

Abstract
It has been well established that populations of RNA viruses transmitted throughout serial bottlenecks suffer from significant fitness declines as a consequence of the accumulation of deleterious mutations by the onset of Muller's ratchet. Bottlenecks are unavoidably linked to different steps of the infectious cycle of most plant RNA viruses, such as vector-mediated transmissions and systemic colonization of new leaves. Here we report evidence for fitness declines by the accumulation of deleterious mutations in the potyvirus Tobacco etch virus (TEV). TEV was inoculated into the nonsystemic host Chenopodium quinoa, and local lesions were isolated and used to initiate 20 independent mutation accumulation lineages. Weekly, a random lesion from each lineage was isolated and used to inoculate the next set of plants. At each transfer, the Malthusian growth rate was estimated. After 11 consecutive transfers, all lineages suffered significant fitness losses, and one even became extinct. The average rate of fitness decline was 5% per day. The average pattern of fitness decline was consistent with antagonistic epistasis between deleterious mutations, as postulated for antiredundant genomes. Temporal fitness fluctuations were not explained by random noise but reflected more complex underlying processes related to emergence and self-organization phenomena.
Due to the lack of proofreading of their replicases, RNA viruses show the highest mutation rates in nature (12). For this reason, along with short generation times and large population sizes, RNA virus populations usually consist of highly heterogeneous mixtures known as quasispecies (11). In a defined environment, the mutant spectrum of a viral population is generally centered on one or several sequences that are fitter, but nonetheless often represent a minor proportion of the mutant distribution (11). Therefore, associated with the inherent stochasticity of transmission or tissue colonization events, viral populations are continuously regenerated from a few individual genomes, and, thus, a finite probability exists that the new population carries an increased mutational load relative to the most-fit members of the ancestral population. Repeated genetic bottleneck events followed by low-fidelity genome replication may lead to substantial fitness losses. In large populations, where purifying selection is a strong force, genomes carrying reversion or second-site compensatory mutations quickly increase in frequency. However, when the effective population size is small, compensatory mutations might not arise (3, 49). Muller (42) predicted that when mutation rates are high and population sizes small, this process occurs in an irreversible ratchet-like manner that leads to the gradual decrease of fitness in a population, particularly in asexual organisms (24). Indeed, the onset of Muller's ratchet further reduces population size, thus accelerating additional mutation accumulation and leading to extinction in a process known as mutational meltdown (23).
The operation of Muller's ratchet in RNA virus populations has been well documented. In a seminal work, Chao (6) found that under demographic conditions in which genetic drift was the major driving force, the mean fitness of segmented RNA bacteriophage φ6 was systematically decreased. Soon after, this observation was extended to Vesicular stomatitis virus (13-15, 17, 43, 44), Foot-and-mouth disease virus (19), Human immunodeficiency virus type 1 (HIV-1) (59, 60), and bacteriophage MS2 (8). Later, the resistance of Foot-and-mouth disease virus populations to extinction after prolonged exposure to periodic bottleneck events was reported (10, 20, 36). Interestingly, these later experiments showed that fitness loss trajectories are biphasic, with a first phase of rapid decline followed by a long stationary phase in which fitness fluctuates around a constant value (10, 36). Furthermore, the magnitude of the fluctuations was not simply reflecting random noise but was the result of a more complex dynamic governed by the sporadic arising of beneficial mutations within a growing plaque and the existence of extinction thresholds below which individual genomes are removed (35, 36, 40).
Unfortunately, similar observations have never been made for a plant virus, despite the fact that bottlenecks are common during vector-mediated transmissions (1, 46) and also during the systemic colonization of new leaves (22, 37, 47), the latter leading to the establishment of genetically diverse populations in different parts of the plant (22, 27). Here, we take a first step toward covering this gap and extend the study of Muller's ratchet to plant virus populations. We report results of an experiment designed to quantitatively explore the fitness losses associated with serial bottleneck transfers of Tobacco etch virus (TEV) on the local lesion host Chenopodium quinoa. In addition to inferring the rate of fitness decline, the existence and sign of epistasis among deleterious mutations, and their relationship, we will also explore whether the temporal pattern of fluctuations in fitness can be trivially explained by random noise or, more interestingly, as a consequence of complex dynamics related to self-organized critical (SOC) phenomena.
MATERIALS AND METHODS
TEV infectious clone and production of a large stock of TEV infectious particles.
The pTEV-7DA infectious clone, kindly provided by James C. Carrington (Oregon State University, Corvallis), was used as an ancestor surrogated wild-type virus (9).
All basic protocols have been described elsewhere (5). In short, pTEV-7DA was linearized with BglII (Fermentas) and transcribed into 5′-capped RNAs using the SP6 mMESSAGE mMACHINE kit (Ambion, Inc.). Transcripts were precipitated, collected, and resuspended in diethyl pyrocarbonate-treated water. RNA integrity and quantity were assessed by gel electrophoresis. Transcripts were mixed with a 1:10 volume of inoculation buffer (100 mg/ml carborundum, 0.5 M K2HPO4) (5). Five 4-week-old Nicotiana tabacum cv. Xanthi plants were inoculated by abrasion with 5 μl of inoculum applied to the third true leaf. Plants were maintained in the greenhouse at 25°C with 16 h of light. Symptoms appeared 4 to 5 days postinoculation (dpi). Eight or 9 dpi, TEV virions were partially purified from symptomatic tissue, as described in reference 5, and pooled in a single large stock.
Inoculation of and virus purification from C. quinoa leaves.
Four different leaves from each one of five different 4-week-old C. quinoa plants were rub inoculated with 10 μl of undiluted, 5-, 10-, and 100-fold-diluted virus, respectively. This randomized inoculation design minimizes plant effects since each dilution is present on each plant, as classically suggested (29, 30). Viral titers, measured as the number of lesion-forming units (LFU) per μl of inoculum, were inferred from the regression of the observed number of local lesions, corrected by a factor also to be estimated from the data, to the dilution factor (29).
TEV particles were purified from local lesions as follows. A well-isolated lesion was cut off from the leaf using the wide side of a 5-μl micropipette tip (approximately 4-mm-diameter disc), immediately frozen with liquid N2, and ground in a mortar with 20 μl of extraction buffer (50 mM phosphate, 3% polyethylene glycol 8000). The extract was carefully collected and centrifuged for 5 min at 13,000 rpm. The supernatant was finally collected, glycerol was added at a final concentration of 20% (vol/vol), and the mixture was frozen at −80°C.
MA experiment.
Mutation accumulation (MA) experiments are a classic genetic approach to the study of deleterious mutations. They are conceptually and mechanistically simple, yet very informative and easy to adapt to widely different experimental systems (38). By imposing a strong bottleneck at each population transfer, the effect of random drift is maximized while the effect of natural selection is minimized, allowing for the accumulation of deleterious mutations that otherwise will be purged from the populations by purifying selection. Figure 1 shows an outline of the MA experiment carried out here with TEV. The TEV stock was serially diluted, and each dilution was inoculated, as described in the previous section, into C. quinoa leaves. Nine days postinoculation, the number of local lesions produced was recorded and 20 lesions were randomly isolated. These lesions constitute the starting point for the 20 independent MA lineages. Virus was extracted from each lesion and serially diluted (0-, 5-, 10-, and 100-fold), and each dilution was used to inoculate a new batch of C. quinoa plants, as described above. Nine days postinoculation, the number of local lesions was recorded for each MA lineage, and a lesion per lineage was randomly chosen and used for the next bottleneck transfer. (Passages 4 and 11 were done after 8 days due to reasons beyond our control.)
FIG. 1.

Scheme of the serial lesion-to-lesion transfer of a TEV population. At each transfer (t), a random lesion was taken, virus purified, and serially diluted. Four dilutions were inoculated on five leaves of each of the next C. quinoa set (t + 1), as described in the text. Only one leaf at each transfer is illustrated here. Nine days postinoculation, newly developed lesions were counted and the number of LFU of the previous lesion was estimated as N(t). Malthusian growth rates were estimated as described in Materials and Methods.
Estimates of Malthusian growth rates.
As a proxy to the fitness of a replicating viral population, we used the per-day Malthusian growth rate. For a lesion sampled at bottleneck transfer t, for the ith (i = 1, 2, … 20) MA lineage, the number of LFU it contained was estimated as the number of visible local lesions produced at the next C. quinoa leaf, Ni(t + 1) (Fig. 1). Since each lesion originated from a single LFU, then the Malthusian growth rate, ri(t), after τ days of viral replication (τ = 8 or 9) can be estimated as ri(t) = 1/τ log Ni(t + 1) day−1.
Population genetics parameters and statistical analyses.
Fitness time series data were analyzed to infer two important population genetics parameters: the rate of fitness decline under MA and the average sign and intensities of the epistatic interactions between accumulated deleterious mutations. To do so, the time series data were fitted to the power fitness function 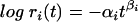 , where αi stands for the per-day rate of fitness decline and βi describes the form of epistasis for each lineage of MA. The per-day rate of fitness decline is directly proportional to the average mutational effect on fitness, s̄ (Appendix). If βi = 1, then mutations act multiplicatively: βi > 1 represents the case of synergistic epistasis where mutational effects are amplified, and 0 < βi < 1 represents the case of antagonistic epistasis where mutational effects suffer of diminishing returns. For a compacted nonredundant RNA genome, an excess of antagonistic epistasis is expected from theoretical considerations (16, 50) and supported by growing empirical evidence from phage φ6 (4), HIV-1 (2), Vesicular stomatitis virus (53), Rous sarcoma virus (48), and the viroids (51).
, where αi stands for the per-day rate of fitness decline and βi describes the form of epistasis for each lineage of MA. The per-day rate of fitness decline is directly proportional to the average mutational effect on fitness, s̄ (Appendix). If βi = 1, then mutations act multiplicatively: βi > 1 represents the case of synergistic epistasis where mutational effects are amplified, and 0 < βi < 1 represents the case of antagonistic epistasis where mutational effects suffer of diminishing returns. For a compacted nonredundant RNA genome, an excess of antagonistic epistasis is expected from theoretical considerations (16, 50) and supported by growing empirical evidence from phage φ6 (4), HIV-1 (2), Vesicular stomatitis virus (53), Rous sarcoma virus (48), and the viroids (51).
To gain further insights into the dynamics of TEV fitness evolution under strong random drift, and particularly seeking scale-free laws characteristic of complex systems self-organized into critical states (SOC), we proceeded to analyze the fluctuations in the time series data. Data from the 20 MA experiments were pooled into a single analysis after expressing the magnitude of fluctuations as the unsigned standardized residuals (USRs) from the fit of the power fitness model to each MA lineage (18). By doing so, deviations from the expected value were put into a common standard deviation scale which makes fluctuation size independent from the actual fitness value measured. Four probability density functions (pdfs) were fitted to the fluctuation data (21). Under the null hypothesis of pure additive sampling error, the USRs should distribute according to a normal pdf, with the parameters the mean, μ, and the standard deviation, σ. The second model fitted was the log-normal pdf. Log-normal distributions arise when errors take place at different steps acting in a multiplicative fashion. Consequently, small differences are amplified, resulting in large fluctuations. The log-normal distribution is characterized by two nonnegative parameters, the median, m, and the shape, σ. The third model fitted was Pareto's pdf, which represents the case of a scale-free distribution belonging to the power-law family of pdfs, and it is usually taken as the most striking signature of SOC. Pareto's pdf is characterized by two nonnegative parameters, the threshold, a, and the shape (or critical exponent), c. Values of 0 < c < 2 are indicative of SOC phenomena (54). It has been suggested (33) that when power laws fail to describe the characteristics of empirical distributions, a good alternative are stretched exponentials such as Weibull's pdf, which is characterized by two nonnegative parameters, the scale, η, and the shape, β < 1. Differences in the goodness of fit for each of these models were assessed by means of the Bayesian information criterion (BIC), which takes into account not only the fit to the model but also its complexity and sample size (26).
All statistical analyses were done using SPSS, version 14.0 (SPSS, Inc., Chicago, IL).
RESULTS
The rate of fitness decline due to the onset of Muller's ratchet.
Figure 2 shows the decline in Malthusian growth rates for all of the MA lineages. The time series data shown in Fig. 2 were fitted by nonlinear regression methods to the power fitness function mentioned above, and the rate of fitness decline was inferred for each MA lineage. All 20 lineages suffered from significant fitness losses even after applying Bonferroni's sequential method for correcting the significance level of multiple tests of the same null hypothesis (t test; in all cases, P ≤ 0.011); and 1 lineage was extinct after three transfers. The per-day rate of fitness decline, α, ranged from 0.020 day−1 to 0.185 day−1, with a median value of 0.051 day−1. The average rate (± standard error) was ᾱ = 0.052 ± 0.008 day−1, a value that is significantly different from zero (one-sample t test; P < 0.001). Thus, we conclude that, on average, TEV fitness declines as much as 5% per day when genetic drift is the main force driving its evolution.
FIG. 2.

Evolution of the Malthusian growth rate for each one of the 20 mutation accumulation lineages. Values were relative to the r value estimated for the ancestral TEV-7DA clone.
Estimate of the sign and intensity of epistasis among deleterious mutations.
After fitting the power fitness function to Fig. 2 data, the epistasis coefficient, β, was also inferred. In 10 cases, β was not significantly different from the null hypothesis of multiplicative fitness effects. In nine cases, β was significantly smaller than 1 (t test; in all cases, P < 0.005), as is the case for antagonistic epistasis. Among those cases showing significant antagonism, β was found in the range 0.418 to 0.931. In only one case was significant synergistic epistasis (β > 1) observed (1.178 ± 0.047; one-sample t test; P = 0.003). However, no case remained significantly different from the null hypothesis after applying the more stringent sequential Bonferroni criterion. Nonetheless, by treating each β estimate as an independent realization of the true statistical distribution, the grand mean of the epistasis coefficient was β̄ = 0.772 ± 0.006, a value which is significantly smaller than the null hypothesis (one-sample t test; P < 0.001). Therefore, we conclude that, on average, deleterious mutations tend to interact antagonistically in modulating TEV fitness.
Interaction between antagonistic epistasis and mutational effect.
It has been ubiquitously observed (51, 56-58) that average mutational effects, s̄, and the strength of antagonistic epistasis are not independent parameters but, instead, are negatively correlated. We sought to investigate if such a negative relationship holds for TEV. As an approximation to s̄, we used α, since, as we have shown in the Appendix, these two parameters are related by definition and therefore a correlation between β and α would necessarily imply a correlation of β with s̄. Figure 3 shows the relationship between β and α for all MA lineages. The value of the Spearman's correlation coefficient was significantly negative (ρS = −0.714; 18 df; P = 0.001). Therefore, we concluded that epistasis and mutational effects are not independent traits but instead may evolve hand in hand: a reduction in the magnitude of mutational effects is associated with a reduction in the magnitude of antagonistic epistasis.
FIG. 3.

Relationship between average mutational effects and the average epistasis coefficient (± standard error).
Analysis of the distribution of fluctuations in the fitness time series data.
After transforming the deviations from the predicted values into USRs, values ranged over 4 orders of magnitude (from 0.0005 to 0.676), a necessary condition when looking for scale-free laws (33). Figure 4A shows the distribution of fitness fluctuation sizes. The average fluctuation size was 0.127, with a standard deviation of 0.137. The distribution was significantly skewed towards small fluctuations (g1 = 1.628 ± 0.182, t178 = 8.964, P < 0.001), and consequently the median fluctuation (0.080) is well below the mean fluctuation. The distribution was also significantly leptokurtic (g1 = 2.467 ± 0.361, t178 = 6.830, P < 0.001), meaning that many values lie near the center, and then the distribution quickly transitions to a long, evenly distributed tail.
FIG. 4.

Panel A shows the distribution of growth rate fluctuation sizes, measured as USRs, during the mutation accumulation experiment. (B) Plots showing the fit of the four alternative empirical pdfs to the observed data (big dots). The dotted line shows the fit to the normal pdf, the short-dashed line shows the fit to the log-normal pdf, the long-dashed line shows the fit to Pareto's pdf, and the continuous line shows the fit to Weibull's pdf.
Table 1 shows the estimated parameters for each of the four distributions fitted to the fluctuation data. Figure 4B shows the fit of the four alternative models to the empirical pdf. Under the null hypothesis of pure sampling error, USRs should distribute according to a normal pdf. As shown in Fig. 4B and Table 1, the fit to the normal pdf explained 95.8% of the observed variability. The second model fitted was the log-normal pdf (Fig. 4B). The fit was significant (Table 1; 97.8% of variability explained) and improved with respect to the normal pdf (lower BIC; Table 1), although it still did not provide the best possible fit among the tested models. The third model fitted was Pareto's pdf (Table 1 and Fig. 4B), which provided the worst fit among the four models tested (largest BIC), explaining only 91.4% of the observed variability. Nonetheless, as expected for SOC, the critical exponent was within the range (0 to 2). The last distribution tested was Weibull's pdf (Table 1 and Fig. 4B). The fit of Weibull's pdf was significant and explained 99.0% of the observed variability in USRs, providing the best fit among the models tested (lowest BIC).
TABLE 1.
Statistics describing the fit of fluctuation size data to alternative pdf modelsa
| Model | R2 | P | Result for parameter (mean ± SE) | BIC |
|---|---|---|---|---|
| Normal | 0.958 | <0.001 | μ = 0.094 ± 0.002 | −1,000.304 |
| σ = 0.096 ± 0.003 | ||||
| Log normal | 0.978 | <0.001 | m = 0.074 ± 0.001 | −1,115.860 |
| σ = 1.320 ± 0.027 | ||||
| Pareto | 0.914 | <0.001 | a = 0.018 ± 0.001 | −874.596 |
| c = 0.566 ± 0.021 | ||||
| Weibull | 0.990 | <0.001 | η = 0.121 ± 0.001 | −1,262.100 |
| β = 0.848 ± 0.012 |
The meanings of the different parameters are described in Materials and Methods.
In conclusion, the pattern of fluctuations observed during fitness decay due to mutation accumulation, rather than being trivially explained by random noise, suggests that at least some of the processes behind viral evolution are related to a broad category of phenomena previously documented in many fields, including physics, biology, and economics, which are related to emerging properties (33, 54).
DISCUSSION
Our first result confirms that whenever genetic drift is the major force driving viral evolution, fitness substantially declines as a consequence of the onset of Muller's ratchet. Indeed, TEV clones lost as much as 5% of their fitness per day, which means that after each bottleneck transfer, they lost 7 × 5% (35%) of their fitness. This result extends previous findings from animal and bacterial viruses (6, 8, 13-15, 17, 19, 20, 36, 43, 44, 59, 60) to plant viruses. This result is particularly relevant given the importance of bottlenecks during the infectious cycle of plant viruses (1, 22, 27, 37, 46, 47). If bottlenecks are so common and their impact on viral fitness so large, then the question is how viruses can continue to keep replicating without heading towards extinction? Three potential reasons can be put forward. In principle, all of these reasons may work for any viral system, although we will make clarifications for the case of plant viruses. First, right after the bottleneck, large population sizes can be generated and, in the process of replication, new genetic variation can be created. This genetic variation may include reversions to the wild-type sequence and second-site compensatory mutations that will spread into the replicating population quite fast. During the systemic infection of a plant, however, the virus suffers from strong bottlenecks (22, 37, 47) which may favor not necessarily the fittest variant. In addition, in contrast to lytic viruses, plant viruses are not released to a common pool in which they compete for resources and, therefore, selection should not be as strong as required for this explanation to work. Second, it has been recently proposed that the entire viral populations may be robust against mutational effects, despite the fact that individual genomes may be highly sensitive to the effect of mutations (16). The reason for this apparent paradox is that the efficiency of natural selection purging out deleterious variability is proportional to the product Nes, where Ne is the effective population size and s is the deleterious fitness effect of a given mutation. In moderate to large populations, such as those typical of RNA viruses, very deleterious mutations (large s) are removed from the population instantaneously, and even those mutations with a mild effect (small s) are removed after short times, preserving a high average population fitness (32). Furthermore, the frequency of lethal mutations has been reported to be quite high (52). In the case of plant viruses, Ne has been estimated to be very low (22, 47), on the order of a few units, and, therefore, the above argument appears to be weak, unless s tends to be very large. Indeed, this is the case for Tobacco mosaic virus, for which the spectrum of mutations is dominated by insertions and deletions rather than by base substitutions (39). Most of these insertions and deletions will be almost immediately lethal (s→∞) and will not contribute to the next viral generation. Third, viruses may turn cellular chaperones to their own benefit to buffer the effect on protein stability of their mutational load. It has been shown that most viruses need cellular chaperones during their life cycle, both to solve their own protein-folding problems and to interfere with cellular processes (41). The fact that mutant misfolded viral proteins elicit the expression of chaperones has been illustrated by Jockush et al. (25). They showed that the presence of Tobacco mosaic virus mutant misfolded coat proteins (CP) triggers the overexpression of Hsp70, with the intensity of the response proportional to the amount of mutant CP. In contrast, the presence of wild-type CP does not elicit such response.
Our second finding is that, on average, deleterious mutations interact in an antagonistic manner, which means that successive mutations have relatively less impact on viral fitness than expected under a multiplicative model. This result is also extending previous findings on animal and bacterial viruses (2, 4, 48, 53) to a plant virus. Given the great heterogeneity in the viral models studied, it seems very plausible that antagonistic epistasis is a general feature of RNA virus genomes. Although epistasis is central for many evolutionary theories, perhaps the most intriguing one is the maintenance of sex and recombination. According to the mutational deterministic hypothesis (31), an excess of synergistic epistasis among deleterious mutations is required to compensate for the twofold advantage of clonal reproduction. In such a situation, recombination would be beneficial for RNA viruses in purging a deleterious mutational load. Hence, the predominance of antagonistic epistasis in TEV invalidates this explanation and raises the question of why positive-stranded viruses, and in particular Potyvirus, have evolved the capacity to recombine (7, 34). In theory, drift can favor the evolution of recombination even for very large populations, as long as there are a sufficient number of loci under selection (28, 45). Whether drift-based explanations for the benefit of recombination are applicable to positive stranded viruses remains to be investigated.
We have also found a negative correlation between mutational effects and the strength of antagonistic epistasis (Fig. 3). This correlation seems to be a ubiquitous phenomenon that has been observed with digital organisms (56), in silico models of bacteriophage T7 infectious cycle (58), and simulated (57) and viroid (51) RNA folding. The dependence of epistasis on mutational effects means that both parameters cannot be evolutionarily optimized independently. If individual genetic robustness has to evolve (i.e., mutational effects become smaller), then the strength of antagonistic epistasis would be relaxed. Antagonistic epistasis is a hallmark of nonredundant genomes where a first mutation has a strong effect on fitness but subsequent mutations, by hitting an already damaged function, may have an increasingly smaller marginal contribution to fitness (16, 50). Therefore, the parasitic runaway lifestyle of RNA viruses, by favoring genomic compaction, is creating antagonistic epistasis and strong sensitivity to deleterious mutations.
Finally, we have observed that fluctuations in fitness during the MA experiments, rather than simply reflecting statistical uncertainty and sampling errors, can be better explained by the stretched exponentials of Weibull's probability distribution. This finding is suggestive of the existence of SOC phenomena associated with viral dynamics, as previously observed for other viruses (18, 36). But, what does it mean that RNA viruses self-organize into a critical state during their evolution? Perhaps the best way of illustrating the meaning of SOC is by using a well-known example—bird flocks. When observing a flock moving on the air, it is not possible to predict its behavior by simply looking at each single bird, but the flock has to be seen as a whole, since it shows a collective behavior which is an emerging property of the system. Small perturbations to the flock may have no impact on its behavior or, with equal probability, have a catastrophic effect, disaggregating it. Similarly, RNA virus populations cannot be understood by simply looking at the individual genome but by examining the entire quasispecies, which shows a series of emerging properties that are the result of the collective behavior of its members. For example, the entire quasispecies may be robustly able to handle to the effect of deleterious mutations as far as all genotypes are connected by neutral mutations (55).
So far, the experimental analysis of viral evolution has been limited, with a few remarkable exceptions, to in vitro cell cultures and mostly to animal and bacterial viruses. As a concluding remark, we would like to suggest that this report extends this very successful approach to a more realistic situation in which the virus replicates and evolves into a real host.
Acknowledgments
We thank the laboratory members and J. A. Daròs for help, comments, and fruitful discussion.
This work has been supported by grants from the Spanish MEC-FEDER (BFU2005-23720-E/BMC and BFU2006-14819-C02-01/BMC), the Generalitat Valenciana (ACOMP06/015), and the EMBO Young Investigator Program to S.F.E.
APPENDIX
The log fitness of a genome carrying n mutations can be described by the power-law fitness function log Wn = −s̄nβ. In this equation, s̄ is the average deleterious fitness effect and β describes the form of epistasis. Our experimental setup, however, did not allow us to directly estimate the number of deleterious mutations, n. Instead, assuming that in any MA lineage the number of mutations at time t, nt, is Poisson distributed with parameter Ut (U stands for the genomic deleterious mutation rate), the mean log fitness of several replicate lineages at time t is given by the following equation:
 |
(1) |
The approximation is valid for large Ut, which is reasonable for an RNA virus, and small deviations from the assumption of multiplicative mutational effects (β ≈ 1). Therefore, the per-day rate of fitness decline, α ≈ s̄Uβ depends on the average mutational effects, the rate of mutation, and the sign and strength of epistasis among mutations.
Footnotes
Published ahead of print on 7 March 2007.
REFERENCES
- 1.Ali, A., H. Li, W. L. Schneider, D. J. Sherman, S. Gray, D. Smith, and M. J. Roossinck. 2006. Analysis of genetic bottlenecks during horizontal transmission of Cucumber mosaic virus. J. Virol. 80:8345-8350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bonhoeffer, S., C. Chappey, N. T. Parkin, J. M. Whitcomb, and C. J. Petropoulos. 2004. Evidence for positive epistasis in HIV-1. Science 306:1547-1550. [DOI] [PubMed] [Google Scholar]
- 3.Burch, C. L., and L. Chao. 2001. Evolution by small steps and rugged landscapes in the RNA virus φ6. Genetics 151:921-927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burch, C. L., and L. Chao. 2004. Epistasis and its relationship to canalization in the RNA virus φ6. Genetics 167:559-567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carrasco, P., J. A. Daròs, P. Agudelo-Romero, and S. F. Elena. 2007. A real-time RT-PCR assay for quantifying the fitness of Tobacco etch virus in competition experiments. J. Virol. Methods 139:181-188. [DOI] [PubMed] [Google Scholar]
- 6.Chao, L. 1990. Fitness of RNA virus decreased by Muller's ratchet. Nature 348:454-455. [DOI] [PubMed] [Google Scholar]
- 7.Chare, E. R., and E. C. Holmes. 2006. A phylogenetic survey of recombination frequency in plant RNA viruses. Arch. Virol. 151:933-946. [DOI] [PubMed] [Google Scholar]
- 8.de la Peña, M. S. F. Elena, and A. Moya. 2000. Effect of deleterious mutation-accumulation on the fitness of RNA bacteriophage MS2. Evolution 54:686-691. [DOI] [PubMed] [Google Scholar]
- 9.Dolja, V. V., H. J. McBride, and J. C. Carrington. 1992. Tagging of plant potyvirus replication and movement by insertion of beta-glucuronidase into the viral polyprotein. Proc. Natl. Acad. Sci. USA 89:10208-10212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Domingo, E., C. Escarmís, E. Lázaro, and S. C. Manrubia. 2005. Quasispecies dynamics and RNA virus extinction. Virus Res. 107:129-139. [DOI] [PubMed] [Google Scholar]
- 11.Domingo, E., V. Martin, C. Perales, A. Grande-Pérez, J. García-Arriaza, and A. Arias. 2006. Viruses as quasispecies: biological implications. Curr. Top. Microbiol. Immunol. 299:51-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drake, J. W., and J. J. Holland. 1999. Mutation rates among RNA viruses. Proc. Natl. Acad. Sci. USA 96:13910-13913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duarte, E. A., D. K. Clarke, A. Moya, E. Domingo, and J. J. Holland. 1992. Rapid fitness losses in mammalian RNA virus clones due to Muller's ratchet. Proc. Natl. Acad. Sci. USA 89:6015-6019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duarte, E. A., D. K. Clarke, A. Moya, S. F. Elena, E. Domingo, and J. Holland. 1993. Many-trillionfold amplification of single RNA virus particles fails to overcome the Muller's ratchet effect. J. Virol. 67:3620-3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duarte, E. A., I. S. Novella, S. Ledesma, D. K. Clarke, A. Moya, S. F. Elena, E. Domingo, and J. J. Holland. 1994. Subclonal components of consensus fitness in an RNA virus clone. J. Virol. 68:4295-4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elena, S. F., P. Carrasco, J. A. Daròs, and R. Sanjuán. 2006. Mechanisms of genetic robustness in RNA viruses. EMBO Rep. 7:168-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elena, S. F., and A. Moya. 1999. Rate of deleterious mutation and the distribution of its effects on fitness in Vesicular stomatitis virus. J. Evol. Biol. 12:1078-1088. [Google Scholar]
- 18.Elena, S. F., and R. Sanjuán. 2005. RNA viruses as complex adaptive systems. Biosystems 81:31-41. [DOI] [PubMed] [Google Scholar]
- 19.Escarmís, C., M. Dávila, N. Charpentier, A. Bracho, A. Moya, and E. Domingo. 1996. Genetic lesions associated with Muller's ratchet in an RNA virus. J. Mol. Biol. 264:255-267. [DOI] [PubMed] [Google Scholar]
- 20.Escarmís, C., G. Gómez-Mariano, M. Dávila, E. Lázaro, and E. Domingo. 2002. Resistance to extinction of low fitness virus subjected to plaque-to-plaque transfers: diversification by mutation clustering. J. Mol. Biol. 315:647-661. [DOI] [PubMed] [Google Scholar]
- 21.Evans, M., N. Hastings, and B. Peacock. 2000. Statistical distributions. Wiley-Interscience, New York, NY.
- 22.French, R., and D. C. Stenger. 2003. Evolution of Wheat streak mosaic virus: dynamics of population growth within plants may explain limited variation. Annu. Rev. Phytopathol. 41:199-214. [DOI] [PubMed] [Google Scholar]
- 23.Gabriel, W., M. Lynch, and R. Bürger. 1993. Muller's ratchet and mutational meltdowns. Evolution 47:1744-1757. [DOI] [PubMed] [Google Scholar]
- 24.Gordo, I., and B. Charlesworth. 2000. The degeneration of asexual haploid populations and the speed of Muller's ratchet. Genetics 154:1379-1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jockusch, H., C. Wiegand, B. Mersch, and D. Rajes. 2001. Mutants of Tobacco mosaic virus with temperature-sensitive coat proteins induce heat shock response in tobacco leaves. Mol. Plant-Microbe Interact. 14:914-917. [DOI] [PubMed] [Google Scholar]
- 26.Johnson, J. B., and K. S. Omland. 2004. Model selection in ecology and evolution. Trends Ecol. Evol. 19:101-108. [DOI] [PubMed] [Google Scholar]
- 27.Jridi, C., J.-F. Martin, V. Marie-Jeanne, G. Labonne, and S. Blanc. 2006. Distinct viral populations differentiate and evolve independently in a single perennial host plant. J. Virol. 80:2349-2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keightley, P. D., and S. P. Otto. 2006. Interference among deleterious mutations favors sex and recombination in finite populations. Nature 443:89-92. [DOI] [PubMed] [Google Scholar]
- 29.Klezckowski, A. 1949. The transformation of local lesion counts for statistical analysis. Ann. Appl. Biol. 36:139-152. [DOI] [PubMed] [Google Scholar]
- 30.Klezckowski, A. 1950. Interpreting relationships between the concentrations of plant viruses and number of local lesions. J. Gen. Microbiol. 4:53-69. [DOI] [PubMed] [Google Scholar]
- 31.Kondrashov, A. S. 1995. Contamination of the genome by very slightly deleterious mutations: why have we not died 100 times over? J. Theor. Biol. 175:583-594. [DOI] [PubMed] [Google Scholar]
- 32.Krakauer, D. C., and J. B. Plotkin. 2002. Redundancy, antiredundancy, and the robustness of genomes. Proc. Natl. Acad. Sci. USA 99:1405-1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laherrère, J., and D. Sornette. 1998. Stretched exponential distributions in nature and economy: “fat tails” with characteristic scales. Eur. Phys. J. B 2:525-539. [Google Scholar]
- 34.Lai, M. M. C. 1992. RNA recombination in animal and plant viruses. Microbiol. Rev. 56:61-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lázaro, E., C. Escarmís, E. Domingo, and S. C. Manrubia. 2002. Modeling viral genome fitness evolution associated with serial bottleneck events: evidence of stationary states of fitness. J. Virol. 76:8675-8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lázaro, E., C. Escarmís, J. Pérez-Mercader, S. C. Manrubia, and E. Domingo. 2003. Resistance of virus to extinction on bottleneck passages: study of a decaying and fluctuating pattern of fitness loss. Proc. Natl. Acad. Sci. USA 100:10830-10835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li, H., and M. J. Roossinck. 2004. Genetic bottlenecks reduce population variation in an experimental RNA virus population. J. Virol. 78:10582-10587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lynch, M., J. Blanchard, D. Houle, T. Kibota, S. Schultz, L. Vassilieva, and J. Willis. 1999. Spontaneous deleterious mutation. Evolution 53:645-663. [DOI] [PubMed] [Google Scholar]
- 39.Malpica, J. M., A. Fraile, I. Moreno, C. I. Obies, J. W. Drake, and F. García-Arenal. 2002. The rate and character of spontaneous mutation in an RNA virus. Genetics 162:1505-1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Manrubia, S. C., C. Escamís, E. Domingo, and E. Lázaro. 2005. High mutation rates, bottlenecks, and robustness of RNA viral quasispecies. Gene 347:273-282. [DOI] [PubMed] [Google Scholar]
- 41.Mayer, M. P. 2005. Recruitment of Hsp70 chaperones: a crucial part of viral survival strategies. Rev. Physiol. Biochem. Pharmacol. 153:1-46. [DOI] [PubMed] [Google Scholar]
- 42.Muller, H. J. 1964. The relation of recombination to mutational advance. Mutat. Res. 1:2-9. [DOI] [PubMed] [Google Scholar]
- 43.Novella, I. S., S. F. Elena, A. Moya, E. Domingo, and J. J. Holland. 1995. Size of genetic bottleneck leading to virus fitness loss is determined by mean initial population fitness. J. Virol. 69:2869-2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Novella, I. S., S. F. Elena, A. Moya, E. Domingo, and J. J. Holland. 1996. Repeated transfer of small RNA virus populations leading to balanced fitness with infrequent stochastic drift. Mol. Gen. Genet. 252:733-738. [DOI] [PubMed] [Google Scholar]
- 45.Otto, S. P., and N. H. Barton. 2001. Selection for recombination in small populations. Evolution 55:1921-1931. [DOI] [PubMed] [Google Scholar]
- 46.Pirone, T. P., and S. Blanc. 1996. Helper-dependent vector transmission of plant viruses. Annu. Rev. Phytopathol. 34:227-247. [DOI] [PubMed] [Google Scholar]
- 47.Sacristán, S., J. M. Malpica, A. Fraile, and F. García-Arenal. 2003. Estimation of population bottlenecks during systemic movement of Tobacco mosaic virus in tobacco plants. J. Virol. 77:9906-9911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sanjuán, R. 2006. Quantifying antagonistic epistasis in a multifunctional RNA secondary structure of the Rous sarcoma virus. J. Gen. Virol. 87:1595-1602. [DOI] [PubMed] [Google Scholar]
- 49.Sanjuán, R., J. M. Cuevas, A. Moya, and S. F. Elena. 2005. Epistasis and the adaptability of an RNA virus. Genetics 170:1001-1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sanjuán, R., and S. F. Elena. 2006. Epistasis correlates to genome complexity. Proc. Natl. Acad. Sci. USA 103:14402-14405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sanjuán, R., J. Forment, and S. F. Elena. 2006. In silico predicted robustness of viroids RNA secondary structures. II. Interaction between mutation pairs. Mol. Biol. Evol. 23:2123-2130. [DOI] [PubMed] [Google Scholar]
- 52.Sanjuán, R., A. Moya, and S. F. Elena. 2004. The distribution of fitness effects caused by single-nucleotide substitutions in an RNA virus. Proc. Natl. Acad. Sci. USA 101:8396-8401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sanjuán, R., A. Moya, and S. F. Elena. 2004. The contribution of epistasis to the architecture of fitness in an RNA virus. Proc. Natl. Acad. Sci. USA 101:15376-15379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Solé, R. V., S. C. Manrubia, M. Benton, S. Kauffman, and P. Bak. 1999. Criticality and scaling in evolutionary ecology. Trends Ecol. Evol. 14:156-160. [DOI] [PubMed] [Google Scholar]
- 55.Wilke, C. O. 2001. Selection for fitness versus selection for robustness in RNA secondary structure folding. Evolution 55:2412-2420. [DOI] [PubMed] [Google Scholar]
- 56.Wilke, C. O., and C. Adami. 2001. Interaction between directional epistasis and average mutational effects. Proc. R. Soc. Ser. B 298:1469-1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wilke, C. O., R. E. Lenski, and C. Adami. 2003. Compensatory mutations cause excess of antagonistic epistasis in RNA secondary structure folding. BMC Evol. Biol. 3:1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.You, L., and J. Yin. 2002. Dependence of epistasis on environment and mutation severity as revealed by in silico mutagenesis of phage T7. Genetics 160:1273-1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yuste, E., C. López-Galíndez, and E. Domingo. 2000. Unusual distribution of mutations associated with serial bottleneck passages of human immunodeficiency virus type 1. J. Virol. 74:9546-9552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yuste, E., S. Sánchez-Palomino, C. Casado, E. Domingo, and C. López-Galíndez. 1999. Drastic fitness loss in human immunodeficiency virus type 1 upon serial bottleneck events. J. Virol. 73:2745-2751. [DOI] [PMC free article] [PubMed] [Google Scholar]