

Abstract
We have studied DNA minicircles containing the ATF/CREB binding site for GCN4 by using a combination of cyclization kinetics experiments and Monte Carlo simulations. Cyclization rates were determined with and without GCN4 for DNA constructs containing the ATF/CREB site separated from a phased A-tract multimer bend by a variable length phasing adaptor. The cyclization results show that GCN4 binding does not significantly change the conformation of the ATF/CREB site, which is intrinsically slightly bent toward the major groove. Monte Carlo simulations quantitate the ATF/CREB site structure as an 8° bend toward the major groove in a coordinate frame near the center of the site. The ATF/CREB site is underwound by 53° relative to the related AP-1 site DNA. The effect of GCN4 binding can be modeled either as a decrease in the local flexibility, corresponding to an estimated 60% increase in the persistence length for the 10-bp binding site, or possibly as a small decrease (1°) in intrinsic bend angle. Our results agree with recent electrophoretic and crystallographic studies and demonstrate that cyclization and simulation can characterize subtle changes in DNA structure and flexibility.
Dimers of bZIP proteins bind dyad symmetric DNA binding sites including the AP-1 site, 5′-ATGACTCAT-3′, and the ATF/CREB site, 5′-ATGACGTCAT-3′. These two sites share the same 5′-ATGA-3′ half-site, but most bZIP proteins can discriminate between them based on the difference in half-site spacing, usually binding weakly or not at all to one of the sites. GCN4, a yeast transcriptional activator, is an exception because it binds the AP-1 and ATF/CREB sites with comparable affinity (1). GCN4, a 281-residue protein from Saccharomyces cerevisiae, activates amino acid biosynthetic pathways under amino acid starvation conditions (2, 3). The 9-bp AP-1 site is the functional binding site in vivo, but mutagenesis studies have shown that the central 7-bp site, 5′-TGACTCA-3′, is the preferentially recognized consensus (4, 5).
Both the circular permutation and phasing gel mobility experiments were done with GCN4 and the AP-1 site (6). These two experiments gave contradictory results with the full-length protein, but the assays with bZIP peptide showed that it does not bend DNA. Recently, the same experiments were done on a bZIP peptide of GCN4 (the 55-aa peptide contains an N-terminal serine followed by residues 228–281) with both the AP-1 and ATF/CREB sites (7). For the AP-1 site, the DNA is straight, whereas for the ATF/CREB site, the DNA was found to be bent toward the major groove.
Two different bZIP peptides derived from GCN4 have been cocrystallized with DNA, one with the AP-1 site and the other with the ATF/CREB site (8–10). The AP-1 site is not appreciably bent in a cocrystal with a 58-aa peptide. In contrast, the ATF/CREB site has an overall bend of about 20° when complexed to a similar 62-aa peptide. Both structures show the same protein–DNA contacts, which are possible because the bending of the ATF/CREB site in the complex creates a contact surface that resembles the AP-1 site.
DNA bending can be studied in solution by DNA ring closure, which can measure the degree of an intrinsic or protein-induced DNA bend (refs. 11 and 12, and references therein). The experiments are sensitive to deformations in DNA that include bending, helical repeat length, and torsional and bending flexibility (12–14). We are able to further interpret the experimental results by using Monte Carlo simulations to refine the DNA structural model (11). The AP-1 site has been shown to be straight in solution by using cyclization kinetics (15). Furthermore, it remains straight when bound to the bZIP proteins Fos/Jun (15, 16) or GCN4 (ref. 17 and unpublished data).
We have studied the ATF/CREB binding site phased against A-tract bends by using cyclization kinetics. In our experiments, the full-length GCN4 protein and the 58-aa bZIP peptide used in cocrystallization (10) behave indistinguishably. The ATF/CREB site molecules show a slight bend toward the major groove, both alone and bound to protein. The optimum cyclization length indicates a significantly decreased DNA torsion relative to the AP-1 site, in both the presence and absence of protein. There is a small but significant decrease in cyclization probability on protein binding, which is more pronounced for the out-of-phase molecules, due apparently to a small protein-induced stiffening of the DNA. Monte Carlo simulations of the ATF/CREB site allow quantitative characterization of a small intrinsic DNA bend and underwinding across the site, relative to the AP-1 site. We conclude that GCN4 is able to bind the existing conformation of the site with no significant distortion, in agreement with Paolella et al. (7).
MATERIALS AND METHODS
GCN4.
GCN4 was purified as described (6) from cells generously provided by Christophe Ampe and Tom Steitz (Yale Univ.) and was stored at −70°C at 25 μM in 20 mM Tris, pH 8.0/1 mM EDTA/50 mM KCl/10 mM DTT/50% glycerol. A 58-residue bZIP peptide containing the C-terminal 56 residues of GCN4 preceded by Met and Lys was generously provided by Tom Ellenberger (Harvard Medical School, Boston) and stored at 4°C at 2 μM in 20 mM Tris, pH 8.0/1 mM EDTA/50 mM KCl/50% glycerol (18).
ATF/CREB Site Minicircles.
The ATF/CREB site molecules were made in the same manner as the yeast AP-1 plasmids (15). The ATF/CREB molecules differ from the AP-1 molecules by one inserted GC bp in the binding site. SalI-EcoRV double-digested plasmids were used as PCR templates for the minicircles. Primer 2 was described (19) and was paired with primer 1a, 1b, or 1c: 1a, 5′-GCAGATATCGATTCCATGGCCACGTTGTAGC-3′; 1b, 5′-GCAGATATCGATTCCATGGCAGCACGTTGTAGC-3′; 1c, 5′-GCAGATATCGATTCCATGGCAAAGCACGTTGTAGC-3′. PCR reactions were as described except the MgCl2 concentration was 4.5 mM (19). PCR products were restricted and purified as described (19) with the exceptions that 3.75 units of ClaI were used per 150 μl of PCR and the DNAs were purified on 10% native gels [40:1, acrylamide:N,N′-methylenebisacrylamide/TBE (50 mM Tris/50 mM boric acid/1 mM EDTA, pH 8.3)].
Cyclization Kinetics Measurements.
Time course experiments were performed essentially as described (19). DNA concentrations were 5–10 nM, and DNAs were incubated with 0 or 30 nM GCN4, either full-length protein or peptide, expressed as the dimer concentration. The reaction buffer was 20 mM Tris, pH 7.5/1 mM EDTA/50 mM KCl/5 mM MgCl2/1 mM ATP/0.05% Nonidet P-40 (Shell)/20 μg/ml gelatin/0.5 mM DTT. All reactions were at 21°C. Poly(dA-dT) was used as competitor DNA at 4 μg/ml. The extent of binding was monitored in each experiment by a gel shift run on an aliquot removed before the addition of ligase [8% native gels (75:1 acrylamide:N,N′-methylenebisacrylamide/50 mM TBE, 1.5 mm thick, 15 × 15 cm) at 25°C, 400 V, 1 hr].
Reactions were initiated by the addition of 7.5 μl of 50,000 units/ml T4 DNA ligase for a final volume of 75 μl (5,000 units/ml ligase, final concentration). Time points were taken over 1–15 min as described (19), and the DNA was analyzed on 6% native gels (40:1 acrylamide:N,N′-methylenebisacrylamide/50 mM TBE).
Because all the molecules undergo both cyclization and bimolecular association, we were able to calculate both rate constants directly (12, 19). In fitting the data, some of the data fit better with a lower effective ligatable DNA concentration than that calculated from the total DNA concentration because of (we assume) the presence of unreactive ends. We performed KINSIM simulations (20) to determine the relationship between the observed J factors and the real J factors based on concentration differences. We corrected the data for this difference. For molecule populations where the effective concentration was between 30 and 50% of the total concentration, J factors were corrected by multiplication by factors ≥1, ranging up to 4. When the effective concentration is >50% of the total, corrections were less than a factor of 2, which corresponds to the estimated experimental error. All of the data were corrected, but 63 of 81 experiments were multiplied by less than a factor of 2, and 18 or 22% were multiplied by factors of 2–4. The corrected results are consistent with the rest of the data with which they were averaged. The J factors reported are average values from multiple experiments, and the error ranges are standard deviations for the averaged experiments.
Gel Shift Competition.
Circular DNAs were prepared from linear DNAs of known specific activity in 200-μl reactions containing the reaction buffer used for cyclization and 500 units/ml T4 DNA ligase. The ligations were incubated overnight at 21°C, phenol-extracted, and gel-purified.
The binding buffer was the same as for cyclization, without the ATP and with the additions of 4 μg/ml poly(dA-dT) and 100 μg/ml BSA. Experiments were performed with the same molecule in both linear and circular forms and either protein or peptide. Linear or circular DNA was incubated with either protein, peptide, or the other DNA for 4 hr, at which time the third component was added, and incubation was continued overnight. The two DNA concentrations were equal in each experiment (5–10 nM), peptide concentrations were 0–128 nM, and protein concentrations were 0–30 nM. The final mixtures, 6 μl, were analyzed on 8% native constant temperature gels as above except at 350 V for 2.5 hr.
Monte Carlo Simulations.
The Monte Carlo simulations are essentially as described (11), with the modifications to be described by J.D.K. and D.M.C. (unpublished data). The simulations were carried out on Silicon Graphics workstations (Mountain View, CA). The DNA sequences were the same as those used in the experiments. The programs recognize A-tracts and give them the junction model characteristics previously defined (11).
The ATF/CREB site is modeled as 9-bp “steps,” which define the 10-bp sequence. The bend is denoted by a flag at the central base pair, and the placement of the bend was changed by altering the twisting into and out of the site, effectively moving the bend center. The unwinding is distributed across the flanking steps on each side and added to the altered twist at the terminal steps. The change in bending flexibility of the site is applied to each of the 9 steps. The helical repeat of the B-form DNA was defined as 10.45 bp/turn, in agreement with the observed optimal cyclization length for molecules with the AP-1 site or catabolite activator protein (15, 19). B-DNA is further characterized by variables for the torsional modulus (C) and the persistence length (P). The chain generation and analysis were performed as described (11, 21–23) with modifications (J.D.K. and D.M.C., unpublished data).
To determine the parameters that best fit the data set, initially we fit Gaussian curves to the simulated length dependence of J for in-phase constructs and compared them with experiment (see Figs. 3 and 4) to identify parameter sets that were consistent with experiment. The out-of-phase molecules were first tested for the experimental relationships of 9A17>11T15 and 11A17>13T15. When these criteria were met, the simulation J factors were compared with the experimental values to make sure they were the same magnitude. To determine the best parameter set and error ranges, an absolute error (ABS) and a relative error (REL) were calculated for each simulated J factor.
 |
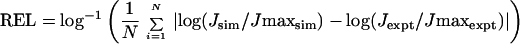 |
where Jexpt is the linear average of the experimental J factors for a particular molecule i, max denotes the best cyclizer (the 15A9 molecule), and N is the number of molecules in the set. The best set of parameters should yield a minimum for both of these criteria, with ABS = 0 and REL = 1 being perfect agreement. Both of these values were calculated for each set of simulation parameters, but REL was favored because it had sharper minima. Values of REL for the complete molecule set are shown in Fig. 1 for two representative parameters.
Figure 3.

Comparison of ATF/CREB site cyclization kinetics data and Monte Carlo simulation results for free DNA. Experimental J factors for in-phase constructs are filled circles with error bars: 7T9 (149 bp), 9T9 (151 bp), 11T9 (153 bp), 13A9 (155 bp), 15A9 (157 bp), 15A11 (158 bp), 17A9 (159 bp), 17A11 (160 bp), and 19A11 (162 bp). The in-phase data (solid line) and simulations are fit to sums of Gaussians as previously described (31). The simulation results are represented by open circles, and their curve fit is the dotted line. J factors for the out-of-phase molecules 9T15 (157 bp), 11T15 (159 bp), 13T15 (161 bp), 9A17 (159 bp), 11A17 (161 bp), and 13A17 (163 bp) fall below the in-phase values. Filled diamonds with error bars are the experimental values, and open squares represent the simulation results.
Figure 4.

Comparison of ATF/CREB site cyclization kinetics data and Monte Carlo simulation results for protein-bound DNA. (a) J factors for in-phase constructs bound to protein, either full-length GCN4 or the bZIP peptide. Experimental values are the filled squares and are fit with the solid line. The curve fit is identical to the one for the DNA alone shown in Fig. 3. In both cases, the optimal cyclization length is 157.7 bp. Open circles and the dash-dot curve fit are the Monte Carlo results from modeling the ATF/CREB site with a decreased bend angle of 7°. Open diamonds and the dashed line are the Monte Carlo results for decreased flexibility of the site, characterized by a P of 144 bp. (b) Results for out-of-phase constructs, showing the fit of the two theoretical models (open symbols as in (a)) to the experimental results (filled squares).
Figure 1.

REL values as a function of representative ATF/CREB site parameters. REL values are determined by the REL equation described under Materials and Methods. The other parameters are held constant at their best fit values whereas the one parameter is varied. The variation of REL with a parameter is used to determine simulation errors. (a) Graph of REL vs. twist angle for the whole molecule set. The twist angle alters the phasing of the ATF/CREB site and the A-tracts. (b) Graph of REL vs. the bend angle for the whole molecule set.
RESULTS
DNA Molecules.
Cyclization kinetics experiments are used to determine J factors, set equal to the ratio of the unimolecular cyclization rate constant to the bimolecular ligation rate constant (24, 25). Cyclization kinetics experiments on protein–DNA complexes are preferably performed on small DNAs (Fig. 2), because differences in cyclization efficiency caused by bending or other DNA structural changes are masked by the increased flexibility of longer molecules. For small molecules J also depends strongly on overall length, because the DNA ends must be torsionally aligned for ligation. The J factors thus have a periodicity corresponding to the DNA helical repeat (13, 14). Our molecules have two phasing adaptor DNA segments, which allow independent variation of the bend phasing between the protein binding site and six phased A-tracts and alteration of the total length. By phasing two bends against each other, the effects of small DNA bends are amplified, and bend direction can be determined. A pair of in-phase bends forms a C-shaped molecule that cyclizes more readily than the S-shaped molecule containing two out-of-phase bends. The two-base ClaI overhang minimizes Ka, the equilibrium constant for bimolecular association; this extends the range of accessible J factors (11, 12).
Figure 2.

ATF/CREB site molecules. The nomenclature specifies the length of adaptor I (center of EcoRI site to center of MspI site), the orientation of the A-tract sequence, and the length of adaptor II, in that order. The A-tract adenines are on the strand that reads 5′–3′ left to right. The bend phasing is described by adaptor II. PCR primers are 1 and 2. The total length of the molecules is 149–163 bp. The sequence of the 47-bp StyI fragment is: 5′-CATGGCAGCACGTTGTAGCTCGAGCAAAAAAAAATGACGTCATCCAC-3′ left to right.
The DNAs were based on those constructed for catabolite activator protein (19) (Fig. 2). Five molecules, 9T9, 15A9, 17A11, 11T15, and 11A17, were cloned. [The nomenclature was previously reported (19); the first number denotes the length of the terminal linker and the second one characterizes the linker between the ATF/CREB site and the A-tracts. The letter represents the orientation of the A-tracts]. Radiolabeled DNA was synthesized by PCR. Three different PCR primers complementary to the binding site end of the DNAs were used: a primer with no mismatches in the region complementary to the plasmid sequence and two primers with either a 2-bp insertion or deletion. The three resulting DNAs, e.g., 7T9, 9T9, and 11T9, maintain the spacing between the A-tracts and the ATF/CREB site while altering the length and torsional alignment of the ends. The DNAs span a length range of 15 bp to cover more than a helical repeat of DNA.
Cyclization Kinetics.
For all of the DNAs, the J factor for the DNA bound to protein was the same as for DNA bound to the 58-aa bZIP peptide of GCN4. This confirms that the bZIP domain contains the important DNA contacts and binds the same DNA structure as the full-length protein.
The optimal length for cyclization, determined from the molecules 17A11, 15A9, and 9T9, is 157.7 bp (from the center of the Gaussians in Figs. 3 and 4a), both with and without bound protein. In order for 157.7 bp to be an integral number of helical repeats, the average helical repeat for the non-A-tract part of the molecule must be about 10.62. The analogous molecules containing the AP-1 site (shorter by 1 GC pair) have an optimum cyclization length of 156 bp (15). We assume that the structural change responsible for unwinding at the ATF/CREB site is localized to the site; the estimated angle corresponds to about 1.7 bp or 60° relative to molecules containing the AP-1 site. Simulation of the cyclization data reduces the unwinding angle estimate to 53° (see below). However, the experiments do not distinguish whether unwinding results from a change in twist or from the presence of intrinsic negative writhe in the site because of two (or more) bends in the 2-fold symmetric sequence.
Molecules that have a half-integral number of helical repeats between the ATF/CREB site and the A-tracts (17A11, 15A9, and 9T9) show similar J factors alone and bound to protein or peptide (Figs. 3 and 4a). Protein-free ATF/CREB site molecules with an integral number of helical repeats between the ATF/CREB site and the A-tracts, 11A17 and 11T15, show approximately a 7-fold decrease in cyclization efficiency compared with the half-integral spaced ATF/CREB site molecules (Tables 1 and 2 and Fig. 3). This difference in J factors between molecules with different phasings is characteristic of an intrinsic DNA bend. The molecules that favor cyclization have the ATF/CREB site centered a half-integral number of helical repeats from the center of the last A-tract bend, implying a bend toward the major groove of the ATF/CREB site, which would then be in-phase relative to the minor groove A-tract bend (26). The simplified analysis previously described (27) yields a bend angle of 13° for our cyclization data (17). The computer simulations (see below) reduce the bend angle estimate to 8°.
Table 1.
Comparison of in-phase and out-of-phase cyclization of ATF/CREB site constructs
| Molecule | Length, bp |
J, nM
|
J(+)/J(−) | |
|---|---|---|---|---|
| DNA alone | +GCN4 | |||
| 15A9 | 157 | 165 | 168 | 1.02 |
| 9T15 | 157 | 35 | 11.5 | 0.33 |
| 17A9 | 159 | 122 | 90 | 0.74 |
| 11T15 | 159 | 11.9 | 4.46 | 0.37 |
| 9A17 | 159 | 18.7 | 9.29 | 0.50 |
| 13T15 | 161 | 3.23 | 1.35 | 0.42 |
| 11A17 | 161 | 5.49 | 2.66 | 0.48 |
Table 2.
Ratios of J factors of molecules of the same length
| Comparison | Molecules |
J ratio
|
|
|---|---|---|---|
| DNA alone | +GCN4 | ||
| In-phase/out-of-phase | 15A9/9T15 | 4.7 | 14.6 |
| 17A9/11T15 | 10.3 | 20.2 | |
| 17A9/9A17 | 6.5 | 9.7 | |
| Out-of-phase/out-of-phase | 9A17/11T15 | 1.6 | 2.1 |
| 11A17/13T15 | 1.7 | 2.0 | |
This table compares molecules with the same or different phasings.
The out-of-phase molecules show an approximately 2-fold difference in the J factors for the DNA alone and bound to protein or peptide, in contrast to the in-phase molecules, for which the difference is significantly smaller (Tables 1 and 2). The results from cyclization kinetics experiments can be compared with competition gel shift experiments on linear and circular DNAs (19) because the effect of protein binding on cyclization should be reflected in the ability of the protein to bind preformed circles compared with linear molecules.
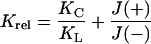 |
For an intrinsic bend, the binding site will most likely be distorted in the outward facing conformation of a preformed circle such that protein binding will be disfavored. Competition gel shifts between linear and circular DNAs corroborate the cyclization results (data not shown). For the in-phase molecule, 15A9, Krel determined from the average of several experiments is 0.87 ± 0.26. This agrees with the absence of an appreciable effect of protein binding on J values. In contrast, for the out-of-phase molecule, 11T15, KC/KL is 0.41 ± 0.18; the average J(+)/J(−) is 0.37. This preference for linear DNA in the competition experiments and decreased J factors for protein-bound DNA can be explained by a stiffening of the site on GCN4 binding. Stiffening the site would make it harder for the out-of-phase molecules to bend the additional amount required for cyclization but would have little effect on the in-phase molecules, for which less distortion is required to cyclize.
Finally, we observe a difference of about a factor of 2 between the J factors for out-of-phase molecules that are the same length (Tables 1 and 2). 9A17 and 11A17 cyclize faster than their equal length isomers 11T15 and 13T15, both with and without bound protein. The simulations show that the relative cyclization probabilities for these isomers are very sensitive to the orientation of the intrinsic bend relative to the A-tract bends. Because of uncertainties in the local helical repeat, sequence information cannot specify precisely the orientation of the center of the binding site relative to the A-tract bend direction. In the simulations, we had to rotate the site by about half a base pair to correctly predict the relative cyclization rates of the A17 and T15 sets.
Monte Carlo Simulations.
Monte Carlo simulations were carried out to further characterize the DNA structure of the ATF/CREB site. Our goal is to define a set of DNA parameters that produce J factors similar to the experimental values. Two sets of simulations were performed to determine the structure of the DNA alone and when bound to GCN4. Simulations of protein-bound DNA reflect the DNA structure when the protein is bound but do not contain a representation of the protein in the calculations.
The DNA molecules are treated as consisting of three types of DNA structures: A-tracts, B-DNA, and the ATF/CREB site. The structure of the A-tract-containing sequences has been defined (11, 28). The B-DNA segments are characterized by parameters for the helical repeat, the C, and the bending flexibility or P. The ATF/CREB site is modeled as a series of base pairs that differ from B-DNA. The site is defined by a bend angle, an unwinding angle across the site, an independent P to allow for a change in local flexibility, and a twist angle that effectively moves the bend center by twisting the site around the DNA. Between the B-DNA and the ATF/CREB site there are seven different parameters, which we determined to be the number necessary to fit most if not all aspects of the experimental data adequately.
Our model includes some parameters that affect the whole site, including the flexibility and twist/bp, and some that are localized to 1 or 2 bp, which include the bend at the center and the twist angle at the two terminal steps. We assume that unwinding is confined to the 10-bp ATF/CREB site, which we model as untwisting, although it could also be because of negative writhe. With unwinding localized to the site, the helical repeat for the non-A-tract DNA is 10.45 bp/turn. We initially modeled the bend center of the ATF/CREB site to the center of the 10-bp sequence as in the cocrystal (8), but during simulations, as discussed above, we also found it necessary to rotate the apparent bend center, moving it slightly toward the A-tracts, to get better agreement with the slight phase differences between the out-of-phase molecules.
The parameters are arbitrarily varied to promote agreement between the simulated and experimentally determined J factors. To determine the best parameter set, we placed more emphasis on the relative J values for the molecules than on their absolute values. In so doing we implicitly acknowledge that there may be a small systematic error in the absolute values of the simulated J values. This choice of optimization criterion is a source of the systematic displacement, about 1.5-fold, of the theoretical curve from the experimental data in Fig. 3.
Once good agreement was found between the experimental and simulated sets of J factors, the parameter set was used as a comparison point for simulations that vary only one parameter, holding the rest at the optimal values. The best parameter set has REL values that are a minimum with respect to all the DNA parameters. The REL, defined under Materials and Methods, is a measure of how well the simulations predict the observed ratio of experimental J values for different molecules. The inability to minimize both REL and ABS simultaneously may mean that yet another parameter is needed, such as an altered C in the binding site. The reported error ranges reflect the change required to produce significant differences in REL on variation of a single parameter. Intermediate values were not tested for all variables.
Our simulation results for the intrinsic structure of the ATF/CREB site include an 8° ± 1° bend toward the major groove rotated 17° ± 3° (≈1/2 bp) toward the A-tracts in the DNA. The bend represents the overall bend angle required to account for the ≈7-fold difference between the J factors for in- and out-of-phase molecules. On this basis and allowing for approximately 2-fold scatter in the J values, we estimate that the sensitivity limit for detecting bends is <5°. The ATF/CREB site has 53° ± 3° of unwinding (relative to the AP-1 site) distributed across it through some unresolved combination of twist and writhe.
One possible effect of protein binding might be to change the local stiffness of the binding site. For the DNA alone it is very difficult to distinguish models having increased local flexibility at the site from those with slightly increased flexibility for the whole molecule. Including an independent local flexibility parameter for the DNA alone slightly improved REL for the whole set of molecules. The best fit result reflects an increased local bending flexibility, expressed as an rms bend angle fluctuation of σθ = 6.125° ± 0.1°/bp, which corresponds to P of 88 bp (12, 22) for the 10-bp binding site, compared with 156 bp for the rest of the molecule. (The simulations use all 4 digits in the bending flexibility angle.) The absolute value of the local flexibility is less important than how it changes when protein is bound (see below), assuming the B-DNA P is constant.
The agreement between the experimental and simulated results is shown in Fig. 3. The shapes of the two curves are virtually identical despite the vertical offset, which is a consequence of the use of REL as the error measure and the requirement to fit the data for the out-of-phase molecules. Possible explanations for the residual difference between the simulation and experimental data sets include the assumed constant P and C for the B-DNA. The simulated unwinding angle is slightly smaller than we predicted from the optimum experimental cyclization length, but it is within the error for the center of the Gaussian.
The structural changes on binding the ATF/CREB site to GCN4 are subtle and represent a slight loss in bending or flexibility of the site. In one solution, the DNA parameters are altered to include a 7° bend angle (reduced from 8° in the DNA alone), thus decreasing the cyclization probability by decreasing the amount of bending across the site. Alternatively, the bending flexibility of the site is decreased to σθ = 4.775°/bp (compared with 6.125°/bp without the protein), which corresponds to a P of 144 bp. The results of these two alternatives are shown compared with the experimental data in Fig. 4. The decreased bend angle fits the out-of-phase data points better, at the expense of the fit to the in-phase molecules. In contrast, the decreased flexibility fits the data set as a whole even better than could be done for DNA alone (Fig. 4). By this interpretation, GCN4 slightly restricts the available DNA conformations of the site. When the site is phased opposite to the A-tracts, on the outside of the cyclized molecule, this decrease in flexibility significantly hinders cyclization, but when the site is on the inside of the circle these effects are not detected experimentally as the site does not have to bend as far away from the lowest energy conformation. The reduction in flexibility that accompanies GCN4 binding corresponds to a 60% increase in the local P.
The B-DNA parameters, which are quite standard, are the same for the DNA alone and when bound to GCN4. We chose the free DNA data set to determine the range of acceptable values for the B-DNA parameters; results for the protein-bound DNA should be very similar. The helical repeat of the non-A-tract portions of the molecules is 10.45 ± <0.05 bp/turn. The bending flexibility of the B-DNA is σθ = 4.584°/bp, which corresponds to a 156-bp P, in accordance with most other values of P. The 5% uncertainty we observe in P is in agreement with earlier studies (11). C is 1.85 ± 0.05 × 10−19 erg⋅cm. This value is on the low end of the reported values for C, corresponding to torsionally flexible DNA.
DISCUSSION
We have shown that, for in-phase molecules, the binding of GCN4 to the ATF/CREB site does not significantly alter the ring closure of DNA. The molecules in which the intrinsically curved ATF/CREB site faces away from the A-tracts show decreased DNA ring closure in the presence of protein. A slight stiffening of the site on protein binding could cause the decrease in J factors while being small enough not to affect cyclization values of the in-phase molecules. (In general, the smaller the value of J, the more logJ is affected by small changes in the DNA parameters.) These observations are further evidence that the native conformation of the ATF/CREB site has an intrinsic bend toward the major groove (7).
We have determined structural parameters for the ATF/CREB molecules by using Monte Carlo simulations. The simple calculation for the bend angle from the cyclization data yielded a 13° bend, slightly larger than the simulated 8° value, a not surprising disagreement given the approximate nature of the simple calculation. The preliminary calculation also slightly overestimated the unwinding across the site, at 60° vs. the simulated result of 53°. On the whole, the simulation results are in good agreement with our experimental values (Fig. 3). The small bend angle of 8° is also in rough agreement with the values from the crystal structure and gel mobility studies. The crystal structure reveals a 20° overall bend, which results primarily from a 9.5° roll angle at the central base pair step and roll angles of −6.1° at two symmetrically placed ApT steps (8, 9). Further, comparison of the AP-1 and ATF/CREB crystal structures (9) shows a 10° difference in bend angle between the DNAs in the two cases. The differences between solution and the crystal packing environment could account for the difference in overall bend angle of 12° between crystal and cyclization results for the ATF/CREB site. (We note that there is full agreement about the bend direction.) The gel studies (7) were performed on DNA constructs in which the protein binding site directly abuts the A-tract bend, enhancing the sensitivity to small bends. Our results are in reasonable agreement with their bend angle of 10–15°, given the difficulty of quantitating absolute bend angles accurately from comparative electrophoresis measurements (15, 29).
There is greater disagreement between our results and the crystal structure with respect to underwinding at the ATF/CREB site. The average twist in the crystal structure is 34.1°, corresponding to a helical repeat of 10.55, not appreciably underwound relative to B DNA. If the estimated underwinding of 53° is confined to untwisting the 10-bp site, the average twist must be reduced to about 30°. A 30° twist per bp has been observed in A′ DNA and RNA structures (30). The A′ form has 12 bp/helical repeat, but most of the other parameters such as tilt and rise are intermediate between A and B forms. The ATF/CREB site in solution may take on some A′ features, a structural distortion that may facilitate protein–DNA interactions similar to those for the shorter B form AP-1 site.
It is possible that the ATF/CREB site adopts significant negative writhe in solution, possibly arising from two symmetrically disposed bends within the site that are not coplanar. For example, the resultant of the two bends 8 bp apart observed in the crystal structure at the ApT steps would yield negative writhe, and their resultant would be of approximately the correct magnitude to correspond to the 8° bend observed in our experiments. We emphasize that our results reflect the global properties of DNA bending and torsion changes and do not yield unequivocal interpretation in terms of local changes in twist or writhe.
Although the agreement between simulation and experimental results is good, there are some discrepancies. DNA sequence effects may contribute to these differences because the DNA molecules differ in their overall lengths and linker sequences. To perform the simulations reliably, we assume that B-DNA has uniform helical repeat, P, and C values. This may not be the case experimentally where conformations may be subject to sequence context.
The ATF/CREB site molecules proved rather challenging to simulate primarily because of the small values of the J factors. Larger J factors produce better statistics in the simulations and lead more quickly to reliable results. The simulations were complicated by the observed J factor differences between the two sets of out-of-phase molecules. These molecules proved to be the most responsive to variation and ended up driving much of the parameter selections. Despite these challenges we believe that the results are reliable and that the derived parameters are consistent with the experimental data. We have demonstrated that the Monte Carlo programs are sensitive enough to distinguish parameters such as flexibility and bend angle that we are unable to measure directly and can be successfully applied to the study of small DNA structural deformations.
Acknowledgments
We thank C. Ampe and T. Steitz for GCN4 cells, R. Beltran and R. Kriwacki for purifying GCN4, T. Ellenberger for the bZIP peptide of GCN4, J. Caradonna and A. Schepartz for computer facilities, and D. Paolella and A. Schepartz for discussions. This work was supported by National Institutes of Health Grant GM 21966.
ABBREVIATIONS
- ABS
absolute error
- REL
relative error
- P
persistence length
- C
torsional modulus
References
- 1.Sellers J W, Vincent A C, Struhl K. Mol Cell Biol. 1990;10:5077–5086. doi: 10.1128/mcb.10.10.5077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arndt K, Fink G R. Proc Natl Acad Sci USA. 1986;83:8516–8520. doi: 10.1073/pnas.83.22.8516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hope I A, Struhl K. Cell. 1985;43:177–188. doi: 10.1016/0092-8674(85)90022-4. [DOI] [PubMed] [Google Scholar]
- 4.Oliphant A R, Brandl C J, Struhl K. Mol Cell Biol. 1989;9:2944–2949. doi: 10.1128/mcb.9.7.2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hill D E, Hope I A, Macke J P, Struhl K. Science. 1986;234:451–457. doi: 10.1126/science.3532321. [DOI] [PubMed] [Google Scholar]
- 6.Gartenberg M R, Ampe C, Steitz T A, Crothers D M. Proc Natl Acad Sci USA. 1990;87:6034–6038. doi: 10.1073/pnas.87.16.6034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paolella D N, Palmer C R, Schepartz A. Science. 1994;264:1130–1133. doi: 10.1126/science.8178171. [DOI] [PubMed] [Google Scholar]
- 8.Konig P, Richmond T J. J Mol Biol. 1993;233:139–154. doi: 10.1006/jmbi.1993.1490. [DOI] [PubMed] [Google Scholar]
- 9.Keller W, König P, Richmond T J. J Mol Biol. 1995;254:657–667. doi: 10.1006/jmbi.1995.0645. [DOI] [PubMed] [Google Scholar]
- 10.Ellenberger T E, Brandl C J, Struhl K, Harrison S C. Cell. 1992;71:1223–1237. doi: 10.1016/s0092-8674(05)80070-4. [DOI] [PubMed] [Google Scholar]
- 11.Koo H-S, Drak J, Rice J A, Crothers D M. Biochemistry. 1990;29:4227–4234. doi: 10.1021/bi00469a027. [DOI] [PubMed] [Google Scholar]
- 12.Crothers D M, Drak J, Kahn J D, Levene S D. Methods Enzymol. 1992;212:3–29. doi: 10.1016/0076-6879(92)12003-9. [DOI] [PubMed] [Google Scholar]
- 13.Shore D, Langowski J, Baldwin R L. Proc Natl Acad Sci USA. 1981;78:4833–4837. doi: 10.1073/pnas.78.8.4833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shore D, Baldwin R L. J Mol Biol. 1983;170:957–981. doi: 10.1016/s0022-2836(83)80198-3. [DOI] [PubMed] [Google Scholar]
- 15.Sitlani A, Crothers D M. Proc Natl Acad Sci USA. 1996;93:3248–3252. doi: 10.1073/pnas.93.8.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sitlani A, Crothers D M. Proc Natl Acad Sci USA. 1998;95:1404–1409. doi: 10.1073/pnas.95.4.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hockings S C. Ph.D. thesis. New Haven, CT: Yale University; 1997. [Google Scholar]
- 18.Weiss M A, Ellenberger T, Wobbe C R, Lee J P, Harrison S C, Struhl K. Nature (London) 1990;347:575–578. doi: 10.1038/347575a0. [DOI] [PubMed] [Google Scholar]
- 19.Kahn J D, Crothers D M. Proc Natl Acad Sci USA. 1992;89:6343–6347. doi: 10.1073/pnas.89.14.6343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barshop B A, Wrenn R F, Frieden C. Anal Biochem. 1983;130:134–145. doi: 10.1016/0003-2697(83)90660-7. [DOI] [PubMed] [Google Scholar]
- 21.Levene S D, Crothers D M. J Biomol Struct Dyn. 1983;1:429–435. doi: 10.1080/07391102.1983.10507452. [DOI] [PubMed] [Google Scholar]
- 22.Levene S D, Crothers D M. J Mol Biol. 1986;189:61–72. doi: 10.1016/0022-2836(86)90381-5. [DOI] [PubMed] [Google Scholar]
- 23.Levene S D, Crothers D M. J Mol Biol. 1986;189:73–83. doi: 10.1016/0022-2836(86)90382-7. [DOI] [PubMed] [Google Scholar]
- 24.Jacobson H, Stockmayer W H. J Chem Phys. 1950;18:1600–1606. [Google Scholar]
- 25.Flory P J, Suter U W, Mutter M. J Am Chem Soc. 1976;98:5733–5739. [Google Scholar]
- 26.Crothers D M, Haran T E, Nadeau J G. J Biol Chem. 1990;265:7093–7096. [PubMed] [Google Scholar]
- 27.Kahn J D, Crothers D M. Cold Spring Harbor Symp Quant Biol. 1993;58:115–122. doi: 10.1101/sqb.1993.058.01.015. [DOI] [PubMed] [Google Scholar]
- 28.Drak J, Crothers D M. Proc Natl Acad Sci USA. 1991;88:3074–3078. doi: 10.1073/pnas.88.8.3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kerppola T K. Proc Natl Acad Sci USA. 1996;93:10117–10122. doi: 10.1073/pnas.93.19.10117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saenger W. Principles of Nucleic Acid Structure. New York: Springer; 1984. [Google Scholar]
- 31.Kahn J D, Yun E, Crothers D M. Nature (London) 1994;368:163–166. doi: 10.1038/368163a0. [DOI] [PubMed] [Google Scholar]