

Abstract
Status epilepticus (SE) triggers neuronal death, reactive gliosis and remodeling of synaptic circuitry, thus leading to profound pathological alterations in CNS physiology. These processes are, in part, regulated by the rapid upregulation of both cytotoxic and cytoprotective genes. One pathway that may couple SE to transcriptionally-dependent alterations in CNS physiology is the CREB (cAMP response element-binding protein)/CRE (cAMP response element) cascade. Here, we utilized the pilocarpine model of SE on a mouse strain transgenic for a CRE-reporter construct (β-galactosidase) to begin to characterize how seizure activity regulates the activation state of the CREB/CRE pathway in both glia and neurons of the hippocampus. SE triggered a rapid (4–8 hrs post SE) but transient increase in CRE-mediated gene expression in the neuronal sublayers. In contrast to neurons, SE induced a lasting increase (up to 20 days) in CRE-mediated transcription in both reactive astrocytes and microglia. CRE-mediated gene expression correlated with expression of the pro-inflammatory enzyme cyclooxygenase-2 (COX-2). To examine the role of CREB in SE-induced COX-2 expression, we generated a transgenic mouse strain that expresses A-CREB, a potent repressor of CREB-dependent transcription. In these animals, the capacity of SE to stimulate COX-2 expression was markedly attenuated, indicating that CREB is a key intermediate in SE-induced COX-2 expression. Collectively these data show that SE triggers two waves of CREB-mediated gene expression, a transient wave in neurons and a long-lasting wave in reactive glial cells, and that CREB couples SE to COX-2 expression.
Keywords: CREB, Cyclooxygenase-2, Astrocyte, Microglia, Pilocarpine, Seizure, Hippocampus, Epilepsy
INTRODUCTION
Seizures are the result of hypersynchronous electrical activity in the brain. A brain region particularly sensitive to the deleterious effects of status epilepticus (SE) is the hippocampal formation, a structure that is essential for advanced cognitive functions such as learning and memory. Individuals suffering from temporal lobe epilepsy often display pronounced cognitive deficits that dramatically diminish their quality of life (Reviewed by Jokeit and Ebner, 2002; Stefan and Pauli, 2002). A well-established constellation of pathological changes resulting from SE have been characterized in the hippocampus, including neuronal death, reactive gliosis, remodeling of synaptic circuitry and a marked increase in neurogenesis (Reviewed by Morimoto et al., 2004; Parent and Lowenstein, 2002; Jankowsky and Patterson, 2001). These profound alterations in the hippocampus follow a stereotypical pattern, from the acute loss of neurons in the CA1 and CA3 subfields, to the reorganization of the mossy fiber collaterals that occurs several weeks after SE.
Many of the SE-induced alterations in hippocampal physiology are mediated by a two-stage process. In the initial stage, the release of the excitatory amino acid neurotransmitter glutamate leads to rapid neuronal cell death (Olney et al., 1983; Ingvar et al., 1988;Covolan and Mello, 2000), and in the second phase, glutamate, in combination with trophic factors and cytokines, drives a wave of transcriptional activity that likely confers both a neuroprotective response and contributes to seizure-induced brain pathology. Along these lines, seizure activity has been shown to stimulate the expression of brain derived neurotrophic factor (BDNF; Isackson et al., 1991; Ernfors et al., 1991), which would function in a neuroprotective manner (Young et al., 1999; Revuelta et al., 2001), cyclooxygenase-2 (COX-2; Tu and Bazan, 2003; Kawaguchi et al., 2005) which may contribute to neuronal cell death (Dore et al., 2003; Rockwell et al., 2004; Kawaguchi et al., 2005), and tissue plasminogen activator (tPA; Qian et al., 1993; Tsirka et al., 1995), which has been shown to elicit neuronal structural changes (Wu et al., 2000). Because of the role gene expression plays in SE-induced alterations in hippocampal physiology, there is a clear need to begin to identify the cellular signaling and inducible transcription events that are activated by synchronous synaptic discharges. Within this context it is of interest to note studies showing a persistent activation of the transcription factor CREB (cAMP response element binding protein) and expression of CREB-regulated immediate early genes such as EGR-1 and c-fos in epileptic foci (Rakhade et al., 2005; Gass et al., 1997; Kiessling and Gass, 1993; Simonato et al., 1991). These findings suggest a role for CREB in the genetic response to seizure activity.
In this study, we examined the regulation of CRE-mediated gene expression in neuronal and non-neuronal cells following pilocarpine-induced status epilepticus (SE). Further, we investigated the potential role of the CREB/CRE transcriptional signaling pathway in hippocampal pathophysiology by assessing its capacity to couple SE to the expression of the pro-inflammatory gene COX-2.
MATERIALS AND METHODS
Animals
Adult male C57BL/6 mice (6–10 weeks of age) were initially intraperitoneally (i.p.) injected with atropine methyl nitrate (1 mg/kg), then 30 min later, with pilocarpine hydrochloride (325 mg/kg, i.p.). Status epilepticus (SE) was defined as a continuous motor seizure over 3 hours following several stage 5 seizures (loss of balance, falling or jumping, Racine, 1972). Only mice that developed status epilepticus after pilocarpine administration were used in this study. SE was elicited in approximately 80% of the mice. Control animals were injected with atropine as above, and then injected with saline, instead of pilocarpine. To facilitate recovery from SE, animals received mouse chow soaked in a high sucrose (10%) saline solution for the first four days following SE. All experimental procedures were in accordance with Ohio State University animal care and use guidelines.
Tissue collection
Mice were anesthetized with ketamine hydrochloride (91 mg/ml) and xylazine (9 mg/ml) and perfused intracardially with a fixative solution of 4% paraformaldehyde (PFA) in 0.1 M sodium phosphate buffer (pH 7.4). Brains were removed, post-fixed in 4% PFA for 4 hours and then cryoprotected with 30% sucrose overnight. Thirty-μm thick coronal sections (−1.8 to −2.5 mm from bregma) were cut on a freezing microtome and processed for immunohistochemistry or Fluoro-jade B labeling.
Immunohistochemistry
Initially, floating sections were washed (3X) in PBS, incubated (20 min) with 0.3% H2O2, then blocked (60 min) with 10% normal goat serum in PBS. Tissue was then incubated (4º C, overnight) with a rabbit polyclonal antibody against either β-galactosidase (1:10,000 dilution; Cortex Biochem., San Leandro CA, USA) or GFP (1:5000 final dilution, Molecular Probes, Eugene OR, USA). Next, the tissue was incubated (1 hr at room temperature) in a biotinylated secondary antibody (1:300, Vector Laboratories, Burlingame CA, USA) and then incubated (45 min at room temperature) in an avidin-biotin-peroxidase complex (Vectastain ABC kit, Vector Laboratories). Nickel-intensified diaminobenzidine was used to visualize the signal. Finally, sections were mounted, dehydrated, and coverslipped with Permount (Sigma). Bright-field images were captured using a Leica DMIRB inverted microscope connected to a 16 bit digital camera (Princeton Instruments; Monmouth Junction NJ, USA).
Immunofluorescent staining was used for sequential triple labeling experiments. On day one, free-floating sections were blocked with 10% horse serum in PBS, then incubated overnight at 4º C with a goat anti-COX-2 antibody (1:200 final dilution, Santa Cruz Biochem. Santa Cruz CA, USA). On day two, the tissue was labeled (2 hrs at room temperature) with an Alexa 546 conjugated donkey anti-goat IgG antibody (1:250 Molecular Probes, Eugene OR, USA). The tissue was then washed (3X) and blocked with 10% normal goat serum in PBS for 1 hour. Next, the tissue was incubated (4º C, overnight) with a primary antibody against β-galactosidase (1: 2,500 Cortex Biochem.) or GFP (1:1000 Molecular Probes). On day three, the tissue was labeled with an Alexa 488-conjugated goat anti-rabbit IgG antibody (1:250), washed (3X) with PBS and then blocked with 10% normal goat serum in PBS for 1 hour. The last primary antibody against GFAP (1:500, Sigma, St. Louis MO, USA) or CD11b (1:250, Chemicon, Temucula CA, USA) was incubated overnight at 4º C. On day four, the tissue was labeled with an Alexa 633 conjugated goat anti-mouse IgG antibody (1:250). The tissue was then mounted and coverslipped using Gelmount (Biomedia, Foster City CA, USA). For double labeling, the primary antibodies against β-galactosidase and GFAP or CD11b were coincubated and labeled with secondary antibodies, as described above. Double labeling against eGFP (rabbit anti-GFP antibody, 1:1 000 final dilution, Molecular Probes) and COX-2, β-galactosidase (1:2,500, Cortex Biochem.) or JunB (1:2500, Santa Cruz Bio.) was performed using the methodology described above. Fluorescent images were captured using a Zeiss 510 Meta confocal microscope (Oberkochen, Germany). Data were analyzed using Metamorph image analysis software (Universal Imaging, Downingtown PA, USA).
Fluoro-jade B histochemistry
Dead or dying cells were labeled using Fluoro-jade B. Initially, tissue was fixed and thin cut as described above. Next, coronal hippocampal sections were washed in distilled water (3X), and then treated with 0.06% potassium permaganate for 10 min at room temperature with gentle shaking. Sections were then washed in distilled water (3X) and incubated with 0.001% Fluoro-jade B (Histo-Chem Inc., Jefferson, AR, U.S.A.) in 0.1% acetic acid for 20 min with gentle shaking. Sections were then washed in distilled water, dried, dipped in xylene for 2 min and coverslipped with DPX.
Generation of transgenic mouse lines
A bitransgenic system was used to generate tetracycline-inducible bicistronic A-CREB-eGFP mice. First, to make the construct that expresses A-CREB and eGFP (enhanced green fluorescence protein), A-CREB was isolated from a pcDNA3 expression vector (provided by Dr. Soren Impey, Oregon Health Sciences University). A-CREB was digested with HindIII and NcoI, blunted with Klenow and inserted into Sma I-digested pIRES2-eGFP (Invitrogen). Primers (Forward: 5′ CGGCGCGGCCGCGCCACCATGGCTAGCATGACTGGTGGACAGC 3′, Reverse: 5′ CGGCGCGGCCGCTTTACTTGTACAGCTCGTCCATGCC 3′) were used to amplify A-CREB/IRES/eGFP. A Kozak sequence (GCCACCATGG) was added upstream of start codon (bold) of A-CREB to improve expression levels and a Not I digestion site (GCGGCCGC) was added to the 5′ end to clone the construct into the pTRE-Tight vector (Clontech, Mountain View CA, USA). The vector was then digested with XhoI to release the TRE promoter, A-CREB/IRES/eGFP and SV 40 polyA sequence. The fragment was then injected into C57BL/6 embryos at the Ohio State University Transgenic Facility. Founders were screened by PCR. To drive transgene expression, the A-CREB-eGFP mice were crossed with CaMKIIα promoter-tTA transgenic mice (Mayford et al., 1996). Bitransgenic offsprings were identified using PCR, and transgene expression was confirmed using immunohistochemistry against eGFP. Of the 5 A-CREB-eGFP::tTA lines isolated, two had transgene expression within the cortex, hippocampus and striatum. Transgene expression was not repressed with the use doxycycline supplementation at any time during this study.
For luciferase reporter gene assays, 293T cells were transfected with a combination of the following vectors: CRE-Luciferase (Lee et al. 2005), TRE-tight A-CREB/IRES/eGFP and pRev-tTA (Clontech, Mountain View, CA). Forty-eight hrs after transfection, cells were stimulated with forskolin (10 μM: 8hrs) and luciferase assays was performed as described (Lee et al., 2005).
CRE-reporter gene expression
CRE-β-galactosidase transgenic mice were obtained from Dr. Daniel Storm and genotyping was performed as previously described (Obrietan et al., 2002). As noted in several studies (Impey et al., 1996; Barth et al., 2000; Obrietan and Hoyt, 2004), β-galactosidase expression was not observed in all animals genotyped as CRE positive. In our analysis, we found that ~35% of the CRE-positive animals lacked detectable β-galactosidase expression. Tissue from mice that did not express the transgene were excluded from analysis. To generate A-CREB-eGFP::tTA::CRE-lacZ transgenic mice, the A-CREB-eGFP::tTA mice were crossed with CRE-β-galactosidase mice and tritransgenic offspring were identified using PCR, and transgene expression was confirmed using immunohistochemistry against eGFP and β-galactosidase.
Western blotting
Mice were anesthetized as described above and then decapitated. Brains were then removed, placed in chilled oxygenation media and then cut into 500 μm sections with a vibratome. Dorsal hippocampal sections (~ −1.8 mm from bregma) were placed on a microscope slides and frozen with dry ice. Tissue from the hippocampus was isolated and then sonicated in 100 μl of RIPA buffer (150 mM NaCl, 50 mM Tris pH 7.4, 1 mM EDTA, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 5 mM NaF, 25 μM sodium vanadate supplemented with protease inhibitor cocktail: complete mini tablet, Roche Diagnostics), total protein levels were determined, and then 30 μg of protein samples were diluted in 3X sample buffer. Extracts were loaded onto a 10% SDS-PAGE gel, electrophoresed using standard procedures, and protein was transblotted onto polyvinylidene fluoride (PVDF) (Immobilon P; Millipore). Following blocking with 10% (wt/vol) powdered milk, membranes were incubated (4°C overnight) with an affinity-purified rabbit polyclonal antibody against Ser133 phosphorylated CREB (1:1000 dilution, Cell Signaling Technology, Beverly MA, USA). Samples were then incubated with an alkaline phosphatase-conjugated anti-rabbit antibody (1:2,500, Perkin-Elmer, Wellesley, MA USA) and the signal was detected using the Western-CDP star chemiluminescent detection system (Perkin-Elmer). As a protein loading control, membranes were probed with a goat polyclonal antibody against total erk-1 and erk-2 (1:2000, Santa Cruz Biochem.). Samples were then incubated with a horseradish peroxidase-conjugated secondary antibody and the signal was detected using a chemiluminescent HRP substrate (Renaissance chemiluminescent detection system: New England Nuclear).
Quantification and data analysis
To determine the number of COX-2 positive non-neuronal cells expressing β-galactosidase within the dentate gyrus, confocal sections were captured (2-μm-thick optical section, ~ −1.8 mm from bregma) and the total number of positive cells were calculated. Data were averaged from 2 sections (bilateral) per animal, and then averaged from three mice for each time point. For quantitation of COX-2 expression in the dentate gyrus granule cell layer of A-CREB-eGFP::tTA transgenic mice, confocal sections were captured as above and COX-2 and eGFP expression were examined in the dentate graule cell layer. To quantitate COX-2, the perinuclear region of the eGFP-positive cells was digitally outlined using Metamorph and used as a template to quantitate COX-2 expression. To quantitate COX-2 expression in non-transgenic cells, the perinuclear region of cells adjacent to transgenic cells was quantitated. COX-2 expression in control transgenic animals (without pilocarpine) was used as a background subtraction to determine the total inducible signal. There was no significant difference in basal COX-2 expression in transgenic versus non-transgenic cells.
For β-galactosidase cell counts, images were captured at 100X magnification and quantitation was performed using MetaMorph software (Universal Imaging). The total number of β-galactosidase positive cells was counted within a digital 200 mm square placed over the GCL, CA3 or CA1. Data were counted from two sections separated by ~ 200 μm (stereotaxic coordinate AP: −1.60 ~ −2.00). Cell counts were analyzed statistically using Student’s t-test, and significance was accepted for P<0.05.All data are expressed as means +/− SEM. All necessary comparisons were carried out using Student’s t-test. A P-value <0.05 was considered as significant.
RESULTS
The muscarinic acetylcholine receptor agonist pilocarpine was used to elicit status epilepticus (SE: Turski et al., 1983 and 1989). Initially, adult mice (C57BL/6) were injected with atropine (1 mg/kg, i.p.), and then injected with pilocarpine (325 mg/kg, i.p.) 30 min later. The seizures were characterized based on Racine’s scale (Racine, 1972). At first, mice typically displayed facial movements (stage 1) and progressed to head nodding (stage 2), forelimb clonus (stage 3), rearing (stage 4), and rearing and falling (stage 5). After the initial stage 5 seizure, a subset of mice developed SE. SE was defined as continuous motor seizures persisting for over 3 hrs. Only mice that developed SE were used. Control animals were injected with atropine, as above, followed by physiological saline (pilocarpine vehicle).
SE stimulates CRE-mediated gene expression in the hippocampus
Representative data from brain sections immunolabeled for the CRE-regulated reporter, β-galactosidase, revealed that SE triggered robust CRE-mediated gene expression 8 hrs post SE onset (Fig. 1A). Induction was observed throughout the central nervous system. Within the hippocampus, expression was detected in the dentate granule cell layer (GCL) and the CA3 and CA1 subfields. This contrasts with the low, heterogeneous, β-galactosidase expression pattern in control mice not injected with pilocarpine (Fig. 1A). By 18 hrs post-seizure onset, β-galactosidase expression had dropped markedly in the excitatory subfields, and by 7 days post-SE, expression had returned to near basal levels. Quantitative data are shown in figure 2C.
Figure 1. Seizure-induced activation of CRE-mediated transcription.
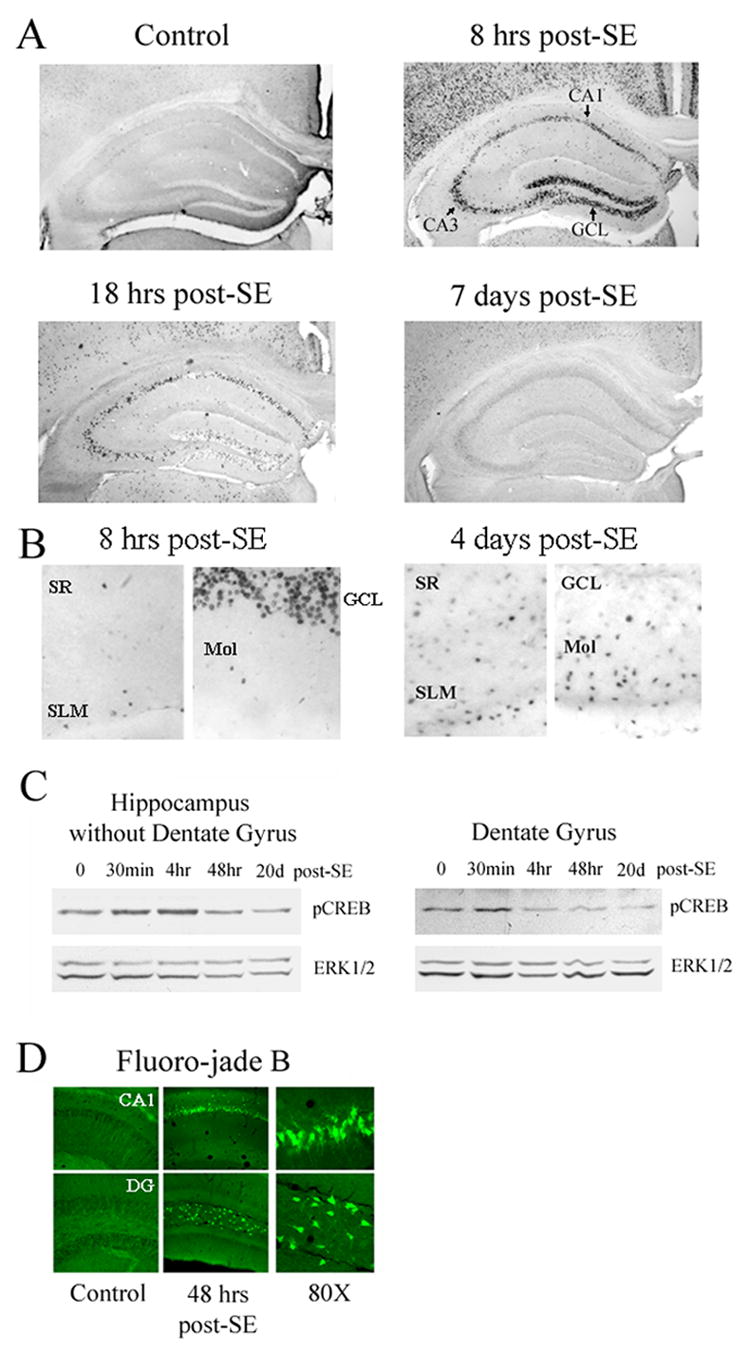
A) Mice transgenic for the CRE-regulated β-galactosidase reporter construct were injected with either pilocarpine (325 mg/kg: i.p.) or vehicle and sacrificed at multiple time points following status epilepticus (SE) onset. Brain sections were immunolabeled for β-galactosidase expression. Small black punctate dots denote cells with a high-level CRE-mediated gene expression. Eight hrs after SE, a marked increase in reporter gene expression was observed in the hippocampus excitatory subfields (areas CA1, CA3 and the granule cell layer), as well is in the cortex. This is in contrast to relatively modest staining pattern in control animals sacrificed 8 hrs post-saline injection. By 18 hrs-post seizure onset, β-galactosidase levels were reduced from the peak level observed at the 8-hr time point, and by 7 days, little CRE-mediated gene expression was observed in the hippocampal neuronal sublayers. B) High magnification images of β-galactosidase staining from animals killed 8 hrs or 4 days post-SE. Relative to 8 hrs post-SE, there was a marked increase in CRE-mediated gene expression outside of the neuronal subfields at 4 days post-SE. GCL: granule cell layer, Mol: molecular cell layer, SR: stratum radiatum, SLM: stratum lacunosum moleculare. C) SE stimulates CREB phosphorylation. Hippocampi from 500 μm-thick coronal brain sections were isolated at multiple time points following SE. At 30 min post-SE, a marked increase in the Ser-133 phosphorylated form of CREB was observed in tissue isolated from the dentate gyrus, as well as non-dentate hippocampus. As a protein loading control, the blots were also probed for total ERK expression. D) Fluoro-jade B labeling was used to detect SE-induced cell death at 48 hrs post-SE. Marked cell death was observed in the CA1 and hilus (Hil). Cell death was not observed in the GCL. DG: dentate gyrus.
Figure 2. Seizure-induced activation of CRE-mediated gene expression in neuronal and non-neuronal cells.
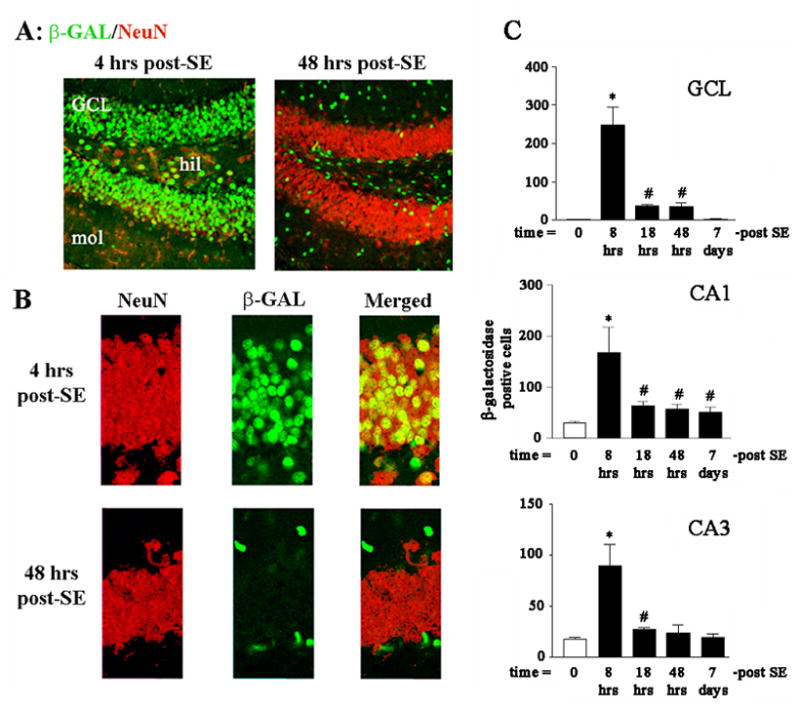
Animals were sacrificed either 4 or 48 hrs after pilocarpine-induced SE. The tissue was then fluorescently immunolabeled for β-galactosidase (green) and for the neuronal nucleus-specific antigen NeuN (red). A) Four hrs after SE, β-galactosidase expression was observed in NeuN positive cells of the granule cell layer (GCL) and the hilus. However, by 48 hrs post-SE onset, β-galactosidase expression was notably absent from the GCL. Furthermore, the remaining β-galactosidase expression did not colocalize with NeuN, indicating that CRE-mediated transcription at the 48 hr time point occurred in non-neuronal cells. B) High magnification of the immunolabeling within the GCL; note the non-overlapping labeling at 48 hrs post-SE. C) Quantitation of β-galactosidase expression in excitatory subregions of the hippocampus. Mean ± SEM data are from 4–6 animals per time point. *: P<0.05 relative to all other data points. #: P<0.05 relative to control (0-post SE) time point.
CREB phosphorylation at Ser 133 is a necessary event in the induction of CRE-mediated gene expression (Gonzalez and Montminy, 1989). Consistent with the effect observed at the level of CRE-mediated transcription, we found that SE triggered a robust increase in the Ser-133 phosphorylated form of CREB in the hippocampus (Fig. 1C). For these experiments, the dentate gyrus was isolated from the rest of the hippocampus and the two samples were processed in parallel. Maximum CREB phospho-activation was observed at 30 min to 4 hrs post-SE. At 48 hrs post-SE, levels of activated CREB were similar to levels observed in control animals (time point 0). These data reveal that SE triggers CREB transactivation which in turn facilitates the induction of CRE-dependent transcription.
Hippocampal neurons are vulnerable to the excitotoxic effects of SE. To assess whether the transient nature of CRE-mediated reporter gene expression within the hippocampal neuronal sublayers correlates with SE-induced cell death, tissue was processed with Fluoro-jade B, a sensitive histochemical marker of dead and dying cells. As expected, at 48 hrs post-SE onset, marked cell death was observed within the CA1 area (Fig. 1D) and limited cell death was observed in area CA3 (data not shown). In contrast, cell death was not detected within the GCL, a cell region shown to be highly resistant to the excitotoxic effects of SE (Fricke and Prince, 1984; Lothman et al., 1991, 1992; Schweitzer et al., 1992; Becker et al., 1999). Given that the GCL was refractory to the excitotoxic effects of SE, these data suggest that the short-lived nature of CREB-dependent transcription was not the result of SE-induced cell death.
In contrast to the transient increase in CRE-mediated gene expression in hippocampal excitatory neurons, marked β-galactosidase expression was observed outside of the neuronal sublayers 4 days post-SE (Fig. 1B). The observation that SE-induced a distinct temporal profile of CRE-mediated gene expression outside of the neuronal sublayers suggests that this transcriptional pathway is differentially regulated within subpopulations of cells. To begin to address this issue, coronal brain sections were double immunolabled for β-galactosidase and the neuronal cell marker, NeuN (Fig. 2A and 2B). Four hours post-seizure onset, β-galactosidase expression was observed in NeuN positive cells in dentate gyrus (Fig. 2A and 2B) as well in the CA3 and CA1 (data not shown). Interestingly, by 48 hrs post-SE, the majority of β-galactosidase-expressing cells in the dentate gyrus were outside of the GCL and were not positive for NeuN (Fig. 2A and 2B).
CRE-mediated transcription in reactive astrocytes and microglia
Next, we examined whether CREB-dependent transcription was induced in reactive astrocytes and microglia. Astrogliosis and microgliosis occur in response to CNS disease and injury. Initially, to test whether our SE model triggered gliosis, hippocampal brain sections were processed for GFAP (glial fibrillary acidic protein). GFAP is expressed specifically by astrocytes and is upregulated following neurotoxic insults such as lesions, ischemia and seizure activity (Orzi et al., 1990; Steward et al., 1991). At 4 hrs post-SE onset (the earliest time point examined), a modest increase in GFAP immunoreactivity was observed, and by 48 hrs post-SE onset, numerous astrocytes with hypertrophied processes and increased soma size were found throughout the hippocampus (Fig. 3A).
Figure 3. CRE-mediated gene expression in reactive astrocytes.
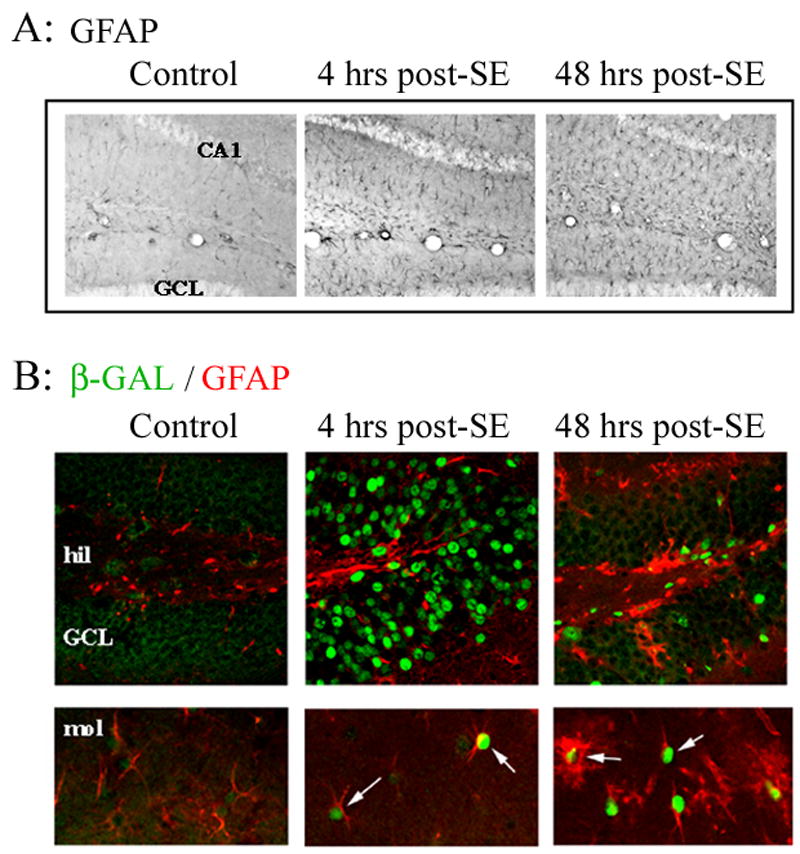
Animals were sacrificed either 4 hrs or 48 hrs after pilocarpine-evoked SE. A) Representative GFAP immunolabeling from the hippocampus reveals that SE triggered a marked upregulation of GFAP labeling relative to control conditions. B) Tissue was fluorescently immunolabeled for β-galactosidase (green) and GFAP (red). Four hrs after SE, β-galactosidase expression was observed in the granule cell layer (GCL). In contrast, limited β-galactosidase expression was detected in GFAP positive cells at this time point. By 48 hrs post-seizure onset, β-galactosidase expression was observed in astrocytes throughout the dentate gyrus. hil: hilus, mol: molecular layer.
To assess whether SE stimulated CRE-mediated gene expression in reactive astrocytes, hippocampal sections were double immunolabled for β-galactosidase and GFAP. At 4 hrs post-SE onset, a modest increase in β-galactosidase expression was detected in astrocytes. This acute induction was coincident with the time period during which robust CRE-mediated gene expression occurred in hippocampal neurons (Fig. 3B). This contrasts with control animals injected with atropine, where low levels of β-galactosidase were observed in astrocytes throughout the hippocampus. By 48 hrs post-SE, robust β-galactosidase expression was observed in reactive astrocytes (Fig. 3B). These data indicate that SE stimulates a rapid and lasting increase in CRE-mediated gene expression in reactive astrocytes.
Next, we examined whether CRE-mediated gene expression was also upregulated in reactive microglia. To identify microglia, tissue was immunohistochemically processed for the expression of the type 3 complement receptor (CD11b). By 4 hrs post SE onset, CD11b expression was observed in the dentate gyrus (Fig. 4A), and by 48 hrs post-SE, marked microgliosis was observed throughout the hippocampus. These findings are consistent with previous studies examining the temporal profile of SE-induced microgliosis (Rizzi et al., 2003; Borges et al., 2003). To determine whether SE stimulates CRE-mediated transcription in microglia, hippocampal brain sections were double labeled for β-galactosidase and CD11b. With our fluorescent immunolabeling technique, it was difficult to detect an increase in CD11b expression 4 hrs post-SE onset. However, by 48 hrs post-SE process-bearing CD11b immunoreactive microglia were detected with colocalized expression of the CRE-regulated reporter gene (Fig. 4B). Quantitative analysis revealed that the relative percentage of neuronal and non-neuronal cells that expressed β-galactosidase was dramatically altered from 4 hrs to 48 hrs post-SE. Thus, at 4 hrs post SE, the majority of β-galactosidase-positive cells were neurons whereas, by 48 hrs, only 16% of the β-galactosidase-positive cells were neurons (Fig. 4C). It should also be noted that the total number of β-galactosidase-positive cells decreased as a function of time post-SE. These data confirm the transient nature of CRE-mediated gene expression in neurons, and that SE triggers a longer-lasting increase in CREB-dependent transcription in non-neuronal cells.
Figure 4. CRE-mediated gene expression in microglia.
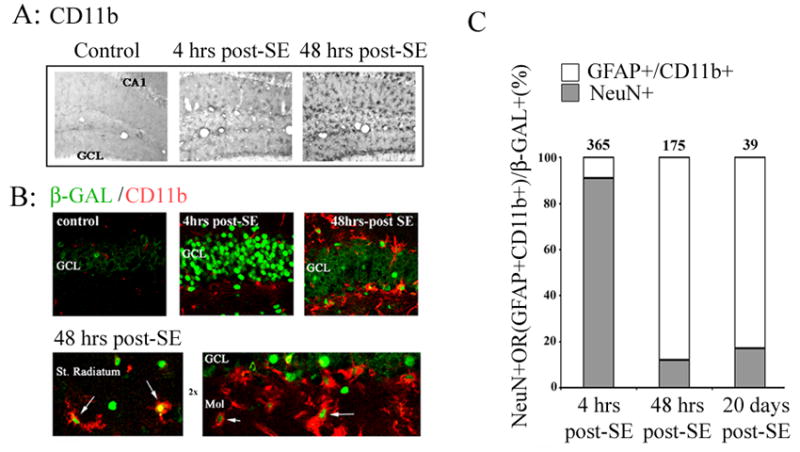
Animals were sacrificed 4 hrs or 48 hrs after pilocarpine-evoked SE. The tissue was then immunolabeled with an antibody against CD11b, a marker for reactive microglia. A) Relative to control animals, SE stimulated robust microglia activation. B) Double immunofluorescent labeling against β-galactosidase (green) and CD11b (red), revealed that seizure activity elicits CRE-dependent transcription in reactive microglia. Four hrs after status epilepticus, β-galactosidase expression was observed primarily in granule cells (GCL) of the dentate gyrus. However, by 48 hrs post-SE onset, a marked increase in β-galactosidase expression was observed in CD11b-positive cells along the GCL boarder, as well as in the stratum (St.) radiatum and the molecular cell layer (mol: arrows). C) Quantitation of β-galactosidase positive cells as a function of both time and cell type. The mean number of β-galactosidase positive neurons (NeuN+) and glia (GFAP+/CD11b+) within the dentate gyrus was determined and expressed as 100% for 3 post-SE time points. Data were collected from three mice per time point. The numbers above bars denote the mean number of β-galactosidase positive cells per dentate gyrus at each time point.
Seizure induced-COX-2 expression
In response to CNS injury there is a rapid upregulation of pro-inflammatory genes such as cyclooxygenase 2 (COX-2), tumor necrosis factor–α (TNF-α), interleukin-1β (IL-1β) and interleukin-6 (IL-6) (Taupin et al., 1993; Shohami et al., 1994 ; Stoll et al., 2002; Turrin and Rivest, 2004). Interestingly, the promoter regions of a number of proinflammatory genes contain CREs, suggesting that they may be regulated by CREB-dependent transcription. To determine whether there is a correlation between CREB-dependent gene transcription and pro-inflammatory gene expression, we examined the temporal and spatial profile of SE-induced COX-2 expression. Four hours after SE, COX-2 expression was dramatically increased in the GCL and CA3 (Fig. 5A). Double labeling experiments revealed a coordinate increase in CRE-mediated gene expression and the induction of COX-2 (Fig. 5B). By 48 hours post-SE onset, high levels of COX-2 were observed in CA3 and CA1, whereas expression within the GCL had decreased from the level observed 4 hours post-SE (Fig. 5A). This expression pattern paralleled the expression pattern of β-galactosidase. Thus, COX-2 expression was first observed at 4 hrs post-SE and early expression (4–48 hrs) was predominantly located in neuronal sublayers. In contrast, delayed expression (2–14 days post-SE) of COX-2 was detected outside of the neuronal sublayers (Fig. 5A: far right panel). Triple immunolabeling experiments revealed that both reactive microglia (CD11b-positive cells) and reactive astrocytes (GFAP-positive cells) that expressed COX-2 also expressed β-galactosidase (5C, 5D and 5E). These data raise the possibility that seizures and cell stress stimulate the expression of the pro-inflammatory enzyme COX-2 via a CREB/CRE-dependent mechanism.
Figure 5. Seizure-induced COX-2 expression.
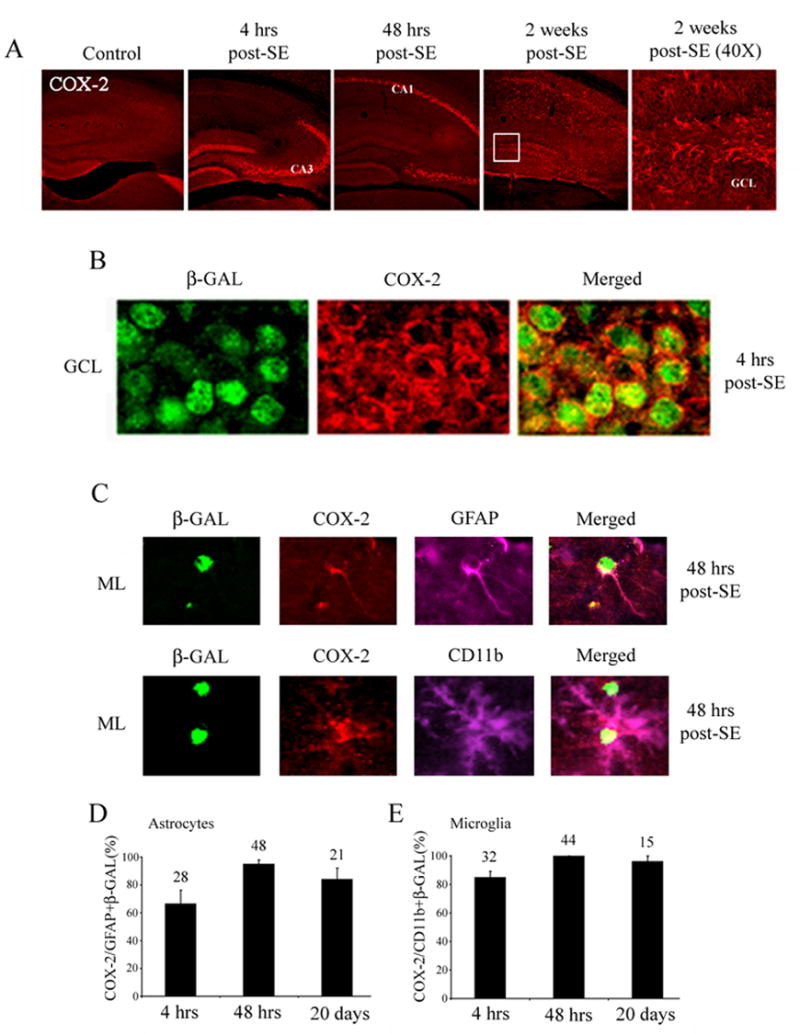
A) Animals were sacrificed 0, 4, 48 hrs and 2 weeks after pilocarpine-evoked SE. The tissue was then fluorescently immunolabeled for COX-2 expression. Four hours after SE, COX-2 expression was dramatically increased in the GCL and area CA3. Forty eight hours after SE, marked COX-2 expression was observed in area CA1. Far right panel shows a high magnification image of the 2 weeks post-SE COX-2 immunolabeling (boxed region). Note that COX-2 expression was detected outside of the GCL. B) High magnification image of the GCL double labeled for COX-2 and β-galactosidase revealed that CRE-mediated gene expression correlates with the expression of COX-2. Data were collected from animals killed 4 hrs post-SE onset. C) Animals were sacrificed 48 hrs after pilocarpine-evoked SE, and the tissue was immunolabeled for glial markers (GFAP and CD11b), the CRE-regulated reporter (β-galactosidase) and COX-2. Representative images from the molecular layer of the dentate gyrus reveal the colocalized expression of β-galactosidase and COX-2 in both reactive astrocytes (GFAP) and microglia (CD11b). D) Cell counts denoting the percentage of β-galactosidase positive cells (astrocytes or microglia) that also express COX-2 in the dentate gyrus. Numbers above each bar indicate the mean number of cells per dentate gyrus. Data were collected from 3 animals per time point. Data were averaged for each animal and then averaged across all animals for each time point. Percentages are expressed as means +/− SEM.
To provide a definite assessment of the role of the CREB/CRE pathway in the induction of COX-2, we generated a transgenic mouse strain that expresses the CREB repressor A-CREB in a tetracycline-inducible manner within forebrain neurons. A-CREB functions as an inhibitor of CREB-dependent transcription by dimerizing with the basic region of CREB and thus blocking its ability to bind to DNA (Ahn et al., 1998). In addition, the construct was engineered to drive the expression of enhanced green fluorescent protein (eGFP) from an internal ribosomal entry site (IRES), thus allowing for identification of the cells that express the transgene construct. Luciferase reporter gene assays revealed that the construct potently repressed forskolin- (an adenylyl cyclase activation: 10 μM) induced CRE-mediated gene expression Figure 6A. Immunohistochemical labeling for eGFP detected heterogeneous transgene expression in neurons within the hippocampus, cortex, and striatum (Fig. 6B). The efficacy of the construct was initially tested by examining SE-mediated, CREB-dependent, gene expression. In A-CREB mice, SE-induced expression of the CREB-regulated gene, JunB, was repressed in cells that express A-CREB-eGFP (Fig. 6C). Furthermore, in A-CREB mice crossed with the CRE-β-galactosidase mice, SE induced β-galactosidase expression was not observed in A-CREB expressing cells (Fig. 6D). Together these data reveal that the A-CREB construct functions as a potent repressor of SE-induced, CRE-dependent, gene expression. To test the role of CREB in SE-induced COX-2 expression, transgenic mice were killed 8 hrs after SE onset and tissue was double immunolabeled for eGFP and COX-2. A cell-by-cell analysis of the GCL revealed that A-CREB expression significantly attenuated SE-induced COX-2 expression (Fig. 7A and 7B). The A-CREB construct is driven by the CaMKII promotor, and as such, is expressed primarily in neuronal cells. In line with this expression pattern, we did not note an effect of transgene expression on SE-induced COX-2 expression in non-neuronal cells (Figure 7C). Collectively, these data indicate that SE drives CRE-mediated gene expression in both neuronal and non-neuronal cells of the hippocampus and that CREB couples seizures to the expression of the proinflammatory gene COX-2.
Figure 6. A-CREB transgenic mice.
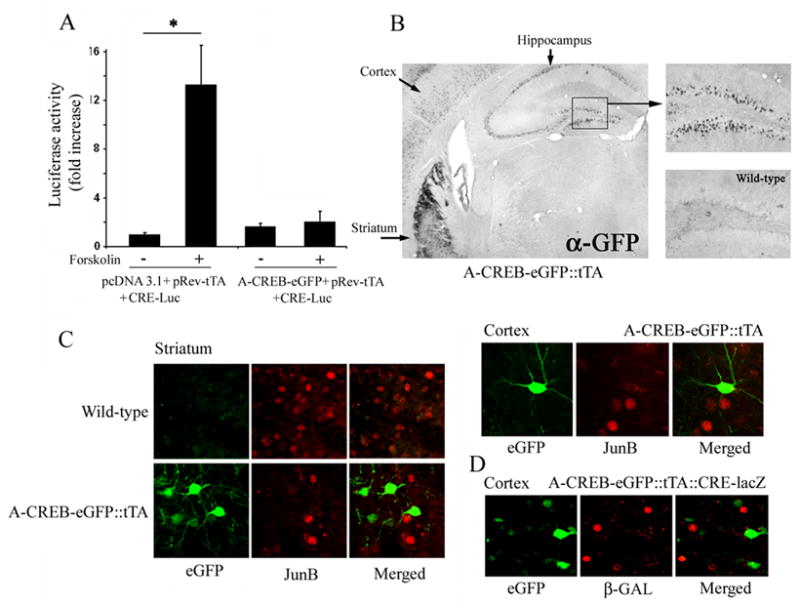
A) 293T cells were transfected with CRE-luciferase, pRev-tTA and either A-CREB/IRES/eGFP or an empty vector (control). Cells were stimulated 48 hrs later with forskolin (10 uM). Data represent the mean ± SEM fold increase in luciferase activity relative to control, untreated cells, which were normalized to a value of 1. B) Immunohistochemistry for eGFP transgene expression. Expression of the A-CREB-eGFP transgene construct was detected in the cortex, striatum and hippocampus. Hippocampal expression is magnified to the right. As a control, tissue from WT animals was also processed for antigenicity against the GFP antibody. C) A-CREB blocks SE-induced JunB expression. A-CREB transgenic mice were injected with pilocarpine, and killed 90 min after SE onset. Double fluorescent immunolabeling revealed that JunB expression did not occur in neurons expressing the A-CREB-eGFP transgene construct. Representative data are shown for the striatum and cortex. Note that the red and green signals do not overlap in the merged images. D) A-CREB blocks SE-induced CRE-mediated gene expression. A-CREB transgenic mice were crossed with CRE-β-galactosidase reporter mice and animals were killed 8 hrs post-SE onset. Fluorescent immunolabeling of cortex for eGFP (green) and β-galactosidase (red) reveals that reporter gene expression did not occur in neurons expressing the A-CREB construct.
Figure 7. A-CREB attenuates SE-induced COX-2 expression.
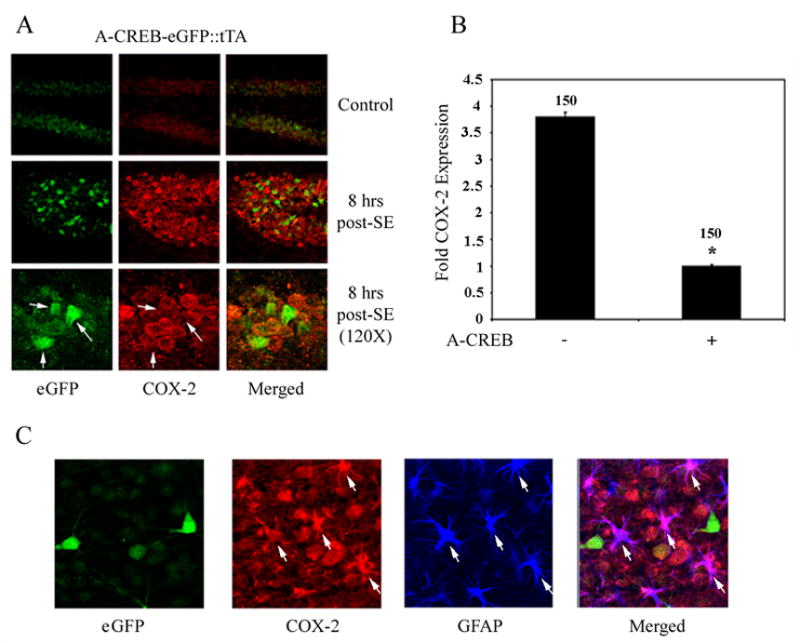
A) Representative images of the dentate GCL double immunolabeled for eGFP (the transgene marker) and COX-2. eGFP was expressed in a limited number of granule cells. High magnification double labeling (bottom) revealed that COX-2 expression was attenuated in transgenic cells (arrows). B) Quantitation of SE-induced COX-2 expression in transgenic and non-transgenic granule cells. COX-2 intensity values were determined as described in the Methods section. Data are expressed as mean fold-induction relative to transgenic cell expression, which was set equal to 1. Error bars denote SEM. Numbers above bars denote the total number of cells examined. *: P<0.005. C) Cortical tissue was triple- labeled for COX-2, A-CREB-eGFP and GFAP. Representative photomicrograph was from a mouse killed 48 hrs-post SE onset. Robust COX-2 expression was observed in GFAP-positive cells (arrows), but not in eGFP-positive neurons.
DISCUSSION
A principal goal of this study was to examine whether SE stimulates CRE-mediated gene expression, and if so to determine both the cell types and temporal regulation of gene induction. The data presented here provide the first detailed information regarding how epileptiform discharges regulate that activation state of the CREB/CRE pathway. Furthermore, the data reveal a divergence in the regulation of CRE-mediated gene expression within neuronal and non-neuronal cells, suggesting that the pathway contributes to distinct cell-type-specific physiological processes.
CREB is a member of the basic leucine zipper family of transcription factors that includes CREM and ATF1 (Lonze and Ginty, 2002; Liang et al., 1996). CREB is highly expressed in both neurons and glia (Lonze and Ginty, 2002; Herdegen et al., 1992), and its transactivation potential is regulated in an activity-dependent manner. Thus, within the nervous system, robust neuronal activity has been shown to trigger the phosphorylation of CREB at Serine 133, an event necessary for CREB-dependent transcription. With respect to seizure models, Moore et al. (1996) showed that pentylenetetrazol triggers the Serine 133 phosphorylated form of CREB. Although phosphorylation is required for CRE-mediated gene expression, it is only one of several essential events that are required to drive CRE-mediated gene expression (Lonze and Ginty, 2002; Shaywitz and Greenberg, 1999; Thiel et al., 2005; Chen et al., 2003; Impey et al., 1996, 1998; Obrietan et al., 1999, 2004; Lee et al., 2005). Thus, the data presented here are the first to examine the dynamics of seizure-induced CRE-dependent transcription.
Beginning 4 hrs post-SE onset, we detected a marked increase in CRE-mediated gene expression within the hippocampal neuronal sublayers. Interestingly, this increase in gene expression was transient. For example, reporter levels in the GCL returned to baseline by ~ 24 hrs post SE onset. This decline in CRE-mediated gene expression within the hippocampal neuronal subfields suggests that there is an uncoupling of CRE-mediated gene expression from an ostensibly robust extracellular excitatory environment.
There are several potential mechanisms that may underlie the transient nature of seizure-induced CRE-mediated gene expression. For example, the duration of CREB phosphorylation appears to be a key event in determining whether CREB effectively couples to the basal transcriptional complex. Work from Hardingham et al., (2002) found that non-excitotoxic levels of NMDA receptor stimulation triggered a lasting form of CREB phosphorylation. Conversely, potentially excitotoxic levels of receptor stimulation led to transient phosphorylation, and abrogation of CRE-mediated gene expression. This effect was found to result from the activation of extrasynaptic NMDA receptors which trigger a discrete set of signaling events that lead to CREB dephosphorylation. In a parallel line of inquiry, we reported that potentially excitotoxic levels of stimulation trigger activation of a phosphatase-dependent shut-off mechanism (Lee et al., 2005). Both of the aforementioned studies were performed with neuronal cultures; additional work will be required to determine the mechanism by which SE triggers transient activation of CRE-mediated gene expression. Thus, within this context, it would appear that the initial bout of neuronal activity elicited by SE was processed as a non-toxic stimulus, and thereby elicits CRE-dependent transcription. This acute induction of SE-induced neuronal activity may be analogous to the level of activity initiated by associative learning or stimuli that elicit long-lasting forms of LTP: two paradigms that have been shown to trigger CRE-mediated gene expression using β-galactosidase transgenic mouse strain (Impey et al., 1996; Impey et al., 1998).
Through its capacity to stimulate the expression of neuroprotective genes, the CREB/CRE transcriptional pathway plays a prominent role in cell health (Walton et al., 1996; Bonni et al., 1999; Walton et al., 1999; Mabuchi, 2001; Lonze and Ginty, 2002; Jaworski et al., 2003). Given this context, it was interesting to find that SE-induced CRE-mediated gene expression was not related to sublayer-specific cell death vulnerability. Along these lines, although both the CA1 area and GCL showed SE-induced increases in CRE-mediated gene expression, only CA1 neurons exhibited marked cell death. However, the approach used here did not delineate whether acute CRE-mediated gene expression occurred in neurons that were refractory to the excitotoxic conditions, thus the role of this pathway in cell viability is not clear. Furthermore, in light of its role in neuroprotection, it was also of interest to find that the CRE pathway also regulated the expression of the proinflammatory gene COX-2.
The enzymatic activity of COX-2 is the rate-limiting step in prostaglandin synthesis, and its rapid upregulation in response to excitotoxic insults has been suggested to contribute to pathophysiological brain damage (reviewed by Consilvio et al., 2004). Along these lines, a number of studies have shown that blockade of COX-2 attenuates excitotoxic cell death ( Ezaki et al., 2005; Kawaguchi et al., 2005). Moreover, transgenic overexpression of COX-2 leads to an increase in cell death resulting from transient focal ischemia (Dore et al., 2003). Conversely, COX-2 and its metabolic products have also been shown to protect neurons against excitotoxic and oxygen/glucose deprivation-induced cell death (Gendron et al., 2005; McCullough et al., 2004; Liu et al., 2005). This dual role of COX-2 (excitotoxic and neuroprotective) is dependent on the model systems and stimulus paradigms used (Gendron et al., 2005).
With respect to the cell types and time course of COX-2 induction, our data are consistent with several recent reports using the pilocarpine mode of SE (Turrin and Rivest, 2004; Voutsinos-Porche et al., 2004). The examination of its inducible regulation has revealed that COX-2 has an array of response elements, including a CRE, an NF-kappaB site, a NF-IL6 motif and a binding site for C/EBPβ, (Tanabe and Tohnai, 2002 ), that would allow for rapid coupling to excitatory stimuli. Interestingly, a number of studies have shown that CREB plays a key role in receptor-dependent coupling to COX-2 (Cho et al., 2004; Yang and Bleich, 2004). Our data are consistent with a role for CREB in COX-2 expression. Along these lines, we show that COX-2 expression parallels the SE-induced expression pattern of the CRE reporter in both neurons and reactive glia. The causal connection was confirmed using a transgenic mouse strain that overexpresses the CREB inhibitor, A-CREB. Thus, transgenic A-CREB repressed SE-induced COX-2 expression. It is interesting to note that the CREB-regulated transcription factor C/EBPβ has also been implicated in COX-2 expression (Wu et al., 2005; Chen et al., 2004). Given that C/EBPβ is induced by neuronal activity in a CREB-dependent manner (Niehof et al., 1997), it is plausible that CREB might stimulate COX-2 expression via both a direct effect and through the indirect induction of C/EBPβ.
Widespread microglial activation and persistent astrogliosis are hallmarks of the pilocarpine model of epilepsy (Borges et al., 2003; Aschner et al., 1999; Hanisch, 2002; Rogove and Tsirka, 1998), and are consistent with the pathological alterations found in human temporal lobe epilepsy. These changes in glial cell physiology are likely mediated by lasting alterations in their transcription profile (Jeon et al., 2004). In this context, it was interesting to find that SE elicited a long-lasting form of CREB-dependent gene expression in reactive astrocytes and reactive microglia. This observation of prolonged CRE-mediated gene expression in glial cells is consistent with pCREB data collected using a number of neuronal injury models. For example, in the hippocampus, increased pCREB levels were observed in reactive glia 7 days following focal stab lesions, and four weeks after kainic acid injection (Carbonell and Mandell, 2003; Ong et al., 2000). Interestingly, in tissue from patients with mesial temporal lobe epilepsy pCREB is also observed in reactive astrocytes (Park et al., 2003). Future experiments will be aimed at addressing the potential role of the CREB/CRE pathway in glial cells as it pertains to SE-induced pathophysiological alterations within the hippocampus. It should be noted that transgenic A-CREB was not expressed in non-neuronal cells, so the connection between CREB and COX-2 expression in glia could not be directly tested. However, given the temporal correlation between β-galactosidase expression and COX-2 expression in reactive glia, it is likely that the CREB/CRE-pathway also regulates COX-2 expression in non-neuronal cells.
In conclusion, this newly identified role of CREB as a positive regulator of SE-induced COX-2 expression is, in some respects, counter to the well established role of CREB as a neuroprotective signaling pathway (Lonze and Ginty, 2002; Tanaka, 2001; Hara et al., 2003). Thus, these data expand the role of CRE-signaling from one that is neuroprotective to one that may contribute to seizure-induced pathological alterations in the CNS.
Acknowledgments
This work was supported by grants from the National Institutes of Health (NS47176 to KO and the Ohio State Neuroscience Center Core 5P30NS045758). YC is a recipient of an Epilepsy Foundation fellowship and BL is a recipient of an American Heart Association fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn S, Olive M, Aggarwal S, Krylov D, Ginty DD, Vinson C. A dominant-negative inhibitor of CREB reveals that it is a general mediator of stimulus-dependent transcription of c-fos. Mol Cell Biol. 1998;18:867–977. doi: 10.1128/mcb.18.2.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschner M, Allen JW, Kimelberg HK, LoPachin RM, Streit WJ. Glial cells in neurotoxicity development. Annu Rev Pharmacol Toxicol. 1999;39:151–173. doi: 10.1146/annurev.pharmtox.39.1.151. [DOI] [PubMed] [Google Scholar]
- Barth AL, McKenna M, Glazewski S, Hill P, Impey S, Storm D, Fox K. Upregulation of cAMP response element-mediated gene expression during experience-dependent plasticity in adult neocortex. J Neurosci. 2000;20:4206–4216. doi: 10.1523/JNEUROSCI.20-11-04206.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker AJ, Gillardon F, Blumcke I, Langendorfer D, Beck H, Wiestler OD. Differential regulation of apoptosis-related genes in resistant and vulnerable subfields of the rat epileptic hippocampus. Brain Res Mol Brain Res. 1999;67:172–176. doi: 10.1016/s0169-328x(99)00060-1. [DOI] [PubMed] [Google Scholar]
- Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science. 1999;286:1358–1362. doi: 10.1126/science.286.5443.1358. [DOI] [PubMed] [Google Scholar]
- Borges K, Gearing M, McDermott DL, Smith AB, Almonte AG, Wainer BH, Dingledine R. Neuronal and glial pathological changes during epileptogenesis in the mouse pilocarpine model. Exp Neurol. 2003;182:21–34. doi: 10.1016/s0014-4886(03)00086-4. [DOI] [PubMed] [Google Scholar]
- Carbonell WS, Mandell JW. Transient neuronal but persistent astroglial activation of ERK/MAP kinase after focal brain injury in mice. J Neurotrauma. 2003;20:327–336. doi: 10.1089/089771503765172282. [DOI] [PubMed] [Google Scholar]
- Chen JC, Huang KC, Wingerd B, Wu WT, Lin WW. HMG-CoA reductase inhibitors induce COX-2 gene expression in murine macrophages: role of MAPK cascades and promoter elements for CREB and C/EBPbeta. Exp Cell Res. 2004;301:305–319. doi: 10.1016/j.yexcr.2004.05.039. [DOI] [PubMed] [Google Scholar]
- Chen Y, Zhuang S, Cassenaer S, Casteel DE, Gudi T, Boss GR, Pilz RB. Synergism between calcium and cyclic GMP in cyclic AMP response element-dependent transcriptional regulation requires cooperation between CREB and C/EBP-beta. Mol Cell Biol. 2003;23:4066–4082. doi: 10.1128/MCB.23.12.4066-4082.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho MK, Cho YH, Lee GH, Kim SG. Induction of cyclooxygenase-2 by bovine type I collagen in macrophages via C/EBP and CREB activation by multiple cell signaling pathways. Biochem Pharmacol. 2004;67:2239–2250. doi: 10.1016/j.bcp.2004.02.024. [DOI] [PubMed] [Google Scholar]
- Consilvio C, Vincent AM, Feldman EL. Neuroinflammation, COX-2, and ALS--a dual role? Exp Neurol. 2004;187:1–10. doi: 10.1016/j.expneurol.2003.12.009. [DOI] [PubMed] [Google Scholar]
- Covolan L, Mello LE. Temporal profile of neuronal injury following pilocarpine or kainic acid-induced status epilepticus. Epilepsy Res. 2000;39:133–152. doi: 10.1016/s0920-1211(99)00119-9. [DOI] [PubMed] [Google Scholar]
- Dore S, Otsuka T, Mito T, Sugo N, Hand T, Wu L, Hurn PD, Traystman RJ, Andreasson K. Neuronal overexpression of cyclooxygenase-2 increases cerebral infarction. Ann Neurol. 2003;54:155–162. doi: 10.1002/ana.10612. [DOI] [PubMed] [Google Scholar]
- Ernfors P, Bengzon J, Kokaia Z, Persson H, Lindvall O. Increased levels of messenger RNAs for neurotrophic factors in the brain during kindling epileptogenesis. Neuron. 1991;7:165–176. doi: 10.1016/0896-6273(91)90084-d. [DOI] [PubMed] [Google Scholar]
- Ezaki Y, Nishihara E, Shibata Y, Matsuo T, Kitagawa N, Nagata I, Shinohara K. Vitamin E prevents the neuronal cell death by repressing cyclooxygenase-2 activity. Neuroreport. 2005;16:1163–1167. doi: 10.1097/00001756-200508010-00006. [DOI] [PubMed] [Google Scholar]
- Fricke RA, Prince DA. Electrophysiology of dentate gyrus granule cells. J Neurophysiol. 1984;51:195–209. doi: 10.1152/jn.1984.51.2.195. [DOI] [PubMed] [Google Scholar]
- Gass P, Bruehl C, Herdegen T, Kiessling M, Lutzenburg M, Witte OW. Induction of FOS and JUN proteins during focal epilepsy: congruences with and differences to [14C]deoxyglucose metabolism. Brain Res Mol Brain Res. 1997;46:177–184. doi: 10.1016/s0169-328x(96)00300-2. [DOI] [PubMed] [Google Scholar]
- Gendron TF, Brunette E, Tauskela JS, Morley P. The dual role of prostaglandin E(2) in excitotoxicity and preconditioning-induced neuroprotection. Eur J Pharmacol. 2005;517:17–27. doi: 10.1016/j.ejphar.2005.05.031. [DOI] [PubMed] [Google Scholar]
- Gonzalez GA, Montminy MR. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell. 1989;59:675–80. doi: 10.1016/0092-8674(89)90013-5. [DOI] [PubMed] [Google Scholar]
- Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;40:140–155. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]
- Hara T, Hamada J, Yano S, Morioka M, Kai Y, Ushio Y. CREB is required for acquisition of ischemic tolerance in gerbil hippocampal CA1 region. J Neurochem. 2003;86:805–814. doi: 10.1046/j.1471-4159.2003.01847.x. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathway. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Herdegen T, Fiallos-Estrada C, Schmid W, Bravo R, Zimmermann M. The transcription factor CREB, but not immediate-early gene encoded proteins, is expressed in activated microglia of lumbar spinal cord following sciatic nerve transection in the rat. Neurosci Lett. 1992;142:57–61. doi: 10.1016/0304-3940(92)90619-i. [DOI] [PubMed] [Google Scholar]
- Impey S, Mark M, Villacres EC, Poser S, Chavkin C, Storm DR. Induction of CRE-mediated gene expression by stimuli that generate long-lasting LTP in area CA1 of the hippocampus. Neuron. 1996;16:973–982. doi: 10.1016/s0896-6273(00)80120-8. [DOI] [PubMed] [Google Scholar]
- Impey S, Smith DM, Obrietan K, Donahue R, Wade C, Storm DR. Stimulation of cAMP response element (CRE)-mediated transcription during contextual learning. Nat Neurosci. 1998;1:595–601. doi: 10.1038/2830. [DOI] [PubMed] [Google Scholar]
- Ingvar M, Morgan PF, Auer RN. The nature and timing of excitotoxic neuronal necrosis in the cerebral cortex, hippocampus and thalamus due to flurothyl-induced status epilepticus. Acta Neuropathol (Berl) 1988;75:362–369. doi: 10.1007/BF00687789. [DOI] [PubMed] [Google Scholar]
- Isackson PJ, Huntsman MM, Murray KD, Gall CM. BDNF mRNA expression is increased in adult rat forebrain after limbic seizures: temporal patterns of induction distinct from NGF. Neuron. 1991;6:937–948. doi: 10.1016/0896-6273(91)90234-q. [DOI] [PubMed] [Google Scholar]
- Jankowsky JL, Patterson PH. The role of cytokines and growth factors in seizures and their sequelae. Prog Neurobiol. 2001;63:125–149. doi: 10.1016/s0301-0082(00)00022-8. [DOI] [PubMed] [Google Scholar]
- Jaworski J, Mioduszewska B, Sanchez-Capelo A, Figiel I, Habas A, Gozdz A, Proszynski T, Hetman M, Mallet J, Kaczmarek L. Inducible cAMP early repressor, an endogenous antagonist of cAMP responsive element-binding protein, evokes neuronal apoptosis in vitro. J Neurosci. 2003;23:4519–4526. doi: 10.1523/JNEUROSCI.23-11-04519.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon GS, Shin DH, Cho SS. Induction of transcription factor c-myb expression in reactive astrocytes following intracerebroventricular kainic acid injection in mouse hippocampus. Neurosci Lett. 2004;360:13–16. doi: 10.1016/j.neulet.2004.01.017. [DOI] [PubMed] [Google Scholar]
- Jokeit H, Ebner A. Effects of chronic epilepsy on intellectual functions. Prog Brain Res. 2002;135:455–463. doi: 10.1016/S0079-6123(02)35042-8. [DOI] [PubMed] [Google Scholar]
- Kawaguchi K, Hickey RW, Rose ME, Zhu L, Chen J, Graham SH. Cyclooxygenase-2 expression is induced in rat brain after kainate-induced seizures and promotes neuronal death in CA3 hippocampus. Brain Res. 2005;1050:130–137. doi: 10.1016/j.brainres.2005.05.038. [DOI] [PubMed] [Google Scholar]
- Kiessling M, Gass P. Immediate early gene expression in experimental epilepsy. Brain Pathol. 1993;3:381–393. doi: 10.1111/j.1750-3639.1993.tb00766.x. [DOI] [PubMed] [Google Scholar]
- Lee B, Butcher GQ, Hoyt KR, Impey S, Obrietan K. Activity-dependent neuroprotection and cAMP response element-binding protein (CREB): kinase coupling, stimulus intensity, and temporal regulation of CREB phosphorylation at serine 133. J Neurosci. 2005;25:1137–1148. doi: 10.1523/JNEUROSCI.4288-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang G, Wolfgang CD, Chen BP, Chen TH, Hai T. ATF3 gene. Genomic organization, promoter, and regulation. J Biol Chem. 1996;271:1695–1701. doi: 10.1074/jbc.271.3.1695. [DOI] [PubMed] [Google Scholar]
- Liu D, Wu L, Breyer R, Mattson MP, Andreasson K. Neuroprotection by the PGE2 EP2 receptor in permanent focal cerebral ischemia. Ann Neurol. 2005;57:758–761. doi: 10.1002/ana.20461. [DOI] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35:605–623. doi: 10.1016/s0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- Lothman EW, Bertram EH, 3rd, Stringer JL. Functional anatomy of hippocampal seizures. Prog Neurobiol. 1991;37:1–82. doi: 10.1016/0301-0082(91)90011-o. [DOI] [PubMed] [Google Scholar]
- Lothman EW, Stringer JL, Bertram EH. The dentate gyrus as a control point for seizures in the hippocampus and beyond. Epilepsy Res Suppl. 1992;7:301–313. [PubMed] [Google Scholar]
- Mabuchi T, Kitagawa K, Kuwabara K, Takasawa K, Ohtsuki T, Xia Z, Storm D, Yanagihara T, Hori M, Matsumoto M. Phosphorylation of cAMP response element binding protein in hippocampal neurons as a protective response after exposure to glutamate in vitro and ischemia in vivo. J Neurosci. 2001;21:9204–9213. doi: 10.1523/JNEUROSCI.21-23-09204.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER. Control of memory formation through regulated expression of a CaMKII transgene. Science. 1996;274:1678–1683. doi: 10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- McCullough L, Wu L, Haughey N, Liang X, Hand T, Wang Q, Breyer RM, Andreasson K. Neuroprotective function of the PGE2 EP2 receptor in cerebral ischemia. J Neurosci. 2004;24:257–268. doi: 10.1523/JNEUROSCI.4485-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore AN, Waxham MN, Dash PK. Neuronal activity increases the phosphorylation of the transcription factor cAMP response element-binding protein (CREB) in rat hippocampus and cortex. J Biol Chem. 1996;271:14214–14220. doi: 10.1074/jbc.271.24.14214. [DOI] [PubMed] [Google Scholar]
- Morimoto K, Fahnestock M, Racine RJ. Kindling and status epilepticus models of epilepsy: rewiring the brain. Prog Neurobiol. 2004;73:1–60. doi: 10.1016/j.pneurobio.2004.03.009. [DOI] [PubMed] [Google Scholar]
- Niehof M, Manns MP, Trautwein C. CREB controls LAP/C/EBP beta transcription. Mol Cell Biol. 1997;17:3600–3613. doi: 10.1128/mcb.17.7.3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obrietan K, Gao XB, van den Pol AN. Excitatory actions of GABA increase BDNF expression via a MAPK-CREB-dependent mechanism--a positive feedback circuit in developing neurons. J Neurophysiol. 2002;88:1005–1015. doi: 10.1152/jn.2002.88.2.1005. [DOI] [PubMed] [Google Scholar]
- Obrietan K, Hoyt KR. CRE-mediated transcription is increased in Huntington's disease transgenic mice. J Neurosci. 2004;24:791–796. doi: 10.1523/JNEUROSCI.3493-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obrietan K, Impey S, Smith D, Athos J, Storm DR. Circadian regulation of cAMP response element-mediated gene expression in the suprachiasmatic nuclei. J Biol Chem. 1999;274:17748–17756. doi: 10.1074/jbc.274.25.17748. [DOI] [PubMed] [Google Scholar]
- Olney JW, deGubareff T, Sloviter RS. “Epileptic” brain damage in rats induced by sustained electrical stimulation of the perforant path”. II Ultrastructural analysis of acute hippocampal pathology. Brain Res Bull. 1983;10:699–712. doi: 10.1016/0361-9230(83)90038-2. [DOI] [PubMed] [Google Scholar]
- Ong WY, Lim HM, Lim TM, Lutz B. Kainate-induced neuronal injury leads to persistent phosphorylation of cAMP response element-binding protein in glial and endothelial cells in the hippocampus. Exp Brain Res. 2000;131:178–186. doi: 10.1007/s002219900329. [DOI] [PubMed] [Google Scholar]
- Orzi F, Zoli M, Passarelli F, Ferraguti F, Fieschi C, Agnati LF. Repeated electroconvulsive shock increases glial fibrillary acidic protein, ornithine decarboxylase, somatostatin and cholecystokinin immunoreactivities in the hippocampal formation of the rat. Brain Res. 1990;533:223–231. doi: 10.1016/0006-8993(90)91343-f. [DOI] [PubMed] [Google Scholar]
- Parent JM, Lowenstein DH. Seizure-induced neurogenesis: are more new neurons good for an adult brain? Prog Brain Res. 2002;135:121–131. doi: 10.1016/S0079-6123(02)35012-X. [DOI] [PubMed] [Google Scholar]
- Park SA, Kim TS, Choi KS, Park HJ, Heo K, Lee BI. Chronic activation of CREB and p90RSK in human epileptic hippocampus. Exp Mol Med. 2003;35:365–370. doi: 10.1038/emm.2003.48. [DOI] [PubMed] [Google Scholar]
- Qian Z, Gilbert ME, Colicos MA, Kandel ER, Kuhl D. Tissue-plasminogen activator is induced as an immediate-early gene during seizure, kindling and long-term potentiation. Nature. 1993;361:453–457. doi: 10.1038/361453a0. [DOI] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation. II Motor seizure Electroencephalogr. Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Rakhade SN, Yao B, Ahmed S, Asano E, Beaumont TL, Shah AK, Draghici S, Krauss R, Chugani HT, Sood S, Loeb JA. A common pattern of persistent gene activation in human neocortical epileptic foci. Ann Neurol. 2005;58:736–747. doi: 10.1002/ana.20633. [DOI] [PubMed] [Google Scholar]
- Revuelta M, Castano A, Venero JL, Machado A, Cano J. Long-lasting induction of brain-derived neurotrophic factor is restricted to resistant cell populations in an animal model of status epilepticus. Neuroscience. 2001;103:955–969. doi: 10.1016/s0306-4522(01)00032-x. [DOI] [PubMed] [Google Scholar]
- Rizzi M, Perego C, Aliprandi M, Richichi C, Ravizza T, Colella D, Veliskova J, Moshe SL, De Simoni MG, Vezzani A. Glia activation and cytokine increase in rat hippocampus by kainic acid-induced status epilepticus during postnatal development. Neurobiol Dis. 2003;14:494–503. doi: 10.1016/j.nbd.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Rockwell P, Martinez J, Papa L, Gomes E. Redox regulates COX-2 upregulation and cell death in the neuronal response to cadmium. Cell Signal. 2004;16:343–353. doi: 10.1016/j.cellsig.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Rogove AD, Tsirka SE. Neurotoxic responses by microglia elicited by excitotoxic injury in the mouse hippocampus. Curr Biol. 1998;8:19–25. doi: 10.1016/s0960-9822(98)70016-8. [DOI] [PubMed] [Google Scholar]
- Schweitzer JS, Patrylo PR, Dudek FE. Prolonged field bursts in the dentate gyrus: dependence on low calcium, high potassium, and nonsynaptic mechanisms. J Neurophysiol. 1992;68:2016–2025. doi: 10.1152/jn.1992.68.6.2016. [DOI] [PubMed] [Google Scholar]
- Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellualr signals. Ann Rev Biochem. 1999;68:821–861. doi: 10.1146/annurev.biochem.68.1.821. [DOI] [PubMed] [Google Scholar]
- Shohami E, Novikov M, Bass R, Yamin A, Gallily R. Closed head injury triggers early production of TNF alpha and IL-6 by brain tissue. J Cereb Blood Flow Metab. 1994;14:615–619. doi: 10.1038/jcbfm.1994.76. [DOI] [PubMed] [Google Scholar]
- Simonato M, Hosford DA, Labiner DM, Shin C, Mansbach HH, McNamara JO. Differential expression of immediate early genes in the hippocampus in the kindling model of epilepsy. Brain Res Mol Brain Res. 1991;11:115–124. doi: 10.1016/0169-328x(91)90113-c. [DOI] [PubMed] [Google Scholar]
- Stefan H, Pauli E. Progressive cognitive decline in epilepsy: an indication of ongoing plasticity. Prog Brain Res. 2002;135:409–417. doi: 10.1016/S0079-6123(02)35038-6. [DOI] [PubMed] [Google Scholar]
- Steward O, Torre ER, Tomasulo R, Lothman E. Neuronal activity up-regulates astroglial gene expression. Proc Natl Acad Sci (USA) 1991;88:6819–6823. doi: 10.1073/pnas.88.15.6819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoll G, Jander S, Schroeter M. Detrimental and beneficial effects of injury-induced inflammation and cytokine expression in the nervous system. Adv Exp Med Biol. 2002;513:87–113. doi: 10.1007/978-1-4615-0123-7_3. [DOI] [PubMed] [Google Scholar]
- Tanabe T, Tohnai N. Prostaglandins Other Lipid Mediat. 2002. Cyclooxygenase isozymes and their gene structures and expression; pp. 68–69.pp. 95–114. [DOI] [PubMed] [Google Scholar]
- Tanaka K. Alteration of second messengers during acute cerebral ischemia - adenylate cyclase, cyclic AMP-dependent protein kinase, and cyclic AMP response element binding protein. Prog Neurobiol. 2001;65:173–207. doi: 10.1016/s0301-0082(01)00002-8. [DOI] [PubMed] [Google Scholar]
- Taupin V, Gogusev J, Descamps-Latscha B, Zavala F. Modulation of tumor necrosis factor-alpha, interleukin-1 beta, interleukin-6, interleukin-8, and granulocyte/macrophage colony-stimulating factor expression in human monocytes by an endogenous anxiogenic benzodiazepine ligand, triakontatetraneuropeptide: evidence for a role of prostaglandins. Mol Pharmacol. 1993;43:64–69. [PubMed] [Google Scholar]
- Thiel G, Al Sarraj J, Vinson C, Stefano L, Bach K. Role of basic region leucine zipper transcription factors cyclic AMP response element binding protein (CREB), CREB2, activating transcription factor 2 and CAAT/enhancer binding protein alpha in cyclic AMP response element-mediated transcription. J Neurochem. 2005;92:321–336. doi: 10.1111/j.1471-4159.2004.02882.x. [DOI] [PubMed] [Google Scholar]
- Tsirka SE, Gualandris A, Amaral DG, Strickland S. Excitotoxin-induced neuronal degeneration and seizure are mediated by tissue plasminogen activator. Nature. 1995;377:340–344. doi: 10.1038/377340a0. [DOI] [PubMed] [Google Scholar]
- Tu B, Bazan NG. Hippocampal kindling epileptogenesis upregulates neuronal cyclooxygenase-2 expression in neocortex. Exp Neurol. 2003;179:167–175. doi: 10.1016/s0014-4886(02)00019-5. [DOI] [PubMed] [Google Scholar]
- Turrin NP, Rivest S. Innate immune reaction in response to seizures: implications for the neuropathology associated with epilepsy. Neurobiol Dis. 2004;16:321–334. doi: 10.1016/j.nbd.2004.03.010. [DOI] [PubMed] [Google Scholar]
- Turski L, Ikonomidou C, Turski WA, Bortolotto ZA, Cavalheiro EA. Review: cholinergic mechanisms and epileptogenesis. The seizures induced by pilocarpine: a novel experimental model of intractable epilepsy. Synapse. 1989;3:154–171. doi: 10.1002/syn.890030207. [DOI] [PubMed] [Google Scholar]
- Turski WA, Cavalheiro EA, Schwarz M, Czuczwar SJ, Kleinrok Z, Turski L. Limbic seizures produced by pilocarpine in rats: behavioural, electroencephalographic and neuropathological study. Behav Brain Res. 1983;9:315–335. doi: 10.1016/0166-4328(83)90136-5. [DOI] [PubMed] [Google Scholar]
- Voutsinos-Porche B, Koning E, Kaplan H, Ferrandon A, Guenounou M, Nehlig A, Motte J. Temporal patterns of the cerebral inflammatory response in the rat lithium-pilocarpine model of temporal lobe epilepsy. Neurobiol Dis. 2004;17:385–402. doi: 10.1016/j.nbd.2004.07.023. [DOI] [PubMed] [Google Scholar]
- Walton M, Sirimanne E, Williams C, Gluckman P, Dragunow M. The role of the cyclic AMP-responsive element binding protein (CREB) in hypoxic-ischemic brain damage and repair. Brain Res Mol Brain Res. 1996;43:21–29. doi: 10.1016/s0169-328x(96)00144-1. [DOI] [PubMed] [Google Scholar]
- Walton M, Woodgate AM, Muravlev A, Xu R, During MJ, Dragunow M. CREB phosphorylation promotes nerve cell survival. J Neurochem. 1999;73:1836–1842. [PubMed] [Google Scholar]
- Wu KK, Liou JY, Cieslik K. Transcriptional Control of COX-2 via C/EBPβ. Arterioscler Thromb Vasc Biol. 2005;25:679–685. doi: 10.1161/01.ATV.0000157899.35660.61. [DOI] [PubMed] [Google Scholar]
- Wu YP, Siao CJ, Lu W, Sung TC, Frohman MA, Milev P, Bugge TH, Degen JL, Levine JM, Margolis RU, Tsirka SE. The tissue plasminogen activator (tPA)/plasmin extracellular proteolytic system regulates seizure-induced hippocampal mossy fiber outgrowth through a proteoglycan substrate. J Cell Biol. 2000;148:1295–1304. doi: 10.1083/jcb.148.6.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Bleich D. Transcriptional regulation of cyclooxygenase-2 gene in pancreatic beta-cells. J Biol Chem. 2004;279:35403–35411. doi: 10.1074/jbc.M404055200. [DOI] [PubMed] [Google Scholar]
- Young D, Lawlor PA, Leone P, Dragunow M, During MJ. Environmental enrichment inhibits spontaneous apoptosis, prevents seizures and is neuroprotective. Nat Med. 1999;5:448–453. doi: 10.1038/7449. [DOI] [PubMed] [Google Scholar]