

Abstract
Hepatitis C virus (HCV)-specific T-cell responses are rarely detected in peripheral blood, especially in the presence of human immunodeficiency virus (HIV) coinfection. Based on recent evidence that T-regulatory cells may be increased in chronic HCV, we hypothesized that functional blockade of regulatory cells could raise HCV-specific responses and might be differentially regulated in the setting of HIV coinfection. Three groups of subjects were studied: HCV monoinfected, HCV-HIV coinfected, and healthy controls. Frequencies of peripheral T cells specific for peptides derived from HCV core, HIV type 1 p24, and recall antigens were analyzed by gamma interferon (IFN-γ) enzyme-linked immunospot assay. HCV-specific T-cell responses were very weak in groups with HCV and HCV-HIV infections. Addition of blocking antibodies against transforming growth factor β1 (TGF-β1), -2, and -3 and interleukin-10 specifically increased the HCV-specific T-cell responses in both infected groups; however, this increase was attenuated in the group with HCV-HIV coinfection compared to HCV infection alone. No increase in recall antigen- or HIV-specific responses was observed. Flow cytometric sorter analysis demonstrated that regulatory-associated cytokines were produced by HCV-specific CD3+CD8+CD25− cells. Enhancement of the IFN-γ effect was observed for both CD4 and CD8 T cells and was mediated primarily by TGF-β1, -2, and -3 neutralization. In conclusion, blockade of TGF-β secretion could enhance peripheral HCV-specific T-cell responses even in the presence of HIV coinfection.
Hepatitis C virus (HCV) is a major health problem worldwide. HCV infection causes chronic hepatitis in up to 80% of infected adults and is associated with steatosis, cirrhosis, and hepatocellular carcinoma. The presence of vigorous and multispecific peripheral immune responses to HCV proteins by both CD8+ and CD4+ T lymphocytes is associated with virus clearance and disease resolution in acute hepatitis C (12, 18, 36, 44). In contrast, frequencies of HCV-specific T cells are very low in the periphery of subjects with chronic hepatitis C (32, 33, 44, 51). This paucity of T-cell responses in the setting of chronic HCV is exquisitely specific to HCV, as responses against recall antigens are preserved (3, 44, 48). Furthermore, compared to other chronic viral infections, such as human immunodeficiency virus (HIV) infection, the magnitude of T-cell responses to HCV is low (2, 31, 49). While HCV-specific T cells are relatively enriched in the liver parenchyma (25, 30, 35), a relative weakness, at least of CD4+ T cell responses, is linked to more rapid disease progression (27). The reasons for the low frequency of HCV-specific T-cell responses in the chronic phase of HCV infection are poorly understood but may involve exhaustion (28, 47), apoptosis of activated T cells (24, 26, 46), or failure of antigen presentation early in infection (5, 37).
Recently, there has been a revival of interest in regulatory T (Treg) cells, which are a heterogeneous population of cells including CD4+CD25+, Tr1, and Th3 cells, but also CD8+ cells (14), γδ T cells, and NK T cells, all of which have been demonstrated to suppress T cells. The potential role of Treg in HCV infection is just beginning to be defined. The majority of published studies have focused on CD4+CD25+ Treg cells (7, 8, 39, 42). HCV persistence has been associated with increased circulating CD4+CD25+ T cells, and their depletion from peripheral blood mononuclear cells (PBMC) enhances the T-cell in vitro capacity to proliferate or to secrete gamma interferon (IFN-γ) in response to HCV and other viruses, such as influenza virus (7), cytomegalovirus (CMV), and Epstein-Barr virus (EBV) (39). Most of these studies also suggested a contact-dependent action of CD4+CD25+ cells (7, 8, 39), while to date, the role of the regulatory-associated cytokines transforming growth factor β (TGF-β) and interleukin-10 (IL-10) in human Treg cell function in HCV infection has not been conclusively defined (9). Some studies found that the suppressive effect of CD4+CD25+ Treg cells on the peripheral T-cell proliferation is TGF-β and IL-10 independent (7, 39). We and others have found that HCV proteins or peptides induced the production of IL-10 by PBMC (21, 22, 34, 47) and liver-infiltrating lymphocytes (1, 20) from patients with chronic HCV, but the functional impact was unclear.
In the present work, we explored the involvement of the regulatory-associated cytokines TGF-β and IL-10 in blocking the peripheral blood HCV-specific T-cell production of IFN-γ. We chose to study subjects with HCV-HIV coinfection and HCV monoinfection to see if the regulatory-cytokine blockade has similar effects on HCV-specific IFN-γ effector responses in both groups, since HIV causes alterations in effector and regulatory-T-cell functions. We also characterized the type of HCV-specific T cells secreting regulatory-associated cytokines. We found that the suppression of HCV core-specific IFN-γ T-cell production in the peripheral blood of subjects with chronic HCV infection was primarily mediated by TGF-β produced by antigen-specific CD8+CD25− T cells.
MATERIALS AND METHODS
Study subjects and samples.
Blood samples were received from 21 subjects with chronic HCV infection who were undergoing routine diagnostic evaluation prior to anti-HCV treatment. Eleven subjects were HCV monoinfected, and 12 subjects were HCV-HIV coinfected (Table 1). All were HCV RNA positive, and no subjects had clinical liver decompensation. Persons with other forms of liver disease, including those caused by hepatitis B virus and alcohol, and other immunosuppressive conditions, including malignancy, chronic renal failure with hemodialysis, organ transplant, or other comorbid diseases requiring immunosuppressive therapy, were excluded. All the coinfected subjects were under treatment for HIV infection. Blood samples were also received from eight healthy individuals. The protocol was reviewed by the Investigational Review Boards of the University of Cincinnati College of Medicine and the Beth Israel Deaconess Medical Center, and all subjects gave informed consent for the collection of samples. PBMC were isolated from EDTA anticoagulated blood with Ficoll-Paque Plus (Amersham Bioscience, Piscataway, NJ) density gradient centrifugation and cryopreserved for later use.
TABLE 1.
Subject demographics
| Demographic | Value
|
P | |
|---|---|---|---|
| HCV monoinfected (n = 11) | HCV-HIV coinfecteda (n = 12) | ||
| Age, median (range) (yr) | 45 (35-61) | 46 (25-61) | NSc |
| Raceb | 6C; 2AA; 2H; 1 other | 7C; 2AA; 1H | NS |
| Male/female | 9/2 | 9/3 | NS |
| CD4+ cell count, median (range) (cells/mm3) | NDd | 540 (104-748) | |
| HIV RNA, median (range) (copies/ml) | NDd | <50 (<50-10,343) | |
| HCV RNA, median (range) (103 IU/ml) | 240 (22-6,110) | 1,600 (240-39,000) | 0.06 |
| HCV genotype (no.) | 1 (10), 3 (1) | 1 (12) | NS |
All coinfected persons were treated for HIV infection.
C, Caucasian; AA, African American; H, Hispanic.
NS, not significant.
ND, not done.
Antigens.
Three sets of synthetic peptides (AIDS Reagent Program, NIH) were used. Set 1 consisted of 29 18-mer peptides spanning the entire HCV core region derived from HCV type 1a strain H77 (10 μg/ml each). Set 2 consisted of 58 15-mer peptides covering the HIV gag-p24 region derived from HIV type 1 consensus B gag (2 μg/ml each). Each set of HCV and HIV peptides was split into three pools. For analysis, the results from the pools were summed as total HCV- or HIV-specific responses. Set 3 (CEF), a pool of 23 major histocompatibility complex class I restricted T-cell 11- to 18-mer peptides from human CMV, EBV, and influenza virus, was used as a control (2 μg/ml each).
IFN-γ and IL-10 ELISPOT assays.
Enzyme-linked immunospot (ELISPOT) assays were performed as previously described (4). Capture and detection antibodies were used at final concentrations of 5 μg/ml and 0.2 μg/ml for IFN-γ (Endogen, Woburn, MA) and 10 μg/ml and 1 μg/ml for IL-10 (Mabtech). PBMC (2 × 105 cells per well in triplicate) were cultured for 20 h with antigens and in the presence or absence of blocking antibodies or their isotype controls. Positive control wells consisted of phytohemagglutinin (5 μg/ml; Sigma-Aldrich, St. Louis, MO). Negative control wells consisted of buffer alone. Antigen-specific spot-forming cell (SFC) frequencies were measured on an automated microscope (Zeiss, Munich, Germany) and expressed after background subtraction (the number of SFCs observed with buffer medium alone). For IFN-γ ELISPOT assays, results were considered positive if a minimum of 50 SFCs/106 cells were detected above background. This threshold was chosen as more than 3 standard deviations above any response observed in the peripheral blood of healthy controls (Fig. 1).
FIG. 1.

IFN-γ ELISPOT assay against HCV core peptides in the presence of isotype controls and the blocking Abs anti-TGF-β and anti-IL-10. Each line corresponds to a subject. The results are shown as SFCs/106 PBMC over background (unstimulated cells). After the addition of blocking Abs, significant enhancement of median HCV-specific T-cell responses was observed in both groups of subjects with HCV monoinfection (n = 11) (P = 0.003) and with HCV-HIV coinfection (n = 12) (P = 02). Similarly, the number of responders to HCV, defined as greater than 50 SFCs/106 cells, was increased from two to nine HCV responders in the HCV-monoinfected group and from one to four responders in the HCV-HIV-coinfected group. No controls had positive responses to HCV. HCV-specific T-cell responses were not different between the two groups, HCV monoinfected and HCV-HIV coinfected, when isotype controls were added, while after the addition of blocking Abs, these responses became significantly higher in the HCV-monoinfected group (P = 0.05).
Blocking assays.
Blocking assays were performed in parallel with the standard ELISPOT assays. Blocking monoclonal antibodies (MAbs) anti-IL-10 and anti-TGF-β1, -2, and -3 (clone DII) or immunoglobulin G1 (IgG1) and IgG2b isotype controls (R&D Systems, Minneapolis, MN) were simultaneously added to the wells at the optimized concentration (10 μg/ml).
Intracytoplasmic cytokine staining.
PBMC were cultured for 6 h with antigens in the presence of Golgi Plug (BD Biosciences Pharmingen, San Diego, CA) added 1 h after the initiation of stimulation. Thereafter, the cells were surface stained with anti-CD3-phycoerythrin-cyanin 5 (PE-Cy5) (BD Biosciences Pharmingen, San Diego, CA), anti-CD4 energy-coupled dye-phycoerythrin-texas red (ECD) (Beckman Coulter, Miami, FL), anti-CD8-phycoerythrin-cyanin 7 (PE-Cy7) (BD Biosciences Pharmingen, San Diego, CA), and anti-CD25-fluorescein isothiocyanate (FITC) (BD Biosciences Pharmingen, San Diego, CA); fixed and permeabilized (fixation/permeabilization solution kit; BD Biosciences Pharmingen, San Diego, CA); and then stained intracellularly with anti-IFN-γ-PE (BD Biosciences Pharmingen, San Diego, CA), anti-TGF-β-PE (Biotest Diagnostics, Denville, NJ), and anti-IL-10-PE (R&D Systems, Minneapolis, MN) MAbs or with an irrelevant isotype-matched control. CD3, CD4, and CD8 surface-stained PBMC were also intracellularly stained with anti-TGF-β-PE and anti-Foxp3-FITC (eBioscience, San Diego, CA). Five-color flow cytometric analysis was performed using an FC500 flow cytometer (BD Biosciences, San Diego, CA); 150,000 events were acquired, 50,000 to 100,000 of which were gated viable cells. The results were expressed as percentages of cytokine-positive cells after background subtraction. Positive controls were obtained by stimulating cells with phorbol myristate acetate (50 ng/ml) and ionomycin (1 μg/ml) (Sigma-Aldrich, St. Louis, MO) or in the presence of both fixed CD3 and soluble CD28 (BD Biosciences Pharmingen, San Diego, CA) and were positive for all samples. The negative control consisted of cells with buffer medium alone.
Depletion assays.
CD25 or CD8 depletion was performed using magnetic Dynabeads (Dynal Biotech) following the kit protocol, which allowed depletion of ≥99% of target cells. In order to keep constant the number of effector T cells producing IFN-γ when comparing results before and after CD8 depletion, in some cases adherent cells were separated by 1 hour of culture to be used as accessory/antigen-presenting cells and were then added back at equal numbers to the untreated or CD8-depleted nonadherent effector cells.
Statistical analysis.
ELISPOT results before and after the addition of blocking MAbs for each subject were compared by a Wilcoxon signed rank test. Results from HCV-HIV and HCV groups were compared using the Mann-Whitney U test. Spearman rank tests were performed for the correlations. Calculations were performed using STATview SAS PC (Cary, NC; version 6.0l), and P values of ≤0.05 were considered significant.
RESULTS
Subject characteristics.
Subject characteristics at study entry are shown in Table 1. All of the coinfected persons were under treatment for HIV infection, and most had undetectable HIV viral loads (VLs) (median, <50 copies/ml; range, <50 to 10,343). CD4+ T-cell counts were done only for the coinfected group and were relatively high in this cohort (median, 540/mm3; range, 104 to 748). As expected, there was a trend toward higher HCV VLs in the coinfected group, with a median HCV VL of >1,600 × 103 IU/ml versus 240 × 103 IU/ml in the HCV-alone group (P = 0.06).
Blocking regulatory cytokines increases peripheral HCV-specific T-cell responses, even in the presence of HIV coinfection.
PBMC were first analyzed by ELISPOT assay for the frequency of T cells producing IFN-γ in response to HCV core, HIV p24, and CEF peptides in the presence and absence of both anti-TGF-β1, -2, and -3 and anti-IL-10 MAbs or their appropriate isotype controls (Fig. 1 and 2). Without HCV stimulation, no effect of TGF-β and IL-10 blockade on IFN-γ T-cell production was observed, and the median numbers of SFC/106 PBMC observed with media, blocking antibodies (Abs) or their isotype controls were similarly low (5, 4, and 5, respectively) (data not shown). The results observed with antigen stimulation for each condition were expressed after subtraction of the number of SFCs observed with no antigen stimulation (i.e., background). No significant difference was observed between the results for cells with media and cells with isotype controls (data not shown), so the results are shown for cells stimulated in the presence of the isotype controls. In subjects with chronic hepatitis, in the absence of blocking Abs, HCV-specific T-cell responses were very weak in both groups (Fig. 1). In subjects with HCV monoinfection, the median was 25 SFCs/106 PBMC (range, 5 to 100), with 2/11 subjects having a positive response (≥50 SFCs/106 PBMC), and in those with HCV-HIV coinfection, the median value was 12 SFCs/106 PBMC (range, 0 to 237), with 1/12 subject having a positive response. Addition of blocking Abs increased the HCV-specific T-cell IFN-γ responses in both groups to a median of 150 SFCs/106 PBMC in those with HCV monoinfection (range, 11 to 333) (P = 0.003) and 36 SFCs/106 PBMC in those with HCV-HIV coinfection (range, 0 to 271) (P = 0.02); there were >50 SFCs/106 PBMC in 9/11 and 4/12 subjects, respectively (Fig. 1). Interestingly, the amplifying effect appeared to be more pronounced in HCV-monoinfected subjects: HCV-specific T-cell responses were not different between the two groups when isotype controls were added, while after the addition of blocking Abs, these responses became significantly higher in the HCV-monoinfected group (P = 0.05).
FIG. 2.

IFN-γ ELISPOT assays against CEF (top) and HIV p24 (bottom) peptides in the presence of isotype controls and anti-TGF-β and anti-IL-10 Abs. Each line corresponds to a subject. The results are shown as SFCs/106 PBMC over background (unstimulated cells). No enhancement of median T-cell responses toward non-HCV antigens was observed after the addition of blocking Abs. Neither controls nor subjects with HCV monoinfection had positive responses to HIV peptides.
In subjects who responded to CEF, specific T cells were detected with high frequencies in all three groups and addition of blocking Abs did not raise the response (Fig. 2). Of note, 6/12 coinfected subjects responded vigorously to CEF (>1,000 SFCs/106 PBMC) compared to the other two groups. This is consistent with our prior experience in another cohort (4) and could be related either to CMV reactivation or more systematic use of influenza virus vaccination in HIV patients. Healthy controls did not have responses against HCV in the presence of isotype or blocking Abs (Fig. 1), and only subjects with HIV infection had detectable HIV-specific responses (Fig. 2). Similarly, addition of blocking MAbs did not significantly increase the T-cell responses to HIV in the group of five HIV-coinfected subjects studied, although HIV-specific responses above 50 SFCs/106 PBMC were found in three subjects before addition and in all five after addition of blocking Abs (Fig. 2).
CD3+CD8+ and CD3+CD4+ T-cell subsets both contributed to HCV-specific production of IFN-γ upon regulatory-cytokine blockade.
In order to determine which cells produced IFN-γ in response to HCV core 18-mer peptides, we performed further experiments in subjects who had adequate PBMC to determine the effect of blocking MAb on intracellular cytokine staining. CD3+CD4+ and CD3+CD8+ cells were analyzed for the production of IFN-γ against a pool of HCV core peptides before and after addition of both anti-TGF-β and anti-IL-10 or their isotype controls. Dot plots for a representative subject with HCV monoinfection are shown (see Fig. 3). Staining with the corresponding PE-conjugated isotype control was less than 0.04% for each stimulation condition (not shown). The frequency of HCV-specific cells was calculated as the percentage of cells responding to HCV peptides minus the background observed with medium alone. The frequency of HCV-specific IFN-γ-producing cells increased from 0.01 to 0.27% after addition of blocking MAbs within CD3+CD4+ cells and from 0 to 0.44% within CD3+CD8+ cells. This was consistent with a large increase observed by ELISPOT assay for this subject: HCV-specific responses of 5 SFCs/106 PBMC before the addition of blocking MAbs, 6 SFCs/106 PBMC with isotype controls, and 333 SFCs/106 PBMC with blocking MAbs. The results for six subjects are shown in Table 2 and demonstrate that both CD3+CD4+ and CD3+CD8+ cells produced IFN-γ upon stimulation of the cells with synthetic peptides, as expected.
FIG. 3.

Frequencies of CD4 and CD8 T cells producing IFN-γ, by intracellular staining, in response to medium alone or to the pool of HCV core peptides before and after the addition of the blocking Abs anti-TGF-β and anti-IL-10 or the appropriate isotype controls in one representative subject, 002P (HCV monoinfected). After 6 h of appropriate stimulation (in the presence of Golgi Plug added 1 h after the start of stimulation), PBMC were stained with anti-CD3-PE-Cy5, anti-CD4-ECD, and anti-CD8-PE-Cy7 and then fixed/permeabilized for intracellular staining with anti-IFN-γ-PE. The dot plots are gated on CD3+ cells. The corresponding isotype control staining was <0.04 (not shown).
TABLE 2.
Both CD4+ and CD8+ T cells produced IFN-γ in response to HCV core peptides after blocking Treg-associated cytokines TGF-β and IL-10
| Subject | Status | Frequencya
|
|||
|---|---|---|---|---|---|
| CD3+ CD4+ IFN-γ+
|
CD3+ CD8+ IFN-γ+
|
||||
| Ab | Isotype | Ab | Isotype | ||
| 002B | HCV-HIV | 0.20 | 0.02 | 1.48 | 0.09 |
| 002P | HCV | 0.27 | 0.01 | 0.44 | 0.07 |
| 0090 | HCV | 0.11 | 0.04 | 0.16 | 0.01 |
| 0055 | HCV | 0.42 | 0.06 | 0.07 | 0.01 |
| 0074 | HCV | 1.74 | 0.00 | 0.77 | 0.06 |
| 00-Q | HCV | 0.36 | 0.09 | 0.32 | 0.00 |
Results are expressed as percent frequencies of CD3+CD4+ and CD3+CD8+ cells that secret IFN-γ in response to HCV, calculated as percent expression following stimulation with HCV minus percent expression of cells in medium alone, in the presence of both Abs (anti-TGF-β and anti-IL-10) and the isotype controls (isotype).
Regulatory-associated cytokines were predominantly produced by CD3+CD8+CD25− cells in response to HCV.
Since the increase in IFN-γ T-cell responses after the blockade of regulatory cytokines was specific for HCV, as increases in the CEF pool or preexisting HIV responses were not seen, we hypothesized that HCV-specific Treg cells were responsible for this increase. HCV-specific cells producing TGF-β and IL-10 were further characterized by flow cytometric analysis for CD3+, CD4+, CD8+, and CD25+ expression from six monoinfected and five coinfected subjects in whom an increase in HCV-specific IFN-γ production was observed when antibodies blocking regulatory cytokines were added. Interestingly, regulatory-associated cytokines were produced in response to HCV core exclusively by CD8+CD25− T cells from 10/11 subjects (Table 3 and Fig. 4B). The exception was PBMC from a single HCV-HIV-coinfected subject (002B), who produced regulatory-associated cytokines exclusively from HCV-specific CD3+CD4+CD25+ cells (Table 3).
TABLE 3.
Treg-associated cytokines predominantly produced by CD8+ T cells in response to HCV core peptides
| Subject | Status | Frequencya
|
|||
|---|---|---|---|---|---|
| CD3+CD8+CD25−
|
CD3+CD4+CD25+
|
||||
| TGF-β± | IL-10+ | TGF-β± | IL-10+ | ||
| 002P | HCV | 0.07 | 0 | 0 | 0 |
| 0090 | HCV | 0.36 | 0.31 | 0.02 | 0.01 |
| 0055 | HCV | 0.08 | 0.01 | 0 | 0 |
| 0074 | HCV | 0.17 | 0.78 | 0 | 0 |
| 00-Q | HCV | 0.38 | 0 | 0 | 0 |
| 9136 | HCV | 0.28 | 0 | 0 | 0.01 |
| 002B | HCV-HIV | 0 | 0 | 0.28 | 0.16 |
| 0186 | HCV-HIV | 0.52 | 0.36 | 0 | 0 |
| 0174 | HCV-HIV | 0.82 | 0.74 | 0 | 0 |
| 002U | HCV-HIV | 0.05 | 0 | 0 | 0 |
| 0992 | HCV-HIV | 0.08 | 0 | 0 | 0 |
Results are expressed as frequencies of TGF-β+CD25− HCV-specific cells within CD3+CD8+ cells and IL-10+CD25+ within CD3+CD4+ cells, after background (medium) subtraction. Neither CD3+CD8+CD25+ nor CD3+CD4+CD25− cells produced Treg cytokines (not shown).
FIG. 4.

Frequencies of T cells producing TGF-β or IL-10, in response to medium alone or to HCV core peptides, by intracellular staining in one representative subject, 0090 (HCV monoinfected). After 6 h of appropriate stimulation (in the presence of Golgi Plug added 1 h after the start of stimulation), PBMC were stained with anti-CD3-PE-Cy5, anti-CD4-ECD, anti-CD8-PE-Cy7, and anti-CD25-FITC and then fixed/permeabilized for intracellular staining with anti-TGF-β-PE, anti-IL-10-PE, or isotype-PE. The dot plots are gated on (A) CD3+CD4+ and (B) CD3+CD8+ cells. Neither CD3+CD8+CD25+ nor CD3+CD4+ cells produced regulatory cytokines.
The enhancement of the HCV-specific T-cell response is mediated by anti-TGF-β.
To determine whether either or both cytokines were critical for the blocking effect, IFN-γ ELISPOT assays were repeated by adding one of each blocking MAb, anti-TGF-β or anti-IL-10, or its isotype control with subjects whose HCV-specific responses were raised by the addition of both anti-cytokine MAbs and who had sufficient cells available. The blocking effect was clearly mediated by TGF-β and was IL-10 independent in six out of seven patients tested (Fig. 5), and the anti-TGF-β blocking effect remained predominant with the remaining patient (subject 0090).
FIG. 5.

IFN-γ ELISPOT assay against HCV core peptides in the presence of medium alone or either both or one of each blocking Ab (anti-TGF-β and/or anti-IL-10) or their corresponding isotype controls. The bars represent the results as SFCs/106 PBMC over background (unstimulated cells), shown for five HCV-monoinfected and two HCV-HIV-coinfected subjects for each stimulation condition. A responder was defined as greater than 50 SFCs/106 PBMC over background, as shown by the horizontal dotted lines. The blocking effect was mediated by TGF-β and was IL-10 independent in six out of seven subjects.
We also performed IL-10 ELISPOT assays for two subjects who had sufficient available cells: 0186 (HCV-HIV coinfected) and 0055 (HCV monoinfected). Addition of TGF-β blocking MAbs also increased the HCV-specific T-cell IL-10 responses in both subjects (Fig. 6), suggesting that blockade of regulatory cytokines can impact the secretion of cytokines other than IFN-γ. However, larger studies would need to be done to draw conclusions on this point.
FIG. 6.

IL-10 ELISPOT assay against the three HCV core pools, P1, P2, and P3, and CEF in the presence of medium, anti-TGF-β blocking MAb, or the corresponding isotype control IgG1. The bars represent the results as SFCs/106 PBMC over background (unstimulated cells), shown for two subjects, 0055 (HCV monoinfected) and 0186 (HIV-HCV coinfected). Addition of the blocking Ab anti-TGF-β enhanced the IL-10 response to HCV pools, but not to CEF.
CD3+CD8+ cells producing TGF-β are Foxp3 negative and are specific for HCV.
In two subjects, 0186 (HCV-HIV coinfected) and 9136 (HCV monoinfected), we further characterized HCV-specific cells producing TGF-β by flow cytometric analysis for CD3+, CD4+, CD8+, and Foxp3 expression, a transcription factor currently considered as perhaps the optimal marker of classic Treg. Interestingly, CD3+CD8+ cells producing TGF-β in response to HCV were Foxp3 negative (Fig. 7).
FIG. 7.

Frequencies of T cells producing TGF-β in response to medium alone or HCV core peptides by intracellular staining in two subjects, 0186 (HCV-HIV coinfected) and 9136 (HCV monoinfected). After 6 h of appropriate stimulation (in the presence of Golgi Plug added 1 h after the start of stimulation), PBMC were stained with anti-CD3-PE-Cy5, anti-CD4-ECD, and anti-CD8-PE-Cy7 and then fixed/permeabilized for intracellular staining with anti-Foxp3-FITC and anti-TGF-β-PE or their isotype controls. The dot plots are gated on (A) CD3+CD4+ and (B) CD3+CD8+ cells. Of note, the staining rates for all the isotype controls were low (<0.02%) (not shown). Only CD4+ cells expressed Foxp3, but they did not produce TGF-β in response to HCV (A). CD3+CD8+ cells producing TGF-β in response to HCV were Foxp3 negative (B).
We also examined the production of TGF-β in response to CEF peptides as an additional control for the HCV specificity of TGF-β production. As shown in Fig. 8, TGF-β production by CD3+CD8+ cells was clearly observed in response to HCV, but not to CEF.
FIG. 8.

Frequencies of T cells producing TGF-β in response to medium alone, to HCV core peptides, or to CEF peptides by intracellular staining in three subjects: 0186 and 0992 with HCV/HIV coinfection and 9136 with HCV monoinfection. After 6 h of appropriate stimulation (in the presence of Golgi Plug added 1 h after the start of stimulation), PBMC were stained with anti-CD3-PE-Cy5, anti-CD4-ECD, and anti-CD8-PE-Cy7 and then fixed/permeabilized for intracellular staining with anti-TGF-β-PE or isotype-PE. The dot plots are gated on CD3+CD8+ cells. CD3+CD4+ cells did not produce the regulatory cytokine, and the isotype controls were low (<0.02). TGF-β production was observed in response to HCV, but not to CEF.
The overall production of TGF-β production by the CD8 cells is shown in Fig. 9 and is significantly higher in response to HCV stimulation than in medium alone (P = 0.01) (Fig. 9A). Of note, there was no significant difference between TGF-β-PE and Iso-PE staining in medium alone (P = 0.12), while after HCV stimulation, TGF-β-PE staining was significantly higher than Iso-PE (P = 0.01) (Fig. 9B). No correlation was observed with HCV viremia, although the sample size of the study was too small to draw definitive conclusions.
FIG. 9.
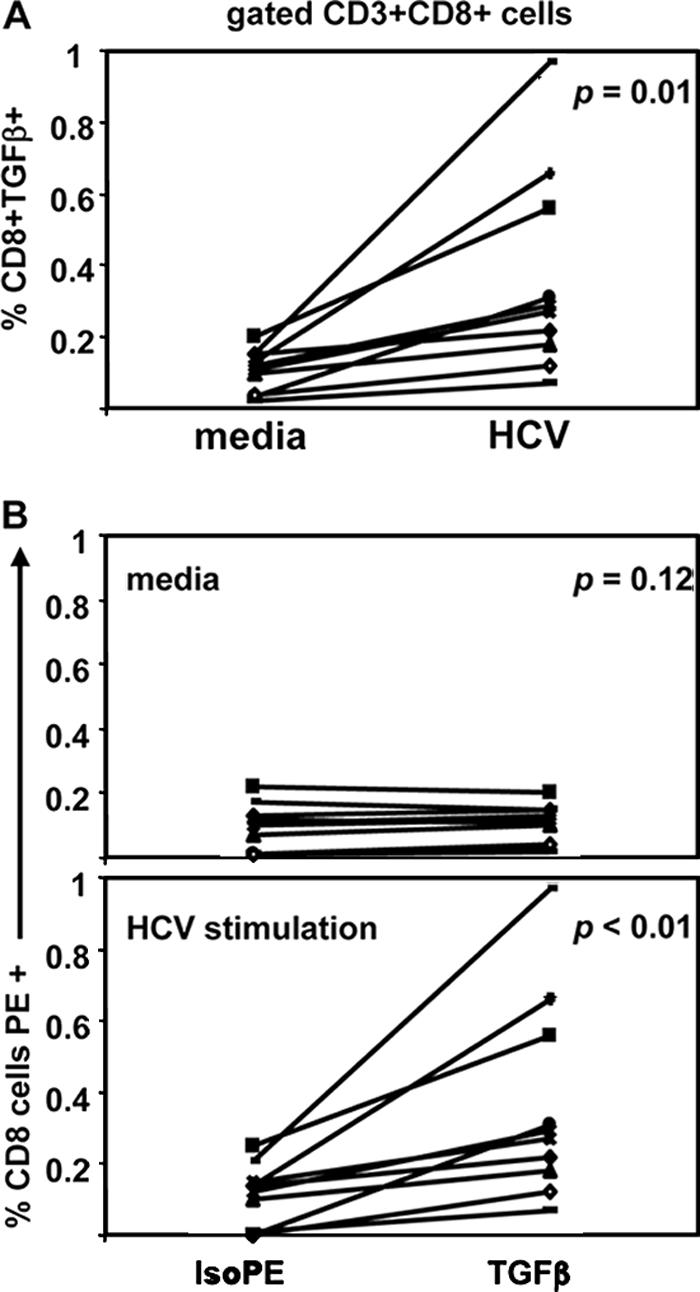
Overall frequencies of CD3+CD8+CD25− cells producing TGF-β by intracellular staining in 10 studied subjects. Each line corresponds to a subject. TGF-β production by CD8 cells was significantly higher in response to HCV stimulation than in medium alone (P = 0.01) (A). There was no significant (P = 0.12) difference between TGF-β-PE and Iso-PE staining in medium alone, while after HCV stimulation, TGF-β-PE staining was significantly (P = 0.01) higher than Iso-PE (B).
Effects of CD8 or CD25 depletion on HCV-specific IFN-γ T-cell responses.
Depletion experiments were performed on the remaining cells from two available subjects in whom TGF-β blockade raised the HCV-specific IFN-γ response (0186, HCV-HIV coinfected, and 00-Q, HCV monoinfected). Depletion of CD25+ cells did not increase the HCV-specific IFN-γ ELISPOT response (data not shown). Depletion of CD8+ cells diminished the HCV-specific IFN-γ ELISPOT response, as well as the IFN-γ ELISPOT response to CEF peptides (data not shown). These results were expected, since CD8+ cell depletion will deplete not only CD8 cells producing TGF-β, but also effector cells producing IFN-γ. Given the lack of a definitive surface marker, we could not specifically deplete functional T cells secreting TGF-β. However, CD8+ cell depletion was also followed by flow cytometric cytokine analysis in the same two subjects, in whom CD4 cells participated in IFN-γ production after regulatory blockade. In an additional subject, 9136 (HCV monoinfected), in order to keep constant the number of effector T cells producing IFN-γ, the same experiment was performed once monocytic antigen-presenting cells were removed via adherence and then added back at equal numbers to the PBMC and CD8-depleted cells. Interestingly, in all these subjects, CD8+ cell depletion increased the HCV-specific intracellular IFN-γ production in CD4 cells (Fig. 10A and B).
FIG. 10.

Effects of CD8 depletion on IFN-γ responses. (A) Frequencies of CD4-producing IFN-γ, by intracellular staining, in response to medium or to the pool of HCV core peptides before and after CD8 depletion in one coinfected subject, 0186. After 6 h of appropriate stimulation (in the presence of Golgi Plug added 1 h after the start of stimulation), PBMC or CD8− cells were stained with anti-CD3-PE-Cy5 and anti-CD4-ECD and then fixed/permeabilized for intracellular staining with anti-IFN-γ-PE and isotype-PE. The dot plots are gated on CD3+CD4+ cells. (B) Frequencies of CD4-producing IFN-γ, by intracellular staining, in response to the pool of HCV core peptides before and after CD8 depletion in three subjects. In subject 9136, the same experiment was performed once antigen-presenting cells were removed and then added back at equal numbers to the PBMC and CD8-depleted cells. The bars represent the results as frequencies of CD4+IFN-γ+ cells gated on CD3+CD4+ cells after background subtraction (unstimulated cells, or medium). The CD8 depletion increased intracellular IFN-γ production in CD4 cells in response to HCV.
In summary, HCV-specific T-cell responses could be markedly and specifically augmented by the neutralization of regulatory cytokines, primarily TGF-β, and regulatory cytokines were produced by HCV-specific CD8+CD25− T cells.
DISCUSSION
Our results provide evidence that functional blockade of regulatory-associated cytokines enhances peripheral HCV-specific T-cell responses, even in the presence of HIV coinfection, although the degree of enhancement was attenuated in persons with HCV-HIV coinfection. We also found that this suppressive activity is primarily mediated by TGF-β, a regulatory-associated cytokine that was predominantly produced in response to stimulation with HCV core peptides by CD8+CD25− T cells.
Persistent HCV infection is associated with very low frequencies of peripheral blood HCV-specific T cells, especially when measured by functional assays, such as lymphoproliferation and ELISPOT assays (44). Our results, together with recent studies (7, 39), suggest that HCV-specific T cells are not completely absent from the periphery of subjects with chronic HCV infection but may be functionally suppressed by Treg cells. There are two previously described subsets of Treg cells, which differ in terms of specificity and effector mechanism: natural Treg cells that develop during the normal process of T-cell maturation in the thymus and adaptive Treg cells that might either develop from mature classical T cells or differentiate from the naturally occurring Treg cell subset under particular conditions of antigen exposure (6). Natural Treg cells, at least in in vitro models, have been shown to function via antigen-independent, contact-dependent, and cytokine-independent mechanisms (40), whereas cytokine-mediated suppression has been established for peripheral adaptive Treg cells in vivo (29). Currently, the most studied marker for Treg cells is CD25, which is also expressed on activated T cells. This surface marker is highly expressed on the natural Treg cells, but its expression on the adaptive Treg cells and other T cells is variable. Previous studies have found an increase in circulating CD4+ CD25+ T cells associated with HCV persistence (7, 8, 42). The suppressive activity of these circulating CD4+CD25+ Treg cells was found to be independent of Treg-associated cytokines and modulated responses to HCV, as well as other antigens (7, 39).
Recently, studies of mice demonstrated that CD8+ cells also contain Treg cells (14, 38). However these CD8+ Treg cells were not antigen specific and mediated the suppression of T-cell responses by IL-10 and not TGF-β (14). Similarly, non-antigen-specific CD8+ Treg cells have been seen in the peripheral blood of patients with chronic inflammatory diseases, such as multiple sclerosis (43), systemic lupus erythematosus (15), or systemic sclerosis (16), and in persons with chronic immune responses, such as HIV and HCV infections (17). Accapezzato and colleagues recently demonstrated antigen-specific CD8+ IL-10-secreting Treg cell lines in the livers of patients with chronic HCV infection (1). To our knowledge, there are no reports of HCV-specific CD8+ T cells producing TGF-β.
In our study, we have provided evidence of the existence of HCV-specific CD8+ T cells secreting TGF-β in the peripheral blood of subjects with chronic HCV infection. Although CD8+ T cells produced both TGF-β and IL-10 in response to HCV, antigen-specific functional assays showed that suppression of HCV-specific IFN-γ response was mediated by TGF-β and was IL-10 independent in most subjects. Accapezzato et al. showed that the addition of neutralizing anti-IL-10 antibodies abrogated the suppression of non-antigen-specific PBMC proliferation by IL-10-producing CD8+ liver-infiltrating lymphocytes (1). However, they found that the non-antigen-specific suppression was also exerted by some CD8+IL-10− liver-infiltrating lymphocytes, suggesting the involvement of further undefined CD8 T cells that could include those producing TGF-β. An inhibitory role for TGF-β1 in suppressing HCV-specific IFN-γ production by PBMC was recently proposed (8). However, the degree of enhancement was modest, and it was not clear which cell type produced TGF-β in this study. The degree of enhancement after TGF-β blockade found in our study could be due to the type of neutralizing TGF-β antibody used, with the previous study using anti-TGF-β1 while we used anti-TGF-β1, -2, and -3 to neutralize all three forms of TGF-β (β1, β2, and β3). A key difference from previous studies of peripheral Treg cells in chronic HCV infection was that we found that regulatory-cytokine-mediated suppression in chronically HCV-infected subjects was restricted to HCV-specific T cells and did not involve other virus-specific T-cells, such as those of CMV or HIV. Therefore, these results suggest that, besides the activation of naturally occurring or induced CD4+CD25+ Treg cells (7, 8, 39), chronic HCV infection may also lead to specific induction of HCV-specific CD8 Treg cells producing TGF-β. Whether this TGF-β might suppress other antigen-specific responses will be the subject of future studies.
Another finding of our study is that the amplifying effect of HCV-specific IFN-γ T-cell responses after the addition of neutralizing Treg cytokine antibodies was more pronounced in HCV-monoinfected subjects than in HIV-HCV-coinfected subjects. This could be explained by a compromised Treg cell activity in HIV coinfection and/or lack of sufficient effector cells. Depletion of T-regulatory cells in the course of HIV infection has been suggested by Eggena et al. and was shown to be more strongly associated with immune activation than with the absolute CD4 count (13). People with HCV-HIV coinfection have more rapid disease progression than is seen in HCV infection alone (19). Taken together, these observations raise the intriguing possibility that Treg cells may protect against liver disease progression.
Although the key role of CD4+ and CD8+ T-cell responses in spontaneous clearance of HCV is accepted based on chimpanzee and human studies (11, 23, 33, 41, 45), the role of T-cell responses in the pathogenesis of liver injury in chronic HCV is unknown. The classic model of the pathogenesis of viral hepatitis invokes the cytotoxic capability of CD8+ T cells. This model is supported by adoptive-transfer experiments with transgenic mice (10, 50). Recently, it has been shown in humans that the frequencies of HCV tetramer-specific IFN-γ-producing intrahepatic CD8+ cells were directly correlated with the hemagglutination inhibition score, whereas the frequency of IL-10-producing tetramer+ CD8+ T cells was inversely associated with the hemagglutination inhibition score (1). Similarly, a higher frequency of intrahepatic HCV-specific IL-10-producing CD4 cells was found in patients with HCV monoinfection than in those with HCV-HIV coinfection (20), who are characterized by more rapid progression to liver disease (6), although that study was too small and was not designed to permit correlations with the histologic outcome and other clinical parameters. Taken together, these studies suggest that Treg cells might play a role in minimizing chronic pathological responses by effector T cells.
In our study, the suppression of HCV-specific IFN-γ-producing peripheral T cells seemed to be primarily mediated by TGF-β, and we identified HCV-specific TGF-β production primarily in CD8+ T cells. TGF-β may be both antiproliferative (37) and profibrogenic (11, 23, 24, 31, 39, 45), so it will be important for future, larger studies to address the role of TGF-β-producing cells in the progression of liver disease and whether similar cells exist in liver-infiltrating lymphocytes. Clearly, prospective studies of individuals with differing degrees of fibrosis progression are needed to understand the roles that different populations of T cells, including Treg cells, have in HCV-related liver disease progression.
Acknowledgments
This work was supported by National Institutes of Health (NIH) grants AI49508 and U19 AI066313 to K.E.S. and M.J.K., DA14495-01 to C.S.G., and DK066917 to M.A.E., and a Harvard University Center for AIDS Research (CFAR) Scholar Award (grant P30 A060354) to N.A.
The authors have no commercial conflicts of interest.
Footnotes
Published ahead of print on 21 March 2007.
REFERENCES
- 1.Accapezzato, D., V. Francavilla, M. Paroli, M. Casciaro, L. V. Chircu, A. Cividini, S. Abrignani, M. U. Mondelli, and V. Barnaba. 2004. Hepatic expansion of a virus-specific regulatory CD8+ T cell population in chronic hepatitis C virus infection. J. Clin. Investig. 113:963-972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alatrakchi, N., V. Di Martino, V. Thibault, and B. Autran. 2002. Strong CD4 Th1 responses to HIV and hepatitis C virus in HIV-infected long-term non-progressors co-infected with hepatitis C virus. AIDS 16:713-717. [DOI] [PubMed] [Google Scholar]
- 3.Alatrakchi, N., V. Di Martino, V. Thibault, Y. Benhamou, C. Katlama, T. Poynard, and B. Autran. 2004. Decreased frequencies of virus-specific T helper type 1 cells during interferon alpha plus ribavirin treatment in HIV-hepatitis C virus co-infection. AIDS 18:121-123. [DOI] [PubMed] [Google Scholar]
- 4.Alatrakchi, N., C. S. Graham, Q. He, K. E. Sherman, and M. J. Koziel. 2005. CD8+ cell responses to hepatitis C virus (HCV) in the liver of persons with HCV-HIV coinfection versus HCV monoinfection. J. Infect. Dis. 191:702-709. [DOI] [PubMed] [Google Scholar]
- 5.Anthony, D. D., N. L. Yonkers, A. B. Post, R. Asaad, F. P. Heinzel, M. M. Lederman, P. V. Lehmann, and H. Valdez. 2004. Selective impairments in dendritic cell-associated function distinguish hepatitis C virus and HIV infection. J. Immunol. 172:4907-4916. [DOI] [PubMed] [Google Scholar]
- 6.Bluestone, J. A., and A. K. Abbas. 2003. Natural versus adaptive regulatory T cells. Nat. Rev. Immunol. 3:253-257. [DOI] [PubMed] [Google Scholar]
- 7.Boettler, T., H. C. Spangenberg, C. Neumann-Haefelin, E. Panther, S. Urbani, C. Ferrari, H. E. Blum, F. von Weizsacker, and R. Thimme. 2005. T cells with a CD4+ CD25+ regulatory phenotype suppress in vitro proliferation of virus-specific CD8+ T cells during chronic hepatitis C virus infection. J. Virol. 79:7860-7867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cabrera, R., Z. Tu, Y. Xu, R. J. Firpi, H. R. Rosen, C. Liu, and D. R. Nelson. 2004. An immunomodulatory role for CD4+CD25+ regulatory T lymphocytes in hepatitis C virus infection. Hepatology 40:1062-1071. [DOI] [PubMed] [Google Scholar]
- 9.Camara, N. O., F. Sebille, and R. I. Lechler. 2003. Human CD4+CD25+ regulatory cells have marked and sustained effects on CD8+ T cell activation. Eur. J. Immunol. 33:3473-3483. [DOI] [PubMed] [Google Scholar]
- 10.Cerny, A., and F. V. Chisari. 1999. Pathogenesis of chronic hepatitis C: immunological features of hepatic injury and viral persistence. Hepatology 30:595-601. [DOI] [PubMed] [Google Scholar]
- 11.Cooper, S., A. L. Erickson, E. J. Adams, J. Kansopon, A. J. Weiner, D. Y. Chien, M. Houghton, P. Parham, and C. M. Walker. 1999. Analysis of a successful immune response against hepatitis C virus. Immunity 10:439-449. [DOI] [PubMed] [Google Scholar]
- 12.Diepolder, H. M., R. Zachoval, R. M. Hoffmann, E. A. Wierenga, T. Santantonio, M. C. Jung, D. Eichenlaub, and G. R. Pape. 1995. Possible mechanism involving T-lymphocyte response to non-structural protein 3 in viral clearance in acute hepatitis C virus infection. Lancet 346:1006-1007. [DOI] [PubMed] [Google Scholar]
- 13.Eggena, M. P., B. Barugahare, N. Jones, M. Okello, S. Mutalya, C. Kityo, P. Mugyenyi, and H. Cao. 2005. Depletion of regulatory T cells in HIV infection is associated with immune activation. J. Immunol. 174:4407-4414. [DOI] [PubMed] [Google Scholar]
- 14.Endharti, A. T., I. M. S. Rifa, Z. Shi, Y. Fukuoka, Y. Nakahara, Y. Kawamoto, K. Takeda, K. Isobe, and H. Suzuki. 2005. Cutting edge: CD8+CD122+ regulatory T cells produce IL-10 to suppress IFN-gamma production and proliferation of CD8+ T cells. J. Immunol. 175:7093-7097. [DOI] [PubMed] [Google Scholar]
- 15.Filaci, G., S. Bacilieri, M. Fravega, M. Monetti, P. Contini, M. Ghio, M. Setti, F. Puppo, and F. Indiveri. 2001. Impairment of CD8+ T suppressor cell function in patients with active systemic lupus erythematosus. J. Immunol. 166:6452-6457. [DOI] [PubMed] [Google Scholar]
- 16.Filaci, G., M. Fravega, S. Negrini, F. Procopio, D. Fenoglio, M. Rizzi, S. Brenci, P. Contini, D. Olive, M. Ghio, M. Setti, R. S. Accolla, F. Puppo, and F. Indiveri. 2004. Nonantigen specific CD8+ T suppressor lymphocytes originate from CD8+CD28− T cells and inhibit both T-cell proliferation and CTL function. Hum. Immunol. 65:142-156. [DOI] [PubMed] [Google Scholar]
- 17.Filaci, G., M. Rizzi, M. Setti, D. Fenoglio, M. Fravega, M. Basso, G. Ansaldo, P. Ceppa, G. Borgonovo, G. Murdaca, F. Ferrera, A. Picciotto, R. Fiocca, G. Torre, and F. Indiveri. 2005. Non-antigen-specific CD8+ T suppressor lymphocytes in diseases characterized by chronic immune responses and inflammation. Ann. N. Y. Acad. Sci. 1050:115-123. [DOI] [PubMed] [Google Scholar]
- 18.Gerlach, J. T., H. M. Diepolder, M. C. Jung, N. H. Gruener, W. W. Schraut, R. Zachoval, R. Hoffmann, C. A. Schirren, T. Santantonio, and G. R. Pape. 1999. Recurrence of hepatitis C virus after loss of virus-specific CD4+ T-cell response in acute hepatitis C. Gastroenterology 117:933-941. [DOI] [PubMed] [Google Scholar]
- 19.Graham, C. S., L. R. Baden, E. Yu, J. M. Mrus, J. Carnie, T. Heeren, and M. J. Koziel. 2001. Influence of human immunodeficiency virus infection on the course of hepatitis C virus infection: a meta-analysis. Clin. Infect. Dis. 33:562-569. [DOI] [PubMed] [Google Scholar]
- 20.Graham, C. S., M. Curry, Q. He, N. Afdhal, D. Nunes, C. Fleming, R. Horsburgh, D. Craven, K. E. Sherman, and M. J. Koziel. 2004. Comparison of HCV-specific intrahepatic CD4+ T cells in HIV/HCV versus HCV. Hepatology 40:125-132. [DOI] [PubMed] [Google Scholar]
- 21.Graham, C. S., A. Wells, E. M. Edwards, T. Herren, S. Tumilty, S. O. Stuver, J. H. Samet, D. Nunes, C. R. Horsburgh, Jr., and M. J. Koziel. 2007. Effect of exposure to injection drugs or alcohol on antigen-specific immune responses in HIV and hepatitis C virus coinfection. J. Infect. Dis. 195:847-856. [DOI] [PubMed] [Google Scholar]
- 22.Graham, C. S., A. Wells, T. Liu, K. E. Sherman, M. Peters, R. T. Chung, A. K. Bhan, J. Andersen, and M. J. Koziel. 2005. Antigen-specific immune responses and liver histology in HIV and hepatitis C coinfection. AIDS 19:767-773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gruner, N. H., T. J. Gerlach, M. C. Jung, H. M. Diepolder, C. A. Schirren, W. W. Schraut, R. Hoffmann, R. Zachoval, T. Santantonio, M. Cucchiarini, A. Cerny, and G. R. Pape. 2000. Association of hepatitis C virus-specific CD8+ T cells with viral clearance in acute hepatitis C. J. Infect. Dis. 181:1528-1536. [DOI] [PubMed] [Google Scholar]
- 24.Hahn, C. S., Y. G. Cho, B. S. Kang, I. M. Lester, and Y. S. Hahn. 2000. The HCV core protein acts as a positive regulator of fas-mediated apoptosis in a human lymphoblastoid T cell line. Virology 276:127-137. [DOI] [PubMed] [Google Scholar]
- 25.He, X. S., B. Rehermann, F. X. Lopez-Labrador, J. Boisvert, R. Cheung, J. Mumm, H. Wedemeyer, M. Berenguer, T. L. Wright, M. M. Davis, and H. B. Greenberg. 1999. Quantitative analysis of hepatitis C virus-specific CD8+ T cells in peripheral blood and liver using peptide-MHC tetramers. Proc. Natl. Acad. Sci. USA 96:5692-5697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iken, K., L. Huang, H. Bekele, E. V. Schmidt, and M. J. Koziel. 2005. Apoptosis of activated CD4+ and CD8+ T cells is enhanced by co-culture with hepatocytes expressing hepatitis C virus (HCV) structural proteins through FasL induction. Virology 346:363-372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kamal, S. M., C. S. Graham, Q. He, L. Bianchi, A. A. Tawil, J. W. Rasenack, K. A. Khalifa, M. M. Massoud, and M. J. Koziel. 2004. Kinetics of intrahepatic hepatitis C virus (HCV)-specific CD4+ T cell responses in HCV and Schistosoma mansoni coinfection: relation to progression of liver fibrosis. J. Infect. Dis. 189:1140-1150. [DOI] [PubMed] [Google Scholar]
- 28.Kantzanou, M., M. Lucas, E. Barnes, H. Komatsu, G. Dusheiko, S. Ward, G. Harcourt, and P. Klenerman. 2003. Viral escape and T cell exhaustion in hepatitis C virus infection analysed using Class I peptide tetramers. Immunol. Lett. 85:165-171. [DOI] [PubMed] [Google Scholar]
- 29.Kingsley, C. I., M. Karim, A. R. Bushell, and K. J. Wood. 2002. CD25+CD4+ regulatory T cells prevent graft rejection: CTLA-4- and IL-10-dependent immunoregulation of alloresponses. J. Immunol. 168:1080-1086. [DOI] [PubMed] [Google Scholar]
- 30.Koziel, M. J., D. Dudley, J. T. Wong, J. Dienstag, M. Houghton, R. Ralston, and B. D. Walker. 1992. Intrahepatic cytotoxic T lymphocytes specific for hepatitis C virus in persons with chronic hepatitis. J. Immunol. 149:3339-3344. [PubMed] [Google Scholar]
- 31.Lauer, G. M., T. N. Nguyen, C. L. Day, G. K. Robbins, T. Flynn, K. McGowan, E. S. Rosenberg, M. Lucas, P. Klenerman, R. T. Chung, and B. D. Walker. 2002. Human immunodeficiency virus type 1-hepatitis C virus coinfection: intraindividual comparison of cellular immune responses against two persistent viruses. J. Virol. 76:2817-2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lechner, F., A. L. Cuero, M. Kantzanou, and P. Klenerman. 2001. Studies of human antiviral CD8+ lymphocytes using class I peptide tetramers. Rev. Med. Virol. 11:11-22. [DOI] [PubMed] [Google Scholar]
- 33.Lechner, F., D. K. Wong, P. R. Dunbar, R. Chapman, R. T. Chung, P. Dohrenwend, G. Robbins, R. Phillips, P. Klenerman, and B. D. Walker. 2000. Analysis of successful immune responses in persons infected with hepatitis C virus. J. Exp. Med. 191:1499-1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.MacDonald, A. J., M. Duffy, M. T. Brady, S. McKiernan, W. Hall, J. Hegarty, M. Curry, and K. H. Mills. 2002. CD4 T helper type 1 and regulatory T cells induced against the same epitopes on the core protein in hepatitis C virus-infected persons. J. Infect. Dis. 185:720-727. [DOI] [PubMed] [Google Scholar]
- 35.Minutello, M. A., P. Pileri, D. Unutmaz, S. Censini, G. Kuo, M. Houghton, M. R. Brunetto, F. Bonino, and S. Abrignani. 1993. Compartmentalization of T lymphocytes to the site of disease: intrahepatic CD4+ T cells specific for the protein NS4 of hepatitis C virus in patients with chronic hepatitis C. J. Exp. Med. 178:17-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Missale, G., R. Bertoni, V. Lamonaca, A. Valli, M. Massari, C. Mori, M. G. Rumi, M. Houghton, F. Fiaccadori, and C. Ferrari. 1996. Different clinical behaviors of acute hepatitis C virus infection are associated with different vigor of the anti-viral cell-mediated immune response. J. Clin. Investig. 98:706-714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Piccioli, D., S. Tavarini, S. Nuti, P. Colombatto, M. Brunetto, F. Bonino, P. Ciccorossi, F. Zorat, G. Pozzato, C. Comar, S. Abrignani, and A. Wack. 2005. Comparable functions of plasmacytoid and monocyte-derived dendritic cells in chronic hepatitis C patients and healthy donors. J. Hepatol. 42:61-67. [DOI] [PubMed] [Google Scholar]
- 38.Rifa'i, M., Y. Kawamoto, I. Nakashima, and H. Suzuki. 2004. Essential roles of CD8+CD122+ regulatory T cells in the maintenance of T cell homeostasis. J. Exp. Med. 200:1123-1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rushbrook, S. M., S. M. Ward, E. Unitt, S. L. Vowler, M. Lucas, P. Klenerman, and G. J. Alexander. 2005. Regulatory T cells suppress in vitro proliferation of virus-specific CD8+ T cells during persistent hepatitis C virus infection. J. Virol. 79:7852-7859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shevach, E. M. 2002. CD4+ CD25+ suppressor T cells: more questions than answers. Nat. Rev. Immunol. 2:389-400. [DOI] [PubMed] [Google Scholar]
- 41.Shoukry, N. H., A. Grakoui, M. Houghton, D. Y. Chien, J. Ghrayeb, K. A. Reimann, and C. M. Walker. 2003. Memory CD8+ T cells are required for protection from persistent hepatitis C virus infection. J. Exp. Med. 197:1645-1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sugimoto, K., F. Ikeda, J. Stadanlick, F. A. Nunes, H. J. Alter, and K. M. Chang. 2003. Suppression of HCV-specific T cells without differential hierarchy demonstrated ex vivo in persistent HCV infection. Hepatology 38:1437-1448. [DOI] [PubMed] [Google Scholar]
- 43.Sun, D., Y. Qin, J. Chluba, J. T. Epplen, and H. Wekerle. 1988. Suppression of experimentally induced autoimmune encephalomyelitis by cytolytic T-T cell interactions. Nature 332:843-845. [DOI] [PubMed] [Google Scholar]
- 44.Takaki, A., M. Wiese, G. Maertens, E. Depla, U. Seifert, A. Liebetrau, J. L. Miller, M. P. Manns, and B. Rehermann. 2000. Cellular immune responses persist and humoral responses decrease two decades after recovery from a single-source outbreak of hepatitis C. Nat. Med. 6:578-582. [DOI] [PubMed] [Google Scholar]
- 45.Thimme, R., D. Oldach, K. M. Chang, C. Steiger, S. C. Ray, and F. V. Chisari. 2001. Determinants of viral clearance and persistence during acute hepatitis C virus infection. J. Exp. Med. 194:1395-1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Toubi, E., A. Kessel, L. Goldstein, G. Slobodin, E. Sabo, Z. Shmuel, and E. Zuckerman. 2001. Enhanced peripheral T-cell apoptosis in chronic hepatitis C virus infection: association with liver disease severity. J. Hepatol. 35:774-780. [DOI] [PubMed] [Google Scholar]
- 47.Ulsenheimer, A., J. T. Gerlach, N. H. Gruener, M. C. Jung, C. A. Schirren, W. Schraut, R. Zachoval, G. R. Pape, and H. M. Diepolder. 2003. Detection of functionally altered hepatitis C virus-specific CD4 T cells in acute and chronic hepatitis C. Hepatology 37:1189-1198. [DOI] [PubMed] [Google Scholar]
- 48.Valdez, H., D. Anthony, F. Farukhi, A. Patki, J. Salkowitz, P. Heeger, D. L. Peterson, A. B. Post, R. Asaad, and M. M. Lederman. 2000. Immune responses to hepatitis C and non-hepatitis C antigens in hepatitis C virus infected and HIV-1 coinfected patients. AIDS 14:2239-2246. [DOI] [PubMed] [Google Scholar]
- 49.Valdez, H., N. L. Carlson, A. B. Post, R. Asaad, P. S. Heeger, M. M. Lederman, P. V. Lehmann, and D. D. Anthony. 2002. HIV long-term non-progressors maintain brisk CD8 T cell responses to other viral antigens. AIDS 16:1113-1118. [DOI] [PubMed] [Google Scholar]
- 50.Wakita, T., A. Katsume, J. Kato, C. Taya, H. Yonekawa, Y. Kanegae, I. Saito, Y. Hayashi, M. Koike, M. Miyamoto, Y. Hiasa, and M. Kohara. 2000. Possible role of cytotoxic T cells in acute liver injury in hepatitis C virus cDNA transgenic mice mediated by Cre/LoxP system. J. Med. Virol. 62:308-317. [DOI] [PubMed] [Google Scholar]
- 51.Wedemeyer, H., X. S. He, M. Nascimbeni, A. R. Davis, H. B. Greenberg, J. H. Hoofnagle, T. J. Liang, H. Alter, and B. Rehermann. 2002. Impaired effector function of hepatitis C virus-specific CD8+ T cells in chronic hepatitis C virus infection. J. Immunol. 169:3447-3458. [DOI] [PubMed] [Google Scholar]