

Abstract
Human T-cell leukemia virus type 1 (HTLV-1) entry into cells is dependent upon the viral envelope glycoprotein-catalyzed fusion of the viral and cellular membranes. Following receptor activation of the envelope, the transmembrane glycoprotein (TM) is thought to undergo a series of fusogenic conformational transitions through a rod-like prehairpin intermediate to a compact trimer-of-hairpins structure. Importantly, synthetic peptides that interfere with the conformational changes of TM are potent inhibitors of membrane fusion and HTLV-1 entry, suggesting that TM is a valid target for antiviral therapy. To assess the utility of TM as a vaccine target and to explore further the function of TM in HTLV-1 pathogenesis, we have begun to examine the immunological properties of TM. Here we demonstrate that a recombinant trimer-of-hairpins form of the TM ectodomain is strongly immunogenic. Monoclonal antibodies raised against the TM immunogen specifically bind to trimeric forms of TM, including structures thought to be important for membrane fusion. Importantly, these antibodies recognize the envelope on virally infected cells but, surprisingly, fail to neutralize envelope-mediated membrane fusion or infection by pseudotyped viral particles. Our data imply that, even in the absence of overt membrane fusion, there are multiple forms of TM on virally infected cells and that some of these display fusion-associated structures. Finally, we demonstrate that many of the antibodies possess the ability to recruit complement to TM, suggesting that envelope-derived immunogens capable of eliciting a combination of neutralizing and complement-fixing antibodies would be of value as subunit vaccines for intervention in HTLV infections.
In some infected individuals, human T-cell leukemia virus type 1 (HTLV-1) causes a rare but aggressive adult T-cell leukemia-lymphoma and a progressive demyelinating disease known as tropical spastic paraparesis or HTLV-associated myelopathy. Despite considerable clinical effort, these virally induced conditions remain difficult to treat. Worldwide, there are approximately 20 million individuals infected with HTLV-1. The virus is endemic in southern Japan, central Africa, the Caribbean islands, and Central and South America, and though rare, HTLV-1 infections have been reported among indigenous and immigrant European populations and among intravenous drug users in Europe and the United States (1, 6, 28a, 70). Given the global distribution of HTLV-1, the impact of infection, and the lack of effective therapy for HTLV-1-associated disease, there is considerable need for improved understanding of the HTLV-1 infection process and the immune response to viral infection.
HTLV-1 primarily infects CD4+ T cells in vivo (1, 6). Infection is initiated by the action of the viral envelope glycoproteins, which are expressed on the surface of the virus or infected cell as a trimer of the gp46 surface glycoproteins (SU) attached to a trimer of the gp21 transmembrane glycoprotein. SU are responsible for the recognition and attachment of viral particles to T cells (32, 33, 55, 72) through the recognition of cell surface molecules such as heparan sulfate glycoproteins (37, 57) and the primary cellular receptor glucose transporter 1 (48). By contrast, the transmembrane glycoprotein (TM) is required to promote fusion of the viral and target cell membranes, thereby allowing viral entry into the host cell (10, 15, 25, 65). By analogy to other retroviruses (63, 65, 66), it is likely that binding of SU to Glut-1 triggers conformational changes within the Env trimer that convert it from a nonfusogenic native state to a fusion-active form (reference 15 and references therein; 44, 65).
A clue to the molecular mechanism of Env-mediated membrane fusion has come from the crystal structure of the HTLV-1 TM ectodomain (7, 44). For each monomer of the homotrimeric TM protein, an amino-terminal fusion peptide is connected via a glycine-rich linker to an α-helical motif that interacts with the equivalent helix of adjacent monomers to form a central triple-stranded coiled coil. At the base of the “core coiled-coil” the peptide backbone folds back on itself in a disulfide-bonded 180° loop referred to as the chain reversal region. The extended C-terminal segment, which includes a short α-helical domain, runs antiparallel to the core coiled-coil and packs into the grooves formed on the surface of the coiled coil. This “trimer-of-hairpins” motif is a highly conserved structure of viral fusion proteins (7, 15, 44) and most likely represents a conformation of TM that is achieved in the late stages of membrane fusion (15, 44).
Accumulating evidence (15, 65) favors a model for fusion in which the insertion of the N-terminal fusion peptide into the target cell membrane results in the formation of a prehairpin intermediate in which the C terminus of TM is anchored in the viral membrane while the amino-terminal fusion peptide is embedded in the membrane of the target cell. Stability of the rod-like prehairpin intermediate is achieved by assembly of the core coiled coil. The prehairpin intermediate then resolves into the trimer-of-hairpins or six-helix bundle structure, which brings the viral and cellular membranes into close proximity, destabilizes the lipid bilayers, and ultimately promotes membrane fusion.
Envelope is a primary target for the adaptive immune response, and HTLV-1-infected individuals develop strong antibody and cytotoxic lymphocyte responses to envelope (3, 13, 20, 22, 46, 47, 59). In primate models, the humoral and cell-mediated immune responses to a recombinant envelope immunogen are sufficiently robust to provide significant long-term protection against HTLV-1 infection (29). Moreover, a variety of antibodies (3, 21, 56), synthetic peptides (5, 35, 58, 62), and soluble polyanions (37, 57) directed at envelope block envelope function and prevent viral infection in vitro. Thus, envelope is an attractive target for small-molecule inhibitors of viral entry, and recombinant envelope appears to be an ideal subunit vaccine candidate.
The immune response to SU is beginning to be understood but, although antibodies to TM are produced in natural infections (21, 46, 47), there have been no studies to specifically examine the immunogenicity and antigenic structure of TM. Consequently, the immunogenic regions of TM have not been comprehensively mapped, the accessibility of viral TM to antibody is unknown, and the neutralization properties of antibodies directed at TM are poorly documented. Given the importance of TM in viral infection and because recombinant TM may be of utility as a component of a gp46/gp21 subunit vaccine, we have begun to address these issues. Here we demonstrate that a recombinant form of TM is immunogenic, and we have used this immunogen to generate a panel of murine anti-TM monoclonal antibodies. The antibodies are profoundly sensitive to the conformational status of TM, and the antibody binding sites are lost on denatured or monomeric antigen. Importantly, the antibodies, which recognize epitopes thought to exist on fusion-active or postfusion forms of TM, are reactive with viral envelope expressed on the surface of infected cells. Investigation of the TM structures that interact with such antibodies will provide insight into envelope function and information of value in vaccine design.
MATERIALS AND METHODS
Animal immunization, hybridoma production, and cell culture.
Antibody responses in mice were generated against the fusion protein MBP-Hairpin (58), corresponding to residues 338 to 425 of the HTLV-1 envelope TM. Six- to 8-week-old BALB/c mice were immunized with approximately 50 μg of the antigen emulsified in 300 μg of adjuvant (Adju-Prime; Pierce). Booster immunizations were administered at 3, 6, and 9 weeks. Mice were bled once for antibody titer determination. In a parallel strategy, CD-1 mice were vaccinated with antigen prepared in TiterMax Gold (CytRx Corporation) following the manufacturer's instructions. Briefly, equal volumes of adjuvant and antigen were mixed to a final antigen concentration of 1 mg/ml. Mice were immunized subcutaneously with 50 μl of the antigen-adjuvant mix. Immunization was repeated three times at 4-week intervals. After the last boost (after 7 days), samples of serum from each animal were collected for antibody titer determination. For both BALB/c and CD-1 mice, the animals were given a final boost by intravenous injection of soluble antigen without adjuvant 5 days prior to fusion. The mice were euthanized and the spleens removed, washed, minced, and gently agitated to release splenocytes. The splenocytes were fused to myeloma cells using polyethylene glycol 4000 (Sigma) and plated out in 96-well plates in RPMI medium supplemented with 20% fetal bovine serum (FBS) and hypoxanthin-aminopterin-thymidine (Sigma) to select for hybridomas. Two weeks later, hybridomas were transferred to HT-supplemented medium, and the culture supernatants were tested by enzyme-linked immunosorbent assay (ELISA) for antibodies with reactivity to the immunogen.
HeLa cells were maintained in Dulbecco's modified Eagle medium supplemented with 10% FBS. Myeloma NS01 cells and the HTLV-1-infected MT-2 and uninfected SUPT-1 cell lines were maintained in RPMI 1640 medium supplemented with 10% FBS.
Plasmids.
Plasmids pHTE-1 (14), pMAL-gp21hairpin (MBP-Hairpin), pMAL-gp21fishhook (MBP-Fishhook), and pMAL-STOP (MBP-Stop) have been described previously (58). Briefly, essential regions of HTLV-1 TM were PCR amplified using pHTE-1 as template and subsequently cloned into the EcoRI and HindIII sites of the vector pMALc2 (New England Biolabs). pMAL-gp21hairpin encodes amino acids Met338 to Thr425 of HTLV-1 TM fused in frame to MBP, and pMAL-gp21fishhook is truncated at amino acid Cys400 of TM. In a similar fashion, plasmids pMAL-N-helix (MBP-N-helix), pMAL-C-helix (MBP-C-helix), and pMAL-CX6CC-loop (MBP-CX6CC-loop) were also generated (51).
Expression and purification of MBP-TM proteins.
Expression and purification of the MBP-TM proteins were carried out as described previously (58). Briefly, Eschericia coli JM109 cells, transformed with pMAL-stop or the MBP-TM expression plasmids, were grown at 37°C in the presence of ampicillin (100 μg/ml) until the optical density at 600 nm reached 0.6. Cells were induced with isopropylthio-β-d-galactoside (IPTG) at a final concentration of 0.5 mM for 4 h at 37°C. Cells were harvested, resuspended in column buffer (20 mM Tris-HCl [pH 7.5], 200 mM NaCl, 1 mM EDTA) supplemented with 1 mM phenylmethylsulfonyl fluoride and aprotinin (1 μg/ml) and subsequently lysed by sonication. Cell debris was pelleted by centrifugation at 9,000 × g for 30 min. The crude lysates were diluted 1:5 in column buffer and loaded onto an amylose column that had been preequilibrated with column buffer. The column was washed with 12 column volumes of column buffer, and the MBP and MBP-TM fusion proteins were eluted with column buffer containing 10 mM maltose. The concentration of the fusion proteins was estimated by the Bradford assay. The recombinant proteins were stored at −80°C in column buffer supplemented with 20% glycerol. The oligomerization states of our MBP-TM chimeras were assessed by Superdex 200 gel filtration chromatography in phosphate-buffered saline (PBS). Gel filtration analyses were calibrated with ferritin (440 kDa), aldolase (158 kDa), albumin (67 kDa), ovalbumin (43 kDa), and RNase A (13.7 kDa; Amersham Biosciences).
ELISA.
Microtiter 96-well plates (NUNC, MAXI-Sorp) were coated overnight at 4°C with the chimeric MBP-TM fusion proteins (MBP-Fishhook, MBP-Hairpin, MBP-N-helix, MBP-C-helix, MBP-CX6CC-loop, or control MBP [all at 10 μg/ml]) in PBS, pH 7.2. Plates were blocked (5% Marvel-PBS-0.2% Tween 20) for 1 h at room temperature and washed (five times), and antibodies at the concentrations indicated were added and incubated with the immobilized target antigen for 2 h at room temperature. Peroxidase-conjugated anti-mouse immunoglobulin G (IgG) (1:10,000 dilution) (Sigma) was added, and the bound antibody was detected using 15 mg/ml fresh ABTS substrate (in 0.1 M citric acid, 0.06% H2O2 [30% wt/wt]). After 10 to 20 min, color development was stopped, and absorbance was read at 415 nm.
For assays comparing antibody reactivity with native versus denatured antigen (as adapted from the method described in reference 52), microtiter 96-well plates were coated and incubated overnight at 4°C with an anti-MBP sheep polyclonal antibody at 10 μg/ml in PBS (pH 7.2). Plates were washed twice with wash buffer (PBS, 0.05% Tween 20) and blocked with blocking buffer (1% bovine serum albumin [BSA] in PBS) for 2 h at room temperature. After further washing, native MBP-Hairpin (10 μg/ml) in Tris-buffered saline (TBS) containing 10% fetal calf serum (TBS-FCS) was added to the wells (100 μl). For comparison, denatured MBP-Hairpin was prepared as described above but also mixed with 50 mM dithiothreitol (DTT) with 1% sodium dodecyl sulfate (SDS) and boiled for 5 min. Nine volumes of TBS-FCS containing 1% Nonidet P-40 were then added, and the solution was cooled to room temperature before addition to the plates. Monoclonal antibodies at the concentrations indicated were added and incubated with the native or denatured target antigen for 2 h at room temperature. Plates were washed five times to remove unbound antibody, and peroxidase-conjugated anti-mouse IgG (1:10,000 dilution) was added and incubated for 1 h, and the bound antibody was detected as described above.
C1q deposition.
Microtiter 96-well plates (NUNC MAXI-Sorp) were coated and incubated overnight at 4°C with 100 μl (10 μg/ml) of MBP-Fishhook or MBP-Hairpin fusion protein in PBS (pH 7.2). Plates were washed twice with wash buffer (PBS, 0.05% Tween 20) and blocked with blocking buffer (1% BSA in PBS) for 2 h at room temperature. Monoclonal antibodies at the concentrations indicated were incubated with the target antigen for 2 h (1 h at 37°C and 1 h at room temperature). Plates were washed 5 times to remove unbound antibody and blocked again for 1 h at room temperature. One hundred microliters of mouse complement sera (diluted 1:10 in veronal buffer) was then added, and plates were tightly covered with Nescofilm and incubated at 37°C for 45 min. Plates were washed again, and 100 μl (100 ng/ml) of biotin anti-mouse complement component C1q monoclonal antibody (Cedarlane) was added in the plates. After 1 h of incubation at room temperature, the plates were washed and incubated for 1 h with 100 μl streptavidin-horseradish peroxidase (HRP; Sigma) (1:10,000 dilution) at room temperature. Plates were washed five times to remove unbound streptavidin-HRP and two times with PBS to remove residual detergent. Finally, bound streptavidin-HRP, and therefore bound C1q, was detected using the ABTS substrate, and the absorbance at 415 nm was read (Bio-Rad plate reader).
Syncytium interference and pseudotyping assays.
Syncytium interference assays were performed by standard methods (32, 33). Briefly, 3 × 105 HeLa cells transfected with the envelope expression vector pHTE-1 were added to 7.0 × 105 untransfected HeLa target cells. The effector and target cells were cocultured in the absence or presence of the P-400-related peptides at the concentrations specified. The cells were incubated for 12 to 15 h at 37°C, washed twice with PBS, and fixed in PBS-3% paraformaldehyde. Assays were performed in triplicate, and the number of syncytia from five low-power fields per replicate was scored by light microscopy.
Pseudotyping assay.
Pseudotyped virus was prepared as previously described (32, 33). Briefly, 293T cells were cotransfected using Fugene-6 with 10 μg of the Env-defective, luciferase-encoding human immunodeficiency virus (HIV) proviral clone pNL4-3.LUC.R−.E− in the presence or absence of 10 μg pHTE-1. Sodium butyrate (20 mM) was added after 18 h, and 48 to 72 h later, the viral supernatants were harvested by centrifugation at 400 × g. For the transduction of HeLa or HOS cells, triplicate samples of 7 × 105 target cells in 1 ml were incubated with 1 ml of undiluted or serially diluted virus stock and plated into wells of a six-well tissue culture dish. Following overnight incubation at 37°C in 5% CO2, cells were harvested, lysed, and assayed for luciferase activity following the manufacturer's instructions using a Luciferase Assay System (Promega, Madison, WI) and a Turner Designs TD-20/20 luminometer (Sunnyvale, CA). For antibody inhibition experiments, 1 ml of HTLV-1 Env-pseudotyped virus was incubated with 1 ml HeLa or HOS cells (4 × 105 cells) in the presence or absence of serial dilutions of purified monoclonal antibody (MAb). After overnight culture, the cells were lysed and assayed for luciferase activity.
Microscopy.
HTLV-1-infected MT-2 cells and noninfected (control) SUPT-1 T cells, at a density of 5 × 106 cells per ml, were plated out on collagen-coated glass coverslips (5 μg/cm2; Sigma) and left to incubate overnight at 37°C in 5% CO2. Cells were washed with PBS and fixed with 3% paraformaldehyde for 15 min at 37°C. Cells were then blocked with PBS-1% BSA. Small square pieces of Parafilm were then placed in the wells of a fresh six-well plate, and 100 μl of hybridoma supernatants or 1 μg/ml purified MAbs were dispensed on the Parafilm. The cell-bearing coverslips were carefully placed on Parafilm with the cell side facing down and left to incubate for 1 h at room temperature. Following several washes with PBS-1% BSA (by carefully reversing the coverslips and adding and aspirating the buffer), cells were incubated in the dark with anti-mouse IgG-fluorescein isothiocyanate (FITC) (1:1,000 dilution; Sigma) at room temperature for 1 h. Coverslips were mounted onto glass slides with Vectashield (Vector Laboratories), sealed with nail varnish, and left to dry in the dark before examination by fluorescence microscopy (Olympus BX60).
Fluorescence-activated cell sorter analysis.
HTLV-1-infected MT-2 and noninfected (control) SUPT-1 cells at a density of 106 cells per ml were mixed with 0.1 ml of hybridoma culture supernatant or 1 μg/ml purified MAbs and incubated at room temperature for 1 h with rotation. The cells were pelleted (1,000 rpm for 5 min in an Eppendorf C5415C microcentrifuge), washed, and incubated with anti-mouse FITC antibody (1:1,000 dilution) in RPMI medium at room temperature for 30 min in the dark. The cells were washed (PBS-0.1% sodium azide) and fixed (0.5% paraformaldehyde in PBS [pH 7.4]). The cells were kept in the dark at 4°C until subjected to FACScan (Becton-Dickinson) analysis as directed by the manufacturer.
RESULTS
Isolation of MAbs recognizing the extracellular domain of HTLV-1 TM.
We have expressed the extracellular domain of HTLV-1 TM (58) lacking the hydrophobic fusion peptide and a panel of TM-derived peptide fragments (51, 58) as fusions to maltose binding protein (Fig. 1a to c). The extracellular domain of TM (Fig. 1a and b), when expressed as a fusion protein, forms stable trimers that recapitulate the important structural features of the trimer-of-hairpins motif of HTLV-1 TM (7, 44, 58). To generate monoclonal antibodies recognizing TM, recombinant trimeric MBP-Hairpin in adjuvant was used to vaccinate BALB/c and CD-1 mice, and secondary vaccinations were performed under identical conditions at 4-week intervals. Finally, the mice were boosted intravenously using MBP-Hairpin without adjuvant. Splenocytes were recovered and fused to NS0-1 myeloma cells, and hybridomas were selected using standard methods.
FIG. 1.

HTLV-1 TM and the recombinant TM fusion proteins. (a) Structure of the trimer-of-hairpins motif of HTLV-1 TM. The central triple-stranded coiled coil is shown in the space-filling form, with the extended antiparallel peptide and the C-helical region shown in color. (b) Model of the TM-derived MBP-Hairpin immunogen used in this study. The structure is displayed with each monomer of TM in ribbon format fused to maltose binding protein (MBP; white space-filling model). (c) Representation of the functional regions of HTLV-1 TM; the N-helical and C-helical regions are indicated as boxes; amino acid coordinates based on the envelope protein precursor are shown. The glycosylation site in viral TM is indicated by the branched structure. Boxed regions below TM highlight the motifs that are fused to MBP. All structures were modeled using MacPymol software using the coordinates provided by Kobe et al. (44), Protein Data Bank, Brookhaven National Laboratory, PDB ID 1MG1.
From a total of 700 hybridomas surveyed, 32 stable hybridoma clones were identified that secreted monoclonal antibodies reactive with the extracellular region of TM. Initial assessments, based on the reactivity of the MAbs with MBP-Hairpin, the mouse strain used, the growth characteristics of the hybridomas, and the antibody class further reduced the MAbs to a panel of 16 that were selected for further study. This selection procedure aimed to reduce the likelihood that multiple hybridomas were derived from the same clonally expanded splenocyte. Examination of the ELISA data indicates that each of the chosen MAbs exhibits strong reactivity with MBP-Hairpin but has little or no reactivity with the MBP-carrier protein; therefore, these MAbs specifically recognize the TM-derived region of the recombinant fusion protein (Fig. 2). To aid characterization and purification of the MAbs, each antibody was isotyped, revealing that the set of hybridomas express multiple classes of immunoglobulin, including seven IgG1, three IgG2a, three IgG2b, and three IgM antibodies (Table 1).
FIG. 2.

Murine monoclonal antibodies bind to the trimer-of-hairpins form. Monoclonal antibodies derived from immunized mice were examined for reactivity to MBP-Hairpin and control MBP. The antigens (MBP and MBP-Hairpin) were coated onto the wells of a 96-well plate and, following incubation with conditioned medium supernatant from each of the hybridomas, bound antibody was detected. The data represent the means and standard deviations of triplicate assays.
TABLE 1.
Origin and isotype of anti-TM MAbs produced in this study
| Hybridoma | Mouse strain | Isotype and subclass |
|---|---|---|
| M17-21 | BALB/c | IgG1 |
| M18-8 | BALB/c | IgG1 |
| M20-25 | BALB/c | IgG1 |
| M16-28 | BALB/c | IgG1 |
| M8-24 | BALB/c | IgG1 |
| M3-4 | CD-1 | IgG1 |
| M6-1 | CD-1 | IgG1 |
| M17-12 | CD-1 | IgG2a |
| M3-18 | CD-1 | IgG2a |
| M15-9 | CD-1 | IgG2a |
| M5-52 | CD-1 | IgG2b |
| M10-19 | CD-1 | IgG2b |
| M9-5 | CD-1 | IgG2b |
| M4-32 | CD-1 | IgM |
| M19-8 | CD-1 | IgM |
| M18-25 | CD-1 | IgM |
Efficiency of MAb binding.
To evaluate the relative affinity of the antibodies for TM, a representative group of the MAbs were purified and examined for concentration-dependent binding to the recombinant trimer-of-hairpins motif (Fig. 3). Nonlinear regression analysis of the binding data (GraphPad Prism 4) and calculation of the concentration of antibody required to achieve half-maximal binding revealed that there is a wide range in the relative affinities of the selected MAbs (Table 2). The highest relative affinities are exhibited by MAbs M18-8, M10-19, and M3-4, which achieve half-maximal binding at 0.02, 0.05, and 0.05 μg/ml, respectively. By contrast, MAbs such as M6-1, M16-28, and M20-1 have considerably lower affinities for TM and require 17- to 30-fold-higher concentrations of antibody to achieve 50% of maximal binding.
FIG. 3.

Dose-dependent binding of MAbs to the trimer-of-hairpins structure. Purified MAbs at the concentrations indicated were incubated with immobilized MBP-Hairpin. Following washing, bound antibody was detected. The data represent the means and standard deviations of triplicate assays.
TABLE 2.
Concentration of antibody calculated to give half-maximal binding
| Hybridoma | MAb concn at 50% binding (μg/ml) |
|---|---|
| M17-21 | 0.094 |
| M18-8 | 0.021 |
| M20-25 | 0.153 |
| M16-28 | 0.601 |
| M8-24 | 0.124 |
| M3-4 | 0.055 |
| M6-1 | 0.672 |
| M20-1 | 0.374 |
| M10-19 | 0.053 |
| M9-5 | 0.073 |
| M5-52 | 0.157 |
Reactivity of MAbs with denatured hairpin.
A principal objective of the vaccination strategy was to generate MAbs that recognize forms of TM that are naturally found on virions or virally infected cells, and the six-helix-bundle form of the HTLV-1 TM was of particular interest. To this end, the trimeric nature of the immunogen was confirmed by size exclusion chromatography prior to immunization (58). We therefore anticipated that some of the MAbs would recognize discontinuous nonlinear epitopes of TM and that recognition of these epitopes would be highly dependent on the conformation of the trimeric MBP-Hairpin. To explore the conformational specificity of the antibodies, we adapted an ELISA-based method (52) to determine the reactivity of the MAbs with native and denatured MBP-Hairpin. For these assays, recombinant MBP-Hairpin was denatured by heating in SDS-Tris buffer containing carrier protein; thereafter the SDS concentration was reduced by diluting the denatured protein 10-fold in SDS-free buffer. Subsequently, native or denatured MBP-Hairpin was captured on the surface of microtiter wells using rabbit anti-MBP polyclonal sera. The captured MBP-Hairpin was subsequently incubated with serial dilutions of each of the test MAbs, and the reactivity of the antibodies was examined by ELISA. As a control for antigen binding, we also examined the ability of an antibody known to recognize a linear epitope of MBP to bind to the immobilized denatured and native MBP fusion proteins.
Using this approach, we found that the anti-MBP MAb 20-1, which recognizes a linear epitope of MBP, bound efficiently to both immobilized native and denatured MBP-Hairpin structures (Fig. 4), indicating that binding of this control MAb is relatively independent of the tertiary structure of the antigen. By contrast, the reactivity of the anti-Hairpin antibodies was exquisitely sensitive to the conformational status of the antigen (Fig. 4). The vast majority of the anti-Hairpin MAbs demonstrated strong reactivity only with native trimeric Hairpin, while the reactivity with denatured Hairpin was either dramatically or partially reduced. Based on estimated half-maximal binding activities, the reactivity of some MAbs with MBP-Hairpin was almost completely lost following antigen denaturation; e.g., MAbs 18-8, 9-5, and 6-1 exhibit a 500- to 1,000-fold loss in binding (Fig. 4). For other MAbs, the loss of binding was relatively modest, e.g., for MAbs 5-52, 3-4, and 16-28 the loss in reactivity against denatured Hairpin was approximately 8-, 10-, and 16-fold, respectively. Thus, the majority of the selected MAbs display a distinct preferential recognition for the native trimer-of-hairpins structure. In keeping with this data, the isolated antibodies exhibit poor reactivity with TM under Western blotting conditions (data not shown).
FIG. 4.

Epitopes recognized by anti-TM MAbs are lost on denatured antigen. Each of the purified monoclonal antibodies at the concentrations indicated was examined for the ability to bind to native (closed squares) or denatured (closed triangles) MBP-Hairpin. Native or denatured (boiled in SDS and DTT and diluted in the presence of a carrier protein) antigen was captured on the solid phase by using polyclonal anti-MBP rabbit sera; subsequently, the anti-TM MAbs were each incubated with the immobilized antigen and following extensive washing, bound MAb was detected. As a control for antigen capture, the native and denatured antigens were also examined for reactivity with the monoclonal antibody M20-1, which binds to a linear epitope of MBP (top left graph). The data represent the means and standard deviations of triplicate assays.
Reactivity with TM fragments.
To begin to map the epitopes recognized by the MAbs, and having demonstrated that the MAbs exhibit strong binding to trimeric but not to denatured recombinant TM, we wished to determine if these MAbs recognized other truncated TM derivatives. The reactivity of each MAb was therefore examined with a panel of TM-derived MBP fusion proteins (Fig. 1c). In addition to the trimeric MBP-Hairpin, which mimics the trimer-of-hairpins form of TM (Fig. 5) (51, 58), the MAbs were also tested for reactivity with a variety of TM-derived MBP-fusion proteins (Fig. 1c), including MBP-Fishhook, representing the exposed but stable trimeric core coiled-coil domain and disulfide-bonded loop of TM (51, 58); MBP-N-helix, a truncated derivative that emulates a monomeric form of the N-helical region (Fig. 5; 51); a monomeric MBP-C-helix (Fig. 5), which mimics the extended peptide chain and short α-helix of the antiparallel C-helical region of TM (Fig. 1; 51); and finally, monomeric MBP-CX6CC-loop that spans the disulfide-bonded loop of TM (Fig. 1c and 5).
FIG. 5.

Oligomerization states of the MBP-TM-derived chimeras. Gel filtration chromatography profiles of the trimeric MBP-hairpin (TH), BSA, and monomeric MBP control proteins are indicated (top trace). The profiles of the MBP-N-helix, MBP-C-helix, and MBP-CX6CC-loop fusion proteins are shown below. The predicted molecular masses of each protein monomer are as follows: MBP, 43 kDa (lane 2); MBP-Hairpin, 53 kDa (lane 3); MBP-N-helix, 48 kDa; MBP-C-helix, 46 kDa; and MBP-CX6CC-loop, 46 kDa. A small amount of aggregated material can be observed in the profile for MBP-CX6CC-loop (approximate elution volume, 10 to 13 ml); this is due to nonspecific intermonomer disulfide bonding and can be reduced in the presence of 5 mM DTT. Calibration markers (arrows above plots) are ferritin, 440 kDa; aldolase, 158 kDa; albumin, 67 kDa; ovalbumin, 43 kDa; and RNase A, 13.7 kDa.
While each of the antibodies efficiently recognized the recombinant trimer-of-hairpins structure, a wide range of reactivity was observed toward the truncated TM derivatives (Fig. 6). All of the MAbs bound to the MBP-Fishhook construct that mimics the stable trimeric core coiled-coil domain of TM (51, 58), indicating that these antibodies do not require the antiparallel C-helical region for the recognition of TM. Consistent with these data, there was little or no binding of the antibodies to the MBP-C-helix, confirming that the epitopes recognized by the MAbs are not contained within the C-helical region of TM. Only one MAb, M9-5, bound to the CX6CC-loop region of TM, indicating that the loop region is not an epitope that is recognized by the majority of the antibodies. Moreover, MAb M9-5 did not bind to Hairpin denatured by boiling in the presence of SDS and DTT (Fig. 4), indicating that disulfide formation likely provides important structural constraints for epitope recognition by M9-5. Surprisingly, the majority of the MAbs also failed to bind to the monomeric N-helix, indicating that trimerization of the coiled coil is a prerequisite for appropriate presentation of the epitopes recognized by these antibodies.
FIG. 6.

Murine MAbs bind to both the trimer-of-hairpins and coiled-coil structures of TM. Purified antibody (a) or antibody in hybridoma-conditioned medium supernatant (b) was examined for binding to a panel of recombinant TM derivatives (shown in Fig. 1). The antigens MBP, MBP-Hairpin, MBP-Fishhook, MBP-N-helix, MBP-C-helix, and MBP-CX6CC-loop were each adsorbed onto the solid phase. Purified antibody (1 μg/ml) or conditioned medium supernatants (50 μl) were incubated for 1 h with each of the immobilized antigens, immobilized complexes were washed, and the bound antibody was detected. The data represent the means and standard deviations of triplicate assays and are typical of multiple independent experiments.
Supporting this view, the MAbs that failed to bind to monomeric forms of TM also failed to bind to a panel of overlapping 30- to 45-amino-acid synthetic peptides that span the entire six-helix-bundle region, including the N- and C-helical regions and the disulfide-bonded chain reversal loop, and these peptides failed to compete with trimeric MBP-Hairpin for MAb binding in competition binding experiments (data not shown).
Recognition of virally expressed envelope.
The accrued data indicate that the MAbs recognize conformational epitopes on the trimer-of-hairpins form of TM. The trimer-of-hairpins structure is generally acknowledged to be a fusion-active or postfusion conformation of retroviral TM. We therefore wished to determine if these MAbs recognized envelope on virally infected cells under nonfusogenic conditions. Since chronic retroviral infection of cells results in receptor interference and downregulation of cell surface receptors (9, 28, 38, 43, 66, 67, 69), little cell-to-cell fusion occurs in chronically infected cell populations in the absence of cocultured, uninfected target cells. Therefore, the chronically HTLV-1-infected T-cell line MT2 was employed to examine the reactivity of the antibodies. Each of the MAbs was examined for reactivity with envelope on the surface of virally infected MT2 cells by flow cytometry. As a control, the antibodies were also tested for binding to the uninfected T-cell line SupT-1. Importantly, none of the anti-TM antibodies displayed any binding to these control cells (data not shown). By contrast, and somewhat surprisingly given that these assays were carried out under nonfusogenic conditions, most of the MAbs exhibited strong binding to the HTLV-1-infected cells (Fig. 7). Thus, many of the antibodies efficiently recognize virally expressed envelope protein even in the absence of overt membrane fusion. While most of the MAbs exhibit efficient recognition of viral TM on infected cells, a few antibodies, for example, M15-9, M17-12, M19-8, and M3-18, displayed only weak fluorescence signals (data not shown) or no fluorescence signal, such as M18-25 (Fig. 7), suggesting that the epitopes recognized by these MAbs may be either partially or completely obscured on native envelope. However, to verify the binding of MAbs to envelope-expressing cells, fluorescence microscopy and immunolabeling were used to examine the localization of antibody bound by infected cells. For all of the MAbs that produced strong flow cytometry signals, we found that the observed pattern of antibody staining was consistent with the recognition of viral envelope as strong peripheral staining of the plasma membrane was observed on the infected cells but not on uninfected controls (Fig. 8).
FIG. 7.

Murine MAbs recognize envelope expressed on virally infected cells. HTLV-1-infected cells (MT2) were incubated with irrelevant primary anti-MBP (M20-1) or the anti-TM MAbs as indicated. Bound antibody was detected using FITC-conjugated anti-mouse antibody and flow cytometry. The solid histograms represent the basal fluorescence of the cell population in the absence of primary antibody, and open histograms represent the fluorescence signal in the presence of the specified primary antibody. As negative controls, binding of the antibodies to uninfected SupT-1 cells was also examined. With SupT-1 cells, no increase in fluorescence of the cell population was observed with any of the test or control antibodies (data not shown). The data are typical of multiple independent experiments.
FIG. 8.
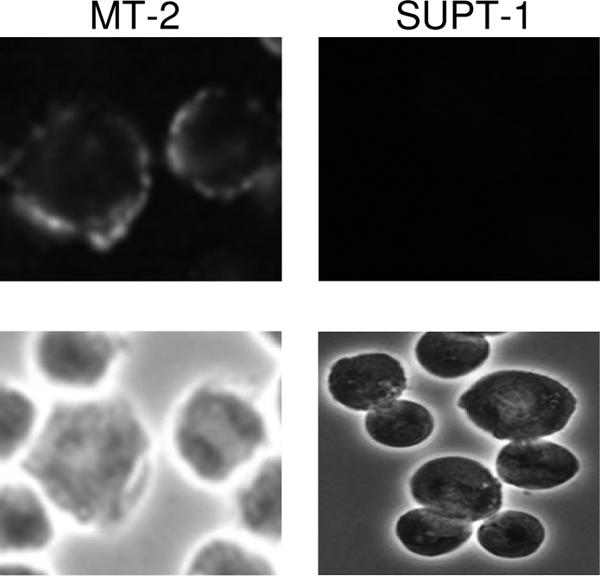
Cell surface expression of epitopes recognized by anti-Hairpin MAbs. HTLV-1-infected (MT2) and uninfected control cells (SupT-1) were incubated with anti-Hairpin MAbs, and bound antibody was detected using FITC-conjugated anti-mouse secondary (upper panels); corresponding phase-contrast images are also shown (lower panels). A typical result for monoclonal antibody M8-24 is shown, demonstrating peripheral staining of the plasma membrane on infected cells but not uninfected cells. All antibodies demonstrating positive staining by flow cytometry analysis also exhibited staining patterns essentially identical to M8-24 by fluorescence microscopy (data not shown).
Neutralizing activity.
A key feature of potential subunit vaccine candidates is the ability to generate neutralizing antibodies. Given the importance of TM in the viral entry process and the observation that peptides directed to HTLV-1 TM are potent inhibitors of membrane fusion, it is possible that recombinant antigens that faithfully mimic the conformational features of viral TM may be useful as vaccine immunogens. Therefore, the ability of anti-Hairpin MAbs to directly neutralize membrane fusion and viral infection was examined. Two complementary assays were used to monitor neutralization of envelope function. First, each MAb was tested for the ability to inhibit syncytium formation between HeLa cells transfected with the envelope expression vector HTE-1 and untransfected HeLa target cells. Second, the MAbs were examined for the ability to block infection of HOS target cells with HIV particles pseudotyped with HTLV-1 envelope. Despite exhaustive analysis, and even at very high antibody concentrations (20 μg/ml) or suboptimal temperatures for Env-mediated membrane fusion (16, 19), no neutralizing activity could be detected for any of the antibodies in either of the neutralization assays. Thus, the monoclonal antibodies generated against the trimer-of-hairpins form of HLTV-1 TM are unable to block envelope-mediated membrane fusion.
Complement fixation.
Our data reveal that antibodies targeting the trimer-of-hairpins motif of HTLV-1 envelope efficiently recognize TM on the surface of infected cells. Nevertheless, these antibodies fail to inhibit envelope-mediated membrane fusion in syncytium-interference assays and in viral infectivity assays using pseudotyped viral particles. However, neutralization of virus by antibody-mediated disruption of envelope function is only one role played by antibody during the humoral response to viral infection. Clearance of virus particles and infected cells is also facilitated by antibody-dependent complement-mediated lysis (8, 26, 53, 60, 75, 76), and for HTLV-1 in particular, infectivity can be inhibited in a complement-dependent manner (30, 49). Therefore, the antibodies raised against the TM-derived immunogen were examined for complement-fixing activity.
An ELISA-based method was employed to compare the ability of the anti-Hairpin antibodies to recruit C1q, the first step of the complement activation cascade. Immobilized MBP-Hairpin complexed with anti-Hairpin antibodies was incubated with whole serum as a source of complement; subsequently, unbound complement was washed away, and the amount of bound C1q was determined. As anticipated, no complement-fixing activity was observed in the absence of antibody or in the presence of irrelevant control antibodies (Fig. 9). However, a wide range of complement activation was observed for the anti-Hairpin antibodies. Several of the anti-Hairpin MAbs, including M6-1, M16-28, M18-25, and M3-18, exhibited little C1q binding activity, whereas eight of the MAbs (M5-52, M8-24, M17-21, M10-19, M3-4, M18-8, M9-5, and M4-32) demonstrated efficient C1q recruitment that ranged between approximately 22- and 35-fold above background. Thus, the recombinant trimeric TM fusion protein is able to stimulate production of antibodies with complement-fixing activity, a property that may be of value in the design of a vaccine targeted against the envelope of HTLV-1.
FIG. 9.

Monoclonal antibodies raised against recombinant TM recruit complement. Immobilized MBP-Hairpin was incubated (1 h) with (a) each of the purified anti-Hairpin antibodies or with (b) hybridoma-conditioned medium supernatants (M17-21p, purified M17-21 control; M17-21s, conditioned medium supernatant control). As controls, antigen was also incubated in the absence of antibody or in the presence of irrelevant antibody (anti-HIV gp120 MAb 178.1 or anti-MBP MAb M20-1 or the irrelevant MAb K18-8). Unbound antibody was washed away, and whole serum (as a source of complement) was added and incubated with the Hairpin-antibody complexes. Subsequently, unbound complement was washed away, and bound C1q was detected. The results represent the means and standard deviations of triplicate assays.
DISCUSSION
Vaccine strategies remain the primary and most effective means of combating viral infections of humans (2, 40, 41, 61), and subunit vaccines in particular have been a recent and dramatic success in providing protection from oncogenic viruses (31, 68). However, despite the broad utility of the vaccine approach and despite considerable promise, effective vaccines targeting retroviral infections have remained elusive. In the case of human immunodeficiency virus, an effective vaccine presents a considerable theoretical and technical challenge, in part due to concerns over the safety of attenuated viral vaccine candidates (11, 27), but also because the tremendous sequence variation of viral isolates results in rapid viral escape from the neutralizing antibody and cytotoxic lymphocyte responses induced by subunit vaccines (4, 45, 74). By contrast, HTLV-1 would appear to be an attractive target for vaccine development. There is relatively little sequence variation between clinical HTLV-1 isolates, even from geographically distinct areas (17, 18, 64); an attenuated strain of the related bovine leukemia virus (BLV) provides long-term protection against experimental BLV infection of cattle (39); and for HTLV-1, an envelope-derived subunit vaccine candidate provides significant protection against virus challenge in a primate model (29). Moreover, it is widely recognized that the envelope-derived SU is a prominent target for neutralizing antibodies and cytotoxic lymphocytes in infected patients (3, 13, 20, 59). The accumulating evidence, therefore, suggests that a subunit vaccine based on viral envelope may be an achievable objective for prophylactic treatment of HTLV-1 infections.
The immune response to HTLV-1 SU is beginning to be understood, but there is currently little information on the immunological properties of TM. We have therefore begun to examine the antigenic properties of TM as a potential component of a subunit vaccine and to provide additional information on the molecular function of TM in the HTLV-1 infection process. Here, we demonstrate that a trimeric form of recombinant TM protein is immunogenic and that MAbs with a wide range of affinities for the immunogen can be readily isolated. Surprisingly, the majority of the antibodies display a remarkable sensitivity to the conformational status of the antigen, and several of the MAbs display greater than 500-fold loss of binding upon denaturation of the antigen. Thus, the tertiary and quaternary structures of TM are important for recognition by these MAbs. While mapping of the epitopes recognized by the MAbs has been hampered by the strict conformational requirements for antibody binding, some important information on recognition of TM has been obtained. The data indicate that the MAbs recognize the trimer-of-hairpins form of TM but fail to recognize denatured TM or monomeric TM fragments that span the N-helical and C-helical regions of gp21. Other than MAb M9-5, the MAbs also fail to bind TM fragments that mimic the disulfide-bonded loop of TM. By contrast, in addition to the trimer-of-hairpins motif, the MAbs strongly bind to the trimeric “fishhook” structure that emulates the exposed core coiled-coil and flexible hinge region of viral TM. Moreover, our unpublished data indicate that synthetic peptides that mimic the looped region of TM fail to compete with the fishhook structure for antibody binding. It is therefore likely, and is consistent with our preliminary data, that the majority of the MAbs recognize epitopes on the triple-stranded coiled-coil region of TM. Importantly, while the trimer-of-hairpins motif is regarded as a late fusion-active or postfusion conformation of TM, current evidence indicates that the core coiled coil is an important structural feature of the prehairpin intermediate of fusion-active TM.
The MAbs isolated in this study recognize and bind to both the six-helix bundle and the central coiled coil. Moreover, the MAbs also recognize virally expressed envelope, as revealed by flow cytometry and immunofluorescence microscopy. Why then do the MAbs fail to block envelope-mediated syncytium formation or infection of cells by pseudotyped viral particles? There are several plausible explanations for this lack of neutralizing activity. First, HTLV-1 TM may present no neutralizing epitopes. However, this possibility seems unlikely given that we have previously demonstrated that synthetic peptides, which specifically bind to the hydrophobic groove on the core coiled coil and efficiently compete with the TM C-helix for binding to this site, potently inhibit envelope-mediated membrane fusion (51, 58). It is therefore anticipated that MAbs, which recognize the surface groove of the coiled coil, should have neutralizing activity. An alternative possibility is that the surfaces of TM recognized by these particular MAbs present an abundance of nonneutralizing epitopes. In this respect, it is worth noting that the crystal structure of TM reveals that only a narrow groove on the coiled coil is involved in interaction with the extended C-helix, and therefore, neutralizing antibodies would have to bind within or overlapping this groove to prevent C-helix docking. However, the antigen used in our studies represents the six-helix-bundle conformation of TM, and the antiparallel C-terminal region of TM masks this groove. Moreover, our data demonstrate that the antibodies bind efficiently to the trimer-of-hairpins form, and therefore the bound antibodies are able to accommodate occupancy of the groove by the C-helix. The data raise the possibility that the MAbs bind to nonneutralizing epitopes on the solvent-exposed surfaces of the coiled coil. We are currently investigating this possibility.
A third, but not mutually exclusive or exhaustive, scenario is that the epitopes available for MAb binding are not accessible on the intact prehairpin intermediate of functional envelope but are obscured by SU. The epitopes are subsequently exposed upon resolution to the hairpin structure. This model assumes that the trimer-of-hairpins structure is a very late fusion-active or postfusion structure and that membrane fusion either is fully committed or has already taken place upon formation of the trimer-of-hairpins structure. In this case, antibodies targeting the six-helix bundle would not neutralize envelope-mediated membrane fusion. This model is compatible with recent data for TM-mediated membrane fusion (15) and is reminiscent of the situation with a well-characterized MAb raised against another retroviral TM protein (34). The MAb NC-1 recognizes the six-helix-bundle structure of HIV-1 TM (34) but, in contrast to our anti-HTLV MAbs, does not bind to the core coiled coil. Antibody NC-1 recognizes viral envelope only following activation by CD4 but, nevertheless, fails to neutralize HIV infectivity (34). Thus, data from two independent retroviral systems indicate that the retroviral six-helix-bundle structure is not an ideal immunogen for the generation of neutralizing antibodies.
Since the prehairpin and six-helix bundle are considered to be fusion-related structures, it was surprising that many MAbs raised against the trimer-of-hairpins structure efficiently recognize envelope on HTLV-1-infected cells even in the absence of membrane fusion. Based on current models, it seems unlikely that the epitopes recognized by these MAbs exist on the prefusogenic HTLV-1 envelope. Instead, it is likely that the epitopes are exposed only upon receptor-mediated activation of the fusogenic properties of envelope. One interpretation of the data is that there are multiple forms of envelope on HTLV-1-infected cells. Since native envelope spikes are thought to exist in a meta-stable conformation (15, 65), they may be readily triggered to yield fusion-active and postfusion TM structures that are efficiently detected by the TM MAbs. Using fluorescence microscopy and antibodies to SU and TM, it has not yet been possible to resolve intact native envelope from fusion-activated forms (our unpublished results). However, it is worth noting that for HIV-1, SU (gp120) is noncovalently linked to TM and is readily shed from envelope spikes or lost by sCD4- or antibody-mediated stripping (23, 42). It is therefore likely that the remaining TM stumps adopt postfusion structures. By comparison, the SU of retroviruses such as avian leukosis-sarcoma virus, murine leukemia virus, BLV, and HTLV are less prone to shedding and are linked to TM via reactive disulfide bridges (24, 36, 73). However, the reactivity of surface-expressed viral envelope with the trimer-of-hairpin MAbs is consistent with the notion that HTLV-1 envelope exists in several forms on infected cells. If further work confirms this view, it will be interesting to determine if alternative envelope structures play a significant role in the pathogenesis of HTLV-1 infections or in viral escape from immune surveillance.
While the anti-Hairpin antibodies failed to block envelope-mediated membrane fusion, many of the antibodies efficiently recruited complement to trimeric TM. Moreover, epitopes capable of binding complement-fixing antibodies are abundantly expressed on the surface of HTLV-1-infected T cells, indicating that antibody-dependent complement-mediated lysis of envelope-expressing cells and viral particles is likely to be an important mechanism for immunological clearance of HTLV-1. This view is supported by the observations that in natural infections, the SU region of envelope is capable of inducing complement-fixing antibodies (49) and that complement can effectively neutralize viral infectivity in vitro (30, 49). Nevertheless, from a vaccine perspective, the ideal immunogen should elicit both neutralizing and complement-fixing antibodies. It therefore remains to be determined if a suitable combination of SU and TM subunits with appropriate conformational structures can be developed that would routinely induce robust neutralizing and complement fixing-antibody responses in vaccinees.
The data presented here imply that it will be technically challenging to generate immunogens capable of eliciting antibodies with the required target specificity and neutralizing activity against HTLV-1 TM. However, the demonstration that synthetic peptides targeting the core coiled coil of HTLV-1 TM potently inhibit TM-mediated membrane fusion (5, 35, 51, 58, 62) and that TM-specific neutralizing antibodies have been identified for HIV-1 (12, 50, 54, 77, 78) suggests that a comprehensive search for neutralizing antibodies reactive with the functional surfaces of HTLV-1 TM is warranted and that the generation of immunogens capable of eliciting such antibodies is achievable. The data presented in this study represent an important first step toward reaching these objectives, and the antibodies produced will be of significant utility to the analysis of TM function in HTLV-1 infections.
Acknowledgments
This work was generously supported by a project grant (LRF-0227) awarded to D.W.B. from the Leukemia Research Fund and, in part, by a grant from the Association for International Cancer Research (AICR).
We thank Helen-Louise Murphy for assistance with chromatography.
Footnotes
Published ahead of print on 21 March 2007.
REFERENCES
- 1.Anonymous. 1996. Human T-cell lymphotropic viruses. IARC Monogr. Eval. Carcinog. Risks Hum. 67:261-334. [PMC free article] [PubMed] [Google Scholar]
- 2.Arvin, A. M., and H. B. Greenberg. 2006. New viral vaccines. Virology 344:240-249. [DOI] [PubMed] [Google Scholar]
- 3.Baba, E., M. Nakamura, Y. Tanaka, M. Kuroki, Y. Itoyama, S. Nakano, and Y. Niho. 1993. Multiple neutralizing B-cell epitopes of human T-cell leukemia virus type 1 (HTLV-1) identified by human monoclonal antibodies. A basis for the design of an HTLV-1 peptide vaccine. J. Immunol. 151:1013-1024. [PubMed] [Google Scholar]
- 4.Borrow, P., H. Lewicki, X. Wei, M. S. Horwitz, N. Peffer, H. Meyers, J. A. Nelson, J. E. Gairin, B. H. Hahn, M. B. Oldstone, and G. M. Shaw. 1997. Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat. Med. 3:205-211. [DOI] [PubMed] [Google Scholar]
- 5.Brighty, D. W., and S. R. Jassal. 2001. The synthetic peptide P-197 inhibits human T-cell leukemia virus type 1 envelope-mediated syncytium formation by a mechanism that is independent of Hsc70. J. Virol. 75:10472-10478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cann, A. J., and I. S. Y. Chen. 1996. Human T-cell leukemia virus type I and II, p. 1501-1527. In B. N. Fields, D. M. Knipe, and P. M. Howley (ed.), Virology, 3rd ed., vol. 2. Lippincott-Raven, Philadelphia, PA. [Google Scholar]
- 7.Center, R. J., B. Kobe, K. A. Wilson, T. Teh, G. J. Howlett, B. E. Kemp, and P. Poumbourios. 1998. Crystallization of a trimeric human-Tcell leukaemia virus type 1 gp21 ectodomain fragment as a chimera with maltose binding protein. Protein Sci. 7:1612-1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cooper, N. R., F. C. Jensen, R. M. Welsh, and M. B. A. Oldstone. 1976. Lysis of RNA tumor viruses by human serum: direct antibody-independent triggering of the classical complement pathway. J. Exp. Med. 144:970-984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dalgleish, A. G., P. C. Beverley, P. R. Clapham, D. H. Crawford, M. F. Greaves, and R. A. Weiss. 1984. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature 312:763-767. [DOI] [PubMed] [Google Scholar]
- 10.Damico, R. L., J. Crane, and P. Bates. 1998. Receptor-triggered membrane association of a model retroviral glycoprotein. Proc. Natl. Acad. Sci. USA 95:2580-2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deacon, N. J., A. Tsykin, A. Solomon, K. Smith, M. Ludford-Menting, D. J. Hooker, D. A. McPhee, A. L. Greenway, A. Ellett, C. Chatfield, V. A. Lawson, S. Crowe, A. Maerz, S. Sonza, J. Learmont, J. S. Sullivan, A. Cunningham, D. Dwyer, D. Dowton, and J. Mills. 1995. Genomic structure of an attenuated quasi species of HIV-1 from a blood transfusion donor and recipients. Science 270:988-991. [DOI] [PubMed] [Google Scholar]
- 12.de Rosny, E., R. Vassell, S. Jiang, K. Kunert, and C. D. Weiss. 2004. Binding of the 2F5 monoclonal antibody to native and fusion-intermediate forms of human immunodeficiency virus type 1 gp41: implications for fusion-inducing conformational changes. J. Virol. 78:2627-2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Desgranges, C., S. Souche, J. C. Vernant, D. Smadja, A. Vahlne, and P. Horal. 1994. Identification of novel neutralization-inducing regions of the human T-cell lymphotropic virus type I envelope glycoproteins with human HTLV-1-seropositive sera. AIDS Res. Hum. Retrovir. 10:163-173. [DOI] [PubMed] [Google Scholar]
- 14.Dokhelar, M. C., H. Pickford, J. Sodroski, and W. A. Haseltine. 1989. HTLV-1 p27rex regulates gag and env protein expression. J. Acquir. Immune Defic. Syndr. 2:431-440. [PubMed] [Google Scholar]
- 15.Eckert, D. M., and P. S. Kim. 2001. Mechanisms of viral membrane fusion and its inhibition. Annu. Rev. Biochem. 70:777-810. [DOI] [PubMed] [Google Scholar]
- 16.Frey, S., M. Marsh, S. Günther, A. Pelchen-Matthews, P. Stephens, S. Ortlepp, and T. Stegmann. 1995. Temperature dependence of cell-cell fusion induced by the envelope glycoprotein of human immunodeficiency virus type 1. J. Virol. 69:1462-1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gessain, A., and G. de The. 1996. Geographic and molecular epidemiology of primate T lymphotropic retroviruses: HTLV-I, HTLV-II, STLV-I, STLV-PP, and PTLV-L. Adv. Virus Res. 47:377-426. [DOI] [PubMed] [Google Scholar]
- 18.Gessain, A., R. C. Gallo, and G. Franchini. 1992. Low degree of human T-cell leukemia/lymphoma virus type I genetic drift in vivo as a means of monitoring viral transmission and movement of ancient human populations. J. Virol. 66:2288-2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Golding, H., M. Zaitseva, E. de Rosny, L. R. King, J. Manischewitz, I. Sidorov, M. K. Gorny, S. Zolla-Pazner, D. S. Dimitrov, and C. D. Weiss. 2002. Dissection of human immunodeficiency virus type 1 entry with neutralizing antibodies to gp41 fusion intermediates. J. Virol. 76:6780-6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hadlock, K. G., J. Rowe, and S. K. Foung. 1999. The humoral immune response to human T-cell lymphotropic virus type 1 envelope glycoprotein gp46 is directed primarily against conformational epitopes. J. Virol. 73:1205-1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hadlock, K. G., J. Rowe, S. Perkins, P. Bradshaw, G. Y. Song, C. Cheng, J. Yang, R. Gascon, J. Halmos, S. M. Rehman, M. S. McGrath, and S. K. Foung. 1997. Neutralizing human monoclonal antibodies to conformational epitopes of human T-cell lymphotropic virus type 1 and 2 gp46. J. Virol. 71:5828-5840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hadlock, K. G., Q. Yang, J. Rowe, and S. K. Foung. 2002. Epitope mapping of human monoclonal antibodies recognizing conformational epitopes within HTLV type 1 gp46, employing HTLV type 1/2 envelope chimeras. AIDS Res. Hum. Retrovir. 18:57-70. [DOI] [PubMed] [Google Scholar]
- 23.Hart, T. K., R. Kirsh, H. Ellens, R. W. Sweet, D. M. Lambert, S. R. Petteway, Jr., J. Leary, and P. J. Bugelski. 1991. Binding of soluble CD4 proteins to human immunodeficiency virus type 1 and infected cells induces release of envelope glycoprotein gp120. Proc. Natl. Acad. Sci. USA 88:2189-2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Helseth, E., U. Olshevsky, C. Furman, and J. Sodroski. 1991. Human immunodeficiency virus type 1 gp120 envelope glycoprotein regions important for association with the gp41 transmembrane glycoprotein. J. Virol. 65:2119-2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hernandez, L. D., R. J. Peters, S. E. Delos, J. A. T. Young, D. A. Agard, and J. M. White. 1997. Activation of a retroviral membrane fusion protein: soluble receptor-induced liposome binding of the ALSV envelope glycoprotein. J. Cell Biol. 39:1455-1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hirsch, R. L. 1982. The complement system: its importance in the host response to viral infection. Microbiol. Rev. 46:71-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hofmann-Lehmann, R., J. Vlasak, A. L. Williams, A. L. Chenine, H. M. McClure, D. C. Anderson, S. O'Neil, and R. M. Ruprecht. 2003. Live attenuated, nef-deleted SIV is pathogenic in most adult macaques after prolonged observation. AIDS 17:157-166. [DOI] [PubMed] [Google Scholar]
- 28.Hoxie, J. A., J. D. Alpers, J. L. Rackowski, K. Huebner, B. S. Haggarty, A. J. Cedarbaum, and J. C. Reed. 1986. Alterations in T4 (CD4) protein and mRNA synthesis in cells infected with HIV. Science 234:1123-1127. [DOI] [PubMed] [Google Scholar]
- 28a.The HTLV-1 European Research Network. 1996. Seroepidemiology of the human T-cell leukaemia/lymphoma viruses in Europe. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 13:68-77. [DOI] [PubMed] [Google Scholar]
- 29.Ibuki, K., S. I. Funahashi, H. Yamamoto, M. Nakamura, T. Igarashi, T. Miura, E. Ido, M. Hayami, and H. Shida. 1997. Long-term persistence of protective immunity in cynomolgus monkeys immunized with a recombinant vaccinia virus expressing the human T cell leukaemia virus type I envelope gene. J. Gen. Virol. 78:147-152. [DOI] [PubMed] [Google Scholar]
- 30.Ikeda, F., Y. Haraguchi, A. Jinno, Y. Iino, Y. Morishita, H. Shiraki, and H. Hoshino. 1998. Human complement component C1q inhibits the infectivity of cell-free HTLV-I. J. Immunol. 161:5712-5719. [PubMed] [Google Scholar]
- 31.Jacobson, I. M., and J. L. Dienstag. 1985. Viral hepatitis vaccines. Annu. Rev. Med. 36:241-261. [DOI] [PubMed] [Google Scholar]
- 32.Jassal, S. R., M. D. Lairmore, A. J. Leigh-Brown, and D. W. Brighty. 2001. Soluble recombinant HTLV-1 surface glycoprotein competitively inhibits syncytia formation and viral infection of cells. Virus Res. 78:17-34. [DOI] [PubMed] [Google Scholar]
- 33.Jassal, S. R., R. G. Pohler, and D. W. Brighty. 2001. Human T-cell leukemia virus type 1 receptor expression among syncytium-resistant cell lines revealed by a novel surface glycoprotein-immunoadhesin. J. Virol. 75:8317-8328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang, S., K. Lin, and M. Lu. 1998. A conformation-specific monoclonal antibody reacting with fusion-active gp41 from the human immunodeficiency virus type 1 envelope glycoprotein. J. Virol. 72:10213-10217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jinno, A., H. H. Shiraki, and H. Hoshino. 1999. Inhibition of cell-free human T-cell leukemia virus type 1 infection at a postbinding step by the synthetic peptide derived from an ectodomain of the gp21 transmembrane glycoprotein. J. Virol. 73:9683-9689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnston, E. R., and K. Radke. 2000. The SU and TM envelope protein subunits of bovine leukemia virus are linked by disulfide bonds, both in cells and in virions. J. Virol. 74:2930-2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jones, K. S., C. Petrow-Sadowski, D. C. Bertolette, Y. Huang, and F. W. Ruscetti. 2005. Heparan sulfate proteoglycans mediate attachment and entry of human T-cell leukemia virus type 1 virions into CD4+ T cells. J. Virol. 79:12692-12702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kawamura, I., Y. Koga, N. Oh-Hori, K. Onodera, G. Kimura, and K. Nomoto. 1989. Depletion of the surface CD4 molecule by the envelope protein of human immunodeficiency virus expressed in a human CD4+ monocytoid cell line. J. Virol. 63:3748-3754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kerkhofs, P., J. S. Gatot, K. Knapen, M. Mammerickx, A. Burny, D. Portetelle, L. Willems, and R. Kettmann. 2000. Long-term protection against bovine leukaemia virus replication in cattle and sheep. J. Gen. Virol. 81:957-963. [DOI] [PubMed] [Google Scholar]
- 40.Kew, O. M., R. W. Sutter, E. M. de Gourville, W. R. Dowdle, and M. A. Pallansch. 2005. Vaccine-derived polioviruses and the endgame strategy for global polio eradication. Annu. Rev. Microbiol. 59:587-635. [DOI] [PubMed] [Google Scholar]
- 41.Kieny, M. P., and M. P. Girard. 2005. Human vaccine research and development: an overview. Vaccine 23:5705-5707. [DOI] [PubMed] [Google Scholar]
- 42.Kirsh, R., T. K. Hart, H. Ellens, J. Miller, S. A. Petteway, Jr., D. M. Lambert, J. Leary, and P. J. Bugelski. 1990. Morphometric analysis of recombinant soluble CD4-mediated release of the envelope glycoprotein gp120 from HIV-1. AIDS Res. Hum. Retrovir. 6:1209-1212. [DOI] [PubMed] [Google Scholar]
- 43.Klatzmann, D., E. Champagne, S. Chamaret, J. Gruest, D. Guetard, T. Hercend, J. C. Gluckman, and L. Montagnier. 1984. T-lymphocyte T4 molecule behaves as the receptor for human retrovirus LAV. Nature 312:767-768. [DOI] [PubMed] [Google Scholar]
- 44.Kobe, B., R. J. Center, B. E. Kemp, and P. Poumbourios. 1999. Crystal structure of human T-cell leukemia virus type 1 gp21 ectodomain crystallized as a maltose-binding protein chimera reveals structural evolution of retroviral transmembrane proteins. Proc. Natl. Acad. Sci. USA 96:4319-4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kwong, P. D., M. L. Doyle, D. J. Casper, C. Cicala, S. A. Leavitt, S. Majeed, T. D. Steenbeke, M. Venturi, I. Chaiken, M. Fung, H. Katinger, P. W. Parren, J. Robinson, D. Van Ryk, L. Wang, D. R. Burton, E. Freire, R. Wyatt, J. Sodroski, W. A. Hendrickson, and J. Arthos. 2002. HIV-1 evades antibody-mediated neutralization through conformational masking of receptor-binding sites. Nature 420:623-624. [DOI] [PubMed] [Google Scholar]
- 46.Loughran, T. P., Jr., K. G. hadlock, Q. Yang, R. Perzova, R. Zambello, G. Semenzato, S. K. Foung, and B. J. Poiesz. 1997. Seroreactivity to an envelope protein of human T-cell leukemia/lymphoma virus in patients with CD3- (natural killer) lymphoproliferative disease of granular lymphocytes. Blood 90:1977-1981. [PubMed] [Google Scholar]
- 47.Loughran, T. P., Jr., K. G. Hadlock, R. Perzova, T. C. Gentile, Q. Yang, S. K. Foung, and B. J. Poiesz. 1998. Epitope mapping of HTLV envelope seroreactivity in LGL leukaemia. Br. J. Haematol. 101:318-324. [DOI] [PubMed] [Google Scholar]
- 48.Manel, N., F. J. Kim, S. Kinet, N. Taylor, M. Sitbon, and J. L. Battini. 2003. The ubiquitous glucose transporter GLUT-1 is a receptor for HTLV. Cell 115:449-459. [DOI] [PubMed] [Google Scholar]
- 49.Matsushita, S., M. Robert-Guroff, J. Cossman, H. Mitsuva, and S. Broder. 1986. Human monoclonal antibody directed against an envelope glycoprotein of human T-cell leukemia virus type I. Proc. Natl. Acad. Sci. USA 83:2672-2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miller, M. D., R. Geleziunas, E. Bianchi, S. Lennard, R. Hrin, H. Zhang, M. Lu, Z. An, P. Ingallinella, M. Finotto, M. Mattu, A. C. Finnefrock, D. Bramhill, J. Cook, D. M. Eckert, R. Hampton, M. Patel, S. Jarantow, J. Joyce, G. Ciliberto, R. Cortese, P. Lu, W. Strohl, W. Schleif, M. McElhaugh, S. Lane, C. Lloyd, D. Lowe, J. Osbourn, T. Vaughan, E. Emini, G. Barbato, P. S. Kim, D. J. Hazuda, J. W. Shiver, and A. Pessi. 2005. A human monoclonal antibody neutralizes diverse HIV-1 isolates by binding a critical gp41 epitope. Proc. Natl. Acad. Sci. USA 102:14759-14764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mirsaliotis, A., K. Nurkiyanova, D. J. Lamb, C. S. Kuo, and D. W. Brighty. 2007. An antibody that blocks HTLV-1 six-selix-bundle formation in vitro identified by a novel assay for inhibitors of envelope function. J. Gen. Virol. 88:660-669. [DOI] [PubMed] [Google Scholar]
- 52.Moore, J. P., and D. D. Ho. 1993. Antibodies to discontinuous or conformationally sensitive epitopes on the gp120 glycoprotein of human immunodeficiency virus type 1 are highly prevalent in sera of infected humans. J. Virol. 67:863-875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morgan, B. P. 1994. Clinical complementology: recent progress and future trends. Eur. J. Clin. Investig. 24:219-228. [DOI] [PubMed] [Google Scholar]
- 54.Muster, T., F. Steindl, M. Purtscher, A. Trkola, A. Klima, G. Himmler, F. Ruker, and H. Katinger. 1993. A conserved neutralizing epitope on gp41 of human immunodeficiency virus type 1. J. Virol. 67:6642-6647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Okuma, K., M. Nakamura, S. Nakano, Y. Niho, and Y. Matsuura. 1999. Host range of human T-cell leukemia virus type I analyzed by a cell fusion-dependent reporter gene activation assay. Virology 254:235-244. [DOI] [PubMed] [Google Scholar]
- 56.Palker, T. J., E. T. Riggs, D. E. Spragion, A. Muir, R. M. Scearce, R. R. Randall, M. W. McAdams, A. McKnight, P. R. Clapham, R. A. Weiss, and B. F. Haynes. 1992. Mapping of homologous, amino-terminal neutralizing regions of human T-cell lymphotropic virus type I and II gp46 envelope glycoproteins. J. Virol. 66:5879-5889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Piñón, J. D., P. J. Klasse, S. R. Jassal, S. Welson, J. Weber, D. W. Brighty, and Q. J. Sattentau. 2003. Human T-cell leukemia virus type 1 envelope glycoprotein gp46 interacts with cell surface heparan sulfate proteoglycans. J. Virol. 77:9922-9930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Piñón, J. D., S. M. Kelly, N. C. Price, J. U. Flanagan, and D. W. Brighty. 2003. An antiviral peptide targets a coiled-coil domain of the human T-cell leukemia virus envelope glycoprotein. J. Virol. 77:3281-3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pique, C., F. Connan, J. P. Levilain, J. Choppin, and M. C. Dokhelar. 1996. Among all human T-cell leukemia virus type 1 proteins, Tax, polymerase, and envelope proteins are predicted as preferential targets for the HLA-A2-restricted cytotoxic T-cell response. J. Virol. 70:4919-4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Porter, R. R., and K. B. M. Reid. 1978. The biochemistry of complement. Nature 275:699-704. [DOI] [PubMed] [Google Scholar]
- 61.Roberts, L. 2004. Polio. Health workers scramble to contain African epidemic. Science 305:24-25. [DOI] [PubMed] [Google Scholar]
- 62.Sagara, Y., Y. Inoue, H. Shiraki, A. Jinno, H. Hoshino, and Y. Maeda. 1996. Identification and mapping of functional domains on human T-cell lymphotropic virus type 1 envelope proteins using synthetic peptides. J. Virol. 70:1564-1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schneider-Schaulies, J. 2000. Cellular receptors for viruses: links to tropism and pathogenesis. J. Gen. Virol. 81:1413-1429. [DOI] [PubMed] [Google Scholar]
- 64.Slattery, J. P., G. Franchini, and A. Gessain. 1999. Genomic evolution, patterns of global, dissemination, and interspecies transmission of human and simian T-cell leukemia/lymphotropic viruses. Genome Res. 9:525-540. [PubMed] [Google Scholar]
- 65.Sodroski, J. G. 1999. HIV-1 inhibitors in the side pocket. Cell 99:243-246. [DOI] [PubMed] [Google Scholar]
- 66.Sommerfelt, M. A. 1999. Retrovirus receptors. J. Gen. Virol. 80:3049-3064. [DOI] [PubMed] [Google Scholar]
- 67.Sommerfelt, M. A., and R. A. Weiss. 1990. Receptor interference groups of 20 retroviruses plating on human cells. Virology 176:58-69. [DOI] [PubMed] [Google Scholar]
- 68.Stanley, M. A. 2006. Human papillomavirus vaccines. Rev. Med. Virol. 16:139-149. [DOI] [PubMed] [Google Scholar]
- 69.Stevenson, M., C. Meier, A. M. Mann, N. Chapman, and A. Wasiak. 1988. Envelope glycoprotein of HIV induces interference and cytolysis resistance in CD4+ cells: mechanism for persistence in AIDS. Cell 53:483-496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Taylor, G. P., and the HTLV-1 European Research Network. 1999. The epidemiology and clinical impact of HTLV infections in Europe. AIDS Rev. 1:195-204. [Google Scholar]
- 71.Reference deleted.
- 72.Trejo, S. R., and L. Ratner. 2000. The HTLV receptor is a widely expressed protein. Virology 268:41-48. [DOI] [PubMed] [Google Scholar]
- 73.Wallin, M., M. Ekstrom, and H. Garoff. 2004. Isomerization of the intersubunit disulphide-bond in Env controls retrovirus fusion. EMBO J. 23:54-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wei, X., J. M. Decker, S. Wang, H. Hui, J. C. Kappes, X. Wu, J. F. Salazar-Gonzalez, M. G. Salazar, J. M. Kilby, M. S. Saag, N. L. Komarova, M. A. Nowak, B. H. Hahn, P. D. Kwong, and G. M. Shaw. 2003. Antibody neutralization and escape by HIV-1. Nature 422:307-312. [DOI] [PubMed] [Google Scholar]
- 75.Welsh, R. M., F. C. Jensen, N. R. Cooper, and M. B. A. Oldstone. 1976. Inactivation and lysis of oncornaviruses by human serum. Virology 74:432-440. [DOI] [PubMed] [Google Scholar]
- 76.Welsh, R. M., N. R. Cooper, F. C. Jensen, and M. B. A. Oldstone. 1975. Human serum lyses RNA tumor viruses. Nature 257:612-614. [DOI] [PubMed] [Google Scholar]
- 77.Zwick, M. B., A. F. Labrijn, M. Wang, C. Spenlehauer, E. O. Saphire, J. M. Binley, J. P. Moore, G. Stiegler, H. Katinger, D. R. Burton, and P. W. Parren. 2001. Broadly neutralizing antibodies targeted to the membrane-proximal external region of human immunodeficiency virus type 1 glycoprotein gp41. J. Virol. 75:10892-10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zwick, M. B., R. Jensen, S. Church, M. Wang, G. Stiegler, R. Kunert, H. Katinger, and B. R. Burton. 2005. Anti-human immunodeficiency virus type 1 (HIV-1) antibodies 2F5 and 4E10 require surprisingly few crucial residues in the membrane-proximal external region of glycoprotein gp41 to neutralize HIV-1. J. Virol. 79:1252-1261. [DOI] [PMC free article] [PubMed] [Google Scholar]