

Abstract
The Kaposi's sarcoma-associated herpesvirus open reading frame 50 (ORF50) protein (called Rta), is necessary and sufficient for reactivation of the virus from latency. We previously demonstrated that a truncated mutant of ORF50 lacking its C-terminal transcriptional activation domain, called ORF50ΔSTAD, formed mixed multimers with wild-type (WT) ORF50 and functioned as a dominant negative inhibitor of reactivation. For this report, we investigated the requirements for multimerization of ORF50/Rta in transactivation and viral reactivation. We analyzed multimerization of WT, mutant, and chimeric ORF50 proteins, using Blue Native polyacrylamide gel electrophoresis and size exclusion chromatography. WT and mutant ORF50 proteins form tetramers and higher-order multimers, but not monomers, in solution. The proline-rich, N-terminal leucine heptapeptide repeat (LR) of ORF50 (amino acids [aa] 244 to 275) is necessary but not sufficient for oligomer formation and functions in concert with the central portion of ORF50/Rta (aa 245 to 414). The dominant negative mutant ORF50ΔSTAD requires the LR to form mixed multimers with WT ORF50 and inhibit its function. In the context of the WT ORF50/Rta protein, mutagenesis of the LR, or replacement of the LR by heterologous multimerization domains from the GCN4 or p53 proteins, demonstrates that tetramers of Rta are sufficient for transactivation and viral reactivation. Mutants of Rta that are unable to form tetramers but retain the ability to form higher-order multimers are reduced in function or are nonfunctional. We concluded that the proline content, but not the leucine content, of the LR is critical for determining the oligomeric state of Rta.
Epidemiologic, serologic, and histopathologic studies have established Kaposi's sarcoma-associated herpesvirus (KSHV; also known as human herpesvirus 8) as the etiologic agent of Kaposi's sarcoma (KS) and primary effusion lymphoma (PEL) (13, 18, 29, 41, 60, 75). Reactivation of KSHV from latency is a crucial step in KS development. The viral load in the peripheral blood is directly proportional to the risk of KS progression and the stage of KS (1, 5, 9, 26, 69, 94), and treatments that reduce the KSHV load are accompanied by KS regression (6, 11, 12, 39, 40, 45, 56, 59, 66, 70, 76, 95). Most of the candidate pathogenic genes of KSHV (encoding proteins with cell growth deregulatory and immunomodulatory functions) are expressed in the delayed early class of the lytic-gene expression program (7, 14, 19, 23, 24, 27, 28, 38, 43, 48, 65, 72, 82). It is likely that reactivation of KSHV contributes to cancer initiation both by facilitating dissemination of the virus and by permitting expression of lytic-cycle genes with direct roles in pathophysiology. Therefore, a complete understanding of KSHV pathogenesis demands elucidation of the mechanisms that regulate viral reactivation and progression through the lytic cycle.
We and others have demonstrated that the KSHV protein Rta (for “replication and transcriptional activator,” expressed from open reading frame 50 [ORF50]) is both necessary and sufficient for reactivation of KSHV in tissue culture models of latency (30, 52, 53, 80, 101). Rta is a transcriptional transactivator that binds directly to the DNA of several KSHV promoters with various affinities and sequence specificities. Rta binds with the highest affinity, among well-studied KSHV promoters, to the PAN promoter, with a dissociation constant (Kd) in the nanomolar range (78, 79).
Rta binds directly to a 16-bp core sequence found in the PAN and kaposin promoters and in ori-Lyt (L) (16, 42, 79, 90). Rta binding to ori-Lyt (L) mediates transcriptional activation and replication complex formation, which are essential for ori-Lyt-dependent DNA replication (90, 91). This site differs significantly from a second Rta binding site shared by the ORF57/Mta and K8 promoters (51) and individual binding sites found in other KSHV promoters (16, 17, 23, 46, 48, 51, 71, 79, 88, 89, 107). In general, Rta binding sites contain A/T-rich trinucleotides similar to interferon-stimulated response elements (103). The trinucleotides are often found in repeating, phased arrays in Rta-responsive promoters (49, 84).
Promoter selection by Rta is not always specified by direct DNA binding. Instead, Rta requires combinatorial interactions with cellular proteins. These cellular proteins include recombination signal binding protein Jk (RBP-Jk; also known as CBF-1 and CSL), AP-1, octamer-1, and CCAAT/enhancer binding protein alpha (C/EBPα) (10, 71, 87-89, 92). Among these proteins, RBP-Jk is essential for KSHV reactivation (47). Rta-mediated transactivation of the promoters of ORF57/Mta, ORF6/single-stranded DNA binding protein, viral G protein-coupled receptor (vGPCR), the KSHV basic leucine zipper (K-bZIP), and Rta itself (auto-activation) (47-49, 94) requires direct interactions with RBP-Jk. A central regulatory role of the RBP-Jk/Rta interaction was demonstrated by the inhibition of KSHV reactivation in murine embryo fibroblasts null for RBP-Jk (47).
Rta promotes DNA binding of RBP-Jk, a mechanism that is fundamentally different from that established for the RBP-Jk-activating proteins, Notch intracellular domain, and Epstein-Barr virus EBNA-2 (10). In the Rta-responsive element (RRE) of the ORF57 promoter, intact DNA binding sites for both proteins are necessary but not sufficient for cooperation; instead, ternary complex formation between Rta, RBP-Jk, and DNA is required. Mutations of the RRE on both sides of the RBP-Jk consensus element inhibit ternary complex formation (10), in agreement with localization of Rta binding sites in both positions (84). In latently infected cells, RBP-Jk is virtually undetectable on the viral ORF57, K-bZIP, vGPCR, and cellular interleukin-6 and HES-1 (for “hairy/enhancer of split 1”) promoters (10). However, during viral reactivation, RBP-Jk is significantly enriched on these promoters in an Rta-dependent fashion. After Rta associates with lytic KSHV promoters, its ability to reactivate the virus from latency requires recruitment of the cellular SWI/SNF chromatin remodeling complex and the TRAP/mediator coactivator (31).
The 691-amino acid (aa) Rta protein shares with its homologs in the gammaherpesviruses two regions of relatively high primary-amino acid homology in the N and C termini (22, 53). The N-terminal 272 amino acids of Rta bind independently to KSHV promoters (51, 79), and Rta's C terminus is a potent transactivator when targeted to promoters by fusion to heterologous DNA binding domains (52, 74, 85). Deletion of the activation domain generates a mutant of Rta (called ORF50ΔSTAD) that forms mixed multimers with wild-type (WT) Rta and functions as an Rta-specific dominant negative inhibitor of transactivation (52). Expression of this dominant negative Rta mutant in PEL cells suppresses viral reactivation induced by multiple signals (52). These data suggest that Rta must form homomultimers to function as the lytic switch protein. Indeed, it has been shown that multimers, but not dimers, of cognate Rta bind to the K-bZIP/ lytic replication-associated protein (RAP) promoter (49). Although previous studies suggested that Rta forms hexamers in solution, the amino acid requirements for Rta multimerization and the functional significance of multimerization for transactivation and viral reactivation remain untested.
In this paper, we demonstrate that Rta tetramers appear to be the preferred oligomeric state for proper transactivation and induction of KSHV reactivation. The proline-rich, N-terminal leucine heptapeptide repeat (LR) of Rta (aa 244 to 275) is an important mediator of Rta's oligomeric state. In addition, multimerization requires a region of the protein C terminal to aa 275. We show that deletion of the LR, site-specific mutation of leucines in the LR, or replacement of the LR with multimerization domains of heterologous proteins alters the extent of Rta multimerization and Rta's functions. Implications of our observations for KSHV replication are discussed.
MATERIALS AND METHODS
Plasmids.
The purification and propagation of all plasmids have been described previously (10). Site-directed mutations and plasmids constructed by using PCR were verified by DNA sequencing.
pcDNA3-FLc50 expresses wild-type ORF50 protein (52); pV5-FLc50 and pV5-50ΔSTAD express WT and C-terminal-truncated ORF50 protein fused to the V5 epitope, respectively (52). pGem3-FLc50 and pGem3-50ΔSTAD express the wild-type and truncated (aa 1 to 514) ORF50 cDNAs, respectively, from the T7 transcription start site (52). pSG-FL50 expresses full-length ORF50/Rta fused at its N terminus to the DNA binding domain of the Saccharomyces cerevisiae protein Gal4 (52).
Plasmid pcDNA3-50ΔLR was constructed by PCR, amplifying Rta nucleotides (nt) 1 to 723, using a reverse primer that introduced an NcoI restriction site. The PCR product was digested with BamHI/NcoI and subcloned into pGem3-50ΔSTAD by replacing the BamHI/NcoI fragment of ORF50ΔSTAD. This generated pGem3-50ΔSTADΔLR. The EagI/NcoI fragment of that plasmid was then subcloned into pGem3-FLc50 by replacing the EagI/NcoI fragment of ORF50. This resulted in an in-frame fusion of ORF50 aa 1 to 238 to aa 273 to 691. The full-length (FL) insert was then cloned into the NheI/EcoRI sites of pcDNA3.1Zeo (Invitrogen) in a two-step procedure in which the 5′ end was PCR amplified to introduce a 5′ Nhe site and the 3′ end was added as a NarI/EcoRI fragment. pV5-50ΔSTADΔLR was constructed by replacing the BsiWI/BstEII fragment of pV5-50ΔSTAD with the same fragment of pGem3-50ΔSTADΔLR.
pcDNA3-50-L3P expresses Rta containing three leucine-to-proline mutations in ORF50 aa 254, 259, and 261. It was constructed using the Quick-Change site-directed mutagenesis kit (Stratagene). The primer sequences, with mutated nucleotides underlined, are 5′-CCAGGAAGCGGTCCCATGCCAGAATCTCCAATTCCGCCAATCCTGGAG-3′ and 5′-CTCCAGGATTGGCGGAATTGGAGATTCTGGCATGGGACCGCTTCCTGG-3′.
pcDNA3-50ΔLR/GCN and pcDNA3-50ΔLR/p53 express Rta proteins in which the LR was replaced with the oligomerization domains of the proteins GCN4 and p53, respectively. These were constructed as follows: the yeast GCN4 dimerization domain was PCR amplified by using forward primer 5′-GGGTCATGAAACAACTTGAAGACAAGG-3′ and reverse primer 5′-CCCCCATGGAGCGTTCGCCAACTAATTTC-3′, with the plasmid pHN516 as a template (a gift from Hilary Nelson, University of Pennsylvania) (25). The p53 tetramerization domain (93) was PCR amplified by using forward primer 5′-GGGTCATGAAGAAGAAACCACTGGATGG-3′ and reverse primer 5′-CCCCCATGGACCCTGGCTCCTTCCCAGC-3′, with the plasmid SN3 wt p53 as a template (a gift from Bert Vogelstein, Johns Hopkins University) (2). The PCR products were digested with NcoI/BspHI (which had been introduced by the primers). Each was cloned into pGem3-FLc50ΔLR, which had been digested with NcoI. The inserts were then transferred to pcDNA3.1 by EcoRI digestion/ligation.
pGem3-50-N1-238 and pGem3-50-N1-272, expressing Rta truncated at the indicated amino acids, were constructed by PCR amplification, using forward primer 5′-GCGGATATCATGGCGCAAGATGAC-3′ and one of the following reverse primers: 50-N1-238, 5′-ATCGTCGACCTATGGCCGGCGTTTCTCAGCAG-3′, or 50-N1-272, 5′-ATGGTCGACCTAGGAAGCCGGCAACAGTCC-3′, respectively. The PCR products were digested with EcoRV and SalI (introduced by the primers), respectively, and ligated to pGem3 that had been digested with SmaI and SalI. pGem3-50ΔAatII was constructed by subcloning the EcoRI/SalI fragment of pcDNA3.1-V5-50ΔAatII into pGem3 digested with the same enzymes.
pGem3-50ΔSTAD-DNGANR expresses ORF50ΔSTAD+PHIL (see Fig. 7A) and was constructed by site-directed mutagenesis of pGem3-50ΔSTAD. The primer sequences, with the mutated nucleotides underlined, are 5′-GGTCTCATGCCAGAATCTGATAATGGTGCCAATCGTGAGCCAGGACTGTTGC-3′ and 5′-GCAACAGTCCTGGCTCACGATTGGCACCATTATCAGATTCTGGCATGAGACC-3′. The plasmid pGem3-50-DNGANR expresses ORF50+PHIL and was constructed by replacing the NdeI/NcoI fragment of pGem3-FLc50 with that from pGem3-50DNGANR. The plasmid pcDNA3.1-50-DNGANR also expresses ORF50+PHIL and was constructed by transferring the EcoRI fragment from pGem3-50-DNGANR to pcDNA3.1.
FIG. 7.

Design and expression of MBP fusions of ORF50. (A) Schematic of ORF50/Rta constructs. The numbers refer to amino acids, and the bars underneath ORF50 represent sequences included in the ORF50 mutants identified in the list at the right. The four amino acid sequences at the bottom were inserted at the LR (aa 244 to 275) in the WT, L3P mutant, or chimeric (GCN4 or p53) proteins. ST, serine/threonine-rich sequence; hyd, hydrophobic; DE, acidic amino acid-rich; +++, basic amino acid-rich sequence; *, amino acid position of the mutations in chimeric proteins. (B) MBP fusions of ORF50 stained with Gel Code Blue stain (Pierce). The indicated proteins were expressed and purified from E. coli, as described in Materials and Methods. Equal amounts of protein were displayed by SDS-PAGE, and the gels were fixed and stained with Gel Code Blue (Pierce).
pMalc2X-FL50 expresses wild-type ORF50 protein fused to the maltose binding protein (MBP) epitope at its N terminus (MBP-50) (10). pMalc2x-50ΔSTAD expresses MBP fused to ORF50 aa 1 to 505 and was constructed by PCR amplification of ORF50 nt 1 to 1516, using primers that introduced EcoRI/SalI sites. The PCR product and pMalc2x were digested with those enzymes and ligated.
pMalc2x-50L3P was constructed by replacing the AatII/BamHI fragment of pMalc2X-FL50 with that from pcDNA3-c50L3P. pMalc2x-50ΔSTADΔLR and pMalc2X-50ΔSTADL3P were constructed by replacing the AatII/BamHI fragment of pMalc2x-50ΔSTAD with that from pcDNA3-c50ΔSTADΔLR and pMalc2X-50ΔSTADΔLR, respectively. pMalc2x-50-DNGANR (expressing MBP-50+PHIL) and pMalc2X-50ΔSTAD-DNGANR (expressing MBP-50ΔSTAD+PHIL) were constructed by replacing the AatII/BamHI fragment of pMalc2x-50ΔSTADΔLR and pMalc2x-FL50ΔLR, respectively, with that from pcDNA3-50DNGANR. pMalc2x-50ΔSTADΔLR/GCN4 and pMalc2x-50ΔSTADΔLR/p53TD were constructed by replacing the AatII/BamHI fragment of Malc2x-50ΔSTADΔLR with those from pcDNA3-50ΔLR/GCN and pcDNA3-50ΔLR/p53, respectively. The reporter plasmids pK-bZIP-GL3 and pNut-1-GL3 were described previously (52, 53).
Cell culture, transfections, and luciferase and β-galactosidase assays.
The human diploid endothelial cell line SLK was propagated and maintained as described previously (52). For transfection, cells were plated at 5 × 104 per well in 6-well plates and transfected with TransIT-LT1 transfection reagent (Mirus), with up to 3 μg DNA used according to the manufacturer's instructions.
The human embryonic-kidney cell line 293T was grown in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum (Sigma), 2 mM L-glutamine, and antibiotics. The cells (1 × 106 per 10-cm plate) were seeded the day prior to transfection to obtain ∼50 to 70% confluence the following day. The medium was changed approximately 4 h before the transfection. A DNA cocktail (10 μg plasmids, 80 μl 10× NTE [150 mM NaCl, 10 mM Tris-HCl, pH 7.4, and 1 mM EDTA, pH 8.0], 100 μl 2 M CaCl2, and water to a final volume of 800 μl) was added dropwise to an equal volume of two times transfection buffer (1 ml 0.5 M HEPES-NaOH [pH 7.1 ±0.05], 8.1 ml water, 0.9 ml 2 M NaCl, and 20 μl 1 M Na2HPO4 for 10 ml buffer), and the mixture was incubated at room temperature for 15 to 30 min. The precipitate was added dropwise to the media, and the plates were swirled gently and returned to the 37°C incubator. The media was changed at 4 to 15 h posttransfection (hpt), and the cell lysates were collected at 48 hpt.
The BCBL-1 cell line was propagated and maintained as described previously (52). Transfections were performed by electroporation of 1 × 107 cells in 0.25 ml incomplete RPMI 1640 media in 0.4-cm electroporation cuvettes. Up to 17 μg DNA were electroporated at 975 uF and 150 V, after which the cells were transferred to 10 ml complete media.
For all transfection experiments, pcDNA3 was cotransfected to normalize the total amount of DNA for each transfection, and pcDNA3.1-His-lacZ was cotransfected as an internal control. Luciferase and β-galactosidase assays were performed using luciferase and β-galactosidase assay kits (Promega) as previously described (52). The data reported here are representative of at least two experiments, with each transfection performed in triplicate.
Viral reactivation assays.
BCBL-1 cells were electroporated in duplicate with the indicated plasmids and harvested 72 h postelectroporation as described previously (53), with the following modifications. The cells (2 × 105) were made to adhere to poly(L-lysine)-coated glass slides within 1-in-diameter circles drawn with a Pap pen (Beckman Coulter). Proteins were detected with rabbit anti-Rta serum and mouse anti-K8.1, fluorescein isothiocyanate-conjugated anti-rabbit, and tetramethyl rhodamine isocyanate-conjugated anti-mouse antibodies. Cells were scored positive if Rta expression was detected by visual inspection; quantitation was done by counting the percentage of Rta-positive cells that were also K8.1 positive (Rta+/K8.1+ divided by Rta+). At least 200 doubly positive cells were counted for each. Cells transfected with an empty expression vector were scored similarly. Results from the empty-vector transfections (spontaneous reactivation) were subtracted from those for each WT or mutant Rta transfection. Transfection efficiency was typically 3 to 8%. Results were normalized to that for cells transfected with the WT Rta expression vector, which was set at 100%.
Protein expression in Escherichia coli and purification.
BL21 CodonPlus RIL/DE3-competent cells (Stratagene) were transformed with the indicated pMalc2x plasmids, and single colonies were inoculated in 25 ml LB media containing 50 μg/μl ampicillin and 50 mg/ml chloramphenicol. Following overnight growth at 37°C with shaking, 5 ml of saturated culture was transferred to 500 ml of fresh LB media containing ampicillin and 1 g of glucose and grown at 37°C for 2 h. The culture was cooled to 16°C, 200 ul IPTG (isopropyl-β-D-thiogalactopyranoside; 180 mg/ml) was added to induce recombinant protein expression, and the cells were shaken at 16°C for 12 to 18 h. The cells were harvested by centrifugation, and the pellet was suspended in 10 ml of 1× column binding buffer (20 mM Tris-HCl, 200 mM NaCl, 1 mM EDTA) supplemented with protease inhibitor cocktail (1:500 [Sigma]) and dithiothreitol (1 mM). Cell lysis, protein purification, and dialysis were performed as previously described (10, 51).
Blue Native-polyacrylamide gel electrophoresis (BN-PAGE).
Following a protocol modified from Schägger et al. (73), Coomassie blue (G250; Serva) was added to individual samples of 1 to 3 μg of protein (approximately 100 ng/μl) to a final concentration of 0.02%. The proteins were loaded onto NuPAGE 4% to 12% Bis-Tris gradient gels (Invitrogen) alongside 5 μg of high-molecular-weight Native Marker (Amersham Biosciences). The cathode buffer contained 50 mM MOPS (morpholinepropanesulfonic acid), 50 mM Tris (pH 7.7), and 0.02% Coomassie blue; the anode buffer contained 50 mM MOPS and 50 mM Tris (pH 7.7). Electrophoresis of the gels was performed at 15 mA (constant) for approximately 6 to 8 h, until the dye front reached the bottom of the gel. The gel was destained with several changes of 25% methanol/10% acetic acid to visualize the proteins.
Gel filtration.
A Superdex200 HR 10/30 column (Amersham Biosciences) was preequilibrated with 1× DNA binding buffer (without glycerol) at 25°C. Forty to one hundred micrograms of each purified protein in 0.5 ml of buffer was loaded onto the column, and the proteins were eluted at a flow rate of 0.5 ml/min. A calibration curve was determined by calculating the log10 molecular mass/elution volume for the protein standards thymoglobulin, ferritin, and catalase (Amersham Biotech) (R2 = 0.9733) (Table 1). The molecular masses of unknown proteins (Rta and derivatives) were determined by comparing the elution volume to the calibration curve.
TABLE 1.
Multimerization of WT, chimeric, and mutant KSHV ORF50/Rta polypeptides
| Protein | Result of gel filtration chromatography for indicated parameter:
|
||||
|---|---|---|---|---|---|
| Predicted molecular mass (kDa)a | Peak no.c | Elution vol (ml) | Calculated molecular mass | No. of subunitsc | |
| Molecular mass standard | |||||
| Thymoglobulin | 669 | NA | 9.5 | 669b | NA |
| Ferritin | 440 | NA | 10.9 | 440b | NA |
| Catalase | 232 | NA | 12.6 | 232b | NA |
| ORF50/Rta | |||||
| MBP-ORF50 | 116.2 | F1 | 8.0 | 1,096 | 10 |
| F2 | 10.6 | 468 | 4 | ||
| MBP-ORF50-L3P | 116.1 | F2 | 10.7 | 457 | 4 |
| MBP-ORF50ΔSTAD | 96.5 | S1 | 8.1 | 1,000 | 10 |
| S2 | 9.8 | 603 | 6 | ||
| S3 | 11.3 | 398 | 4 | ||
| MBP-50ΔSTADΔLR | 93.1 | S1 | 8.2 | 1,000 | 10 |
| S1.5 | 9.3 | 794 | 8 | ||
| S3 | 11.4 | 398 | 4 | ||
| MBP-50ΔSTAD-L3P | 96.4 | S1 | 8.1 | 1,000 | 10 |
| S2 | 10.0 | 631 | 6 | ||
| S3 | 11.4 | 398 | 4 | ||
Calculated using ExPASY.
Supplied by the manufacturer.
NA, not applicable.
GST fusion protein interaction assays.
Glutathione S-transferase (GST)-RBP-Jk and GST proteins were expressed and purified exactly as previously described (10). One milliliter of crude E. coli lysate containing GST-RBP-Jk was incubated with 30 μl of preswollen glutathione-Sepharose beads (1:1 [wt/vol] in 1× NETN+ [20 mM Tris, pH 7.5, 100 mM NaCl, 1 mM EDTA, and 0.5% Nonidet P-40 supplemented with protease inhibitors; Sigma]) at 4°C for 2 h. GST moieties alone (amounts equal to that of GST-RBP-Jk, as determined by Bradford analyses; Bio-Rad) were bound to the beads independently. The beads were washed three times in 1× NETN+ to remove unbound proteins. Rta and its derivatives were expressed in rabbit reticulocyte lysates (RRL; TNT-coupled transcription/translation system) in the presence of L-[35S]methionine to label proteins according to the manufacturer's (Promega) directions, and 0.5 μl was put aside for visualization as 5% input. Ten microliters of each programmed RRL was mixed with the bead-bound GST fusion protein in 250 μl of 1× NETN+ and then incubated for 2 h at 4°C with nutation. The complexes were washed extensively, the beads were boiled in 2× Laemmli buffer, and the bound proteins were displayed by sodium dodecyl sulfate (SDS)-PAGE. The proteins were visualized by autoradiography after their signals were amplified in 0.5 M salicylic acid.
EMSAs.
Electrophoretic mobility shift assays (EMSAs) were performed exactly as previously described (10), using annealed oligonucleotides labeled with 32P:K-bZIPwt:F (5′-GATCTATTTGTGAAACAATAATGATTAAAGGGGA-3′) and R (5′-GATCTCCCCTTTAATCATTATTGTTTCACAAATA-3′) and nut-1/PAN F (5′-GATCTTTCCAAAAATGGGTGGCTAACCTGTCCAAAATATGGGAACACTGGAA-3′) and R (5′-GATCTTCCAGTGTTCCCATATTTTGGACAGGTTAGCCACCCATTTTTGGAAA-3′).
Coimmunoprecipitations.
For analyses in RRLs (TNT T7 Quick Transcription/Translation kit; Promega), RRLs were programmed with WT and mutant Rta's expressed from pGem3. Each reaction mixture contained a total of 3 μg plasmid DNA and L-[35S]methionine to label the products. One-fifteenth of each lysate was put aside as input to identify the translated proteins, and the volume of each lysate was increased to 150 μl by adding 10s buffer (50 mM HEPES [pH 7.2], 10 mM NaPO4 [pH 7.0], 250 mM NaCl, 0.2% NP-40, 0.1% Triton X-100, 0.005% SDS, and 2.5 mM β-mercaptoethanol) supplemented with protease inhibitor cocktail (8). Four microliters of ORF50 antibody (which was generated versus the C terminus of Rta, aa 525 to 691) (53) (see Fig. 6A) was incubated with each lysate for 2 h at 4°C with nutation. Protein A agarose was added, and incubation was continued for 1 h. The beads were washed five times with 0.5 ml 10s buffer and boiled in Laemmli sample buffer, and the immunoprecipitated proteins were separated by SDS-PAGE. The gels were fixed for 30 min in 50% methanol/10% acetic acid. The gels were incubated in 0.5 M sodium salicylate to increase the intensity of the radioactive signals and then dried and analyzed by autoradiography.
FIG. 6.

The LR is necessary but not sufficient for homomultimerization of ORF50/Rta. (A) Schematic of ORF50-truncation proteins. The numbers refer to amino acids, and the bars underneath ORF50 represent sequences included in the ORF50-truncated mutants identified in the list at the right. The bottom bar indicates the region of ORF50/Rta used as antigen for generation of the Rta-specific antiserum (53) used for the experiments for panel B. ST, serine/threonine-rich sequence; hyd, hydrophobic; DE, acidic amino acid-rich; +++, basic amino acid-rich sequence. (B) Immunoprecipitation of RRLs. RRLs were programmed with the indicated plasmids and analyzed as for Fig. 5B. The panel to the far right shows a longer exposure for lanes 13 to 16. I.P., immunoprecipitation; +, transfected plasmid.
For analyses in transfected cell extracts, 293T cells were transfected with plasmids pcDNA3, pcDNA3-FLc50, pcDNA3-50-L3P, pV5-c50ΔSTAD, and pV5-50ΔSTADΔLR. The cells were harvested 48 h posttransfection and lysed in 10s buffer supplemented with protease inhibitor cocktail. One-tenth of each lysate was put aside as input to confirm protein expression, and the remainder of each lysate was incubated with anti-V5 antibody (Bethyl) or anti-Gal4 DBD antibody (Santa Cruz) for 2 h at 4°C with rotation. Forty microliters of protein A agarose was added, and incubation was continued for 1 h. The beads were washed three times with 0.5 ml 10s buffer and boiled in Laemmli sample buffer. Immunoprecipitated proteins were separated by SDS-PAGE and detected by Western blotting with anti-ORF50 antibody (53) or anti-V5 antibody.
Western blotting.
Western blotting was performed as described previously (51), with the following modifications. The antibodies were diluted as follows: anti-ORF50, 1:5,000; anti-V5, 1:1,000; and horseradish peroxidase-conjugated secondary antibodies, 1:5,000. All washes were performed with phosphate-buffered saline containing 0.1% Tween 20. Horseradish peroxidase was detected by enhanced chemiluminescence (Pierce) according to the manufacturer's recommendations.
Immunofluorescence.
Immunofluorescence was performed exactly as previously described (53).
Protein cross-linking.
BCBL-1 cells were left untreated or were treated with tetradecanoyl phorbol acetate (TPA; 20 ng/ml) for 1 to 24 h. Extracts of total cellular protein were prepared, and equal amounts of protein were treated with increasing amounts of the cross-linking reagent disuccinimidyl suberate (DSS; Pierce) as previously described (61), using cell lysis buffer at pH 7.5. The proteins were analyzed by SDS-PAGE/Western blotting, using a 5% gel in Tris-glycine buffer. The Hi-Mark Protein Ladder (Invitrogen) was used as a marker for apparent molecular masses. The calibration curve was determined by calculating the log10 molecular mass/elution volume for the protein standards (R2 = 0.9608).
RESULTS
The LR is necessary for ORF50/Rta-mediated transactivation.
To identify the requirements for Rta homomultimerization, we focused initially on the LR of Rta that lies within aa 244 to 275 (Fig. 1A). The LR is contained within the domain of Rta required for interaction with the cellular proteins K-RBP, RBP-Jk, and C/EBPα (10, 46, 86, 88, 89) (Fig. 1A). Figure 1B shows the alignment of Rta's LR with its gammaherpesvirus homologs and with the well-characterized leucine zipper (LZ) of the S. cerevisiae protein GCN4. The GCN4 LZ forms parallel coiled coils in solution and mediates dimerization (36, 63, 64). Rta LR and GCN4 LZ both contain four leucine residues spaced at 7-aa intervals (Fig. 1B). Three of these phased leucines are conserved among the Rta homologs of the gamma-2-herpesviruses of Old World primates (Fig. 1B). However, the divergence at the other positions of the heptad pattern predicts significant structural differences between the Rta LR and the GCN4 LZ. In particular, the LR of ORF50/Rta is not predicted to form a coiled coil (58) due to the presence of five proline amino acids, which are absent from the GCN4 LZ (Fig. 1B).
FIG. 1.

Primary amino acid sequence of KSHV ORF50/Rta protein. (A) Structure/function map of ORF50/Rta. A schematic of the primary amino acid sequence of the FL ORF50/Rta protein is shown. The numbers on the diagram refer to amino acid positions; the numbers to the right in parentheses indicate references. The bars with arrowheads indicate the positions of single-amino acid mutations that eliminate ubiquitin E3 ligase activity and binding to C/EBPα and DNA. The bars underneath ORF50 represent domains with functions identified in the list at the right. See the text for the relevant references. Abbreviations: NLS, nuclear localization sequence; AatII, position of truncation of cDNA by restriction digestion by the AatII enzyme; ST, serine/threonine-rich sequence; hyd, hydrophobic; DE, acidic amino acid-rich; HDAC1, histone deacetylase 1. Symbols: *, location of single-amino-acid mutation that eliminates binding to C/EBPα; +++, basic amino acid-rich sequence. (B) Alignment of KSHV ORF50/Rta LR with other gammaherpesvirus ORF50 proteins. Numbers refer to the animo acid position. Arrows point to leucines in the heptapeptide repeat. White lettering on a black background indicates amino acid identity. The bar under the viral sequences indicates the core of highest identity among all the indicated proteins. “heptad” indicates the lettering of canonical amino acid positions in heptad repeats. Abbreviations: M. mulatta, Macaca mulatta (either RRV/H26-95 or RRV/17577 isolates); M. fuscata, Macaca fuscata (GenBank accession number NC_007016) (S. G. Hansen, N. Avery, M. K. Axthelm, and S. W. Wong, unpublished data); HVS, herpesvirus saimiri; MHV-68, murine gammaherpesvirus 68; EHV-2, equine herpesvirus 2; AHV-1, alcelaphine herpesvirus 1; EBV, Epstein-Barr virus; GCN4, S. cerevisiae GCN4 leucine zipper.
To determine the requirement for the LR for transcriptional activation by the FL ORF50 protein, we deleted the LR in the clone ORF50ΔLR. We previously investigated the mechanism of ORF50 transactivation by transiently cotransfecting mammalian cells with an ORF50 expression vector and reporter plasmids in which various KSHV promoters drive expression of the luciferase gene. We and others have shown that, among viral promoters, the ORF57, K-bZIP, and nut-1/PAN promoters are strongly transactivated by ORF50 in these assays (10, 15-17, 46, 51-53, 77-79, 86-88, 92, 103).
Figure 2 shows the results of transfections of the human endothelial cell line SLK with expression vectors for WT Rta or the ΔLR mutant. As expected, the K-bZIP (Fig. 2A) and Nut-1/PAN (Fig. 2B) promoters were activated by WT ORF50 (maximally to about 100- to 140-fold), but the mutant ORF50ΔLR protein failed to transactivate both promoters (Fig. 2A and B). Since these promoters have been demonstrated to be activated by Rta, using different mechanisms (46, 78), the LR must be generally required for Rta to function as a transactivator. To exclude the possibility that deletion of the LR affected the stability or nuclear localization of the ORF50 protein, we performed Western blotting (Fig. 2C) and immunofluorescence (Fig. 2D) analyses of the transfected SLK cells. These assays demonstrated that the expression and nuclear localization levels of the WT, mutant, and 50ΔLR proteins were similar.
FIG. 2.
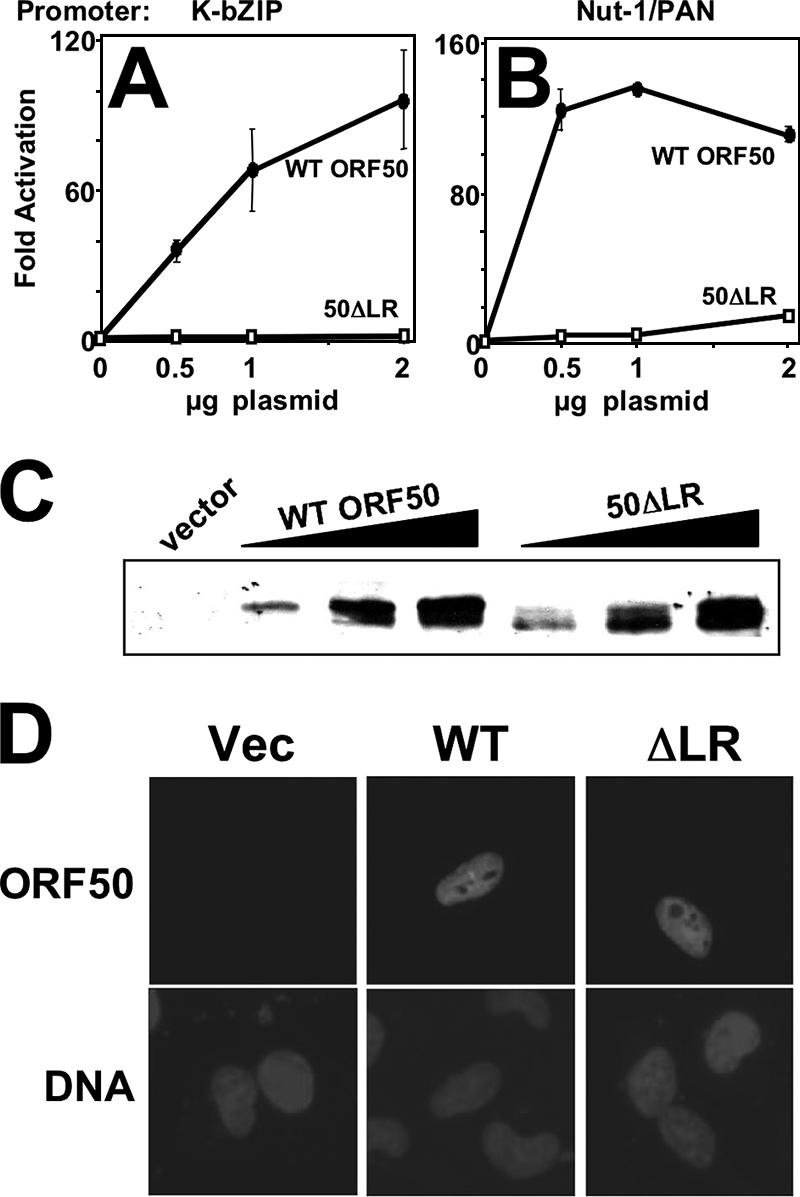
The LR is required for KSHV Rta to activate transcription. Human endothelial SLK cells were cotransfected with the indicated amounts of vector expressing WT ORF50 or ORF50ΔLR and a luciferase reporter vector for the K-bZIP (A) or Nut-1/PAN (B) promoter. Total DNA was normalized by the addition of empty expression vectors. The cells were harvested and lysed at 40 h posttransfection. Luciferase activity was normalized to β-galactosidase activity, and n-fold activation is given relative to that of the reporter vector transfected alone (0 μg plasmid). Vertical bars indicate standard deviations. Data represent results of triplicate transfection experiments repeated at least twice. See Materials and Methods for details. (C) Equivalent amounts of WT ORF50 and ORF50ΔLR were expressed in transfected SLK cells. The extracts used for the experiments shown in panel A were analyzed by SDS-PAGE/Western blotting, using anti-Rta serum. (D) WT ORF50 and ORF50ΔLR are both expressed in SLK cell nuclei. SLK cells were transfected as for panel A and analyzed by immunofluorescence 40 h posttransfection, using anti-Rta primary serum, fluorescein isothiocyanate-conjugated secondary serum, and DAPI (4′,6′-diamidino-2-phenylindole) to stain nuclear DNA. Vec, vector.
Deletion of the LR does not eliminate interaction of ORF50/Rta with RBP-Jk/CSL or with DNA.
Interactions of ORF50/Rta with the cellular protein RBP-Jk/CSL and DNA are required for Rta to transactivate the K-bZIP (92; K. D. Carroll, F. Khadim, S. Spadavecchia, D. Palmeri, and D. M. Lukac, unpublished data) and Nut-1/PAN promoters (79). Since neither promoter was activated by ORF50ΔLR (Fig. 2), we asked whether deletion of the LR eliminated either of these interactions. We expressed RBP-Jk as a fusion to the glutathione binding domain of the glutathione-S-transferase (GST) protein and tested its binding to WT and mutant ORF50 proteins generated in RRLs. As shown in Fig. 3A, WT ORF50, ORF50ΔSTAD, and ORF50ΔLR all interacted with GST-RBP-Jk but not with the GST moiety alone.
FIG. 3.

Deletion of the LR does not eliminate interaction of ORF50/Rta with RBP-Jk or promoter DNA. (A) GST-RBP-Jk or the GST moiety alone was purified and tested for binding to RRLs programmed with plasmids expressing the indicated proteins. The proteins were labeled cotranslationally by addition of L-[35S]-methionine to the RRLs. Bound proteins were visualized by autoradiography after being displayed by SDS-PAGE. Five percent of the input protein was also analyzed as a size reference. (B) WT ORF50 (50WT) or ORF50ΔLR (50ΔLR) was expressed and purified as a fusion to the MBP and tested in increasing amounts for binding to 32P-labeled oligos of the Rta binding sites from the indicated promoters. 0, lanes containing labeled DNA mixed with buffer in the absence of protein.
We tested the ability of WT ORF50 or ORF50ΔLR to bind to the Nut-1/PAN or K-bZIP promoter, using electrophoretic mobility shift assays. As shown in Fig. 3B, both WT ORF50 and ORF50ΔLR bound strongly to 32P-labeled oligos encoding Rta's binding sites in both promoters (51, 77). Interestingly, the major WT Rta/DNA complex was slightly reduced with ORF50ΔLR, and two new complexes appeared (Fig. 3B). One of the new ORF50ΔLR/DNA complexes appeared in the wells of the gel. These data suggest that ORF50 DNA binding, per se, is not inhibited by deleting the LR, but the patterns of Rta/DNA complexes are altered.
The LR is necessary for the dominant negative mutant ORF50ΔSTAD to interact with and inhibit transactivation by WT ORF50.
We previously demonstrated that a mutant of Rta truncated C-terminally at aa 531 (ORF50ΔSTAD) (Fig. 1A) specifically inhibited Rta-mediated transactivation and KSHV reactivation (52). ORF50ΔSTAD and WT Rta proteins bound to each other directly, suggesting that ORF50ΔSTAD functions trans-dominantly by forming inactive, mixed multimers with wild-type ORF50/Rta protein (52). We reasoned that defining the mechanism by which ORF50ΔSTAD functions would provide important insights into the molecular regulation of reactivation governed by multimerization. To determine whether the LR was necessary for the trans-dominant phenotype of ORF50ΔSTAD (hereafter called 50ΔSTAD), we tested the ability of 50ΔSTAD with the LR deleted (50ΔSTADΔLR) to inhibit WT ORF50-mediated transactivation of the K-bZIP promoter. SLK cells were transfected with a constant amount of WT ORF50 expression vector together with increasing amounts of 50ΔSTAD or 50ΔSTADΔLR plasmids. The transactivation of the viral K-bZIP promoter by WT ORF50 was dramatically inhibited by the dominant negative mutant 50ΔSTAD in a dose-dependent manner (Fig. 4A). However, 50ΔSTADΔLR was completely unable to inhibit ORF50-mediated transactivation at any amount of cotransfected plasmid. This confirms that the LR is required for ORF50ΔSTAD to inhibit WT ORF50 transactivation.
FIG. 4.

The LR is required for ORF50ΔSTAD to function as a dominant negative transcriptional inhibitor of WT ORF50/Rta. (A) SLK cells were cotransfected with the indicated plasmids. The cells were harvested, and extracts were analyzed as for Fig. 2. (B) Top panel, equivalent amounts of V5-ORF50ΔSTAD and V5-ORF50ΔSTADΔLR were expressed in transfected SLK cells. Bottom panel, WT ORF50 expression levels did not change in the presence of ORF50ΔSTAD or ORF50ΔSTADΔLR. Extracts from the transfection experiments for panel A were analyzed by SDS-PAGE/Western blotting, using anti-V5 primary serum (top panel) or anti-Rta primary serum (bottom panel). The bands seen in the vector-only lane (Vec) represent background bands in the cell extracts.
The top panel of Figure 4B shows that 50ΔSTAD and 50ΔSTADΔLR were expressed at equivalent levels in transfected SLK cells. Both proteins were also correctly and identically localized to the nuclei of transfected cells (data not shown). The bottom panel of Figure 4B shows that the WT ORF50 protein was expressed at high levels regardless of the amount of coexpression of 50ΔSTAD and 50ΔSTADΔLR. Since Rta can mark heterologous proteins for degradation by ubiquitin ligation (102), these data suggest that the dominant negative phenotype of ORF50ΔSTAD is unrelated to degradation of WT ORF50.
To determine whether the LR was required for the ORF50ΔSTAD-ORF50 interaction, we transfected human embryonic-kidney 293 cells with plasmids expressing WT ORF50, 50ΔSTAD, and 50ΔSTADΔLR, both alone and together. The 50ΔSTAD and 50ΔSTADΔLR proteins were expressed as C-terminal fusions to the V5 epitope tag. As expected, all proteins were localized to the nuclei of the 293 cells (data not shown). Following immunoprecipitation with an anti-V5 antibody, Western blotting (Fig. 5A) showed that WT FL ORF50 protein was coimmunoprecipitated with 50ΔSTAD but not with 50ΔSTADΔLR. Importantly, WT ORF50 was not precipitated when expressed alone. Thus, the LR is required for the dominant negative ORF50ΔSTAD protein to interact with WT ORF50.
FIG. 5.

The LR is required for ORF50ΔSTAD to interact with WT ORF50/Rta. (A) 293 cells were cotransfected with the indicated plasmids, and total cellular extracts were prepared 48 h posttransfection. Equal amounts of protein were immunoprecipitated, using anti-V5 serum, and the proteins were analyzed by SDS-PAGE/Western blotting, using anti-Rta primary serum. Ten percent of the total cell extract for each immunoprecipitation was analyzed for the input on the Western blot (Input). (B) RRLs were programmed with the indicated plasmids in the presence of [35S]methionine to label synthesized proteins. Equal amounts of RRL were immunoprecipitated, using anti-Rta serum, and the proteins were analyzed by SDS-PAGE/autoradiography. Ten percent of the RRL for each immunoprecipitation was analyzed for the input lanes (10% Input). (C) 293 cells were cotransfected with the indicated plasmids, and immunoprecipitation was performed as for panel A, using anti-Gal4 primary antibody. Western blotting was performed using anti-V5 primary antibody. I.P., immunoprecipitation; Ig, immunoglobulin band; +, transfected plasmid.
To confirm the above result, RRLs were programmed with vectors expressing WT ORF50 alone or together with either 50ΔSTAD (aa 1 to 514) or 50ΔSTADΔLR. [35S]-methionine was included to label the products. The lysates were incubated with anti-ORF50 antibody (53), which recognizes only the full-length protein, aa 525 to 691, and the immunoprecipitates were collected with protein A agarose. After being washed, the proteins bound to the full-length ORF50 were examined by SDS-PAGE. Figure 5B shows that the dominant negative 50ΔSTAD protein was coimmunoprecipitated with full-length WT ORF50, confirming that the two proteins form stable heterodimers in vitro. However, similar to the results for extracts of transfected cells, 50ΔSTADΔLR was unable to interact with full-length WT ORF50. Importantly, the truncated proteins were not precipitated when expressed alone. Taken together, the data shown in Fig. 4 and 5 demonstrate that the LR is required for 50ΔSTAD to interact with WT ORF50 and inhibit its function in a dominant negative fashion.
To confirm that FL ORF50/Rta can interact with itself, we coexpressed ORF50 as a fusion to either the V5 epitope or the DNA binding domain of the S. cerevisiae protein Gal 4. Following immunoprecipitation with a Gal4-specific antibody, Western blotting (Fig. 5C) showed that FL, V5-tagged ORF50 protein was coimmunoprecipitated with Gal4-ORF50. Importantly, V5-ORF50 was not precipitated when expressed alone. These data show that full-length WT ORF50/Rta forms homomultimers in mammalian cell extracts.
The LR is necessary but not sufficient for mediating multimerization of ORF50/Rta.
The data described above show that the LR is required for formation of mixed multimers of 50ΔSTAD and 50ΔSTADΔLR. We were interested in whether the LR was also sufficient for multimerization. We programmed RRLs with cDNAs for WT ORF50 alone or for three additional C-terminal-truncation mutants of ORF50. Shown in Fig. 6A, these mutants encoded aa 1 to 414 (ORF50ΔAat II), 1 to 272 (ORF50-N1-272), or 1 to 238 (ORF50-N1-238). We performed coimmunoprecipitation using our polyclonal ORF50/Rta-specific antiserum, which was generated using the C terminus of Rta (aa 525 to 691) as the antigen (Fig. 6A). Therefore, the antiserum detects only the WT ORF50 protein but none of the mutants diagrammed in Fig. 6A. Coimmunoprecipitation experiments (Fig. 6B) show that only ORF50ΔAatII was capable of forming multimers with WT ORF50 when cotranslated in RRL. Even though the ORF50 aa 1 to 272 mutant contains the LR, it was unable to bind detectably to WT ORF50. These data suggest that the LR region plus the region lying just C-terminally to it, aa 276 to 414 (Fig. 1A), encompass Rta's multimerization domain.
Purified recombinant ORF50 proteins form oligomers in vitro.
The data above show that the LR region of Rta is necessary for (i) transactivation by WT Rta, (ii) dominant negative function of the mutant ORF50ΔSTAD, and (iii) heteromeric interactions of WT Rta with ORF50ΔSTAD.
To determine the extent of multimerization required for ORF50/Rta to function as the KSHV lytic switch protein, we tested the self-association properties of a series of MBP-ORF50 fusions (Fig. 7A) expressed in E. coli. In Fig. 7B, SDS-PAGE analyses of MBP-50 (full-length WT ORF50, left panel), MBP-50ΔSTAD (right panel), and derivatives of each showed that the proteins were solubly expressed in intact form and were purified to near homogeneity.
For initial visualization of ORF50 multimers, we employed BN-PAGE, a high-resolution method requiring relatively little protein (73). As detailed in Materials and Methods, 0.5, 1, 2, and 3 μg of each protein was loaded onto a 4- to 12%-gradient gel in nondenaturing/nonreducing conditions. Figure 8A, lanes 2 to 5, shows that WT MBP-50 forms a single band that migrates with an apparent molecular mass slightly greater than 440 kDa. Figure 8A, lanes 6 to 9, shows that MBP-50ΔSTAD forms multiple bands in the BN gel, indicating the existence of at least four multimeric species in the preparation. These species ranged from about 350 kDa to >1,000 kDa in molecular mass (i.e., in the gel wells).
FIG. 8.

Multimerization of MBP-ORF50 and MBP-50ΔSTAD. (A) BN-PAGE. One to three micrograms of MBP-50 or MBP-50ΔSTAD was analyzed by BN-PAGE on 4% to 12% Bis-Tris gradient gels, as described in Materials and Methods. When the dye front reached the bottom, the gel was destained and analyzed by capturing a digital image in visible light. The migration position of the molecular mass markers is shown at the left. The numbered arrows on the right show migration positions of the MBP fusion protein complexes; apparent molecular masses were determined by gel exclusion chromatography as for panels B and C. (B and C) Gel filtration chromatography. MBP-50 (B) or MBP-50ΔSTAD (C) was separated by gel filtration chromatography, using a Superdex200 HR 10/30 column, as described in Materials and Methods. Fractions (0.5 ml) were collected, and protein content was measured by optical density at 280 nm. The peaks are labeled F1 and F2 for full-length ORF50 and S1, S2, and S3 for ORF50ΔSTAD. The molecular mass of each peak is shown above each elution profile; the molecular mass was determined by comparison to the elution profile of thymoglobulin, ferritin, and catalase (Table 1). MBP fusion proteins, or MBP alone, was identified in the indicated peaks by Western blotting, using anti-MBP antibody (data not shown).
We compared the concentration dependence of multimer formation of each MBP fusion protein with that of the MBP moiety alone. The MBP moiety alone remained monomeric up to 0.5 μg/μl but formed dimers above that concentration (data not shown). Importantly, multimerization of all of the MBP-ORF50 fusion proteins (shown in Fig. 8 and thereafter) was independent of the concentration over the range tested (0.1 μg/μl to 1.0 μg/μl). We concluded that the MBP moiety does not participate in multimerization of the MBP fusion proteins. When we cleaved the MBP moiety from the MBP-Rta fusion protein using Factor Xa, little soluble, full-length Rta remained (data not shown). Therefore, we could not formally test the influence of MBP on the fusion protein.
To more accurately determine the molecular masses of the proteins visualized by BN-PAGE, we subjected the samples to size exclusion chromatography using Superdex200. Figure 8B shows a representative elution profile for MBP-50. The major protein peaks were compared to those from a calibration curve and corresponded to apparent molecular masses of 1,096 and 468 kDa, respectively. These sizes are consistent with MBP-50 being a decamer (10 times) and a tetramer (4 times) (for a summary, see Table 1). The corresponding tetrameric BN-PAGE complex is indicated in Fig. 8A; the decameric complex remains in the well on the BN-PAGE gel (Fig. 8A).
Figure 8C shows that MBP-50ΔSTAD elutes in three major peaks: 1,000 kDa (10 subunits), 603 kDa (6 subunits), and 398 kDa (4 subunits) (for a summary, see Table 1). The 603- and 398- kDa peaks correspond to the migration of the major complexes observable on BN-PAGE (Fig. 8A). The decamer of MBP-50ΔSTAD (1,000 kDa) visualized by chromatography remains in the well on BN-PAGE. The relatively high “shoulder” apparent in Fig. 8C corresponds to the two minor complexes migrating between the wells and the hexamer band on BN-PAGE (Fig. 8A, lanes 6 to 9).
Overall, BN-PAGE and gel filtration were in excellent agreement for both proteins. A predominant species for both MBP-50 and MBP-50ΔSTAD is a tetramer. The hexameric complex appears to be unique to MBP-50ΔSTAD; however, we note that there is a relatively high shoulder between peaks F1 (4 subunits) and F2 (10 subunits) in Fig. 8B. This shoulder corresponds to a region in which a hexamer would elute. In agreement, BN-PAGE shows a very faint complex observable in the expected range for a hexamer of MBP-50 (Fig. 8A, lane 5). Western blotting confirmed that MBP-50 and MBP-50ΔSTAD eluted in the peak fractions shown in Fig. 8B and C, respectively, and were absent from samples with a relative absorbance of less than 2 (data not shown). Western blotting also showed that the MBP moiety alone was found in the indicated peaks on Fig. 8B and C (ca.15 ml). Both BN-PAGE and gel filtration indicated that MBP-ORF50 does not form monomers.
We tested the multimerization properties of ORF50/Rta expressed as an N-terminal fusion to a histidine-thioredoxin epitope (Thio-His-Rta; data not shown). This Rta fusion protein was expressed solubly and primarily formed tetramers and minor populations of hexamers and decamers identical to MBP-Rta in Fig. 8A. Thus, the type of epitope used for soluble expression and purification of Rta did not influence the multimerization properties of the fusion protein.
Two LR substitution mutants form tetramers and function in transcription similarly to WT Rta.
To determine the effect of deleting the LR on the native multimeric state of WT Rta, we subjected MBP-50ΔLR (Fig. 7) to BN-PAGE and gel filtration. Instead of forming monomers, MBP-50ΔLR failed to enter the BN-PAGE gel and eluted from the gel filtration column as a single peak at the upper limit of resolution (eluting at ca. 5 ml; data not shown). These observations suggested that MBP-50ΔLR was forming decamers, higher order multimers, or insoluble aggregates.
As an alternative to deleting the LR, we attempted to disrupt oligomer formation by introducing dramatic amino acid substitutions within the LR. Besides containing the leucine heptapeptide repeat, the LR also contains five prolines (Fig. 1B). Three of the phased leucines in the LR and all of the prolines are conserved among the Rta homologs in the gamma-2-herpesviruses of Old World primates. Although alpha helices can incorporate single prolines as kinks in the protein secondary structure, the abundance of prolines in the LR would be expected to prohibit formation of a typical helix (3, 36, 54, 62, 67, 97, 98). We hypothesized that the LR contributes to Rta multimerization without forming a typical LZ coiled-coil structure.
Our mutations targeted the region of the LR containing the highest conservation, the central 14 aa (Fig. 1B). We substituted three of the leucine residues with prolines to ensure ablation of alpha helix formation (Fig. 7A). The mutant protein was expressed as an MBP fusion in E. coli and was analyzed by gel filtration. As shown in Fig. 9A, MBP-50-L3P eluted in a single major peak of 457 kDa in a position nearly identical to the tetrameric peak of WT MBP-50 (Fig. 9B). Peak F2 in Fig. 9A also had a small shoulder corresponding to the expected elution of a hexamer (ca. 9.5 ml). Unlike WT MBP-ORF50, MBP-ORF50-L3P did not form a decamer (Fig. 9B). These data suggest that the L3P mutation in the LR stabilizes the Rta tetramer and inhibits decamer formation. The data demonstrate that the leucine heptapeptide repeat is not required for Rta multimerization. The gel filtration data were supported by BN-PAGE analyses (data not shown).
FIG. 9.

The MBP-50-L3P mutant primarily forms tetramers in solution. MBP-50-L3P (A) and MBP-ORF50 (B) were analyzed by gel filtration chromatography as described in the legend to Fig. 8. ORF50 and ORF50-L3P were analyzed for transactivation of the K-bZIP promoter (C) and expression (D) as described for Fig. 2.
To test the functional consequences of the L3P mutation, we compared WT ORF50 to 50-L3P for transactivation of the K-bZIP promoter in SLK cells. To our surprise, 50-L3P transactivated the viral promoter to a magnitude similar to that of WT Rta (Fig. 9C). 50-L3P was well expressed in the nuclei of transfected cells (Fig. 9D and data not shown). These data suggest that tetramerization of Rta is sufficient for its function as a transactivator. Moreover, the leucine heptapeptide repeat, per se, is not required for proper Rta function.
Although not predicted to form an alpha helix, the LR might nonetheless contribute to Rta multimerization through hydrophobic interactions mediated by the leucine side chains. In that scenario, since the proline side chain is also hydrophobic, the proline substitutions of the L3P mutant would not be expected to dramatically alter the overall hydrophobicity of the LR. Therefore, we designed a second LR substitution mutant that converted the hydrophobic LR core to a net hydrophile. As shown in Fig. 7A, four of six amino acids in the LR core were converted from hydrophobic (I, L) to hydrophilic (D, N, R) in the ORF50+PHIL mutant. This mutant LR contains an extended hydrophilic patch, since the substituted amino acids are flanked by additional amino acids with hydrophilic side chains (E, S) (Fig. 7A).
In the context of either MBP-ORF50 or MBP-ORF50ΔSTAD, the +PHIL mutants were difficult to express in E. coli. Only MBP-ORF50ΔSTAD+PHIL could be expressed in amounts sufficient for BN-PAGE analyses. We compared the multimerization properties of that mutant to MBP-ORF50ΔSTAD. Figure 10A shows that both proteins form indistinguishable patterns of multimers (though analyzed at different concentrations). In both cases, tetramers (ca. 398 kDa) appear to be the predominant species, similar to WT ORF50. Furthermore, WT ORF50+PHIL was not impaired in its ability to transactivate the K-bZIP promoter in transiently transfected SLK cells (Fig. 10B). Figure 10C confirms expression of the mutant protein. Therefore, neither disruption of the leucine heptapeptide repeat nor increasing the hydrophilicity of the LR impairs ORF50's ability to transactivate.
FIG. 10.
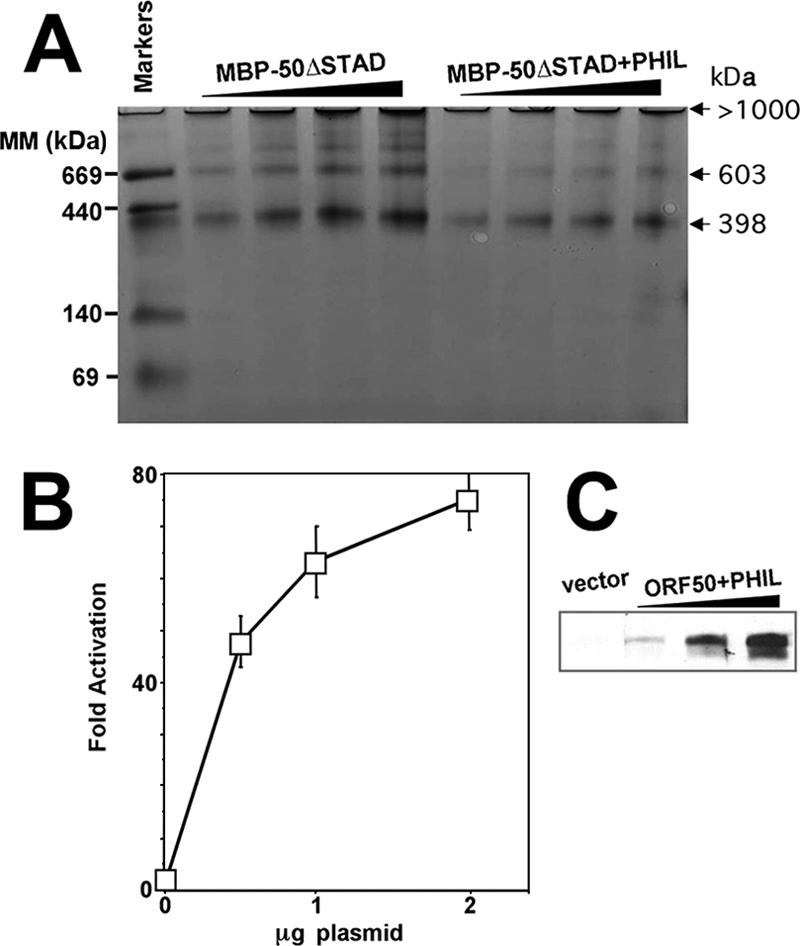
ORF50+PHIL functions with WT ORF50 transcriptional activity. (A) MBP-50ΔSTAD and MBP-50ΔSTAD+PHIL were analyzed by BN-PAGE as described for Fig. 8A. (B) ORF50+PHIL transactivates transcription with WT ORF50 activity. The indicated plasmids were cotransfected into SLK cells with the K-bZIP/RAP reporter plasmid, extracts were generated, and transactivation was analyzed as described in the legend to Fig. 2A. (C) Equivalent amounts of WT ORF50 and ORF50+PHIL were expressed in transfected SLK cells. Extracts from the experiments for panel A were analyzed by SDS-PAGE/Western blotting, using anti-Rta serum.
Tetramers of ORF50/Rta are sufficient for inducing KSHV lytic reactivation and replication.
We previously demonstrated that the ability of WT and mutant Rta to transactivate transcription of promoter/reporter plasmids in uninfected cells corresponded directly with the ability of ectopically expressed Rta to reactivate the complete viral lytic cascade (52, 53). Our marker for induction of reactivation by Rta was the K8.1 protein, which is expressed in PEL cells with true late kinetics (i.e., K8.1 expression requires viral DNA replication) (50, 53, 99). To test the LR mutants for their abilities to reactivate the KSHV lytic cycle, we scored reactivation at the single-cell level 72 h after electroporating BCBL-1 cells. Cells were scored positive if Rta expression was detected by visual inspection by fluorescence microscopy; for quantitation, we counted the percentage of Rta-positive cells that were also K8.1 positive (Rta+/K8.1+ divided by Rta+). At least 200 doubly positive cells were counted for each. Cells transfected with an empty expression vector were scored similarly. Results from the empty vector transfections (spontaneous reactivation) were subtracted from those of each WT or mutant Rta transfection. Results were normalized to those for cells transfected with the WT Rta expression vector, which was set at 100%. As shown in Fig. 11A, ORF50-L3P reactivated KSHV identically to WT ORF50/Rta. Conversely, ORF50ΔLR was severely impaired, but not completely deficient, for reactivation, with about 25% of the activity of WT Rta. These data suggest that tetramers of ORF50/Rta are sufficient for reactivating the complete KSHV lytic cycle. Conversely, higher-order multimers, as formed by ORF50ΔLR, are incapable of reactivating the KSHV lytic cycle.
FIG. 11.
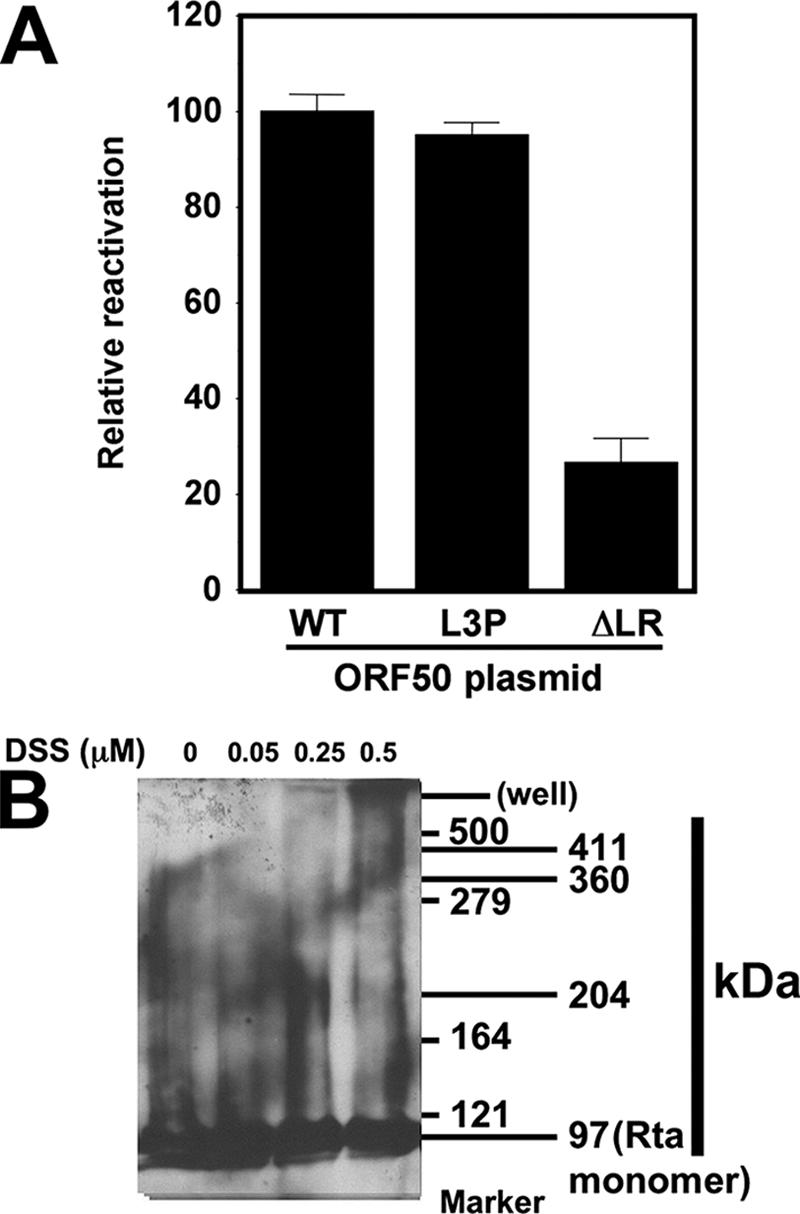
Tetramers of ORF50/Rta are sufficient to reactivate KSHV from latency. (A) BCBL-1 cells were electroporated in duplicate with plasmids expressing the indicated proteins and analyzed for reactivation by indirect immunofluorescence 72 h later. The cells were scored by fluorescent microscopic visualization as the percentage of Rta single-positive cells that also expressed K8.1 (Rta/K8.1 double-positive cells; no cells expressed K8.1 in the absence of Rta). To eliminate spontaneously reactivating cells, the identical calculation was performed on cells electroporated with an empty control vector, pcDNA3.1; that percentage was subtracted from the values for all other transfections. Those corrected percentages were then normalized to that of WT ORF50-transfected cells, which was adjusted to 100%. (B) Total cellular extracts from uninduced or TPA-induced (24 h) BCBL-1 cells were treated with the cross-linker DSS and then analyzed by SDS-PAGE/Western blotting, using Rta-specific antiserum as the primary antibody. The first column of numbers on the right shows the migration positions of the protein molecular mass markers. The second column shows the migration positions of Rta-containing complexes.
To attempt to determine whether Rta formed tetramers in infected cells, total cellular protein extracts were prepared from TPA-induced cells and treated with the cross-linking agent DSS. Reactions were then analyzed by SDS-PAGE and Western blotting with anti-Rta serum. As shown in Fig. 11B, monomers of Rta migrate at an apparent molecular mass of 97 kDa in this reaction/gel system. As increasing amounts of DSS were incubated with the extracts, a series of Rta-containing complexes formed (evaluated by observing the Western blot from left to right). In order of their appearance, these complexes were 204, 360, and 411 kDa. As this is a complex, crude protein lysate, it is not clear whether heterologous proteins can be cross-linked to Rta under these conditions. Nonetheless, the 204- and 411-kDa complexes are consistent with Rta cross-linked in extracts as dimers and tetramers, respectively.
Figure 11B also shows a prominent band representing Rta monomers; however, the physiologic relevance of monomeric Rta is unclear. Since the efficiency of DSS for cross-linking highly purified, recombinant proteins is typically only 30 to 40% (81, 100), the monomeric band might represent single units of Rta that were not cross-linked in the crude, unfractionated PEL extracts. Alternatively, eukaryotic-specific posttranslational modifications might favor monomers of Rta that are not observed when Rta is expressed in prokaryotic hosts. Figure 11B shows the results for BCBL-1 cells treated with TPA for 24 h; at earlier time points, a similar pattern of DSS dose-dependent multimers of Rta was observable in protein extracts (data not shown).
ORF50-L3P is inhibited by the dominant negative mutant ORF50ΔSTAD.
Since the L3P mutant of WT Rta retained WT function (Fig. 9C and 11), we also expected it to be inhibited by the trans-dominant ORF50ΔSTAD mutant. As shown in Fig. 12A, 50ΔSTAD inhibited ORF50-L3P-mediated transactivation in a dose-responsive fashion, similar to its effect on WT ORF50. To determine whether 50-L3P and 50ΔSTAD could directly bind to each other, we transfected 293 cells with plasmids expressing WT ORF50, ORF50L3P, V5-50ΔSTAD, and V5-50ΔSTADΔLR, either alone or in combination. V5 anti-serum was used to precipitate the V5-tagged proteins, and coimmunoprecipitated proteins were detected by Western blotting with ORF50 anti-serum. As shown in Fig. 12B, WT ORF50 and mutant ORF50-L3P were coimmunoprecipitated with V5-50ΔSTAD but were not coimmunoprecipitated with V5-50ΔSTADΔLR. These data affirm that the LR-dependent interaction of 50ΔSTAD with WT ORF50 is required for dominant negative transcriptional function and suggest that the phased leucines of the LR heptapeptide repeat are not essential for ORF50 multimerization.
FIG. 12.

ORF50ΔSTAD inhibits ORF50-L3P transactivation using a mechanism similar to that of WT ORF50. (A) ORF50ΔSTAD inhibits ORF50-L3P transactivation in a dominant negative fashion. SLK cells were cotransfected with the indicated plasmids. The cells were harvested, and extracts were analyzed as described in the legend to Fig. 2. (B) ORF50ΔSTAD binds directly to ORF50-L3P in transfected cells. 293 cells were cotransfected with the indicated plasmids, and total cellular extracts were prepared 48 h posttransfection. Equal amounts of protein were immunoprecipitated, using anti-V5 serum, and the proteins were analyzed by SDS-PAGE/Western blotting, using anti-Rta primary serum. Ten percent of the total cell extract for each immunoprecipitation was analyzed for the input Western blots (Input). Antibodies used for the input Western blots are indicated to the left of the bottom panels. IP, immunoprecipitation.
The LR is required for ORF50 to form tetramers and hexamers.
The 50-L3P mutant retained WT ORF50 activity (Fig. 9 and 11), suggesting that the form of Rta that functions transcriptionally and in viral reactivation is a tetramer. Deletion of the LR-ablated ORF50's transcriptional and reactivation functions but did not convert Rta to a monomer. Therefore, it was critical to determine the oligomeric state of ORF50ΔLR. As mentioned above, we could not distinguish between decamers and misfolded aggregates of FL MBP-ORF50-ΔLR. We reasoned that the smaller size of MBP50-ΔSTADΔLR might facilitate such analysis. Therefore, we compared the multimeric profiles of MBP-ORF50-ΔSTAD, MBP50-ΔSTADΔLR, and MBP50-ΔSTAD-L3P, using BN-PAGE and size exclusion chromatography.
As depicted in Fig. 13A and B, MBP-50ΔSTADΔLR formed very little of the tetrameric or hexameric complexes that are formed in abundance by MBP-50ΔSTAD (Fig. 13A and D). Instead, the major species formed by MBP-50ΔSTADΔLR is the decamer (1,000 kDa) and a unique peak corresponding to an octamer (794 kD; summarized in Table 1). The conclusions from both techniques were in excellent agreement. These data suggest that the LR favors formation of tetramers and hexamers by Rta and that deletion of the LR converts these forms to octamers and decamers.
FIG. 13.

The LR is required for ORF50 to form tetramers. The indicated proteins were analyzed by BN-PAGE (A) or gel filtration chromatography (B, C, and D) as described in the legend to Fig. 8.
As shown in Fig. 13A and C, MBP-50ΔSTAD-L3P preferentially forms tetramers, a smaller population of hexamers, and a minor population of decamers. This profile is similar to the multimers formed by FL MBP-50-L3P (Fig. 9A) and affirms that the L3P mutation stabilizes the tetrameric form of Rta.
Chimeric proteins reveal that Rta transcriptional activity is reduced as multimerization increases.
The LZ of the yeast protein GCN4 and a C-terminal region of the human protein p53 mediate dimerization and tetramerization, respectively, of those two proteins (21, 37, 63, 64). When those domains are fused to heterologous proteins, each can function dominantly to mediate dimerization and tetramerization, respectively, of the chimeras (25, 93). We replaced the ORF50 LR with the GCN4 LZ (ORF50ΔLR-GCN4) or the p53 tetramerization domain (ORF50ΔLR-p53TD) and tested the chimeras for multimerization and transactivation. Notably, the GCN4 LZ and the ORF50 LR share the heptapeptide repeat containing four leucines, but the GCN4 LZ lacks the prolines characteristic of the LRs from the Old World primate gamma-2-herpesviruses (Fig. 7A). The p53 tetramerization domain (TD) contains little sequence conservation with the ORF50 LR, despite the fact that both mediate tetramerization. To determine the oligomeric states of ORF50ΔLR-GCN4 and ORF50ΔLR-p53TD, we expressed each as MBP fusions. The chimeric proteins were purified from E. coli and analyzed by BN-PAGE. As shown in Fig. 14A, much of MBP-50ΔSTADΔLR-GCN4 remained in the wells of the gel, suggesting that it formed decamers, higher order oligomers, or aggregates. A very small amount migrated with apparent molecular masses consistent with those of octamers and hexamers. MBP-50ΔSTADΔLR-p53TD surprisingly did not form tetramers but instead formed a prominent band equivalent to the hexamers of MBP-50ΔSTAD and MBP-50ΔSTAD-L3P. The inability of the GCN4 LZ and p53 TD to impart their typical multimerization properties on ORF50ΔLR agreed with the data shown in Fig. 6B: the LR is not the only region of ORF50 that contributes to Rta multimerization. To determine the transcriptional consequences of replacing the ORF50 LR with the GCN4 LZ and the p53 TD, we expressed each transiently in SLK cells and compared them to WT ORF50 for transactivation of the K-bZIP reporter plasmid. As shown in Fig. 14B, ORF50ΔLR-GCN4 was unable to transactivate the reporter at all input plasmid amounts. This suggests that octamers and higher-order multimers of ORF50 are transcriptionally nonfunctional.
FIG. 14.
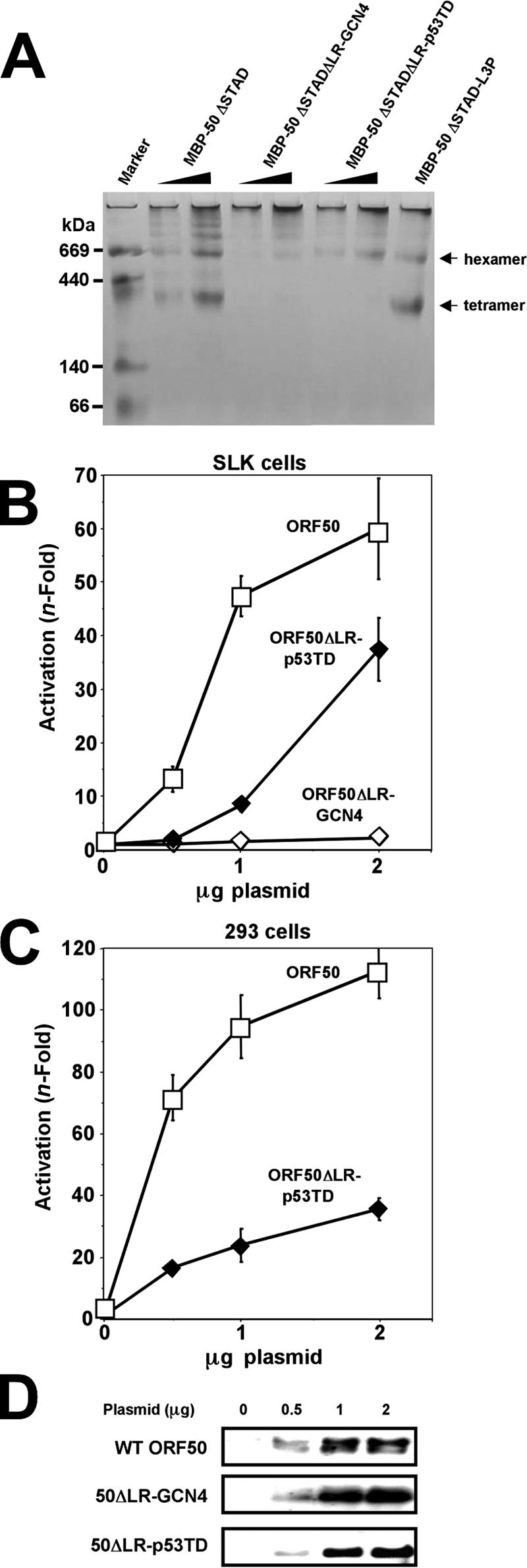
Tetramerization is required for full ORF50 transcriptional activity. (A) Multimerization of ORF50 chimeric proteins. The indicated proteins were analyzed by BN-PAGE as described in the legend to Fig. 8A. The arrows to the right indicate migration of tetramers and hexamers of MBP-50ΔSTAD and MBP50ΔSTADΔLR, which were included as molecular mass controls. (B and C) Transactivation by ORF50 decreases as the extent of multimerization increases. SLK (B) and 293 (C) cells were transfected with the indicated amounts of plasmids together with the K-bZIP/RAP reporter vector. Extracts were prepared and analyzed as described in the legend to Fig. 2. (D) Equivalent amounts of WT ORF50, 50ΔLR-GCN4, and 50ΔLR-p53TD were expressed in SLK cells. Extracts from the transfection experiment for panel B were analyzed by SDS-PAGE/Western blotting, using anti-Rta primary serum.
ORF50ΔLR-p53TD was severely debilitated in transactivation at low amounts of input plasmid (Fig. 14B). Increasing amounts of transfected ORF50ΔLR-p53TD showed a dose-responsive increase in transactivation. Maximal transactivation by this ORF50 chimera never exceeded 66% of WT levels in SLK cells (Fig. 14B and data not shown). In 293 cell transfections, ORF50ΔLR-p53TD was more debilitated than in SLK cells, with a maximal transactivation of ca. 30% of WT levels. These data suggest that the p53 TD was not sufficient to fully replace the WT transcriptional function of the ORF50 LR, possibly because of its inability to form tetramers. Hexamers of ORF50ΔLR-p53TD thus appear to be partially functional in transactivation. As a control, Fig. 14D shows that both ORF50 chimeras were well expressed in SLK cells.
DISCUSSION
In this report, we have demonstrated that tetramers of ORF50/Rta are sufficient to transactivate transcription and induce lytic viral reactivation of KSHV from latency. WT ORF50 protein forms tetramers and decamers and a minor amount of hexamers in solution (Fig. 8). The LR (aa 244 to 275) (Fig. 1) is one component of Rta that contributes to the extent of Rta multimerization and is required for Rta tetramerization (Fig. 13). Deletion of the LR abolishes transactivation by Rta (Fig. 2) and severely impairs induction of reactivation (Fig. 11). Deletion of the LR does not eliminate interaction of ORF50/Rta with RBP-Jk or with DNA (Fig. 3). Mutation of the LR by site-directed mutagenesis (Fig. 9 and 10) or complete replacement with heterologous multimerization domains (Fig. 14) alters the multimerization state of Rta. Replacement of three central leucine residues with prolines in the Rta LR generates the protein ORF50-L3P (Fig. 7A), which primarily forms tetramers and, importantly, retains WT function in transactivation and viral reactivation (Fig. 9 and 11). Since reactivation stimulated by ORF50/Rta was not completely abrogated by deletion of the LR (Fig. 11), the mutant retains some of its function in infected cells. In extracts from reactivating PEL cells, Rta is cross-linked into complexes consistent with dimerization and tetramerization, though a significant portion of Rta remains monomeric under these conditions (Fig. 11B).
Our study demonstrates that, as the extent of Rta multimerization increases, the ability of Rta to transactivate transcription decreases. Hexamers of Rta are partially active, but higher-order multimers are inactive (Fig. 14). Neither WT Rta nor any of our Rta variants formed monomers in solution.
We were limited in our visualization of higher-order multimers of WT Rta because their molecular masses approached and exceeded the limits of resolution of both BN-PAGE and size exclusion chromatography (Fig. 8 and Table 1). Therefore, we took advantage of a C-terminal-truncation mutant of Rta, called ORF50ΔSTAD (Fig. 1), that we had previously shown to inhibit Rta-mediated transactivation and KSHV reactivation (52). In that study, ORF50ΔSTAD and WT Rta proteins bound to each other directly, suggesting that ORF50ΔSTAD functions trans-dominantly by forming inactive mixed multimers with WT Rta protein. Expressed as a fusion to MBP, ORF50ΔSTAD (aa 1 to 505) formed tetramers, similar to WT ORF50 (Fig. 8). However, solutions of ORF50ΔSTAD had a significantly greater hexamer content than those of WT ORF50 and a substantially lower decamer content. This suggested that the C terminus of Rta, deleted in ORF50ΔSTAD, contributes to decamer formation of the WT protein. Chang, et al. recently showed that the C terminus of ORF50 inhibits DNA binding and the stability of the cognate protein and proposed a model in which the C terminus binds directly to the Rta N terminus to mediate this inhibition (15). Mutations in a KKRK motif, located between aa 527 and 530, reversed this effect (15) (Fig. 1A). Although we have not formally tested this interaction, our present data, as listed below, support the hypothesis of Chang et al. (15): the ORF50ΔSTAD truncation (i) eliminates the KKRK motif, (ii) increases the tetrameric content, and (iii) decreases the decameric content of the resulting protein. In the context of the cognate ORF50 protein, we predict that this increase in ratio of tetramer to decamer will be found to be consistent with an increase in transcriptional activity and stimulation of reactivation.
We reasoned that characterizing the trans-dominant mechanism of ORF50ΔSTAD would provide important insights into the function of the WT ORF50 protein and molecular regulation of reactivation. We showed that deletion of the LR in ORF50ΔSTAD ablated its ability to inhibit the WT ORF50 protein (Fig. 4) and eliminated direct interaction between the two proteins (Fig. 5). This strongly supported our hypothesis that the dominant negative ORF50ΔSTAD protein functioned by forming mixed multimers with WT ORF50. However, the data does not exclude alternative mechanisms for ORF50ΔSTAD's trans-dominant phenotype. ORF50ΔSTAD protein contains all of the domains required for (i) Rta promoter association (by binding DNA or interacting with heterologous DNA binding proteins) (51, 79), (ii) stimulation of RBP-Jk DNA binding activity (10), (iii) binding to histone deacetylase 1 (32), and (iv) E3 ubiquitin ligase activity (102) (Fig. 1A). In many cases, single-amino acid substitutions within each domain ablate each of these functions (Fig. 1A). Conceivably, the trans-dominant phenotype of ORF50ΔSTAD might not result from forming inactive heteromers with WT Rta. Instead, ORF50ΔSTAD's dominant negative phenotype might result from forming homomultimers that compete with homomultimeric, WT Rta for any of these functions. Similarly, the inability of LR mutants in the context of the WT ORF50 protein to reactivate KSHV might result from inhibiting direct and necessary interactions of the LR with heterologous proteins or DNA. Although our experiments show that the LR is not required for Rta to bind RBP-Jk, the K-bZIP promoter, or the Nut-1/PAN promoter (Fig. 3), Rta contacts other proteins and promoters not tested here. Furthermore, our experiments do not distinguish those putative mechanisms from LR's role in Rta tetramerization; the LR deletion might eliminate homomultimeric interactions of Rta that are, in turn, required for heteromeric interactions of Rta with DNA or heterologous proteins. The Rta LR (aa 244 to 275) contains four heptapeptide repeats with leucines in every seventh position (Fig. 1B). The LR can therefore be aligned with LZ domains, such as that of the yeast protein GCN4 (Fig. 1B). LZs fold into alpha helices that mediate multimerization by forming coiled-coil structures with other LZs. In coiled coils, the amino acid positions of “a” and “d” (Fig. 1B) are typically hydrophobic residues, and “d” is most often leucine (34-36, 63, 64, 83, 97, 104, 105). The amino acid positions of “g” and “e” are frequently charged residues. Although the Rta LR partially satisfies these rules by containing leucines in its “d” positions, only two of the “a,” “g,” and “e” amino acids follow the conventions for typical coiled coils. Furthermore, the LR contains five “helix-breaking” prolines (3, 36, 54, 62, 67, 97, 98). Therefore, it is unlikely that the LR forms a coiled-coil structure. In support of this prediction, substitution of the core leucines of the LR did not affect Rta's function (Fig. 9 and 10). Substitution of three leucines with hydrophilic amino acids did not affect ORF50's multimerization (Fig. 10), and substitutions with prolines “stabilized” the Rta tetramer (mutant L3P) (Fig. 9). We were also unable to generate a transcriptionally active Rta protein by replacing its LR with derivatives of the regulated dimerization motif from the FK506 binding protein (20) (data not shown), suggesting that the Rta LR does not mediate homodimeric interactions.
Instead, our data support an essential role for proline residues in determining the extent of Rta multimerization. In this way, prolines in the LR region seem to limit the extent of multimerization, while leucines seem to promote formation of higher-order multimers. The substitution of three central leucines in the LR region with prolines in the ORF50-L3P mutant (Fig. 7A) vastly favored formation of tetramers that had WT activity in transcription and reactivating KSHV from latency (Fig. 9 and 11). Conversely, substitution of the LR with the GCN4 LZ, a four-membered leucine heptapeptide repeat that is completely devoid of prolines (Fig. 7A), yielded a chimeric protein that formed decamers or higher-order multimers that were transcriptionally inactive (Fig. 14). Phylogenetic comparisons of the LR also support the notion that the leucine repeat per se is not the key feature of aa 244 to 275 required for WT ORF50 function. Four of the leucines and all five prolines in the KSHV Rta LR are conserved in its homologs from gamma-2-herpesviruses of Old World primates (Fig. 1A). However, only the central PXXL peptide is conserved in all of Rta's gammaherpesvirus homologs (Fig. 1A).
We hypothesize that prolines outside of the LR (aa 244 to 275) also play a role in Rta oligomerization. The LR was required for the transcriptional functions of both the WT Rta and ORF50ΔSTAD proteins (Fig. 2 and 4). However, it was not sufficient to mediate the interaction between WT Rta and Rta aa 1 to 272 (Fig. 6). Instead, the smallest truncation of Rta that interacts with WT Rta contained aa 1 to 414 (ORF50ΔAatII) (Fig. 4). These data implicate a requirement for the LR plus C-terminal amino acids (276 to 414) in self-interactions of Rta and suggest that LR plus N-terminal amino acids (1 to 243) are not sufficient. Our observations that the GCN4 LZ, the heat shock factor trimerization domain, and the p53 TD were unable to confer dimerization, trimerization, or tetramerization, respectively, on Rta when substituted for the LR (Fig. 14 and data not shown) also support the notion that regions of Rta located outside the LR are critical for determining the multimerization state of the protein. A striking difference between the N and C termini flanking the LR is their respective proline contents. Amino acids 1 to 243 of Rta contain 4.1% proline (10 residues), while aa 276 to 691 contain 13.2% proline (55 residues). Both leucines and prolines commonly participate in protein-protein interactions (57, 83, 96). Our data do not distinguish between a cis-acting role for prolines in the structure of an individual Rta monomer or a direct participation of prolines in interactions between Rta monomers. Interestingly, ORF50ΔSTADΔLR could not interact with and inhibit WT ORF50 (Fig. 4 and 5) but retained the ability to form homodecamers (Fig. 13). These data suggest that the LR confers on ORF50 a lower relative dissociation constant that is not overcome by mixing WT homomultimers with LR-mutated homomultimers.
Wang, et al., addressed Rta multimer formation using an EMSA approach with Rta generated in RRLs and a labeled probe from the nut-1/PAN promoter (88, 89). That study found that Rta amino acids 1 to 377 were sufficient to bind DNA and form a heterodimeric complex with WT Rta. Deletion of aa 11 to 112 impaired heterodimer formation. However, that mutation also impaired DNA binding of Rta to the nut-1/PAN promoter (88, 89), so it is difficult to compare to our data since ours was DNA independent. Nonetheless, that report suggests that a region of Rta N-terminal to the LR is also important for oligomerization, and dimers of Rta might be sufficient to activate the PAN promoter. Epstein-Barr virus Rta forms DNA-independent dimers dependent on its N terminus, as well (55). Song et al. also noted that KSHV Rta (aa 1 to 320) appears to form multiple oligomers in EMSA (78).
The multimerization state of Rta likely affects multiple aspects of Rta's function at different promoters. Liao et al. have identified a phased repeat of A/T-rich trinucleotides that is conserved in multiple Rta-responsive KSHV promoters (49). The ability of Rta to transactivate transcription increases proportionally to the number of A/T repeats in a reporter plasmid (49). Multimers but not dimers of Rta bound to the K-bZIP/RAP promoter in that study. Liao et al. thus proposed that Rta makes long-range contacts along promoter DNA that are facilitated by its ability to multimerize (49). Our data prove that tetramerization is essential for Rta-mediated transactivation of the K-bZIP/RAP promoter; accordingly, hexamers of Rta chimeras were less transcriptionally active. Using sucrose gradient centrifugation, Liao et al. concluded that semipure Rta primarily formed hexamers in solution; however, chemical cross-linking combined with EMSAs in that study demonstrated that tetramers of Rta were the largest complexes that bound to DNA (49). These EMSA data agree with our functional analyses.
We have recently reported that maximal transactivation of the RRE of the ORF57/Mta promoter corresponds with ternary complex formation among Rta, the cellular protein RBP-Jk, and DNA (10). Mutations of the ORF57 RRE that reduced or eliminated Rta transactivation and ternary complex formation mapped to both sides of the RBP-Jk site. Furthermore, a C-terminal truncation of Rta that retained its ability to bind to the Mta RRE and to RBP-Jk in solution was nonetheless unable to form the ternary complex. Although WT Rta independently formed three different complexes on the Mta RRE in EMSAs, suggestive of multimerization, only two of these complexes interacted with DNA-bound RBP-Jk to form the ternary complex (10). Based on these data together with those of the present study, we propose that Rta activates transcription of some promoters by forming multimers that bind to and straddle cellular promoter binding proteins. Indeed, we and others have found that full activation of the K-bZIP promoter by Rta requires DNA binding of Rta, RBP-Jk (10, 92), C/EBPα (88, 89), and octamer-1 (K. Carroll, F. Khadim, D. Palmeri, and D. M. Lukac, unpublished data).
Less than 10% of PEL cells transfected with an Rta expression vector express viral lytic proteins (44, 52, 53, 68; D. Palmeri, S. Spadavecchia, K. Carroll, and D. M. Lukac, unpublished data), so it is likely that regulation of Rta's expression and function are critical for lytic reactivation. We propose that the multimerization of Rta is an important regulatory point for the lytic switch. Under the conditions used in this study, we did not detect monomers of purified Rta in solution. Since oligomerization of Rta was independent of concentration even at 50 ng/μl, it suggests that the Kd for Rta self-association is probably very small and not biologically meaningful. However, since hexamers (and higher-order multimers) appeared to be nonfunctional, this also suggests an upper limit to Rta concentration that is permissive for reactivation. As we tested Rta purified from E. coli, our study did not address the role of posttranslational modifications in Rta multimerization. Indeed, Rta has been shown to be modified in vivo by phosphorylation and poly(ADP- ribosylation) (33, 52). Our data strongly support prolines as a major determinant of Rta multimerization. Protein domains containing prolines are well-established targets of kinases and hydroxylases, and prolines make significant contributions to protein structure by their rigid aromatic side chains and isomerization (4, 96, 106). Proline-dependent multimerization would thus be an attractive target for regulating Rta function.
Acknowledgments
This work was supported by the New Jersey Commission on Cancer Research, the American Cancer Society, and the American Heart Association.
We thank the Lukac lab members and Vivian Bellofatto for helpful discussions and critical readings of the manuscript, Shuishu Wang for advice on gel exclusion chromatography, Anoop Kaviriyani for assorted technical contributions, and Hilary Nelson and Bert Vogelstein for reagents.
Footnotes
Published ahead of print on 28 March 2007.
REFERENCES
- 1.Ambroziak, J., D. Blackbourn, B. Herndier, R. Glogan, J. Gullet, A. McDonald, E. Lennette, and J. Levy. 1995. Herpesvirus-like sequences in HIV-infected and uninfected Kaposi's sarcoma patients. Science 268:582-583. [DOI] [PubMed] [Google Scholar]
- 2.Baker, S. J., S. Markowitz, E. R. Fearon, J. K. Willson, and B. Vogelstein. 1990. Suppression of human colorectal carcinoma cell growth by wild-type p53. Science 249:912-915. [DOI] [PubMed] [Google Scholar]
- 3.Barlow, D. J., and J. M. Thornton. 1988. Helix geometry in proteins. J. Mol. Biol. 201:601-619. [DOI] [PubMed] [Google Scholar]
- 4.Berra, E., A. Ginouves, and J. Pouyssegur. 2006. The hypoxia-inducible-factor hydroxylases bring fresh air into hypoxia signalling. EMBO Rep. 7:41-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boneschi, V., L. Brambilla, E. Berti, S. Ferrucci, M. Corbellino, C. Parravicini, and S. Fossati. 2001. Human herpesvirus 8 DNA in the skin and blood of patients with Mediterranean Kaposi's sarcoma: clinical correlations. Dermatology 203:19-23. [DOI] [PubMed] [Google Scholar]
- 6.Bourboulia, D., D. Aldam, D. Lagos, E. Allen, I. Williams, D. Cornforth, A. Copas, and C. Boshoff. 2004. Short- and long-term effects of highly active antiretroviral therapy on Kaposi sarcoma-associated herpesvirus immune responses and viraemia. AIDS 18:485-493. [DOI] [PubMed] [Google Scholar]
- 7.Bowser, B. S., S. M. DeWire, and B. Damania. 2002. Transcriptional regulation of the K1 gene product of Kaposi's sarcoma-associated herpesvirus. J. Virol. 76:12574-12583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boyer, T. G., and A. J. Berk. 1993. Functional interaction of adenovirus E1A with holo-TFIID. Genes Dev. 7:1810-1823. [DOI] [PubMed] [Google Scholar]
- 9.Campbell, T. B., M. Borok, L. Gwanzura, S. MaWhinney, I. E. White, B. Ndemera, I. Gudza, L. Fitzpatrick, and R. T. Schooley. 2000. Relationship of human herpesvirus 8 peripheral blood virus load and Kaposi's sarcoma clinical stage. AIDS 14:2109-2116. [DOI] [PubMed] [Google Scholar]
- 10.Carroll, K. D., W. Bu, D. Palmeri, S. Spadavecchia, S. J. Lynch, S. A. Marras, S. Tyagi, and D. M. Lukac. 2006. Kaposi's sarcoma-associated herpesvirus lytic switch protein stimulates DNA binding of RBP-Jk/CSL to activate the Notch pathway. J. Virol. 80:9697-9709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Casper, C., W. G. Nichols, M. L. Huang, L. Corey, and A. Wald. 2004. Remission of HHV-8 and HIV-associated multicentric Castleman disease with ganciclovir treatment. Blood 103:1632-1634. [DOI] [PubMed] [Google Scholar]
- 12.Cattelan, A., M. Calabro, P. Gasperini, S. Aversa, M. Zanchetta, F. Meneghetti, A. A. De Rossi, and L. Chieco-Bianchi. 2001. Acquired immunodeficiency syndrome-related Kaposi's sarcoma regression after highly active antiretroviral therapy: biologic correlates of clinical outcome. J. Natl. Cancer Inst. Monogr. 28:44-49. [DOI] [PubMed] [Google Scholar]
- 13.Cesarman, E., Y. Chang, P. S. Moore, J. W. Said, and D. M. Knowles. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 332:1186-1191. [DOI] [PubMed] [Google Scholar]
- 14.Cesarman, E., and E. Mesri. 2000. Viral G protein-coupled receptor and Kaposi's sarcoma: a model of paracrine neoplasia? J. Exp. Med. 191:417-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chang, P. J., and G. Miller. 2004. Autoregulation of DNA binding and protein stability of Kaposi's sarcoma-associated herpesvirus ORF50 protein. J. Virol. 78:10657-10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang, P.-J., D. Shedd, L. Gradoville, M.-S. Cho, L.-W. Chen, J. Chang, and G. Miller. 2002. Open reading frame 50 protein of Kaposi's sarcoma-associated herpesvirus directly activates the viral PAN and K12 genes by binding to related response elements. J. Virol. 76:3168-3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chang, P. J., D. Shedd, and G. Miller. 2005. Two subclasses of Kaposi's sarcoma-associated herpesvirus lytic cycle promoters distinguished by open reading frame 50 mutant proteins that are deficient in binding to DNA. J. Virol. 79:8750-8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang, Y., E. Cesarman, M. S. Pessin, F. Lee, J. Culpepper, D. M. Knowles, and P. S. Moore. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865-1869. [DOI] [PubMed] [Google Scholar]
- 19.Chen, J., K. Ueka, S. Sakakibara, T. Okuno, and K. Yamanishi. 2000. Transcriptional regulation of the Kaposi's sarcoma-associated herpesvirus viral interferon regulatory factor gene. J. Virol. 74:8623-8634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clackson, T., W. Yang, L. Rozamus, M. Hatada, J. Amara, C. Rollins, L. Stevenson, S. Magari, S. Wood, N. Courage, X. Lu, F. Cerasoli, M. Gilman, and D. Holt. 1998. Redesigning an FKBP-ligand interface to generate chemical dimerizers with novel specificity. Proc. Natl. Acad. Sci. USA 95:10437-10442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clore, G. M., J. G. Omichinski, K. Sakaguchi, N. Zambrano, H. Sakamoto, E. Appella, and A. M. Gronenborn. 1994. High-resolution structure of the oligomerization domain of p53 by multidimensional NMR. Science 265:386-391. [DOI] [PubMed] [Google Scholar]
- 22.Damania, B., J. H. Jeong, B. S. Bowser, S. M. DeWire, M. R. Staudt, and D. P. Dittmer. 2004. Comparison of the Rta/Orf50 transactivator proteins of gamma-2-herpesviruses. J. Virol. 78:5491-5499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deng, H., M. J. Song, J. T. Chu, and R. Sun. 2002. Transcriptional regulation of the interleukin-6 gene of human herpesvirus 8 (Kaposi's sarcoma-associated herpesvirus). J. Virol. 76:8252-8264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dourmishev, L. A., A. L. Dourmishev, D. Palmeri, R. A. Schwartz, and D. M. Lukac. 2003. Molecular genetics of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) epidemiology and pathogenesis. Microbiol. Mol. Biol. Rev. 67:175-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Drees, B. L., E. K. Grotkopp, and H. C. Nelson. 1997. The GCN4 leucine zipper can functionally substitute for the heat shock transcription factor's trimerization domain. J. Mol. Biol. 273:61-74. [DOI] [PubMed] [Google Scholar]
- 26.Engels, E. A., R. J. Biggar, V. A. Marshall, M. A. Walters, C. J. Gamache, D. Whitby, and J. J. Goedert. 2003. Detection and quantification of Kaposi's sarcoma-associated herpesvirus to predict AIDS-associated Kaposi's sarcoma. AIDS 17:1847-1851. [DOI] [PubMed] [Google Scholar]
- 27.Ensoli, B., M. Sturzl, and P. Monini. 2000. Cytokine-mediated growth promotion of Kaposi's sarcoma and primary effusion lymphoma. Sem. Cancer Biol. 10:367-381. [DOI] [PubMed] [Google Scholar]
- 28.Fakhari, F. D., and D. P. Dittmer. 2002. Charting latency transcripts in Kaposi's sarcoma-associated herpesvirus by whole-genome real-time quantitative PCR. J. Virol. 76:6213-6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao, S. J., L. Kingsley, M. Li, W. Zheng, C. Parravicini, J. Ziegler, R. Newton, C. R. Rinaldo, A. Saah, J. Phair, R. Detels, Y. Chang, and P. S. Moore. 1996. KSHV antibodies among Americans, Italians and Ugandans with and without Kaposi's sarcoma. Nat. Med. 2:925-928. [DOI] [PubMed] [Google Scholar]
- 30.Gradoville, L., J. Gerlach, E. Grogan, D. Shedd, S. Nikiforow, C. Metroka, and G. Miller. 2000. Kaposi's sarcoma-associated herpesvirus open reading frame 50/Rta protein activates the entire lytic cycle in the HH-B2 primary effusion lymphoma cell line. J. Virol. 74:6207-6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gwack, Y., H. J. Baek, H. Nakamura, S. H. Lee, M. Meisterernst, R. G. Roeder, and J. U. Jung. 2003. Principal role of TRAP/mediator and SWI/SNF complexes in Kaposi's sarcoma-associated herpesvirus RTA-mediated lytic reactivation. Mol. Cell. Biol. 23:2055-2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gwack, Y., H. Byun, S. Hwang, C. Lim, and J. Choe. 2001. CREB-binding protein and histone deacetylase regulate the transcriptional activity of Kaposi's sarcoma-associated herpesvirus open reading frame 50. J. Virol. 75:1909-1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gwack, Y., H. Nakamura, S. H. Lee, J. Souvlis, J. T. Yustein, S. Gygi, H. J. Kung, and J. U. Jung. 2003. Poly(ADP-ribose) polymerase 1 and Ste20-like kinase hKFC act as transcriptional repressors for gamma-2 herpesvirus lytic replication. Mol. Cell. Biol. 23:8282-8294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hodges, R. S., P. D. Semchuk, A. K. Taneja, C. M. Kay, J. M. Parker, and C. T. Mant. 1988. Protein design using model synthetic peptides. Pept Res. 1:19-30. [PubMed] [Google Scholar]
- 35.Hodges, R. S., N. E. Zhou, C. M. Kay, and P. D. Semchuk. 1990. Synthetic model proteins: contribution of hydrophobic residues and disulfide bonds to protein stability. Pept. Res. 3:123-137. [PubMed] [Google Scholar]
- 36.Hu, J. C., E. K. O'Shea, P. S. Kim, and R. T. Sauer. 1990. Sequence requirements for coiled-coils: analysis with lambda repressor-GCN4 leucine zipper fusions. Science 250:1400-1403. [DOI] [PubMed] [Google Scholar]
- 37.Jeffrey, P. D., S. Gorina, and N. P. Pavletich. 1995. Crystal structure of the tetramerization domain of the p53 tumor suppressor at 1.7 angstroms. Science 267:1498-1502. [DOI] [PubMed] [Google Scholar]
- 38.Jenner, R., M. Alba, C. Boshoff, and P. Kellam. 2001. Kaposi's sarcoma-associated herpesvirus latent and lytic gene expression as revealed by DNA arrays. J. Virol. 75:891-902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jones, J. L., D. L. Hanson, S. Y. Chu, J. W. Ward, and H. W. Jaffe. 1995. AIDS-associated Kaposi's sarcoma. Science 267:1078-1079. [DOI] [PubMed] [Google Scholar]
- 40.Jones, J. L., D. L. Hanson, M. S. Dworkin, and H. W. Jaffe. 2000. Incidence and trends in Kaposi's sarcoma in the era of effective antiretroviral therapy. J. Acquir. Immune Defic. Syndr. 24:270-274. [DOI] [PubMed] [Google Scholar]
- 41.Kedes, D., E. Operskalski, M. Busch, R. Kohn, J. Flood, and D. Ganem. 1996. The seroepidemiology of human herpesvirus 8 (Kaposi's sarcoma-associated herpesvirus): distribution of infection in KS risk groups and evidence for sexual transmission. Nat. Med. 2:918-924. [DOI] [PubMed] [Google Scholar]
- 42.Kirshner, J. R., D. M. Lukac, J. Chang, and D. Ganem. 2000. Kaposi's sarcoma-associated herpesvirus open reading frame 57 encodes a posttranscriptional regulator with multiple distinct activities. J. Virol. 74:3586-3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kirshner, J. R., K. Staskus, A. Haase, M. Lagunoff, and D. Ganem. 1999. Expression of the open reading frame 74 (G-protein-coupled receptor) gene of Kaposi's sarcoma (KS)-associated herpesvirus: implications for KS pathogenesis. J. Virol. 73:6006-6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lagunoff, M., D. Lukac, and D. Ganem. 2001. Immunoreceptor tyrosine-based activation motif-dependent signaling by Kaposi's sarcoma-associated herpesvirus K1 protein: effects on lytic viral replication. J. Virol. 75:5891-5898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lebbé, C., L. Blum, C. Pellet, G. Blanchard, O. Verola, P. Morel, O. Danne, and F. Calvo. 1998. Clinical and biological impact of antiretroviral therapy with protease inhibitors on HIV-related Kaposi's sarcoma. AIDS 12:F45-F49. [DOI] [PubMed] [Google Scholar]
- 46.Liang, Y., J. Chang, S. Lynch, D. M. Lukac, and D. Ganem. 2002. The lytic switch protein of KSHV activates gene expression via functional interaction with RBP-Jk, the target of the Notch signaling pathway. Genes Dev. 16:1977-1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liang, Y., and D. Ganem. 2003. Lytic but not latent infection by Kaposi's sarcoma-associated herpesvirus requires host CSL protein, the mediator of Notch signaling. Proc. Natl. Acad. Sci. USA 100:8490-8495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liang, Y., and D. Ganem. 2004. RBP-J (CSL) is essential for activation of the K14/vGPCR promoter of Kaposi's sarcoma-associated herpesvirus by the lytic switch protein RTA. J. Virol. 78:6818-6826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liao, W., Y. Tang, Y. L. Kuo, B. Y. Liu, C. J. Xu, and C. Z. Giam. 2003. Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 transcriptional activator Rta is an oligomeric DNA-binding protein that interacts with tandem arrays of phased A/T-trinucleotide motifs. J. Virol. 77:9399-9411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lu, M., J. Suen, C. Frias, R. Pfeiffer, M. H. Tsai, E. Chuang, and S. L. Zeichner. 2004. Dissection of the Kaposi's sarcoma-associated herpesvirus gene expression program by using the viral DNA replication inhibitor cidofovir. J. Virol. 78:13637-13652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lukac, D., L. Garibyan, J. Kirshner, D. Palmeri, and D. Ganem. 2001. DNA binding by the Kaposi's sarcoma-associated herpesvirus lytic switch protein is necessary for transcriptional activation of two viral delayed early promoters. J. Virol. 75:6786-6799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lukac, D. M., J. R. Kirshner, and D. Ganem. 1999. Transcriptional activation by the product of open reading frame 50 of Kaposi's sarcoma-associated herpesvirus is required for lytic viral reactivation in B cells. J. Virol. 73:9348-9361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lukac, D. M., R. Renne, J. R. Kirshner, and D. Ganem. 1998. Reactivation of Kaposi's sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology 252:304-312. [DOI] [PubMed] [Google Scholar]
- 54.MacArthur, M. W., and J. M. Thornton. 1991. Influence of proline residues on protein conformation. J. Mol. Biol. 218:397-412. [DOI] [PubMed] [Google Scholar]
- 55.Manet, E., A. Rigolet, H. Gruffat, J.-F. Giot, and A. Sergeant. 1991. Domains of the Epstein-Barr virus (EBV) transcription factor R required for dimerization, DNA binding and activation. Nucleic Acids Res. 19:2661-2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martin, D. F., B. D. Kuppermann, R. A. Wolitz, A. G. Palestine, H. Li, and C. A. Robinson. 1999. Oral ganciclovir for patients with cytomegalovirus retinitis treated with a ganciclovir implant. N. Engl. J. Med. 340:1063-1070. [DOI] [PubMed] [Google Scholar]
- 57.Matsushima, N., N. Tachi, Y. Kuroki, P. Enkhbayar, M. Osaki, M. Kamiya, and R. H. Kretsinger. 2005. Structural analysis of leucine-rich-repeat variants in proteins associated with human diseases. Cell. Mol. Life Sci. 62:2771-2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McDonnell, A. V., T. Jiang, A. E. Keating, and B. Berger. 2006. Paircoil2: improved prediction of coiled coils from sequence. Bioinformatics 22:356-358. [DOI] [PubMed] [Google Scholar]
- 59.Mocroft, A., O. Kirk, N. Clumeck, P. Gargalianos-Kakolyris, H. Trocha, N. Chentsova, F. Antunes, H. J. Stellbrink, A. N. Phillips, and J. D. Lundgren. 2004. The changing pattern of Kaposi sarcoma in patients with HIV, 1994-2003: the EuroSIDA study. Cancer 100:2644-2654. [DOI] [PubMed] [Google Scholar]
- 60.Nador, R. G., E. Cesarman, A. Chadburn, D. B. Dawson, M. Q. Ansari, J. Sald, and D. M. Knowles. 1996. Primary effusion lymphoma: a distinct clinicopathologic entity associated with the Kaposi's sarcoma-associated herpes virus. Blood 88:645-656. [PubMed] [Google Scholar]
- 61.Nitta, E., K. Izutsu, Y. Yamaguchi, Y. Imai, S. Ogawa, S. Chiba, M. Kurokawa, and H. Hirai. 2005. Oligomerization of Evi-1 regulated by the PR domain contributes to recruitment of corepressor CtBP. Oncogene 24:6165-6173. [DOI] [PubMed] [Google Scholar]
- 62.O'Neil, K. T., and W. F. DeGrado. 1990. A thermodynamic scale for the helix-forming tendencies of the commonly occurring amino acids. Science 250:646-651. [DOI] [PubMed] [Google Scholar]
- 63.O'Shea, E. K., J. D. Klemm, P. S. Kim, and T. Alber. 1991. X-ray structure of the GCN4 leucine zipper, a two-stranded, parallel coiled coil. Science 254:539-544. [DOI] [PubMed] [Google Scholar]
- 64.O'Shea, E. K., R. Rutkowski, and P. S. Kim. 1989. Evidence that the leucine zipper is a coiled coil. Science 243:538-542. [DOI] [PubMed] [Google Scholar]
- 65.Paulose-Murphy, M., N.-K. Ha, C. Xiang, Y. Chen, L. Gillim, R. Yarchoan, P. Meltzer, M. Bittner, J. Trent, and S. Zeichner. 2001. Transcription program of human herpesvirus 8 (Kaposi's sarcoma-associated herpesvirus). J. Virol. 75:4843-4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pellet, C., S. Chevret, L. Blum, C. Gauville, M. Hurault, G. Blanchard, F. Agbalika, C. Lascoux, D. Ponscarme, P. Morel, F. Calvo, and C. Lebbe. 2001. Virologic and immunologic parameters that predict clinical response of AIDS-associated Kaposi's sarcoma to highly active antiretroviral therapy. J. Investig. Dermatol. 117:858-863. [DOI] [PubMed] [Google Scholar]
- 67.Piela, L., G. Nemethy, and H. A. Scheraga. 1987. Proline-induced constraints in alpha-helices. Biopolymers 26:1587-1600. [DOI] [PubMed] [Google Scholar]
- 68.Polson, A., L. Huang, D. Lukac, J. Blethrow, D. Morgan, A. Burlingame, and D. Ganem. 2001. Kaposi's sarcoma-associated herpesvirus K-bZIP protein is phosphorylated by cyclin-dependent kinases. J. Virol. 75:3175-3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Quinlivan, E. B., C. Zhang, P. W. Stewart, C. Komoltri, M. G. Davis, and R. S. Wehbie. 2002. Elevated virus loads of Kaposi's sarcoma-associated human herpesvirus 8 predict Kaposi's sarcoma disease progression, but elevated levels of human immunodeficiency virus type 1 do not. J. Infect. Dis. 185:1736-1744. [DOI] [PubMed] [Google Scholar]
- 70.Robles, R., D. Lugo, L. Gee, and M. A. Jacobson. 1999. Effect of antiviral drugs used to treat cytomegalovirus end-organ disease on subsequent course of previously diagnosed Kaposi's sarcoma in patients with AIDS. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 20:34-38. [DOI] [PubMed] [Google Scholar]
- 71.Sakakibara, S., K. Ueda, J. Chen, T. Okuno, and K. Yamanishi. 2001. Octamer-binding sequence is a key element for the autoregulation of Kaposi's sarcoma-associated herpesvirus ORF50/Lyta gene expression. J. Virol. 75:6894-6900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sarid, R., O. Flore, R. A. Bohenzky, Y. Chang, and P. S. Moore. 1998. Transcription mapping of the Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) genome in a body cavity-based lymphoma cell line (BC-1). J. Virol. 72:1005-1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schägger, H., W. A. Cramer, and G. von Jagow. 1994. Analysis of molecular masses and oligomeric states of protein complexes by blue native electrophoresis and isolation of membrane protein complexes by two-dimensional native electrophoresis. Anal. Biochem. 217:220-230. [DOI] [PubMed] [Google Scholar]
- 74.Seaman, W., D. Ye, R. Wang, E. Hale, M. Weisse, and E. Quinlivan. 1999. Gene expression from the ORF50/K8 region of Kaposi's sarcoma-associated herpesvirus. Virology 263:436-449. [DOI] [PubMed] [Google Scholar]
- 75.Simpson, G. R., T. F. Schulz, D. Whitby, P. M. Cook, C. Boshoff, L. Rainbow, M. R. Howard, S. J. Gao, R. A. Bohenzky, P. Simmonds, C. Lee, A. de Ruiter, A. Hatzakis, R. S. Tedder, I. V. Weller, R. A. Weiss, and P. S. Moore. 1996. Prevalence of Kaposi's sarcoma associated herpesvirus infection measured by antibodies to recombinant capsid protein and latent immunofluorescence antigen. Lancet 348:1133-1138. [DOI] [PubMed] [Google Scholar]
- 76.Sirianni, M. C., L. Vincenzi, S. Topino, A. Giovannetti, F. Mazzetta, F. Libi, D. Scaramuzzi, M. Andreoni, E. Pinter, S. Baccarini, G. Rezza, P. Monini, and B. Ensoli. 2002. NK cell activity controls human herpesvirus 8 latent infection and is restored upon highly active antiretroviral therapy in AIDS patients with regressing Kaposi's sarcoma. Eur. J. Immunol. 32:2711-2720. [DOI] [PubMed] [Google Scholar]
- 77.Song, M., H. Brown, T.-T. Wu, and R. Sun. 2001. Transcription activation of polyadenylated nuclear RNA by Rta in human herpesvirus 8/Kaposi's sarcoma-associated herpesvirus. J. Virol. 75:3129-3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Song, M. J., H. Deng, and R. Sun. 2003. Comparative study of regulation of RTA-responsive genes in Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8. J. Virol. 77:9451-9462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Song, M. J., X. Li, H. J. Brown, and R. Sun. 2002. Characterization of interactions between RTA and the promoter of polyadenylated nuclear RNA in Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8. J. Virol. 76:5000-5013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sun, R., S. F. Lin, L. Gradoville, Y. Yuan, F. Zhu, and G. Miller. 1998. A viral gene that activates lytic cycle expression of Kaposi's sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA 95:10866-10871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tellinghuisen, T. L., R. Perera, and R. J. Kuhn. 2001. In vitro assembly of Sindbis virus core-like particles from cross-linked dimers of truncated and mutant capsid proteins. J. Virol. 75:2810-2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ueda, K., K. Ishikawa, K. Nishimura, S. Sakakibara, E. Do, and K. Yamanishi. 2002. Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) replication and transcription factor activates the K9 (vIRF) gene through two distinct cis elements by a non-DNA-binding mechanism. J. Virol. 76:12044-12054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vinson, C., M. Myakishev, A. Acharya, A. A. Mir, J. R. Moll, and M. Bonovich. 2002. Classification of human B-ZIP proteins based on dimerization properties. Mol. Cell. Biol. 22:6321-6335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang, J., J. Zhang, L. Zhang, W. Harrington, Jr., J. T. West, and C. Wood. 2005. Modulation of human herpesvirus 8/Kaposi's sarcoma-associated herpesvirus replication and transcription activator transactivation by interferon regulatory factor 7. J. Virol. 79:2420-2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang, S., S. Liu, M. Wu, Y. Geng, and C. Wood. 2001. Kaposi's sarcoma-associated herpesvirus/human herpesvirus-8 ORF50 gene product contains a potent C-terminal activation domain which activates gene expression via a specific target sequence. Arch. Virol. 146:1415-1426. [DOI] [PubMed] [Google Scholar]
- 86.Wang, S., S. Liu, M. H. Wu, Y. Geng, and C. Wood. 2001. Identification of a cellular protein that interacts and synergizes with the RTA (ORF50) protein of Kaposi's sarcoma-associated herpesvirus in transcriptional activation. J. Virol. 75:11961-11973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang, S. E., F. Y. Wu, H. Chen, M. Shamay, Q. Zheng, and G. S. Hayward. 2004. Early activation of the Kaposi's sarcoma-associated herpesvirus RTA, RAP, and MTA promoters by the tetradecanoyl phorbol acetate-induced AP1 pathway. J. Virol. 78:4248-4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang, S. E., F. Y. Wu, M. Fujimuro, J. Zong, S. D. Hayward, and G. S. Hayward. 2003. Role of CCAAT/enhancer-binding protein alpha (C/EBPα) in activation of the Kaposi's sarcoma-associated herpesvirus (KSHV) lytic-cycle replication-associated protein (RAP) promoter in cooperation with the KSHV replication and transcription activator (RTA) and RAP. J. Virol. 77:600-623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang, S. E., F. Y. Wu, Y. Yu, and G. S. Hayward. 2003. CCAAT/enhancer-binding protein- α is induced during the early stages of Kaposi's sarcoma-associated herpesvirus (KSHV) lytic cycle reactivation and together with the KSHV replication and transcription activator (RTA) cooperatively stimulates the viral RTA, MTA, and PAN promoters. J. Virol. 77:9590-9612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang, Y., H. Li, M. Y. Chan, F. X. Zhu, D. M. Lukac, and Y. Yuan. 2004. Kaposi's sarcoma-associated herpesvirus ori-Lyt-dependent DNA replication: cis-acting requirements for replication and ori-Lyt-associated RNA transcription. J. Virol. 78:8615-8629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang, Y., Q. Tang, G. G. Maul, and Y. Yuan. 2006. Kaposi's sarcoma-associated herpesvirus ori-Lyt-dependent DNA replication: dual role of replication and transcription activator. J. Virol. 80:12171-12186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang, Y., and Y. Yuan. 2007. Essential role of RBP-Jkappa in activation of the K8 delayed-early promoter of Kaposi's sarcoma-associated herpesvirus by ORF50/RTA. Virology 359:19-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Waterman, M. J., J. L. Waterman, and T. D. Halazonetis. 1996. An engineered four-stranded coiled coil substitutes for the tetramerization domain of wild-type p53 and alleviates transdominant inhibition by tumor-derived p53 mutants. Cancer Res. 56:158-163. [PubMed] [Google Scholar]
- 94.Whitby, D., M. R. Howard, M. Tenant-Flowers, N. S. Brink, A. Copas, C. Boshoff, T. Hatzioannou, F. E. Suggett, D. M. Aldam, A. S. Denton, et al. 1995. Detection of Kaposi sarcoma associated herpesvirus in peripheral blood of HIV-infected individuals and progression to Kaposi's sarcoma. Lancet 346:799-802. [DOI] [PubMed] [Google Scholar]
- 95.Wilkinson, J., A. Cope, J. Gill, D. Bourboulia, P. Hayes, N. Imami, T. Kubo, A. Marcelin, V. Calvez, R. Weiss, B. Gazzard, C. Boshoff, and F. Gotch. 2002. Identification of Kaposi's sarcoma-associated herpesvirus (KSHV)- specific cytotoxic T-lymphocyte epitopes and evaluation of reconstitution of KSHV-specific responses in human immunodeficiency virus type 1-infected patients receiving highly active antiretroviral therapy. J. Virol. 76:2634-2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Williamson, M. P. 1994. The structure and function of proline-rich regions in proteins. Biochem. J. 297:249-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Woolfson, D. N., and T. Alber. 1995. Predicting oligomerization states of coiled coils. Protein Sci. 4:1596-1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Woolfson, D. N., and D. H. Williams. 1990. The influence of proline residues on alpha-helical structure. FEBS Lett. 277:185-188. [DOI] [PubMed] [Google Scholar]
- 99.Wu, L., R. Renne, D. Ganem, and B. Forghani. 2000. Human herpesvirus 8 glycoprotein K8.1: expression, post-translational modification and localization analyzed by monoclonal antibody. J. Clin. Virol. 17:127-136. [DOI] [PubMed] [Google Scholar]
- 100.Wu, S. M., V. P. Mutucumarana, S. Geromanos, and D. W. Stafford. 1997. The propeptide binding site of the bovine gamma-glutamyl carboxylase. J. Biol. Chem. 272:11718-11722. [DOI] [PubMed] [Google Scholar]
- 101.Xu, Y., D. P. AuCoin, A. R. Huete, S. A. Cei, L. J. Hanson, and G. S. Pari. 2005. A Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 ORF50 deletion mutant is defective for reactivation of latent virus and DNA replication. J. Virol. 79:3479-3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yu, Y., S. E. Wang, and G. S. Hayward. 2005. The KSHV immediate-early transcription factor RTA encodes ubiquitin E3 ligase activity that targets IRF7 for proteosome-mediated degradation. Immunity 22:59-70. [DOI] [PubMed] [Google Scholar]
- 103.Zhang, J., J. Wang, C. Wood, D. Xu, and L. Zhang. 2005. Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 replication and transcription activator regulates viral and cellular genes via interferon-stimulated response elements. J. Virol. 79:5640-5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhou, N. E., C. M. Kay, and R. S. Hodges. 1994. The role of interhelical ionic interactions in controlling protein folding and stability. De novo designed synthetic two-stranded alpha-helical coiled-coils. J. Mol. Biol. 237:500-512. [DOI] [PubMed] [Google Scholar]
- 105.Zhou, N. E., C. M. Kay, and R. S. Hodges. 1992. Synthetic model proteins: the relative contribution of leucine residues at the nonequivalent positions of the 3-4 hydrophobic repeat to the stability of the two-stranded alpha-helical coiled-coil. Biochemistry 31:5739-5746. [DOI] [PubMed] [Google Scholar]
- 106.Zhou, X. Z., P. J. Lu, G. Wulf, and K. P. Lu. 1999. Phosphorylation-dependent prolyl isomerization: a novel signaling regulatory mechanism. Cell. Mol. Life Sci. 56:788-806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ziegelbauer, J., A. Grundhoff, and D. Ganem. 2006. Exploring the DNA binding interactions of the Kaposi's sarcoma-associated herpesvirus lytic switch protein by selective amplification of bound sequences in vitro. J. Virol. 80:2958-2967. [DOI] [PMC free article] [PubMed] [Google Scholar]