggered the loss of Ser-2P RNAP II staining, whereas the ORF63 protein did not appear to do so (data not shown). The negative result seen for ORF63 did not appear to be due to the lower level of expression of ORF63, because even when the amount of transfected US1.5 plasmid was reduced 16-fold to compensate for its higher expression efficiency, US1.5 still efficiently triggered loss of Ser-2P RNAP II staining (Fig. 6D, panels a to c), but ORF63 did not (panels d to f). We conclude that similar to the case with ICP22, the HSV-1 US1.5 protein can trigger loss of Ser-2P RNAP II in transfected cells. In contrast, the VZV ORF63 protein does not appear to possess this activity.
DISCUSSION
Two distinct viral pathways trigger the loss of Ser-2P RNAP II during HSV-1 infection.
In the standard eukaryotic mRNA transcription cycle, the RNAP II CTD becomes phosphorylated on Ser-2 at a postinitiation stage of transcription. This modification is associated with the transition of RNAP II to an elongation-competent form and is critical for the recruitment of polyadenylation and possibly splicing factors to the transcription complex. Given the importance of Ser-2P RNAP II to cellular gene transcription, it is intriguing that HSV-1-infected cells rapidly lose this form of RNAP II (10, 16). In this study, we have investigated the viral factors that trigger this disappearance. Our major finding is that HSV-1 possesses two separate pathways that trigger this loss. One is mediated by the IE protein ICP22 and is operative at early times after infection. The other is ICP22 independent, and depends on L gene expression. Although the identification of ICP22 as a triggering factor is novel, the finding that L-phase events induce Ser-2P loss is consistent with the recent work from Sandri-Goldin's laboratory (10).
Our new findings explain our past results, which appeared to simultaneously implicate and exclude IE proteins in the loss of Ser-2 RNAP II (16). That is, we previously found that IE protein synthesis is both required and sufficient, in the context of HSV-1 infection, for the loss of Ser-2 RNAP II. However, using IE mutants, we were unable to implicate a single known IE regulatory protein (including ICP22) in the effect. Our discovery of redundant functions explains our past results: ICP22 can trigger the loss of Ser-2P RNAP II under IE conditions (e.g., in cells infected with an ICP4 mutant), whereas the viral L factor can trigger the loss in ICP22 mutant-infected cells.
The fact that HSV-1 has evolved redundant pathways for triggering the loss of Ser-2 RNAP II suggests that optimal viral replication or persistence in the human host is favored by this change. HSV-1 is known to encode redundant functions that target other host cell processes. For example, multiple viral gene products can inhibit virus-induced apoptosis (2) or counteract the cellular PKR system (28). What general function could the loss of Ser-2P RNAP II serve? We have previously suggested that HSV-1 gene transcription may not generate or require Ser-2P RNAP II (16). If so, virus-induced loss of Ser-2P RNAP II could serve as a mechanism by which HSV-1 specifically inhibits host cell transcription. Alternatively, Dai-Ju et al. have suggested that HSV-1 genome transcription may generate Ser-2P RNAP II but that Ser-2P RNAP II complexes from opposing viral strands could collide and stall on the HSV-1 genome, ultimately requiring the proteasome for their resolution (10). In this scenario, Ser-2P RNAP II loss may be needed for the productive release of viral transcripts. Obviously, further work is needed to explore these and other possibilities. We expect that that the d120/22 mutant will be helpful in these studies, since it is the first mutant which has been found to be unable to trigger any loss of Ser-2P RNAP II.
HSV-1-induced redistribution of Ser-2P RNAP II to splicing speckles.
In our previous study, we provided evidence that some residual Ser-2P RNAP II is relocalized after HSV-1 infection into nuclear speckles (16). This conclusion was based on immunofluorescence experiments using the H5 antibody. However, the H5 antibody has been reported to cross-react with cellular SR splicing proteins, which localize to splicing speckles, under conditions where the levels of Ser-2P RNAP II are low (14). Thus, Dai-Ju et al. suggested that the apparent redistribution of residual Ser-2P RNAP II to speckles in HSV-1-infected cells could be an artifact driven by the loss of Ser-2P RNAP II (10). However, in this work, we observed that H5 stains speckles in the d120-22 infection, under conditions where there is minimal or no loss of Ser-2P RNAP II. This argues that HSV-1 infection does indeed result in a redistribution of Ser-2P RNAP II into nuclear speckles. The biological significance of this phenomenon is currently unknown, as are the viral factors which are responsible.
ICP22 triggers loss of Ser-2P RNAP II.
Our studies indicate that ICP22 mediates loss of Ser-2 RNAP II during infection and furthermore that ICP22 can mediate this activity even in transfected cells. The latter result indicates that no other viral factors are required for this function. The finding that ICP22 effects the loss of Ser-2P RNAP II is perhaps not surprising, since we have previously found that ICP22 is required during infection for the appearance of the intermediately phosphorylated RNAP III form (45), which lacks Ser-2 phosphorylation (16). One could thus hypothesize that ICP22's primary function is to trigger loss of Ser-2 phosphorylation from RNAP IIO, leading to the appearance of an intermediately migrating form. Indeed, this has been observed in cells treated with a drug that blocks cdk9 activity (26). However, the ability of ICP22 to induce RNAP III is clearly separable from its ability to trigger Ser-2P RNAP II loss, since the former but not the latter requires the HSV-1 UL13 protein as a cofactor (24). Therefore, the most straightforward interpretation of our results is that ICP22 possesses two separate (but possibly related) regulatory activities that affect RNAP II phosphorylation.
ICP22 homologs are conserved among the alphaherpesviruses but are not found in beta- or gammaherpesviruses (48). Our work adds to the list of known functions for HSV-1 ICP22. Although the ICP22 gene is not essential for viral growth in Vero cells and some other cell lines (40), it is required for efficient replication in other cells, such as human embryonic lung cells (39, 52) and primary human fibroblasts (K. Fraser, P. Southern, and S. Rice, unpublished). Furthermore, ICP22 is required for acute replication and virulence in animal models of HSV-1 infection (38, 52). The primary defect of ICP22 mutants in cells where ICP22 is required for efficient growth appears to be a reduction in the expression of a subset of L genes (39, 52). In addition to effects on viral gene expression, ICP22 is responsible for altering the levels and activities of some cell cycle proteins during infection (1, 6, 30). Consistent with such cell cycle effects, an ICP22 mutant has been shown to replicate poorly in normally permissive Vero cells when they are synchronized and infected in S phase (30). Finally, it was recently shown that ICP22 plays a role in determining the protein composition of the viral tegument (31). In regard to this last function, it is worth mentioning that our ICP22 mutant virus stocks are grown in V22 cells and should have the normal complement of tegument proteins.
As noted above, many of ICP22's regulatory functions require the HSV-1 virion protein kinase UL13, which directly or indirectly mediates some of ICP22's complex phosphorylation (42). These functions include ICP22's cell-type-dependent stimulation of viral growth and L gene expression (24, 41), its ability to induce degradation of cyclins A and B (1), and its capacity to induce RNAP III (24). Our results with both infected and transfected cells indicate that ICP22's ability to trigger loss of Ser-2P RNAP II does not require UL13. In fact, in some experiments, we have observed that loss of Ser-2P RNAP in infected cells occurs more rapidly in UL13 mutant infections than it does in WT infections (data not shown). This suggests that UL13 may negatively regulate this function of ICP22. Previous studies have shown that ICP22 phosphorylation is mediated by newly expressed UL13, as opposed to UL13 introduced from the virion (41). Thus, it is possible that early in infection, prior to UL13 expression, ICP22's primary effect on RNAP II could be to trigger the loss of Ser-2 phosphorylation. Later, expression of new UL13 may serve as a switch to alter ICP22's activity so that it mediates induction of the RNAP III form.
Our data indicate that the HSV-1 ICP22-related protein US1.5 also possesses the ability to cause loss of Ser-2P RNAP II. This indicates that the ability of ICP22 to cause loss of Ser-2P RNAP II maps to the C-terminal two-thirds of ICP22 (i.e., residues 147 to 420), which is the portion shared with US1.5. However, we found that the ICP22 homolog of VZV, ORF63, does not trigger the loss of Ser-2P RNAP II, indicating that this function of ICP22 is not conserved in an alphaherpesvirus homolog. We are currently using a plasmid transfection strategy to more precisely map the portion of ICP22/US1.5 which mediates the loss of Ser-2P RNAP II. Given our results with ORF63, it will be interesting to see whether the relevant sequences include those which are widely conserved among ICP22 homologs (51).
Although the mechanism by which ICP22 induces the loss of Ser-2P RNAP II is currently unknown, two potential mechanisms are worth considering. First, ICP22 could promote the specific proteolysis of this form of RNAP II. There is some evidence for this, since Sandri-Goldin and colleagues showed that some Ser-2P RNAP II is degraded during infection by a proteasome-dependent process (10). Second, it is possible that ICP22 affects Ser-2 CTD phosphorylation directly by inhibiting P-TEFb. There is evidence supporting this, as well, since it was recently shown that ICP22 physically interacts with the cdk9 subunit of P-TEFb (15). This interaction could inhibit CTD kinase activity, thus explaining our results. Alternately, it is possible that ICP22 modifies the substrate specificity of P-TEFb, causing it to phosphorylate Ser-5 rather than Ser-2. This would be consistent with experiments showing that ICP22-cdk9 complexes have elevated CTD kinase activity in vitro (15). There are precedents in the literature for the alteration of the CTD specificity of P-TEFb by viral proteins. In one example, the human immunodeficiency virus Tat protein was shown to modify cdk9 activity so that it phosphorylates Ser-5 in addition to Ser-2 (63). In another, the Epstein-Barr virus protein EBNA 2 was found to stimulate CTD phosphorylation at Ser-5 via a cdk9-dependent mechanism (3).
Does expression of ICP22 globally affect cellular transcription?
In several eukaryotic systems, the presence of Ser-2P RNAP II, as assayed by H5 immunoreactivity, has been used as a marker for active RNAP II transcription (reviewed in reference 33). Given this, it was surprising that ICP22-transfected cells are virtually devoid of this antigen 2 days following transfection. This raises the question of whether RNAP II transcription is globally inhibited in transfected cells expressing ICP22. Although we do not yet know the answer, it is worth noting that the ICP22-expressing cells appear morphologically normal, suggesting that they are not dead or dying. Although the lack of Ser-2P RNAP II is unusual for eukaryotic cells and is thought to reflect transcriptional quiescence, it has been documented in several instances. For instance, loss of Ser-2P RNAP II occurs during the early embryonic development of several organisms, including Drosophila, Caenorhabditis elegans, mice, and frogs and is associated with a global but transient repression of zygotic transcription (32, 53). Moreover, some of the first analyses of the H5 antibody indicated that a fraction of cycling mammalian cells do not stain for this antigen, nor do growth-arrested cells (4). Thus, eukaryotic cells can tolerate a lack of Ser-2P RNAP II, at least in certain circumstances, some of which may be cell cycle dependent. We are currently attempting to develop a cell line that harbors an inducible ICP22 gene so that we can study the effect of ICP22 on host cell transcription and physiology under well-controlled conditions.
Acknowledgments
We thank Andrew Davison for the VZV ORF63 plasmid and John Blaho for generously supplying ICP22 antisera. Tran Huynh and Linse Lahti are gratefully acknowledged for construction of plasmids pCMVTag22 and pCMVTagorf63, respectively, and for initial characterization of their effects on RNAP II. We also appreciate the expert technical assistance of Oksana Goldman. Finally, thanks are due to Wade Bresnahan, Tommy Bastian, and Laura Okagaki for helpful comments on the manuscript.
This research was supported by a grant to S.A.R. from the NIH (RO1-AI50127). K.A.F. was supported by an NIH predoctoral training award (T32-AI07421).
Footnotes
Published ahead of print on 7 March 2007.
REFERENCES
- 1.Advani, S. J., R. Brandimarti, R. R. Weichselbaum, and B. Roizman. 2000. The disappearance of cyclins A and B and the increase in activity of the G2/M-phase cellular kinase cdc2 in herpes simplex virus 1-infected cells require expression of the α22/US1.5 and UL13 viral genes. J. Virol. 74:8-15. [PMC free article] [PubMed] [Google Scholar]
- 2.Aubert, M., and J. A. Blaho. 2001. Modulation of apoptosis during herpes simplex virus infection in human cells. Microbes Infect. 3:859-866. [DOI] [PubMed] [Google Scholar]
- 3.Bark-Jones, S. J., H. M. Webb, and M. J. West. 2006. EBV EBNA 2 stimulates CDK9-dependent transcription and RNA polymerase II phosphorylation on serine 5. Oncogene 25:1775-1785. [DOI] [PubMed] [Google Scholar]
- 4.Bregman, D. B., L. Du, Y. Li, S. Ribisi, and S. L. Warren. 1994. Cytostellin distributes to nuclear regions enriched with splicing factors. J. Cell Sci. 107:387-396. [DOI] [PubMed] [Google Scholar]
- 5.Bregman, D. B., L. Du, S. van der Zee, and S. L. Warren. 1995. Transcription-dependent redistribution of the large subunit of RNA polymerase II to discrete nuclear domains. J. Cell Biol. 129:287-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bruni, R., and B. Roizman. 1998. Herpes simplex virus 1 regulatory protein ICP22 interacts with a new cell cycle-regulated factor and accumulates in a cell cycle-dependent fashion in infected cells. J. Virol. 72:8525-8531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cai, W., T. L. Astor, L. M. Liptak, C. Cho, D. M. Coen, and P. A. Schaffer. 1993. The herpes simplex virus type 1 regulatory protein ICP0 enhances virus replication during acute infection and reactivation from latency. J. Virol. 67:7501-7512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carter, K. L., and B. Roizman. 1996. The promoter and transcriptional unit of a novel herpes simplex virus 1 alpha gene are contained in, and encode a protein in frame with, the open reading frame of the alpha 22 gene. J. Virol. 70:172-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen, J., and S. Silverstein. 1992. Herpes simplex viruses with mutations in the gene encoding ICP0 are defective in gene expression. J. Virol. 66:2916-2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dai-Ju, J. Q., L. Li, L. A. Johnson, and R. M. Sandri-Goldin. 2006. ICP27 interacts with the C-terminal domain of RNA polymerase II and facilitates its recruitment to herpes simplex virus 1 transcription sites, where it undergoes proteasomal degradation during infection. J. Virol. 80:3567-3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davison, A. J., and J. E. Scott. 1986. The complete DNA sequence of varicella-zoster virus. J. Gen. Virol. 67:1759-1816. [DOI] [PubMed] [Google Scholar]
- 12.DeLuca, N. A., A. M. McCarthy, and P. A. Schaffer. 1985. Isolation and characterization of deletion mutants of herpes simplex virus type 1 in the gene encoding immediate-early regulatory protein ICP4. J. Virol. 56:558-570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DeLuca, N. A., and P. A. Schaffer. 1988. Physical and functional domains of the herpes simplex virus transcriptional regulatory protein ICP4. J. Virol. 62:732-743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doyle, O., J. L. Corden, C. Murphy, and J. G. Gall. 2002. The distribution of RNA polymerase II largest subunit (RPB1) in the Xenopus germinal vesicle. J. Struct. Biol. 140:154-166. [DOI] [PubMed] [Google Scholar]
- 15.Durand, L. O., S. J. Advani, A. P. Poon, and B. Roizman. 2005. The carboxyl-terminal domain of RNA polymerase II is phosphorylated by a complex containing cdk9 and infected-cell protein 22 of herpes simplex virus 1. J. Virol. 79:6757-6762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fraser, K. A., and S. A. Rice. 2005. Herpes simplex virus type 1 infection leads to loss of serine-2 phosphorylation on the carboxyl-terminal domain of RNA polymerase II. J. Virol. 79:11323-11334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holland, L. E., K. P. Anderson, C. Shipman, Jr., and E. K. Wagner. 1980. Viral DNA synthesis is required for the efficient expression of specific herpes simplex virus type 1 mRNA species. Virology 101:10-24. [DOI] [PubMed] [Google Scholar]
- 18.Honess, R. W., and B. Roizman. 1974. Regulation of herpesvirus macromolecular synthesis. I. Cascade regulation of the synthesis of three groups of viral proteins. J. Virol. 14:8-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hughes, R. G., Jr., and W. H. Munyon. 1975. Temperature-sensitive mutants of herpes simplex virus type 1 defective in lysis but not in transformation. J. Virol. 16:275-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jordan, R., and P. A. Schaffer. 1997. Activation of gene expression by herpes simplex virus type 1 ICP0 occurs at the level of mRNA synthesis. J. Virol. 71:6850-6862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kemp, L. M., and D. S. Latchman. 1988. Induction and repression of cellular gene transcription during herpes simplex virus infection are mediated by different viral immediate-early gene products. Eur. J. Biochem. 174:443-449. [DOI] [PubMed] [Google Scholar]
- 22.Kobor, M. S., and J. Greenblatt. 2002. Regulation of transcription elongation by phosphorylation. Biochim. Biophys. Acta 1577:261-275. [DOI] [PubMed] [Google Scholar]
- 23.Lamond, A. I., and D. L. Spector. 2003. Nuclear speckles: a model for nuclear organelles. Nat. Rev. Mol. Cell Biol. 4:605-612. [DOI] [PubMed] [Google Scholar]
- 24.Long, M. C., V. Leong, P. A. Schaffer, C. A. Spencer, and S. A. Rice. 1999. ICP22 and the UL13 protein kinase are both required for herpes simplex virus-induced modification of the large subunit of RNA polymerase II. J. Virol. 73:5593-5604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mayman, B. A., and Y. Nishioka. 1985. Differential stability of host mRNAs in Friend erythroleukemia cells infected with herpes simplex virus type 1. J. Virol. 53:1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Medlin, J., A. Scurry, A. Taylor, F. Zhang, B. M. Peterlin, and S. Murphy. 2005. P-TEFb is not an essential elongation factor for the intronless human U2 snRNA and histone H2b genes. EMBO J. 24:4154-4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meinhart, A., T. Kamenski, S. Hoeppner, S. Baumli, and P. Cramer. 2005. A structural perspective of CTD function. Genes Dev. 19:1401-1415. [DOI] [PubMed] [Google Scholar]
- 28.Mulvey, M., J. Poppers, D. Sternberg, and I. Mohr. 2003. Regulation of eIF2α phosphorylation by different functions that act during discrete phases in the herpes simplex virus type 1 life cycle. J. Virol. 77:10917-10928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ogle, W. O., and B. Roizman. 1999. Functional anatomy of herpes simplex virus 1 overlapping genes encoding infected-cell protein 22 and US1.5 protein. J. Virol. 73:4305-4315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Orlando, J. S., T. L. Astor, S. A. Rundle, and P. A. Schaffer. 2006. The products of the herpes simplex virus type 1 immediate-early US1/US1.5 genes downregulate levels of S-phase-specific cyclins and facilitate virus replication in S-phase Vero cells. J. Virol. 80:4005-4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Orlando, J. S., J. W. Balliet, A. S. Kushnir, T. L. Astor, M. Kosz-Vnenchak, S. A. Rice, D. M. Knipe, and P. A. Schaffer. 2006. ICP22 is required for wild-type composition and infectivity of herpes simplex virus type 1 virions. J. Virol. 80:9381-9390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Palancade, B., S. Bellier, G. Almouzni, and O. Bensaude. 2001. Incomplete RNA polymerase II phosphorylation in Xenopus laevis early embryos. J. Cell Sci. 114:2483-2489. [DOI] [PubMed] [Google Scholar]
- 33.Palancade, B., and O. Bensaude. 2003. Investigating RNA polymerase II carboxyl-terminal domain (CTD) phosphorylation. Eur. J. Biochem. 270:3859-3870. [DOI] [PubMed] [Google Scholar]
- 34.Perkins, K. D., J. Gregonis, S. Borge, and S. A. Rice. 2003. Transactivation of a viral target gene by herpes simplex virus ICP27 is posttranscriptional and does not require the endogenous promoter or polyadenylation site. J. Virol. 77:9872-9884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peterlin, B. M., and D. H. Price. 2006. Controlling the elongation phase of transcription with P-TEFb. Mol. Cell 23:297-305. [DOI] [PubMed] [Google Scholar]
- 36.Phatnani, H. P., and A. L. Greenleaf. 2006. Phosphorylation and functions of the RNA polymerase II CTD. Genes Dev. 20:2922-2936. [DOI] [PubMed] [Google Scholar]
- 37.Phelan, A., M. Carmo-Fonseca, J. McLaughlan, A. I. Lamond, and J. B. Clements. 1993. A herpes simplex virus type 1 immediate-early gene product, IE63, regulates small nuclear ribonucleoprotein distribution. Proc. Natl. Acad. Sci. USA 90:9056-9060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Poffenberger, K. L., A. D. Idowu, E. B. Fraser-Smith, P. E. Raichlen, and R. C. Herman. 1994. A herpes simplex virus type 1 ICP22 deletion mutant is altered for virulence and latency in vivo. Arch. Virol. 139:111-119. [DOI] [PubMed] [Google Scholar]
- 39.Poffenberger, K. L., P. E. Raichlen, and R. C. Herman. 1993. In vitro characterization of a herpes simplex virus type 1 ICP22 deletion mutant. Virus Genes 7:171-186. [DOI] [PubMed] [Google Scholar]
- 40.Post, L. E., and B. Roizman. 1981. A generalized technique for deletion of specific genes in large genomes: alpha gene 22 of herpes simplex virus 1 is not essential for growth. Cell 25:227-232. [DOI] [PubMed] [Google Scholar]
- 41.Purves, F. C., W. O. Ogle, and B. Roizman. 1993. Processing of the herpes simplex virus regulatory protein alpha 22 mediated by the UL13 protein kinase determines the accumulation of a subset of alpha and gamma mRNAs and proteins in infected cells. Proc. Natl. Acad. Sci. USA 90:6701-6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Purves, F. C., and B. Roizman. 1992. The UL13 gene of herpes simplex virus 1 encodes the functions for posttranslational processing associated with phosphorylation of the regulatory protein alpha 22. Proc. Natl. Acad. Sci. USA 89:7310-7314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quinlan, M. P., L. B. Chen, and D. M. Knipe. 1984. The intranuclear location of a herpes simplex virus DNA-binding protein is determined by the status of viral DNA replication. Cell 36:857-868. [DOI] [PubMed] [Google Scholar]
- 44.Rice, S. A., and D. M. Knipe. 1990. Genetic evidence for two distinct transactivation functions of the herpes simplex virus alpha protein ICP27. J. Virol. 64:1704-1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rice, S. A., M. C. Long, V. Lam, P. A. Schaffer, and C. A. Spencer. 1995. Herpes simplex virus immediate-early protein ICP22 is required for viral modification of host RNA polymerase II and establishment of the normal viral transcription program. J. Virol. 69:5550-5559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rice, S. A., M. C. Long, V. Lam, and C. A. Spencer. 1994. RNA polymerase II is aberrantly phosphorylated and localized to viral replication compartments following herpes simplex virus infection. J. Virol. 68:988-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roizman, B., and D. M. Knipe. 2001. Herpes simplex virus and their replication, p. 2399-2460. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 4th ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 48.Roizman, B., and P. E. Pellett. 2001. The family Herpesviridae: a brief introduction, p. 2381-2398. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 4th ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 49.Sacks, W. R., C. C. Greene, D. P. Aschman, and P. A. Schaffer. 1985. Herpes simplex virus type 1 ICP27 is an essential regulatory protein. J. Virol. 55:796-805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saunders, A., L. J. Core, and J. T. Lis. 2006. Breaking barriers to transcription elongation. Nat. Rev. Mol. Cell Biol. 7:557-567. [DOI] [PubMed] [Google Scholar]
- 51.Schwyzer, M., U. V. Wirth, B. Vogt, and C. Fraefel. 1994. BICP22 of bovine herpesvirus 1 is encoded by a spliced 1.7 kb RNA which exhibits immediate early and late transcription kinetics. J. Gen. Virol. 75:1703-1711. [DOI] [PubMed] [Google Scholar]
- 52.Sears, A. E., I. W. Halliburton, B. Meignier, S. Silver, and B. Roizman. 1985. Herpes simplex virus 1 mutant deleted in the alpha 22 gene: growth and gene expression in permissive and restrictive cells and establishment of latency in mice. J. Virol. 55:338-346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seydoux, G., and M. A. Dunn. 1997. Transcriptionally repressed germ cells lack a subpopulation of phosphorylated RNA polymerase II in early embryos of Caenorhabditis elegans and Drosophila melanogaster. Development 124:2191-2201. [DOI] [PubMed] [Google Scholar]
- 54.Smibert, C. A., and J. R. Smiley. 1990. Differential regulation of endogenous and transduced beta-globin genes during infection of erythroid cells with a herpes simplex virus type 1 recombinant. J. Virol. 64:3882-3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Spencer, C. A., M. E. Dahmus, and S. A. Rice. 1997. Repression of host RNA polymerase II transcription by herpes simplex virus type 1. J. Virol. 71:2031-2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stenberg, R. M., and L. I. Pizer. 1982. Herpes simplex virus-induced changes in cellular and adenovirus RNA metabolism in an adenovirus type 5-transformed human cell line. J. Virol. 42:474-487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Watson, R. J., and J. B. Clements. 1980. A herpes simplex virus type 1 function continuously required for early and late virus RNA synthesis. Nature 285:329-330. [DOI] [PubMed] [Google Scholar]
- 58.Yang, F., Q. Jiang, J. Zhao, Y. Ren, M. D. Sutton, and J. Feng. 2005. Parkin stabilizes microtubules through strong binding mediated by three independent domains. J. Biol. Chem. 280:17154-17162. [DOI] [PubMed] [Google Scholar]
- 59.Young, R. A. 1991. RNA polymerase II. Annu. Rev. Biochem. 60:689-715. [DOI] [PubMed] [Google Scholar]
- 60.Zabierowski, S., and N. A. DeLuca. 2004. Differential cellular requirements for activation of herpes simplex virus type 1 early (tk) and late (gC) promoters by ICP4. J. Virol. 78:6162-6170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zeng, C., E. Kim, S. L. Warren, and S. M. Berget. 1997. Dynamic relocation of transcription and splicing factors dependent upon transcriptional activity. EMBO J. 16:1401-1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou, C., and D. M. Knipe. 2002. Association of herpes simplex virus type 1 ICP8 and ICP27 proteins with cellular RNA polymerase II holoenzyme. J. Virol. 76:5893-5904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhou, M., M. A. Halanski, M. F. Radonovich, F. Kashanchi, J. Peng, D. H. Price, and J. N. Brady. 2000. Tat modifies the activity of CDK9 to phosphorylate serine 5 of the RNA polymerase II carboxyl-terminal domain during human immunodeficiency virus type 1 transcription. Mol. Cell. Biol. 20:5077-5086. [DOI] [PMC free article] [PubMed] [Google Scholar]