

Abstract
Mutational activation of BRAF is a frequent event in human malignant melanomas suggesting that BRAF-dependent signaling is conducive to melanoma cell growth and survival. Previously published work reported that melanoma cells exhibit constitutive anti-apoptotic nuclear factor κB (NF-κB) transcription factor activation triggered by proteolysis of its inhibitor IκB. IκB degradation is dependent upon its phosphorylation by the IκB kinase (IKK) complex and subsequent ubiquitination facilitated by β-Trcp E3 ubiquitin ligase. Here, we report that melanocytes expressing a conditionally oncogenic form of BRAFV600E exhibit enhanced β-Trcp expression, increased IKK activity and a concomitant increase in the rate of IκBα degradation. Conversely, inhibition of BRAF signaling using either a broad-spectrum Raf inhibitor (BAY 43-9006) or by selective knock-down of BRAFV600E expression by RNA interference in human melanoma cells leads to decreased IKK activity and β-Trcp expression, stabilization of IκB, inhibition of NF-κB transcriptional activity and sensitization of these cells to apoptosis. Taken together, these data support a model in which mutational activation of BRAF in human melanomas contributes to constitutive induction of NF-κB activity and to increased survival of melanoma cells.
Keywords: BRAF, melanoma, β-Trcp, NF-κB, IκB kinase
Malignant melanoma is the most lethal skin cancer, whose ability to rapidly metastasize often prevents surgical cure. Furthermore, the systemic treatment of melanoma is largely ineffective due to the intrinsic resistance of melanoma cells to numerous anticancer agents. Increased survival of melanoma cells is primarily attributed to the constitutive activation of the transcription factor nuclear factor κB (NF-κB), which regulates the expression of many anti-apoptotic, pro-proliferative and pro-metastatic genes (Amiri and Richmond, 2005; Ueda and Richmond, 2006). Canonical activation of the NF-κB pathway occurs when NF-κB switches its localization from the cytoplasm, where it is maintained inactive by assembly with the inhibitor IκB protein, to the nucleus, where NF-κB regulates gene expression. NF-κB activation relies upon the phosphorylation-dependent ubiquitination and degradation of IκB mediated by the IκB kinase (IKK) complex and β-Trcp E3 ubiquitin ligases (reviewed by Karin and Ben-Neriah, 2000). Consequently, both IKK activity and the levels of β-Trcp regulate the extent of IκB degradation and hence NF-κB activation (Fuchs et al., 2004).
The genetic basis that underlies the elevated NF-κB activity in malignant melanoma largely remains elusive. Constitutively active IKK has been demonstrated to sustain NF-κB activation in human melanoma cells, resulting in induction of the chemokine CXCL1. CXCL1, in turn, is capable of activating IKK and NF-κB and promoting cell survival and tumorigenesis (Yang and Richmond, 2001). However, the original genetic alterations that initiate this feed-forward mechanism in melanoma remain unclear. One of the major oncogenic events described in the genesis of malignant melanoma is constitutive activation of the Ras-regulated RAF→MEK→ERK mitogen-activated protein kinase (MAPK) pathway. This is achieved most frequently by activating mutations in either BRAF (e.g. V600E substitution) or, less frequently, in N-RAS (reviewed by Smalley, 2003; Smalley and Herlyn, 2005). Recent evidence indicates that oncogenic BRAF activity is essential for human melanoma cell growth and survival (Hoeflich et al., 2006). However, despite prior reports that RAF can activate NF-κB (Norris and Baldwin, 1999), the mechanism(s) by which BRAFV600E (BRAFVE) may elicit NF-κB signaling in melanoma cells have not yet been elucidated.
Activation of the canonical NF-κB pathway depends on both IKK activity, which has been shown to be elevated in human melanomas (Amiri and Richmond, 2005; Ueda and Richmond, 2006), and the levels of β-Trcp, which functions as the substrate recognition subunit of the E3 ubiquitin ligase that facilitates IκB ubiquitination (Fuchs et al., 2004). Previously, we reported that β-Trcp2 mRNA and protein levels are induced by the mitogenic stimuli via the ERK MAPK pathway in a transcription-dependent manner in fibroblasts and myocytes (Spiegelman et al., 2002a). Here, we examined the levels of β-Trcp in human melanoma cell lines that often harbor either BRAF or N-RAS mutations and were reported to exhibit the constitutive activation of the ERK MAPK pathway (Satyamoorthy et al., 2003). As shown in Figure 1a, most of the melanoma cell lines express higher levels of β-Trcp than normal human melanocytes. To determine whether the expression of oncogenic BRAFVE leads to induction of β-Trcp, we employed Melan-A mouse melanocytes that stably express a BRAFVE:ERT2 fusion protein (Figure 1b; see Supplementary Information for details). The kinase activity of the BRAFVE:ERT2 fusion protein is activated following treatment of cells with the synthetic estrogen analog 4-hydroxytamoxifen (4- OHT). Using this model we found that BRAFVE:ERT2 activation resulted in both elevated ERK1/2 MAPK activity (as assessed by ERK1/2 phosphorylation) and induction of β-Trcp expression (Figure 1c). Treatment of parental cells not expressing BRAFVE:ER with 4- OHT neither promoted ERK1/2 activity nor increased β-Trcp levels. These data therefore indicate that oncogenic BRAFVE is capable of inducing β-Trcp expression in mouse melanocytes. This induction is most likely mediated by the activation of the MAPK pathway given our result that treatment of cells with specific MEK1 inhibitor (U0126) attenuated an increase in β-Trcp levels (Figure 1d).
Figure 1.
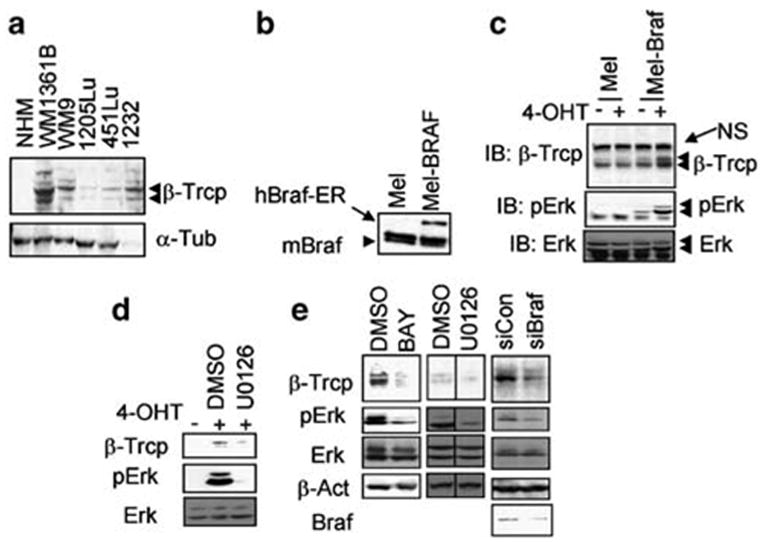
BRAF activity maintains the expression levels of β-Trcp. (a) Immunoblotting analysis of β-Trcp (upper panel) and α-tubulin (lower panel, loading control) levels in normal human melanocytes (NHM) and human melanoma cell lines. Immunoblotting was carried out as described earlier (Spiegelman et al., 2002b). (b) Mouse Melan-A melanocytes (‘Mel’) or derivative cells expressing BRAFVE:ERT2 (‘Mel-BRAF’) were analysed for BRAF levels using F7 monoclonal antibody (Santa Cruz, Santa Cruz, CA, USA) that recognizes both human and mouse BRAF. ‘NS’ denotes nonspecific band, used as a loading control. (c) Mouse Melan-A melanocytes (‘Mel’) or derivative cells expressing BRAFVE:ERT2 (‘Mel-BRAF’) were pretreated with 4-OHT to induce the activity of BRAFV600E and were analysed for the levels of β-Trcp expression, phosphorylation of ERK1/2 and the total levels of ERK1/2 as indicated. ‘NS’ indicates nonspecific band. (d) Mel-BRAF cells were treated with 4-OHT and either dimethylsulfoxide (DMSO) or U0126 as indicated and analysed by immunoblotting with indicated antibodies. (e) Immunoblotting analyses with the indicated antibodies were carried out in 1205Lu human melanoma cells treated with DMSO or BAY 43-9006 (1 μM) or U0126 (5 μM) or co-transfected with shRNA against either BRAFV600E (‘siBRAF’) or irrelevant target (‘siCON’) as indicated. Transfection of 1205Lu cells was carried out using Lipofectamin Plus (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s recommendations.
In order to exclude that effects of activated BRAF in melanocytes might be attributed solely to its overexpression, we next determined whether inhibition of BRAFVE→MEK→ERK signaling in human melanoma cells that harbor an activated allele of BRAF may affect β-Trcp expression. To this end, we used pharmacologic inhibitors as well as the RNA interference approach to knock down the oncogenic BRAFVE. Both treatments decreased the activity of a β-Trcp2 promoter-driven luciferase reporter in human melanoma cells (data not shown), which was reported earlier to be stimulated by mitogenic stimuli including viral Ras (Spiegelman et al., 2002a). Treatment of 1205Lu human melanoma cells with either an inhibitor of Raf kinases (BAY 43-9006, Figure 1e) or with specific MEK1 inhibitors (U0126, Figure 1e or PD 098059, data not shown) reduced both constitutive ERK activation and the levels of β-Trcp expression. In addition, transfection of 1205Lu melanoma cells with an small hairpin RNA (shRNA) construct that specifically suppresses expression of the BRAFVE mutant allele without affecting wild-type BRAF (Hingorani et al. 2003; Figure 1e, lower panel) led to decreased ERK activation and expression of β-Trcp (Figure 1e). Taken together, these data provide both pharmacologic and genetic evidence that oncogenic BRAFVE-mediated activation of the MAPK pathway contributes to the maintenance of β-Trcp expression in human melanoma cells.
As modulation of β-Trcp levels affects the rate of proteolytic turnover of its phosphorylated substrates (Fuchs et al., 2004), we sought to determine whether the expression of oncogenic BRAF affects the stability of IκBα. Previous reports indicated that forced expression of BRAF mutants in COS and NIH 3T3 cells leads to NF-κB activation, which parallels the activation of ERK and which can be inhibited by non-degradable IκB super-repressor (Ikenoue et al., 2003, 2004). Pretreatment of Melan-A cells that harbor the BRAFVE:ERT2 fusion protein with 4-OHT led to an accelerated degradation of IκBα (measured by a cycloheximide (CHX) chase) that was not seen in parental mouse Melan-A cells (Figure 2a). Remarkably, this increased IκBα turnover, which occurred concomitantly with increased ERK activation, was observed in cells that were not treated with known activators of IKK, such as tumor necrosis factor alpha (TNFα). Given that β-Trcp-driven ubiquitination depends on IκB phosphorylation by IKK, we next investigated the effect of oncogenic BRAFVE activation on the activation status of endogenous IKK. As shown in Figure 2b, activation of BRAFVE:ERT2 led to an increase in IKK activity, measured by an immunokinase assay using GST-IκBα as a substrate. Importantly, treatment of oncogenic BRAF-containing melanocytes with MEK1 inhibitor U0126 attenuated the increase in both IKK catalytic activity (Figure 2c) and κB luciferase reporter activity (Figure 2d), indicating that the effects of BRAF are mediated by the MAPK pathway. In all, these results implicate oncogenic BRAF in the accelerated degradation of IκBα and activation of NF-κB; the former event is in turn likely to be stimulated by both IKK activation and β-Trcp induction.
Figure 2.
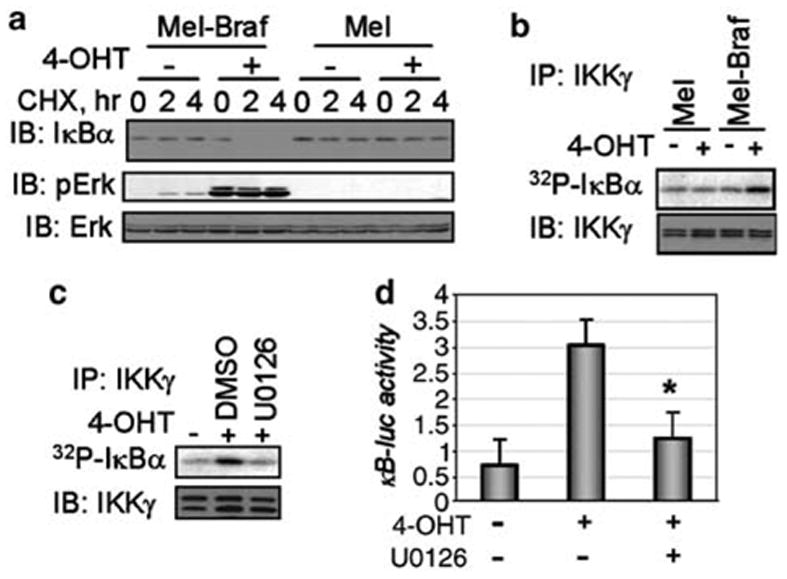
BRAF activates IKK and promotes IκB degradation in mouse melanocytes. (a) Stability of endogenous IκBα measured by CHX chase in Melan-A melanocytes (‘Mel’) or derivative cells expressing BRAFVE:ERT2 (pretreated with 4-OHT before adding CHX as indicated) is shown in the upper panel. The lower panels depict the immunoblotting analysis of phosphorylation and levels of ERK1/2 in these cells. (b) IKK immunokinase activity measured in the presence of 32P-γ-ATP and GST-IκBα by the endogenous IKK complex immunopurified (using IKKγ antibody as described elsewhere; Dejardin et al., 2002) from Melan-A cells or Melan-A-BRAFVE: ERT2 (pretreated with 4-OHT as indicated) is analysed by autoradiography and depicted in the upper panel. Immunoblotting analyses of aliquots from IKKγ immunoprecipitates using antibodies against IKKγ are shown in the lower panels. (c) Melan-A-BRAFVE: ERT2 cells treated with 4-OHT and U0126 as indicated were analysed for endogenous IKK activity as described in panel b. (d) NF-κB transcriptional activity in BRAF-expressing melanocytes treated as indicated was measured by the activity of κB-driven firefly luciferase reporter normalized per Renilla luciferase activity and depicted in arbitrary units. Standard deviation from three experiments is shown. Asteriskdenotes P<0.05 in t-test compared to the group not treated with U0126.
These conclusions were further supported by the observation that treatment of 1205Lu human melanoma cells with BAY 43-9006 led to a marked decrease in the efficiency of IκBα turnover (Figure 3a) and the activity of the endogenous IKK complex (Figure 3b). In addition, the degradation of coexpressed Flag-tagged IκBα was dramatically impaired in these cells transfected with shRNA against BRAFVE (Figure 3c). Furthermore, a moderate decrease in the activity of coexpressed Flag-tagged IKKβ was observed upon knock-down of oncogenic BRAF (Figure 3d). A limited extent of inhibition here can be attributed to the findings that, under the conditions of overexpression, IKKβ activity is partly independent of upstream signaling events (Zandi et al., 1997). These data suggest that oncogenic BRAFVE-dependent signaling contributes to constitutive IKK activity and IκB degradation in human melanoma cells.
Figure 3.
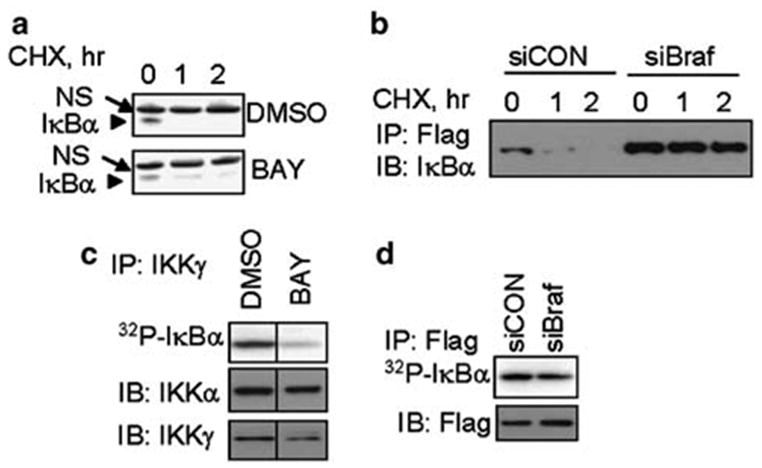
BRAF regulates IKK activity and IκB degradation in human melanoma cells. (a) Stability of endogenous IκBα measured by CHX chase in 1205Lu human melanoma cells treated with DMSO or BAY 43-9006 (as indicated) measured by immunoblotting with IκBα antibody. ‘NS’ indicates nonspecific band, used here as a loading control. (b) The endogenous IKK activity in 1205Lu human melanoma cells treated with DMSO or BAY 43-9006 (as indicated) is measured by immunokinase assay as described in Figure 2b. (c) Stability of exogenous Flag-tagged IκBα co-transfected into 1205Lu human melanoma cells along with either siCON or siBRAF measured by immunoprecipitation with Flag antibody followed by immunoblotting with IκBα antibody. (d) The activity of Flag-IKKβ coexpressed in 1205Lu human melanoma cells along with either siCON or siBRAF (as indicated) is measured by immunokinase assay as described in Figure 2b. An aliquot from each Flag-immunoprecipitate was analysed by immunoblotting using antibodies against Flag tag (lower panel).
Given that IκB stability is regulated by BRAF, we predicted that signaling initiated by oncogenic BRAF should play an important role in the maintenance of constitutive NF-κB activity and perhaps, affect the survival of melanoma cells. Consistent with this hypothesis, we indeed found that inhibition of BRAFVE by either mutant-specific shRNA construct or treatment of melanoma cells with BAY 43-9006 resulted in decreased NF-κB transcriptional activity, measured by κB-driven luciferase reporter (Figure 4a). A similar inhibition of NF-κB activity was achieved by treating the cells with U0126 inhibitor (Figure 4a), indicating that oncogenic BRAF is likely to maintain the constitutive NF-κB activity via the MAPK pathway in human melanoma cells. This mode of regulation is dissimilar to MEK1-independent activation of NF-κB by viral mutant RAF in Jurkat cells (Baumann et al., 2000).
Figure 4.
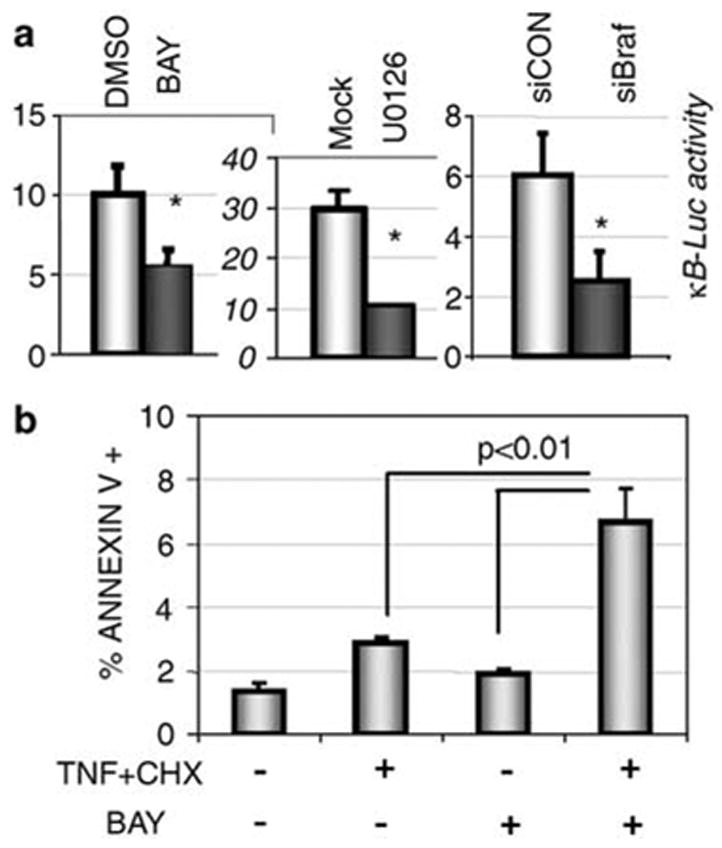
BRAF regulates NF-κB activity and sensitivity of human melanoma cells to apoptosis. (a) NF-κB transcriptional activity in 1205Lu human melanoma cells that underwent indicated treatments or transfections is measured by the activity of κB-driven firefly luciferase reporter normalized per Renilla luciferase activity and depicted in arbitrary units. Standard deviation from three experiments is shown. Asteriskdenotes P<0.05 in t-test compared to either DMSO or siCON group. (b) Percent of early apoptotic (Annexin V-positive) 1205Lu cells induced by TNFα (20 ng/ml) and CHX (10 μg/ml) within 5 h of treatment alone or in combination with BAY 43-9006 (1 μM) as indicated. Standard deviation from three experiments is shown. The rate of apoptosis was measured by flow cytometric analysis upon the cell staining with allophycocyanin conjugate (APC)-labeled-Annexin V with or without 7-amino-actinomycin D (7-AAD) using the Annexin V kit (BD Biosciences Pharmingen, San Diego, CA, USA).
To determine the functional significance of oncogenic BRAF-dependent maintenance of basal NF-κB activity in human melanoma cells, we assessed the rate of apoptosis in these cells using an experimental model in which we combined treatment with TNFα and CHX. Under these conditions (which were shown to induce apoptosis in melanoma cells in a β-Trcp activity-dependent manner; Soldatenkov et al., 1999) TNFα-inducible NF-κB activation does not upregulate the anti-apoptotic proteins because protein synthesis is inhibited by CHX. Hence, the basal NF-κB activity that exists in cells before treatment determines their resistance to apoptosis. Pretreatment of 1205Lu human melanoma cells with BAY 43-9006 led to a significant increase in the number of apoptotic cells (Figure 4b). These data are consistent with the recently demonstrated apoptotic effects of prolonged treatment with BAY 43- 9006 (Panka et al., 2006) and with regression in cell and tumor growth upon RNAi-mediated knock-down of BRAF (Sharma et al., 2005; Hoeflich et al., 2006). They also indicate that mutationally activated BRAF plays an important role in the survival of melanoma cells.
Evidence presented herein strongly suggests that oncogenic BRAF plays an important role in the maintenance of constitutive NF-κB activity in human melanoma cells as well as in their survival. These results are in agreement with the data obtained from forced expression of BRAF mutants in COS or NIH 3T3 cells (Ikenoue et al., 2003, 2004). Given that BRAF mutations tend to occur early in melanomogenesis and are often found in benign nevi (reviewed by Smalley, 2003; Smalley and Herlyn, 2005), it is tempting to speculate about the role of oncogenic BRAF-mediated NF-κB induction in the progression of malignant melanoma. Furthermore, BRAF inhibitors that decrease NF-κB activity might be useful adjuvants for combined chemotherapy of human melanoma. Indeed, one IKK inhibitor has already demonstrated dramatic efficacy against melanoma cells in pre-clinical settings (Yang et al., 2006).
Early results of clinical trials on monotherapy with BAY 43-9006 (Sorafenib) in human melanoma suggested that it should be combined with other anticancer agents (Smalley and Herlyn, 2005). Whereas BAY 43- 9006 was shown to downregulate the expression of Bcl-XL (an NF-κB target protein), the mechanisms by which this agent promotes apoptosis in human melanoma cells are complex (Panka et al., 2006) and cannot be attributed solely to NF-κB inhibition. Future basic and translational research efforts are warranted to delineate additional mechanisms by which inhibition of oncogenic BRAF, on the one hand, and effects of available Raf and IKK inhibitors, on the other hand, mediate melanoma cell death and tumor regression. A comprehensive understanding of the mechanisms regulated by oncogenic BRAF will be required to optimize the therapeutic effects of combined therapy against malignant melanomas.
Acknowledgments
We are indebted to Michael May and Dave Tuveson for the discussion, reagents and critical comments. We thank D Ballard, DC Bennett M Karin and C Smith for the reagents. This workwas supported in part by NIH Grants CA092900 (to SYF), CA108972 (to MM), CA 102709 (to ATT) and CA25874 and CA93372 (to MH).
References
- Amiri KI, Richmond A. Role of nuclear factor-kappa B in melanoma. Cancer Metast Rev. 2005;24:301–313. doi: 10.1007/s10555-005-1579-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann B, Weber CK, Troppmair J, Whiteside S, Israel A, Rapp UR, et al. Raf induces NF-kappaB by membrane shuttle kinase MEKK1, a signaling pathway critical for transformation. Proc Natl Acad Sci USA. 2000;97:4615–4620. doi: 10.1073/pnas.080583397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejardin E, Droin NM, Delhase M, Haas E, Cao Y, Makris C, et al. The lymphotoxin-beta receptor induces different patterns of gene expression via two NF-kappaB pathways. Immunity. 2002;17:525–535. doi: 10.1016/s1074-7613(02)00423-5. [DOI] [PubMed] [Google Scholar]
- Fuchs SY, Spiegelman VS, Kumar KG. The many faces of beta-TrCP E3 ubiquitin ligases: reflections in the magic mirror of cancer. Oncogene. 2004;23:2028–2036. doi: 10.1038/sj.onc.1207389. [DOI] [PubMed] [Google Scholar]
- Hingorani SR, Jacobetz MA, Robertson GP, Herlyn M, Tuveson DA. Suppression of BRAF(V599E) in human melanoma abrogates transformation. Cancer Res. 2003;63:5198–5202. [PubMed] [Google Scholar]
- Hoeflich KP, Gray DC, Eby MT, Tien JY, Wong L, Bower J, et al. Oncogenic BRAF is required for tumor growth and maintenance in melanoma models. Cancer Res. 2006;66:999–1006. doi: 10.1158/0008-5472.CAN-05-2720. [DOI] [PubMed] [Google Scholar]
- Ikenoue T, Hikiba Y, Kanai F, Aragaki J, Tanaka Y, Imamura J, et al. Different effects of point mutations within the B-Raf glycine-rich loop in colorectal tumors on mitogen-activated protein/extracellular signal-regulated kinase kinase/extracellular signal-regulated kinase and nuclear factor kappaB pathway and cellular transformation. Cancer Res. 2004;64:3428–3435. doi: 10.1158/0008-5472.CAN-03-3591. [DOI] [PubMed] [Google Scholar]
- Ikenoue T, Hikiba Y, Kanai F, Tanaka Y, Imamura J, Imamura T, et al. Functional analysis of mutations within the kinase activation segment of B-Raf in human colorectal tumors. Cancer Res. 2003;63:8132–8137. [PubMed] [Google Scholar]
- Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol. 2000;18:621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- Norris JL, Baldwin AS., Jr Oncogenic Ras enhances NF-kappaB transcriptional activity through Raf-dependent and Raf-independent mitogen-activated protein kinase signaling pathways. J Biol Chem. 1999;274:13841–13846. doi: 10.1074/jbc.274.20.13841. [DOI] [PubMed] [Google Scholar]
- Panka DJ, Wang W, Atkins MB, Mier JW. The Raf inhibitor BAY 43-9006 (Sorafenib) induces caspase-independent apoptosis in melanoma cells. Cancer Res. 2006;66:1611–1619. doi: 10.1158/0008-5472.CAN-05-0808. [DOI] [PubMed] [Google Scholar]
- Satyamoorthy K, Li G, Gerrero MR, Brose MS, Volpe P, Weber BL, et al. Constitutive mitogen-activated protein kinase activation in melanoma is mediated by both BRAF mutations and autocrine growth factor stimulation. Cancer Res. 2003;63:756–759. [PubMed] [Google Scholar]
- Sharma A, Trivedi NR, Zimmerman MA, Tuveson DA, Smith CD, Robertson GP. Mutant V599EB-Raf regulates growth and vascular development of malignant melanoma tumors. Cancer Res. 2005;65:2412–2421. doi: 10.1158/0008-5472.CAN-04-2423. [DOI] [PubMed] [Google Scholar]
- Smalley KS. A pivotal role for ERK in the oncogenic behaviour of malignant melanoma? Int J Cancer. 2003;104:527–532. doi: 10.1002/ijc.10978. [DOI] [PubMed] [Google Scholar]
- Smalley KS, Herlyn M. Targeting intracellular signaling pathways as a novel strategy in melanoma therapeutics. Ann NY Acad Sci. 2005;1059:16–25. doi: 10.1196/annals.1339.005. [DOI] [PubMed] [Google Scholar]
- Soldatenkov VA, Dritschilo A, Ronai Z, Fuchs SY. Inhibition of homologue of Slimb (HOS) function sensitizes human melanoma cells for apoptosis. Cancer Res. 1999;59:5085–5088. [PubMed] [Google Scholar]
- Spiegelman VS, Tang W, Chan AM, Igarashi M, Aaronson SA, Sassoon DA, et al. Induction of homologue of Slimb ubiquitin ligase receptor by mitogen signaling. J Biol Chem. 2002a;277:36624–36630. doi: 10.1074/jbc.M204524200. [DOI] [PubMed] [Google Scholar]
- Spiegelman VS, Tang W, Katoh M, Slaga TJ, Fuchs SY. Inhibition of HOS expression and activities by Wnt pathway. Oncogene. 2002b;21:856–860. doi: 10.1038/sj.onc.1205132. [DOI] [PubMed] [Google Scholar]
- Ueda Y, Richmond A. NF-kappaB activation in melanoma. Pigment Cell Res. 2006;19:112–124. doi: 10.1111/j.1600-0749.2006.00304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Amiri KI, Burke JR, Schmid JA, Richmond A. BMS-345541 targets inhibitor of kappaB kinase and induces apoptosis in melanoma: involvement of nuclear factor kappaB and mitochondria pathways. Clin Cancer Res. 2006;12:950–960. doi: 10.1158/1078-0432.CCR-05-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Richmond A. Constitutive IkappaB kinase activity correlates with nuclear factor-kappaB activation in human melanoma cells. Cancer Res. 2001;61:4901–4909. [PubMed] [Google Scholar]
- Zandi E, Rothwarf DM, Delhase M, Hayakawa M, Karin M. The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell. 1997;91:243–252. doi: 10.1016/s0092-8674(00)80406-7. [DOI] [PubMed] [Google Scholar]