

Abstract
Proteins involved in cholesterol trafficking are known to contribute to the pathogenesis of atherosclerosis and Alzheimer's disease. Allelic variants in the cholesterol transporters apolipoprotein E and ATP-binding cassette protein A1 (ABCA1) have recently been associated with susceptibility to age-related macular degeneration (AMD). Histopathological analyses of eyes with AMD demonstrate the presence of cholesterol and cholesteryl ester deposits beneath the retinal pigment epithelium (RPE), implicating abnormal cholesterol trafficking in disease progression. Here, we show that A2E, a quaternary amine and retinoid by-product of the visual cycle, causes the accumulation of free and esterified cholesterol in RPE cells. The mechanism involves neither generalized alterations in late endosomal/lysosomal pH nor a direct inhibition of acid lipase activity. Rather, A2E prevents cholesterol efflux from these organelles, which in turn indirectly inhibits acid lipase, leading to a subsequent accumulation of cholesteryl esters. Transcriptional activation of the ABCA1 cholesterol transporter by agonists of the liver X receptor/peroxisome proliferator-activated receptor pathway relieves the A2E-induced block on cholesterol efflux and restores cholesterol homeostasis in RPE cells. Our data also demonstrate that A2E, which is a cone-shaped lipid, increases the chemical activity and displacement of cholesterol from model membranes, providing a biophysical mechanism for cholesterol sequestration in A2E-loaded cells. Although endogenously produced A2E in the RPE has been associated with macular degeneration, the precise mechanisms are unclear. Our results provide direct evidence that A2E causes aberrant cholesterol metabolism in RPE cells which could likely contribute to AMD progression.
Keywords: drusen, lysosome, macular degeneration, phagocytosis, retina
Cholesterol modulates the biophysical properties of membranes and participates in lipid and protein trafficking. In the vertebrate retina, cholesterol stabilizes rhodopsin in photoreceptor outer segment (OS) disks and slows visual cycle kinetics (1). Photoreceptors continuously renew their OS by shedding distal disks in a circadian rhythm (2). Daily phagocytosis and processing of shed photoreceptor OS places a heavy metabolic burden on the underlying postmitotic retinal pigment epithelium (RPE) cells. Over time, incomplete digestion of internalized OS disks leads to the accumulation of autofluorescent lipid-protein aggregates called lipofuscin in RPE lysosomes. A major fluorophore of RPE lipofuscin is a hydrophobic quaternary amine called A2E (Fig. 1A), formed as a bis-retinal/ethanolamine Schiff base adduct (3, 4). A2E is a minor by-product of the visual cycle and progressively accumulates in RPE late endosomes/lysosomes (LE/Ly) [supporting information (SI) Fig. 7] throughout life (4, 5). With age, the RPE becomes less efficient in coping with the increasing burden of lysosomal A2E and its attendant toxicity. A causal role for A2E in RPE and photoreceptor damage was delineated in the abcr−/− mouse model of Stargardt's macular degeneration, which shows accelerated A2E deposition in the RPE and subsequent vision loss (6, 7). A2E also accumulates in other animal models of inherited retinal degenerations (4).
Fig. 1.

A2E and RPE cholesterol and lipid levels. (A) A2E is a cone-shaped molecule with a small polar headgroup and large hydrophobic retinoid side chains. (B) Quantification of total cellular phospholipids (PL), sphingomyelin (SM), free cholesterol (FC), and CE 48 h after A2E loading in ARPE-19 cells. Significantly different from controls, ∗, P < 0.05; ∗∗, P < 0.001. (C) Filipin staining (blue) of intracellular cholesterol pools in control (a) and A2E-loaded (b) (red) cells. (Right) High-magnification images of area denoted by box in b. (Scale bar, 5 μm.) (D) Total cellular cholesterol levels after OS phagocytosis normalized to cholesterol levels in cells not fed OS. Significantly different from controls at corresponding time, ∗, P < 0.05; ∗∗, P < 0.001. (E) Total cellular phospholipid levels after OS phagocytosis normalized to phospholipid levels in cells not fed OS. Significantly different from controls at 24 h, ∗, P < 0.05.
Several hypotheses (reviewed in ref. 3) have been proposed to explain how A2E might damage the RPE, including lysosomal dysfunction and blue-light mediated toxicity. We have previously demonstrated that A2E specifically delays the metabolism of phagocytosed OS lipids by the RPE, whereas lysosomal proteolysis is unaffected (8). Progressive lipid and cholesterol accumulation in the Bruch's membrane beneath the RPE has been identified as a contributing factor to age-related macular degeneration (AMD) (5). Apolipoproteins, cholesterol and cholesteryl esters (CE) are abundant in both drusen (lipid-protein aggregates in the Bruch's membrane) and other subRPE deposits in AMD eyes (9–11), suggesting compromised cholesterol homeostasis in the RPE. Moreover, cardiovascular disease and allelic variants in apolipoprotein E and ATP-binding cassette protein A1 (ABCA1) have been linked to AMD susceptibility in humans (12, 13).
However, little is known about factors that regulate cholesterol levels in the RPE and how these are altered in pathological conditions. Major subcellular sites of cholesterol metabolism are the endoplasmic reticulum, where it is synthesized, and LE/Ly, where CE internalized as part of lipoproteins or OS are degraded by acid lipase and free cholesterol is effluxed out by specific transporters. The objective of the present study was to investigate whether A2E interferes with cholesterol metabolism in the RPE and to identify the mechanism by which this might occur. We show that A2E, at levels similar to those found in aging human eyes, interferes with cholesterol efflux from LE/Ly, which in turn inhibits acid lipase and results in a feed-forward cycle of cholesterol and CE accumulation in RPE cells.
Results
A2E Causes Endogenous Cholesterol Accumulation in RPE Cells.
To ensure the physiological relevance of our experiments, we established extracellular A2E concentrations required for RPE cells to accumulate A2E in the range found in vivo (14). In ARPE-19 cells, a single 15-μM dose or three 5-μM doses over 7 days resulted in intracellular A2E amounts in this range (SI Table 2). Thus, A2E accumulation is an additive process, and RPE cells lack the capacity to either degrade or efflux it out. Cells exposed to A2E had increased levels of both free cholesterol and CE compared with controls, whereas phospholipids and sphingomyelin were unchanged (Fig. 1B). Analysis of intracellular cholesterol distribution using the sterol-binding probe filipin showed a dramatic increase in filipin fluorescence in A2E-loaded cells (Fig. 1C). Brightly fluorescent filipin puncta colocalized with A2E, indicative of enlarged LE/Ly cholesterol pools. We observed a 1.56-fold increase in free cholesterol levels in the presence of A2E (Fig. 1B). Because LE/Ly contain ≈10% of total cell cholesterol (15) and assuming that ≥50% of the A2E-induced cholesterol accumulation occurs in LE/Ly, this translates to a 4- to 8-fold increase in the free cholesterol content of LE/Ly (see SI Text for detailed calculations).
To test whether A2E disrupts lipid metabolism after phagocytosis, we measured phospholipid and cholesterol levels after feeding bovine OS to ARPE-19 cells with or without A2E. We have previously shown that A2E affects neither the binding nor internalization of OS by RPE cells (8). Immediately after phagocytosis, total cholesterol levels in both control and A2E-loaded cells increased 3-fold relative to cells that were not fed OS. At 6 and 24 h after phagocytosis, there was significantly more cholesterol in cells with A2E compared with controls (Fig. 1D). We also observed an increase in total phospholipids in A2E-loaded cells 24 h after phagocytosis (Fig. 1E), in agreement with our published data that A2E delays the clearance of phagocytosed lipids (8).
A2E Does Not Alter LE/Ly pH.
It has been suggested that A2E increases lysosomal pH and thus inactivates lysosomal enzymes (16). Because acid lipase has a pH optimum of 4–5, an increase in lysosomal pH would lead to inhibition of enzyme activity and an accumulation of both CE and cholesterol (17). However, Holz et al. (16) did not quantify the intracellular concentration of A2E, which might have accumulated at nonphysiological levels. We used two independent assays to determine whether A2E, at physiological levels, increases LE/Ly pH. First, a qualitative estimate of LE/Ly pH using Lysosensor blue showed no difference in probe fluorescence between control and A2E-loaded ARPE-19 cells, whereas a short exposure to bafilomycin A1 decreased fluorescence, signifying an increase in pH (Fig. 2A). Second, quantification of LE/Ly pH with the ratiometric dye Lysosensor yellow/blue gave similar values for both control and A2E-laden cells (Fig. 2B), whereas treatment with bafilomycin A1 increased the pH by 1 unit. That A2E, at intracellular levels similar to that found in vivo, does not alter LE/Ly pH is in accordance with its fixed positive charge and lack of exchangeable protons (Fig. 1A) and supports in vitro data showing that A2E does not inhibit the activity of several lysosomal enzymes (3).
Fig. 2.

LE/Ly pH and acid lipase activity are unaffected by A2E. (A) Qualitative estimate of LE/Ly pH with Lysosensor blue in ARPE-19 cells. Baf, bafilomycin A1. (Scale bar, 10 μm.) (B) Quantification of LE/Ly pH using Lysosensor yellow/blue. (C) Acid lipase activity using soluble protein extracts from either control or A2E-loaded cells as the enzyme source and TFMUO micelles as the substrate. (D) Acid lipase activity in soluble protein extracts from untreated cells as the enzyme source and TFMUO micelles with or without A2E as the substrate.
Acid Lipase Activity Is Not Directly Inhibited by A2E.
We next tested the possibility that A2E might promote CE and cholesterol accumulation by directly inhibiting acid lipase activity (17). Acid lipase is a soluble LE/Ly enzyme that transiently associates with membranes to hydrolyze ester bonds at the lipid–water interface. Upon partitioning into LE/Ly membranes, lipophilic A2E could interfere with acid lipase activity by directly interacting with the enzyme's catalytic domain or by preventing enzyme–substrate interaction via steric hindrance. To test this hypothesis, we used the fluorogenic substrate trifluoromethyl umbelliferyloleate (TFMUO) to measure acid lipase activity in d407 protein lysates (18). Wolman's disease fibroblasts that lack acid lipase activity and age-matched normal human fibroblasts were used as controls. Lack of substrate hydrolysis at pH 7 validated the assay. A2E did not affect the expression or degradation of acid lipase because 10 μg of soluble protein from either vehicle- or A2E-treated cells hydrolyzed TFMUO with similar kinetics (Fig. 2C). Further, A2E did not interfere with enzyme–substrate interactions, because there was no change in the hydrolysis rate when substrate micelles contained A2E (Fig. 2D). Thus, CE accumulation in RPE cells is not because of a direct inhibition of acid lipase by A2E.
Late Endosomal Cholesterol Levels Control CE Hydrolysis in RPE Cells.
It has been proposed that CE hydrolysis in LE/Ly is biphasic; initial hydrolysis occurs until endosomal membranes become saturated with free cholesterol and further hydrolysis depends on desorption of cholesterol onto other membranes/carriers (19). Indeed, we found that increasing the cholesterol content of TFMUO micelles caused a marked decrease in acid lipase activity (Fig. 3A), indicating that the enzyme is susceptible to feedback inhibition and/or a cholesterol-induced reduction in membrane fluidity, which decreases substrate diffusion and enzyme–substrate interactions.
Fig. 3.

A2E-induced inhibition of CE metabolism is overcome by increasing cholesterol efflux. (A) Acid lipase activity as a function of cholesterol content in substrate micelles. Significantly different from 0% cholesterol, ∗, P < 0.01. (B) Degradation of Bodipy-CE in control and A2E-loaded ARPE-19 cells. ∗, significantly different from controls; P < 0.05. (C) Effect of the hydrophobic amine U18666A on Bodipy-CE hydrolysis. ∗, P < 0.05. (D) Effect of A2E and the PPARγ agonist pioglitazone on Bodipy-CE hydrolysis in ARPE-19 and primary bovine RPE cells. ∗, significantly different from vehicle-treated cells, P < 0.01; ∗∗, significantly different from A2E-loaded cells, P < 0.05.
This finding prompted us to further investigate the relationship between LE/Ly cholesterol levels and CE metabolism in RPE cells. To follow CE hydrolysis, we used Bodipy-labeled cholesteryl dodecanoate (Bodipy-CE), because it is specifically metabolized by acid lipase (20). Control and A2E-loaded ARPE-19 cells internalized Bodipy-CE vesicles with similar kinetics and Bodipy-CE was transported to A2E-containing LE/Ly (SI Fig. 8). However, significantly more undegraded Bodipy-CE was recovered from cells with A2E compared with controls (Fig. 3B), confirming that A2E interferes with CE metabolism. Blocking cholesterol efflux by treatment with the hydrophobic amine U18666A (19) also inhibited Bodipy-CE hydrolysis (Fig. 3C), indicating that late endosomal cholesterol efflux is necessary for optimal acid lipase activity.
Activation of the Liver X Receptor (LXR)/Peroxisome Proliferator-Activated Receptor (PPAR) Pathway Restores Cholesterol Homeostasis in A2E-Laden RPE Cells.
We then asked whether A2E-induced cholesterol and CE accumulation can be overcome by pharmacologically increasing cholesterol efflux. First, treatment with a PPARγ agonist pioglitazone, a transcriptional activator of the cholesterol transporter ABCA1 via the LXR-PPAR pathway (21), significantly increased Bodipy-CE hydrolysis in both A2E-laden ARPE-19 and primary bovine RPE cells (Fig. 3D).
Next, confocal microscopy confirmed that treatment of A2E-laden cells with either pioglitazone or the LXR agonist TO901317 decreased intracellular filipin staining to a level that was comparable to controls (Fig. 4 A and B). Both agonists significantly increased ABCA1 protein expression (Fig. 4 D and E) but did not alter cellular A2E levels (Fig. 4C), indicating that they specifically induced ABCA1-mediated cholesterol efflux. Because LXR/PPAR agonists decreased free cholesterol levels and restored CE metabolism, we conclude that A2E primarily causes cholesterol accumulation in RPE LE/Ly, which then inhibits acid lipase activity, leading to CE storage.
Fig. 4.

LXR/PPAR agonists restore cholesterol homeostasis in A2E-laden cells. (A) Filipin staining of endogenous cholesterol in polarized ARPE-19 cells treated with vehicle (Control), A2E, A2E and pioglitazone (A2E+Pio), or A2E and the LXR agonist TO901317 (A2E+TO). (B) Quantification of intracellular filipin fluorescence. Data were normalized to vehicle-treated control cells whose intracellular fluorescence is set to 100%. Significantly different from all other conditions, ∗, P < 0.01. For Control, A2E and A2E+Pio, n ≥ 200; for A2E+TO, n = 80. (C) Quantification of A2E levels after treatment with either Pio or TO compared with A2E alone. (D) Representative ABCA1 immunoblot of A2E-loaded cells treated with either Pio or TO. (E) Quantification of ABCA1 protein levels normalized to β-actin; ∗, significantly different from cells exposed to A2E alone, P < 0.05.
To ascertain whether cholesterol accumulation in response to A2E was because of an RPE-specific mechanism, we examined other cell types. Polarized Madin–Darby canine kidney (MDCK) and nonpolarized BHK-21 cells both showed increased filipin fluorescence in the presence of A2E compared with controls (Fig. 5), indicating that A2E-induced cholesterol accumulation is not cell-type-specific but may depend on a biophysical interaction between A2E and cholesterol.
Fig. 5.
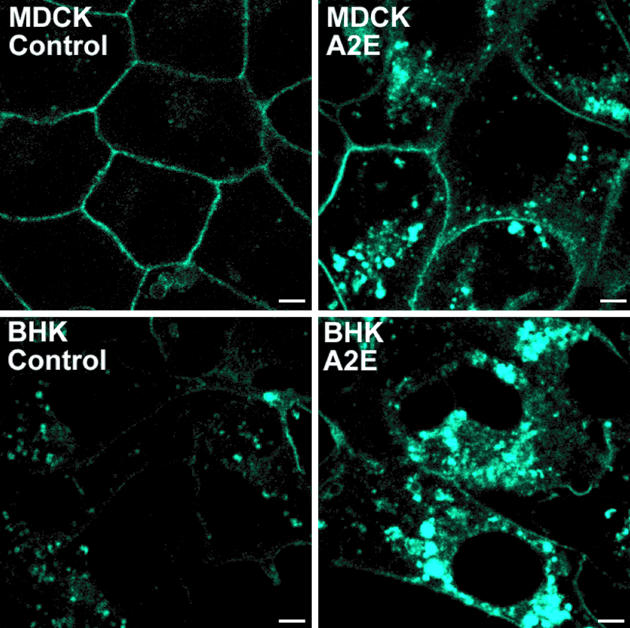
Filipin staining of endogenous cholesterol in control or A2E-laden polarized MDCK and BHK-21 cells. (Scale bar, 5 μm.)
A2E Accelerates Cholesterol Dissociation from Model Membranes.
To identify the mechanism by which A2E causes cholesterol accumulation, we studied cholesterol dynamics in lipid bilayers with or without A2E. Cholesterol has a small polar headgroup (the OH), which provides inadequate protection for its hydrophobic sterol rings from exposure to water. Lipids with bulky headgroups (e.g., phosphatidylcholine) form an “umbrella” and shield neighboring sterol molecules from unfavorable interactions (22). Ceramides and other cone-shaped lipids compete with cholesterol for space under the umbrella and displace cholesterol from the bilayer (23). A2E is also a cone-shaped lipid, with a small polar headgroup relative to its retinoid side chains (3) (Fig. 1A). We speculated that, when present in the same bilayer, A2E might compete with cholesterol for tight packing with neighboring phospholipids and thus displace cholesterol from the membrane. To test this hypothesis, we used accessibility of membrane cholesterol to cholesterol oxidase as a measure of cholesterol displacement in bilayers with A2E. Because cholesterol oxidase is sensitive to slight changes in membrane organization, enzyme activity is a reliable indicator of cholesterol stability within the bilayer (24).
The rate of cholesterol oxidation was significantly greater in liposomes with A2E compared with control liposomes (Fig. 6A and Table 1). This was not due to A2E's positive charge, because liposomes containing another cationic lipid, dioleoyltrimethyl ammonium propane (DOTAP), instead of A2E, failed to increase cholesterol oxidation. The inverted cone-shaped lipid lysophosphatidylcholine (LysoPC), which expands the membrane phospholipid pool (23), abolished the A2E-induced increase in cholesterol oxidation (Fig. 6A). Furthermore, ≈4% of total membrane cholesterol was oxidized after 4 h (Table 1), indicating that A2E does not permeabilize lipid bilayers or cause other gross alterations in membrane architecture. Liposomes with A2E also increased cholesterol transfer to methyl-β-cyclodextrin (Fig. 6B), providing additional proof that A2E displaces membrane cholesterol and thus disturbs bilayer cholesterol organization.
Fig. 6.

A2E accelerates cholesterol displacement from lipid bilayers. (A) Influence of A2E on the accessibility of bilayer cholesterol to cholesterol oxidase. Liposomes (20 mol% cholesterol and 0.04 mol% of A2E, LysoPC or DOTAP) were incubated with cholesterol oxidase for the indicated times at 37°C. ∗, significantly different from all other liposomes at the corresponding time point; P < 0.01. (B) Kinetics of cholesterol transfer from control or A2E liposomes to cyclodextrin.
Table 1.
Cholesterol dissociation rates from lipid bilayers
| Liposome | Koff, h−1 | t1/2, h | Percent cholesterol oxidized |
|---|---|---|---|
| Control | 0.0134 ± 0.0003 | 51.66 | 1.95 ± 0.20 |
| A2E | 0.0222 ± 0.0003 | 31.20 | 3.98 ± 0.86 |
| Control + LysoPC | 0.0109 ± 0.0004 | 63.40 | 1.80 ± 0.39 |
| A2E + LysoPC | 0.0116 ± 0.0004 | 59.65 | 1.89 ± 0.33 |
| DOTAP | 0.0120 ± 0.0006 | 57.69 | 1.96 ± 0.21 |
Discussion
Phagocytosis and degradation of shed OS is an indispensable function performed by the RPE to maintain photoreceptor health and vision. Several lines of evidence suggest that OS phagocytosis is tightly coupled to RPE lipid and cholesterol metabolism: (i) Phagocytosis triggers expression of the AP-2 transcription factor (25), which regulates acid lipase expression (26); (ii) OS phagocytosis induces PPARγ (25), which increases transcription of cholesterol transporters (21); and (iii) selective metabolism of OS lipids by the RPE leads to the conservation and recycling of docosahexaenoic acid (27). In addition, RPE cells express several receptors and cholesterol transporters (28) that participate in lipoprotein uptake and cholesterol efflux.
Because OS degradation takes place mainly in LE/Ly, and because our published data indicate that the lipofuscin fluorophore A2E in RPE LE/Ly interferes with the clearance of OS lipids (8), we investigated the effect of A2E on endogenous cholesterol and lipid metabolism in the RPE. Our results show that A2E, at levels similar to those found in vivo, specifically causes cholesterol and CE accumulation in RPE cells. We did not observe an increase in LE/Ly pH in A2E-loaded cells, which is expected, because A2E is a quaternary amine that lacks exchangeable protons (Fig. 1A). Unlike classical lysosomotropic amines, the accumulation of A2E in LE/Ly is not because of “proton trapping” (29). As protons are neither consumed nor leaked out because of passive diffusion of the protonated base in A2E-loaded cells, there is no driving force for alkalinization. Moreover, a nonspecific increase in LE/Ly pH would lead to widespread inhibition of lysosomal enzymes, which has not been observed either in vitro or in vivo in the presence of A2E (3).
Using model lipid bilayers, we found that cone-shaped A2E competes with cholesterol for space under the phospholipid umbrella (22), displacing it from membranes. To undergo efflux from LE/Ly, cholesterol located in late endosomal internal membranes needs to be shuttled to the limiting membrane by nonvesicular mechanisms (30). Our data show that in RPE cells, cholesterol accumulates in the same organelles as A2E, indicating a block in LE/Ly cholesterol efflux. It is conceivable that once cholesterol displaced from A2E-containing membranes reaches a critical concentration, it is excluded from the pool destined for nonvesicular transport to the limiting membrane and thus gets trapped within the LE/Ly. Increased LE/Ly cholesterol levels result in an indirect inhibition of RPE acid lipase, in agreement with previous studies showing that late endosomal cholesterol efflux is necessary for acid lipase activity (19). The subsequent accumulation of CE along with free cholesterol generates a feed-forward cycle of further cholesterol accumulation, inhibition of acid lipase, and increased CE in the cell.
Cholesterol efflux from LE/Ly to the plasma membrane is preferentially mediated by ABCA1 (31). Our data show that LRX/PPAR agonists induce ABCA1 expression, decrease cholesterol accumulation, and restore CE metabolism in RPE cells. Cholesterol accumulation in LE/Ly may also interfere with other degradative functions, such as the processing of OS lipids, as previously demonstrated by us (8) and confirmed in this study. Indeed, increased membrane cholesterol has been found to inhibit lysosomal phospholipase A1 (32).
The presence of cholesterol and CE deposits in the Bruch's membrane of AMD eyes (9, 10) and the correlation of genetic variations in cholesterol transporters with AMD susceptibility (12, 13) point to an age-related alteration in cholesterol homeostasis in the RPE that may contribute to AMD progression. In this context, it is noteworthy that cholesterol and CE deposits in the Bruch's membrane are thought to originate from the RPE, including incompletely processed OS and additional sources such as the RPE itself (9). Because our data demonstrate that A2E causes a >4-fold increase in LE/Ly free cholesterol (apart from CE accumulation), and because membranous debris beneath the RPE appear to originate as blebs and/or exocytosed organelles from the RPE basal surface (33, 34), it is tempting to speculate that the source of subRPE cholesterol/CE deposits could be LE/Ly internal membranes extruded from A2E-laden cells.
Our study provides evidence in support of the hypothesis that loss of cholesterol homeostasis in the RPE may play an important role in the pathogenesis of retinal dystrophy. Biochemical and histological analyses of cholesterol and phospholipid homeostasis in abcr−/− mice would be useful in providing an in vivo correlate for our observations on A2E-induced cholesterol accumulation in RPE cells. However, these experiments are beyond the scope of the present study, because the main objective here was to identify the mechanism by which A2E can derail intracellular cholesterol trafficking pathways. At the cellular level, impaired cholesterol efflux from RPE LE/Ly may interfere with rhodopsin activation and photoreceptor membrane remodeling. Excess cholesterol, either within the RPE or extruded into the Bruch's membrane, could function as an atypical chronic inflammatory stimulus (35) that contributes to AMD. Finally, strategies that increase cholesterol efflux from the RPE may be a promising therapeutic approach in circumventing disease progression.
Materials and Methods
Cell Culture.
Human ARPE-19 cells [American Type Culture Collection (ATCC), Bethesda, MD] were cultured in RPMI medium 1640 + 1% FBS for ≥6 weeks to establish polarity. Human d407 cells (a gift of R. C. Hunt, University of South Carolina, Columbia, SC) were grown in DMEM + 3% FBS for 48 h before A2E loading. Primary bovine RPE cells were isolated from freshly obtained eyes and cultured on laminin-coated dishes in DMEM/1% FBS (ATCC). Cells were plated at a density of 40,000 cells per cm2 on 24-well plates for biochemical experiments. For confocal microscopy, cells were plated on laminin-coated Transwell filters (BD Biosciences, San Jose, CA) for ARPE-19 and bovine RPE or alcian blue-coated coverslips for d407 cells. Each experiment was performed in at least two different RPE cell types. Wolman's disease fibroblasts and age-matched normal human fibroblasts (Coriell Cell Repositories, Camden, NJ) were cultured in DMEM/15% FBS. Madin–Darby canine kidney Type II cells were cultured on Transwell filters in DMEM/5% FBS, and BHK-21 cells were cultured on coverslips in alpha minimum essential medium (AMEM)/10% FBS.
A2E Synthesis and Loading.
A2E was synthesized according to a published protocol (36) and purified by HPLC (>97%, electrospray ionization–MS). Cells were exposed to A2E (50 μM for d407 cells and 15 μM for ARPE-19 and bovine RPE cells) for 6 h in growth medium and chased overnight in fresh medium.
Lipid and Sterol Quantification.
Cellular lipids were extracted in chloroform/methanol, and total lipid phosphorous was determined by a colorimetric assay after acid digestion (37). Sphingomyelin content was determined by the Amplex Red Sphingomyelinase assay (Molecular Probes, Eugene, OR). Samples were incubated with 100 μM Amplex red reagent, 2 units/ml HRP, 0.2 units/ml choline oxidase, 8 units/ml alkaline phosphatase, and 1 milliunit neutral sphingomyelinase. Free cholesterol and CE were quantified by the Amplex Red Cholesterol assay (Molecular Probes).
OS Phagocytosis.
ARPE-19 cells were fed bovine OS for 4 h (8) and chased in fresh medium. After lipid extraction, total phospholipids and free cholesterol were quantified as above.
Bodipy-CE Hydrolysis.
Vesicles with egg phosphatidylcholine, brain phosphatidylserine, cholesterol (Avanti Polar Lipids, Alabaster, AL), and Bodipy-CE (Molecular Probes) (molar ratio 42:42:15:0.5) were prepared by sonicating on ice for 10 3-min cycles (Branson Sonifier 350; Branson Ultrasonics, Danbury, CT). Cells were incubated with Bodipy-CE vesicles (45 min, 37°C) and chased in fresh medium. Undegraded Bodipy-CE was quantified by TLC in a mobile phase of hexane/isopropanol/acetic acid (93:6:1). Cells were treated with an acyl CoA: cholesterol acyl transferase inhibitor FR179254 (400 nM; Calbiochem, San Diego, CA) for a true estimate of Bodipy-CE accumulation. Other drugs used were U18666A (500 nM; Calbiochem), pioglitazone (10 μM; Calbiochem), and TO901317 (1 μM; Cayman, Ann Arbor, MI).
Filipin Staining and Quantification.
Paraformaldehyde-fixed cells were stained with 50 μg/ml filipin for 45 min followed by confocal microscopy. Intracellular filipin fluorescence was quantified by using Leica (Leica Microsystems, Wetzlar, Germany) software. For each cell, a region of interest (ROI) was drawn just within the plasma membrane, and the fluorescence intensity per pixel of the ROI per plane of the confocal image stack was quantified.
Late Endosomal/Lysosomal pH.
Control or A2E-loaded cells were incubated with 7.5 μM Lysosensor blue (Molecular Probes, 5 min, 37°C), rinsed to remove excess dye, and imaged immediately. For quantitative determination of pH, cells were trypsinized, resuspended in recording medium (20 mM Hepes/4.5 g/liter glucose/1% FBS in Hanks' balanced salt solution) and incubated with 7.5 μM Lysosensor yellow/blue, as above. After centrifugation, cells were resuspended in Mes buffer, pH 7.0, and the fluorescence ratio at 436 and 525 nm was measured (λex 380 nm). Calibration curves were established by using Mes buffers of defined pH values, and nigericin and monensin were used to dissipate intracellular pH gradients. Measured ratios for known pH values were fitted to a Boltzmann equation, and LE/Ly pH of control and A2E-loaded cells was calculated. Bafilomycin A1 (50 nM; Sigma, St. Louis, MO) was used as a positive control.
Acid Lipase Activity.
Substrate micelles with 2 mM TFMUO (Sigma) and 3.25 mM egg phosphatidylcholine (EPC) were made in 2.5 ml of 2.4 mM sodium taurocholate by sonicating on ice for 5 min (18). Under these conditions, the assay was linear up to 30 μg/ml protein for ≥2 h at 37°C. A2E-containing micelles were prepared by adding 2.6 μg of A2E per milligram of lipid to the TFMUO/EPC mixture. The final assay solution contained 0–500 μM TFMUO, 150 μl of 0.2 M acetate buffer (pH 4), 10 μg of soluble protein extract from RPE cells, or Wolman's disease fibroblasts or normal human fibroblasts, in a total volume of 600 μl. After 1 h at 37°C, umbelliferone released upon hydrolysis of TFMUO was measured at 502 nm (λex 385 nm). For experiments on cholesterol content and acid lipase activity, TFMUO micelles were prepared as above with increasing amounts of cholesterol (keeping TFMUO concentration in the micelles constant), and the assay was performed with 300 μM substrate.
Confocal Microscopy.
Cells were imaged on a Leica TCS-SP2 laser-scanning confocal microscope using the 63 × 1.4 N.A. oil immersion objective. The argon/argon–krypton laser was used for Bodipy-CE, and the green helium–neon laser was used for A2E. In experiments with filipin, the 405 laser was used to excite both filipin and A2E, because A2E has an excitation peak at 418 nm, and images were captured at different emission windows (420–440 for filipin and 590–610 for A2E). For each set of experiments, the laser power, voltage, and offset were identical for a given fluorophore. All images were acquired with Leica software and processed identically for control and treated conditions.
Immunoblotting.
Whole-cell lysates were prepared in buffer containing 1% Triton X-100, and proteins (≈50 μg) were electrophoresed on 7.5% SDS-polyacrylamide gels and transferred to nitrocellulose membranes. Blots were probed with rabbit polyclonal antibodies to either ABCA1 (Abcam, Cambridge, MA; 1:1,000) or actin (Santa Cruz Biotechnology, Santa Cruz, CA; 1:1,000). After chemiluminescence development, bands were quantified by using ImageJ (NIH, Bethesda, MD), and ABCA1 levels were normalized to that of β-actin.
A2E Quantitation.
Cells were treated with A2E and agonists as above. Lipids were harvested, and A2E levels were quantified by TLC by using a mobile phase of chloroform/methanol/water (84:15:1, vol/vol).
Liposomes.
Liposomes with a lipid composition similar to that of late endosomes (38) (dioleoylphosphatidylcholine, dioleoylphosphatidylethanolamine, sphingomyelin, phosphatidylserine, phosphatidylinositol, and cholesterol; 50:20:9:2:4:20 mol%) were prepared by extrusion in 10 mM Hepes/150 mM NaCl buffer (7.5 μmol of total lipid per milliliter). When required, A2E, lysophosphatidylcholine, or dioleoyltrimethyl ammonium propane (0.04 mol%) was added to the lipid mixture. Based on our data, ≈200 ng of A2E per milligram of total lipid is present in RPE cells; of total cellular lipid, ≈67 μg is present in LE/Ly (15). Because A2E is predominantly present in LE/Ly, this amounts to 200 ng of A2E/67 μg of total lipid or 0.04 mol%.
Cholesterol Oxidase Assay.
Liposomes were diluted to 750 μM in buffer (0.5 M potassium phosphate/0.25 M NaCl, pH 7.4) and incubated with 2 units/ml HRP, 2 units/ml cholesterol oxidase, and 300 μM Amplex red reagent at 37°C. Fluorescence was measured at 590 nm (λex 540 nm) on a Spectramax Gemini microplate reader (Molecular Devices, Sunnyvale, CA). Dissociation rates were calculated by fitting kinetic data to a first-order exponential equation (Prism; GraphPad, San Diego, CA).
Cholesterol Transfer to Cyclodextrin.
Control or A2E-containing liposomes (375 μM) were incubated with 2.5 mM methyl-β-cyclodextrin at 37°C. Samples were filtered through Microcon YM-30 membranes (Millipore, Billerica, MA, 1,300 × g, 15 min) to separate liposomes from cyclodextrin–cholesterol complexes (39). The amount of cholesterol transferred was quantified by the Amplex red cholesterol assay after chloroform/methanol extraction of filtrates and retentates.
Statistical Analysis.
Data were analyzed by using either a two-tailed t test or one-way ANOVA followed by the Bonferroni multiple comparisons post-test (Prism). Unless otherwise stated, data are presented as mean ± SEM of three or more independent experiments, with three to four replicates per condition per experiment.
Supplementary Material
Acknowledgments
We thank Dr. Larry Leung for assistance with A2E synthesis; Dena Almeida for help with bovine RPE cultures; and Drs. Anant K. Menon, Robert G. Thorne, Arun Deora, and Emeline Nandrot for helpful discussions. This work was supported by a National Research Service Award (F32 EY015363) to A.L. and National Institutes of Health Grants EY08538 (to E.R.-B.) and EY13295 (to S.C.F.) as well as by the Dyson Foundation and the Research to Prevent Blindness Foundation. S.C.F. is the Research to Prevent Blindness William and Mary Greve Scholar and the recipient of an Irma T. Hirschl Career Scientist Award.
Abbreviations
- AMD
age-related macular degeneration
- RPE
retinal pigment epithelium
- ABCA1
ATP-binding cassette protein A1
- CE
cholesteryl esters
- Bodipy-CE
Bodipy-labeled cholesteryl dodecanoate
- LE/Ly
late endosomes/lysosomes
- LXR
liver X receptor
- OS
outer segment
- PPAR
peroxisome proliferator-activated receptor
- TFMUO
trifluoromethyl umbelliferyloleate
- DOTAP
dioleoyltrimethyl ammonium propane
- LysoPC
lysophosphatidylcholine.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0702504104/DC1.
References
- 1.Albert AD, Boesze-Battaglia K. Prog Lipid Res. 2005;44:99–124. doi: 10.1016/j.plipres.2005.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bok D. J Cell Sci Suppl. 1993;17:189–195. doi: 10.1242/jcs.1993.supplement_17.27. [DOI] [PubMed] [Google Scholar]
- 3.Sparrow JR, Boulton M. Exp Eye Res. 2005;80:595–606. doi: 10.1016/j.exer.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 4.Travis GH, Golczak M, Moise AR, Palczewski K. Annu Rev Pharmacol Toxicol. 2007;47:469–512. doi: 10.1146/annurev.pharmtox.47.120505.105225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rattner A, Nathans J. Nat Rev Neurosci. 2006;7:860–872. doi: 10.1038/nrn2007. [DOI] [PubMed] [Google Scholar]
- 6.Weng J, Mata NL, Azarian SM, Tzekov RT, Birch DG, Travis GH. Cell. 1999;98:13–23. doi: 10.1016/S0092-8674(00)80602-9. [DOI] [PubMed] [Google Scholar]
- 7.Radu RA, Mata NL, Nusinowitz S, Liu X, Sieving PA, Travis GH. Proc Natl Acad Sci USA. 2003;100:4742–4747. doi: 10.1073/pnas.0737855100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Finnemann SC, Leung LW, Rodriguez-Boulan E. Proc Natl Acad Sci USA. 2002;99:3842–3847. doi: 10.1073/pnas.052025899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Curcio CA, Presley JB, Malek G, Medeiros NE, Avery DV, Kruth HS. Exp Eye Res. 2005;81:731–741. doi: 10.1016/j.exer.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 10.Li CM, Chung BH, Presley JB, Malek G, Zhang X, Dashti N, Li L, Chen J, Bradley K, Kruth HS, et al. Invest Ophthalmol Vis Sci. 2005;46:2576–2586. doi: 10.1167/iovs.05-0034. [DOI] [PubMed] [Google Scholar]
- 11.Malek G, Li C-M, Guidry C, Medeiros NE, Curcio CA. Am J Pathol. 2003;162:413–425. doi: 10.1016/S0002-9440(10)63836-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tikellis G, Sun C, Gorin MB, Klein R, Klein BE, Larsen EK, Siscovick DS, Hubbard LD, Wong TY. Arch Ophthalmol. 2007;125:68–73. doi: 10.1001/archopht.125.1.68. [DOI] [PubMed] [Google Scholar]
- 13.Zareparsi S, Buraczynska M, Branham KE, Shah S, Eng D, Li M, Pawar H, Yashar BM, Moroi SE, Lichter PR, et al. Hum Mol Genet. 2005;14:1449–1455. doi: 10.1093/hmg/ddi154. [DOI] [PubMed] [Google Scholar]
- 14.Sparrow JR, Parish CA, Hashimoto M, Nakanishi K. Invest Ophthalmol Vis Sci. 1999;40:2988–2995. [PubMed] [Google Scholar]
- 15.Gennis RB. Biomembranes–Molecular Structure and Function. New York: Springer; 1989. pp. 20–23. [Google Scholar]
- 16.Holz FG, Schutt F, Kopitz J, Eldred GE, Kruse FE, Volcker HE, Cantz M. Invest Ophthalmol Vis Sci. 1999;40:737–743. [PubMed] [Google Scholar]
- 17.Kuriyama M, Yoshida H, Suzuki M, Fujiyama J, Igata A. J Lipid Res. 1990;31:1605–1612. [PubMed] [Google Scholar]
- 18.Sheriff S, Du H, Grabowski GA. J Biol Chem. 1995;270:27766–27772. doi: 10.1074/jbc.270.46.27766. [DOI] [PubMed] [Google Scholar]
- 19.Wang Y, Castoreno AB, Stockinger W, Nohturfft A. J Biol Chem. 2005;280:11876–11886. doi: 10.1074/jbc.M414676200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reaven E, Tsai L, Azhar S. J Lipid Res. 1995;36:1602–1617. [PubMed] [Google Scholar]
- 21.Li AC, Glass CK. J Lipid Res. 2004;45:2161–2173. doi: 10.1194/jlr.R400010-JLR200. [DOI] [PubMed] [Google Scholar]
- 22.Huang J, Feigenson GW. Biophys J. 1999;76:2142–2157. doi: 10.1016/S0006-3495(99)77369-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lange Y, Ye J, Steck TL. J Biol Chem. 2005;280:36126–36131. doi: 10.1074/jbc.M507149200. [DOI] [PubMed] [Google Scholar]
- 24.Ahn KW, Sampson NS. Biochemistry. 2004;43:827–836. doi: 10.1021/bi035697q. [DOI] [PubMed] [Google Scholar]
- 25.Ershov AV, Bazan NG. J Neurosci Res. 2000;60:328–337. doi: 10.1002/(SICI)1097-4547(20000501)60:3<328::AID-JNR7>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 26.Reis S, Buchler C, Langmann T, Fehringer P, Aslanidis C, Schmitz G. J Lipid Res. 1998;39:2125–2134. [PubMed] [Google Scholar]
- 27.Rodriguez de Turco EB, Parkins N, Ershov AV, Bazan NG. J Neurosci Res. 1999;57:479–486. [PubMed] [Google Scholar]
- 28.Tserentsoodol N, Gordiyenko NV, Pascual I, Lee JW, Fliesler SJ, Rodriguez IR. Mol Vis. 2006;12:1319–1333. [PubMed] [Google Scholar]
- 29.Ohkuma S, Poole B. Proc Natl Acad Sci USA. 1978;75:3327–3331. doi: 10.1073/pnas.75.7.3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holtta-Vuori M, Ikonen E. Biochem Soc Trans. 2006;34:392–394. doi: 10.1042/BST0340392. [DOI] [PubMed] [Google Scholar]
- 31.Chen W, Sun Y, Welch C, Gorelik A, Leventhal AR, Tabas I, Tall AR. J Biol Chem. 2001;276:43564–43569. doi: 10.1074/jbc.M107938200. [DOI] [PubMed] [Google Scholar]
- 32.Piret J, Schanck A, Delfosse S, Van Bambeke F, Kishore BK, Tulkens PM, Mingeot-Leclercq MP. Chem Phys Lipids. 2005;133:1–15. doi: 10.1016/j.chemphyslip.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 33.Anderson DH, Mullins RF, Hageman GS, Johnson LV. Am J Ophthalmol. 2002;134:411–431. doi: 10.1016/s0002-9394(02)01624-0. [DOI] [PubMed] [Google Scholar]
- 34.Sarks S, Cherepanoff S, Killingsworth M, Sarks J. Invest Ophthalmol Vis Sci. 2007;48:968–977. doi: 10.1167/iovs.06-0443. [DOI] [PubMed] [Google Scholar]
- 35.Bok D. Proc Natl Acad Sci USA. 2005;102:7053–7054. doi: 10.1073/pnas.0502819102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parish CA, Hashimoto M, Nakanishi K, Dillon J, Sparrow J. Proc Natl Acad Sci USA. 1998;95:14609–14613. doi: 10.1073/pnas.95.25.14609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rouser G, Fkeischer S, Yamamoto A. Lipids. 1970;5:494–496. doi: 10.1007/BF02531316. [DOI] [PubMed] [Google Scholar]
- 38.Kobayashi T, Beuchat MH, Chevallier J, Makino A, Mayran N, Escola JM, Lebrand C, Cosson P, Gruenberg J. J Biol Chem. 2002;277:32157–32164. doi: 10.1074/jbc.M202838200. [DOI] [PubMed] [Google Scholar]
- 39.Niu SL, Mitchell DC, Litman BJ. J Biol Chem. 2002;277:20139–20145. doi: 10.1074/jbc.M200594200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.