


Abstract
Oxazole-containing macrocycles represent a promising class of anticancer agents that target G-quadruplex DNA. We report the results of spectroscopic studies aimed at defining the mode, energetics and specificity with which a hexaoxazole-containing macrocycle (HXDV) binds to the intramolecular quadruplex formed by the human telomeric DNA model oligonucleotide d(T2AG3)4 in the presence of potassium ions. HXDV binds solely to the quadruplex nucleic acid form, but not to the duplex or triplex form. HXDV binds d(T2AG3)4 with a stoichiometry of two drug molecules per quadruplex, with these binding reactions being coupled to the destacking of adenine residues from the terminal G-tetrads. HXDV binding to d(T2AG3)4 does not alter the length of the quadruplex. These collective observations are indicative of a nonintercalative ‘terminal capping’ mode of interaction in which one HXDV molecule binds to each end of the quadruplex. The binding of HXDV to d(T2AG3)4 is entropy driven, with this entropic driving force reflecting contributions from favorable drug-induced alterations in the configurational entropy of the host quadruplex as well as in net hydration. The ‘terminal capping’ mode of binding revealed by our studies may prove to be a general feature of the interactions between oxazole-containing macrocyclic ligands (including telomestatin) and intramolecular DNA quadruplexes.
INTRODUCTION
Telomeres are nucleoprotein structures located at the ends of eukaryotic chromosomes. Maintenance of these structures is essential for the prevention of chromosome degradation and illegitimate recombination (1). Telomerase, a ribonucleoprotein complex with reverse transcriptase activity, is responsible for the maintenance of telomeres (2,3). Inhibition of telomerase activity is associated with telomere shortening and subsequent activation of DNA damage responses that include cell cycle arrest, senescence and apoptosis (4–10). These effects have rendered telomerase an appealing target for anticancer therapeutics (11–15). The appeal of telomerase as an anticancer drug target is bolstered by the fact that its activity is found in 85–90% of all human tumors, but not in normal cells (6,7,10,16). As an example, telomerase-targeted therapy has been suggested for the treatment of malignant gliomas, since they are telomerase-positive, while normal brain tissue has no telomerase activity (17).
Like telomerase inhibition, deprotection of telomeric DNA can also cause the activation of DNA damage responses (8,18–22). In some human tumor cells, telomere deprotection induced by over-expression of a dominant-negative form of the telomeric DNA-binding protein TRF2 results in ATM-dependent apoptosis or senescence (8,9). Furthermore, inactivation of the telomeric DNA-binding protein Cdc13p in yeast results in RAD9-dependent G2 arrest and MEC1-dependent apoptosis (18,19). Since MEC1 is the functional yeast homolog of ATM, the latter result suggests that deprotection of telomeres activates a common ATM signaling pathway in both yeast and humans.
The 3′-ends of human telomeric DNA contain G-rich single-stranded overhangs (G-tails) with 16–35 tandem repeats of the 5′-T2AG3-3′ hexameric sequence (23,24). These G-tails can readily adopt thermodynamically stable G-quadruplex structures both in vitro (25–32) and in vivo (33,34). Compounds that stabilize G-quadruplex DNA have the potential for inhibiting the activities of both telomerase and telomeric DNA-binding proteins by denying them access to the telomere (11–15,35). To date, a diverse array of G-quadruplex-stabilizing compounds has been identified, including macrocyclic oxazoles (20,36–43), anthraquinones (44,45), acridines (46–50), cationic porphyrins (51–60), bistriazoles (61), perylenes (62–64), ethidium derivatives (65,66), fluorenones (67), pentacyclic acridinium salts (68–70) and fluoroquinophenoxazines (71,72). Among these compounds, the macrocyclic oxazoles and the bistriazoles exhibit a particularly high degree of selectivity for G-quadruplex relative to duplex DNA (37,43,61).
Telomestatin (a natural product isolated from Streptomyces anulatus) has proven to be one of the more potent and selective inhibitors of telomerase (36,37). This compound is a macrocyclic torand consisting of seven oxazole rings and one thiazoline ring. It exhibits cytotoxicity (IC50) versus neuroblastoma cell lines ranging from 0.8 to 4.0 μM (73). In this connection, oxazole-containing macrocycles represent a promising new class of G-quadruplex-targeting anticancer agents. We have previously shown that a macrocyclic hexaoxazole (HXDV, see structure in Figure 1) inhibits the growth of human lymphoblastoma RPMI 8402 cells with an IC50 of 0.4 μM (43). Our studies also revealed that HXDV binds and thermally stabilizes the structure formed in the presence of potassium ions by an oligomer [d(T2AG3)4] that contains four repeats of the multiply repeated sequence in the G-tails of human telomeric DNA (Figure 1) (43). Recent studies indicate that the potassium-stabilized structure adopted by d(T2AG3)4 in solution is an intramolecular (3 + 1) G-quadruplex in which three strands are oriented in one direction and the fourth strand is oriented in the opposite direction (29–32). While binding and stabilizing the d(T2AG3)4 quadruplex, HXDV did not interact with duplex DNA derived from salmon testes (ST DNA) (43).
Figure 1.

(A) The chemical structure of HXDV. (B) Base sequences of the seven DNA oligomers used in this study. In these sequences, AP denotes the fluorescent base 2-aminopurine, AF denotes the fluorescent dye Alexa Fluor 546 and Dab denotes the quencher dabcyl.
Although oxazole-containing macrocycles like telomestatin and HXDV have been shown to bind and stabilize G-quadruplex DNA, the mode and energetics associated with the G-quadruplex binding of such compounds remains to be delineated. In the current study, we define the mode, energetics and specificity with which HXDV binds human telomeric quadruplex DNA, which we model with the d(T2AG3)4 sequence. Our results indicate that HXDV binds solely to the quadruplex and not to the duplex or triplex form of nucleic acids. In addition, our studies also reveal that HXDV binds to d(T2AG3)4 via an entropically driven ‘terminal capping’ mode of interaction. To the best of our knowledge, these results provide the first experimental demonstration for this mode of interaction by an oxazole-containing macrocycle, which hitherto has been only theorized based on computational simulations (37,43).
MATERIALS AND METHODS
Ligand and nucleic acid molecules
HXDV was synthesized as described previously (43). Stock solutions of this compound were prepared in dimethyl sulfoxide (DMSO) and stored at −20°C. All DNA oligomers were obtained in their HPLC-purified forms from Integrated DNA Technologies (Coralville, IA, USA). The concentrations of all unlabeled and 2-aminopurine (AP)-labeled DNA solutions were determined spectrophotometrically using the following extinction coefficients at 260 nm and 90°C (ɛ260–90°C) [in units of (mol strand/l)−1 · cm−1]: 231 100 ± 2300 for d(T2AG3)4; 231 540 ± 26 700 for 9AP; 236 800 ± 4700 for 15AP; 223 100 ± 4200 for 21AP and 208 300 ± 5100 for 15,21AP. These extinction coefficients were determined by enzymatic digestion and subsequent colorimetric phosphate assay using previously established protocols (74). The concentrations of all Alexa Fluor 546-labeled DNA solutions were also determined spectrophotometrically. However, these determinations were based on the extinction coefficient for the Alexa Fluor dye at 554 nm and 25°C (ɛ554–25°C = 112 000 M−1 · cm−1) reported in the 2005 Molecular Probes Handbook. The r(UG4U) oligomer was obtained in its PAGE-purified form from Dharmacon Research Inc. (Lafayette, CO, USA). The concentrations of r(UG4U) solutions were determined using an ɛ260–90°C of 70 000 ± 7500 (mol strand/l)−1 · cm−1, with this ɛ260–90°C value being derived as described above for the unlabeled and AP-labeled DNA oligomers.
Salmon testes DNA, as well as all DNA and RNA polynucleotides, were obtained from Sigma (St. Louis, MO, USA), and used without further purification. Polynucleotide solution concentrations were determined using the following extinction coefficients in units of (mol nucleotide/l)−1 · cm−1: ɛ260–25°C = 6000 for p(dA) · p(dT); ɛ265–25°C = 8700 for p(dT); ɛ258–25°C = 9800 for p(rA) and ɛ260–25°C = 9350 for p(rU).
Temperature-dependent spectrophotometry
Temperature-dependent absorption experiments were conducted on an AVIV Model 14DS Spectrophotometer (Aviv Biomedical, Lakewood, NJ, USA) equipped with a thermoelectrically controlled cell holder. Quartz cells with a pathlength of 1.0 cm were used for all the absorbance studies. Temperature-dependent absorption profiles were acquired at either 260 (for duplex and triplex) or 295 (for quadruplex) nm with a 5 s averaging time. The temperature was raised in 0.5°C increments, and the samples were allowed to equilibrate for 1.5 min at each temperature setting. In the quadruplex melting studies, the concentrations of d(T2AG3)4, 9AP, 15AP, 21AP and 15,21AP were 5 μM in strand (120 μM in nucleotide), while the concentration of r(UG4U) was 20 μM in strand (120 μM in nucleotide). When present in these quadruplex studies, the HXDV concentration was 20 μM. In the duplex and triplex melting studies, the nucleic acid concentration was 15 μM in base pair (30 μM in nucleotide) or 15 μM in base triple (45 μM in nucleotide) and the HXDV concentration, when present, was 15 μM. The buffer for all the UV melting experiments contained 10 mM EPPS (pH 7.5). In addition, sufficient KCl was added to each solution to bring the total K+ concentration to either 150 mM for d(T2AG3)4 and p(rA) · p(dT), 2 mM for r(UG4U), 50 mM for ST DNA, 250 mM for p(dA) · 2p(dT) or 20 mM for p(rA) · 2p(rU). Prior to their use in UV melting experiments, all nucleic acid solutions were preheated at 90°C for 5 min and slowly cooled to room temperature over a period of 4 h.
Circular dichroism (CD) spectroscopy
Circular dichroism experiments were conducted at 35°C on an AVIV Model 202 spectropolarimeter equipped with a thermoelectrically controlled cell holder. A quartz cell with a 1 cm pathlength was used for all the CD studies. CD spectra were recorded from 340 to 240 nm in 1 nm increments with an averaging time of 2 s. The buffer conditions were 10 mM EPPS (pH 7.5) and sufficient KCl to bring the total K+ concentration to 150 mM. The concentration of d(T2AG3)4 was 8 μM in strand (192 μM in nucleotide), and, when present, the osmolalities of glycerol and ethylene glycol were 2.0.
Steady-state fluorescence spectroscopy
All steady-state fluorescence experiments were conducted on an AVIV model ATF105 spectrofluorometer equipped with a thermoelectrically controlled cell holder. The solution conditions for the steady-state fluorescence experiments were identical to those described above for the CD experiments.
Fluorescence quantum yield values (Φ) for 9AP, 15AP, 21AP and their HXDV complexes were determined relative to quinine sulfate in 1 N H2SO4, which was assigned a value of 0.546 at 25°C (75). In these measurements, the excitation wavelength was set at 310 nm, with the excitation and emission bandwidths being set 5 nm. The pathlengths of the quartz cell used in these measurements were 0.2 cm in the excitation direction and 1 cm in the emission direction. The 9AP, 15AP and 21AP concentrations were 18 μM in strand (432 μM in nucleotide), while the concentration of quinine sulfate was 20 μM. When present, the HXDV concentration (90 μM) was chosen so as to ensure that essentially all the DNA was ligand-bound under the solution conditions employed. Fluorescence emission spectra were acquired at 25°C from 605 to 320 nm in 1 nm increments with a 1 s averaging time. All fluorescence emission spectra were corrected by subtraction of the corresponding spectra of buffer alone or buffer plus HXDV.
The characterizations of AF and AF-Dab were conducted with the excitation wavelength set at 540 nm and the excitation and emission bandwidths set at 5 nm. The pathlength of the quartz cell used in these measurements was 1 cm in both the excitation and emission directions. Fluorescence emission spectra were acquired at either 25 or 90°C from 720 to 545 nm in 1 nm increments with a 1 s averaging time. These emission spectra were corrected by subtraction of the corresponding spectra of buffer alone. Temperature-dependent characterizations of AF-Dab were conducted with the emission wavelength set at 579 nm and the averaging time set at 10 s. The temperature was raised in 1°C increments, and the samples were allowed to equilibrate for 1 min at each temperature setting. The concentrations of AF and AF-Dab were always 1 μM in strand (25 μM in nucleotide), while the concentration of HXDV, when present, was 50 μM.
For the HXDV titration experiments, which were conducted at temperatures ranging from 15 to 45°C, 1–10 μl aliquots of ligand (at concentrations ranging from 500 μM to 2 mM in DMSO) were sequentially added to solutions of 15,21AP that were 10 μM in strand (240 μM in nucleotide). After each addition, the sample was left to equilibrate for 3 min, whereupon the emission spectrum from 450 to 320 nm was recorded. These emission spectra were corrected by subtraction of the corresponding spectra of buffer alone or buffer plus the appropriate amount of HXDV. In these measurements, the excitation wavelength was set at 310 nm, with the excitation and emission bandwidths being set 5 nm. The pathlengths of the quartz cell used in these measurements were 1 cm in the excitation direction and 0.2 cm in the emission direction.
Concentrations of free and bound HXDV ([HXDV]free and [HXDV]bound) were derived from the HXDV-induced changes in 15,21AP fluorescence in the following manner. Fluorescence intensities at infinite HXDV concentration (I∞) were determined by fitting semilogarithmic plots of fluorescence intensity (I) versus total HXDV concentration ([HXDV]total) with the following sigmoidal relationship:
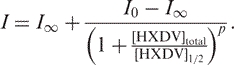 |
1 |
In this relationship, I0 is the fluorescence intensity in the absence of HXDV, p the Hill slope and [HXDV]½ is the HXDV concentration at which half of the AP-labeled DNA sites are bound. The I∞ values obtained from fits with Equation (1) were used to determine values of [HXDV]bound by proportionality. Values of [HXDV]free were then determined by subtraction of [HXDV]bound from [HXDV]total.
Time-resolved fluorescence anisotropy
Time-resolved fluorescence anisotropy measurements were conducted at 25°C on a PTI EasyLife LS fluorescence lifetime system fitted with a 310 nm LED light source, film polarizers in both the excitation and emission directions, and a 350 nm long pass filter (Chroma Technology Corp., Rockingham, VT, USA) in the emission direction. The slit width in the excitation direction was set at 6 mm. The fluorescence decay curves were acquired logarithmically in 400 channels with a 0.5 s integration time. The start and end delays of the acquisitions were 70 and 115 ns, respectively. The instrument response function was detected using light scattered by a dilute suspension of nondairy creamer, with the emission polarizer oriented parallel to the excitation polarization and no cut-on filter in place. The DNA concentration was 10 μM in strand (240 μM in nucleotide), and, when present, DNA-saturating concentrations of HXDV (90 μM) were employed. Each final decay profile reflected an average of six independent scans and was deconvolved in the Felix32 program (PTI Inc., London, Ontario, CANADA). The solution conditions for these time-resolved fluorescence experiments were identical to those described above for the CD experiments.
Fluorescence intensity decays measured with the emission polarizer oriented either parallel {IVV(t)} or perpendicular {IVH(t)} to the excitation polarization were best fit by the following sum of three exponentials:
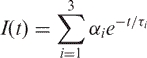 |
2 |
In this relationship, the values of αi are the amplitudes of each component and the values of τi are the corresponding fluorescence lifetimes. IVV(t) and IVH(t) were then deconvolved simultaneously with a magic angle decay {D(t)} using the following relationships:
| 3a |
| 3b |
In these relationships, G is the instrumental correction factor and r(t) is the anisotropy decay, which was best fit by the following sum of two exponentials:
| 4 |
The limiting anisotropy at time zero {r(0)} was derived from the sum of β1 and β2. Values of G ranged from 0.99 to 1.03, and were determined using the following relationship:
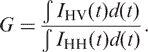 |
5 |
RESULTS AND DISCUSSION
HXDV binding is specific for quadruplex relative to duplex or triplex nucleic acid forms
We sought to evaluate the ability of HXDV to bind and thermally stabilize the duplex, triplex and quadruplex forms of nucleic acids. Toward this end, we monitored the UV absorbances of the nucleic acids as a function of temperature in the absence and presence of HXDV. The melting of duplex and triplex nucleic acids is generally associated with a hyperchromic shift at 260 nm (76,77), while the melting of quadruplex nucleic acids is associated with a hypochromic shift at 295 nm (26,78). Thus, the temperature-dependent absorbances of duplexes and triplexes were monitored at 260 nm, with corresponding quadruplex absorbances being monitored at 295 nm. ST DNA, p(rA) · p(rU), p(rA) · p(dT), p(dA) · 2p(dT), p(rA) · 2p(rU), d(T2AG3)4 and r(UG4U) were used as representative models of a DNA duplex, an RNA duplex, a hybrid DNA · RNA duplex, a DNA triplex, an RNA triplex, a DNA quadruplex and an RNA quadruplex, respectively. All the UV melting studies were conducted at pH 7.5 in the presence of potassium ions.
Figure 2 shows the UV melting profiles (depicted in their first-derivative forms) of d(T2AG3)4, ST DNA, p(dA) · 2p(dT), r(UG4U), p(rA) · p(dT) and p(rA) · 2p(rU) in the absence (filled circles) and presence (open circles) of HXDV. Note that the presence of HXDV does not impact the thermal transitions of the ST DNA (B) and p(rA) · p(dT) (E) duplexes. Furthermore, it does not alter the Watson–Crick (WC) thermal transitions [which reflect the conversion of duplex and single strand to all single strand (77)] of the p(dA) · 2p(dT) (C) and p(rA) · 2p(rU) (F) triplexes. These results suggest that neither ST DNA, p(rA) · p(dT), p(dA) · p(dT), nor p(rA) · p(rU) interacts with HXDV. In other words, HXDV does not appear to interact with DNA · DNA, RNA · RNA or RNA · DNA hybrid duplexes. Not only does the presence of HXDV fail to alter the WC thermal transitions of the p(dA) · 2p(dT) and p(rA) · 2p(rU) triplexes, it also fails to impact the corresponding Hoogsteen (HG) thermal transitions [which reflect the conversion of the triplex to duplex and single strand (77)] of these two triplexes. Thus, neither the DNA nor the RNA triplex form appears to be capable of serving as a binding substrate for HXDV.
Figure 2.

First derivatives of the UV melting profiles of d(T2AG3)4 (A), ST DNA (B), p(dA) · 2p(dT) (C), r(UG4U) (D), p(rA) · p(dT) (E) and p(rA) · 2p(rU) (F) in the absence (filled circles) and presence (open circles) of HXDV. The UV melting profiles for the quadruplexes formed by d(T2AG3)4 and r(UG4U) were acquired at 295 nm, while the UV melting profiles for the duplexes formed by ST DNA and p(rA) · p(dT) and the triplexes formed by p(dA) · 2p(dT) and p(rA) · 2p(rU) were acquired at 260 nm. For the UV melting profiles of the p(dA) · 2p(dT) and p(rA) · 2p(rU) triplexes, HG denotes the Hoogsteen transition of the triplex into duplex plus single strand, while WC denotes the Watson–Crick transition of the remaining duplex into single strands.
Although the presence of HXDV does not alter the thermal stabilities of the duplex and triplex nucleic acids noted above, it increases the thermal stabilities of the d(T2AG3)4 and r(UG4U) quadruplexes (Figure 2A and D, respectively). These HXDV-induced thermal enhancements are indicative of ligand binding to the host quadruplexes. The transition temperature (Ttran) corresponding to the minimum of the d(T2AG3)4 melting profile increases from 63.0°C in the absence of HXDV to 80.5°C in the presence of the ligand (Figure 2A). HXDV increases the thermal stability of r(UG4U) to a lesser extent than d(T2AG3)4, with the Ttran of r(UG4U) increasing from 103.0 to 104.0°C (Figure 2D). It is tempting to suggest that the reduced extent of thermal enhancement afforded the tetramolecular quadruplex formed by r(UG4U) (79,80) relative to the monomolecular quadruplex formed by d(T2AG3)4 (27,32) may reflect a comparatively reduced affinity of HXDV for the RNA quadruplex versus the DNA quadruplex. However, it is also possible that the differential impact of HXDV binding on the thermal stabilities of r(UG4U) and d(T2AG3)4 reflects the markedly differing Ttran-values of the two quadruplexes.
In the aggregate, our UV melting results are consistent with HXDV binding solely to quadruplex and not to duplex and triplex nucleic acid forms. With the exception of telomestatin, few other compounds approach this degree of quadruplex-binding specificity (38,39,61,81).
Steady-state fluorescence studies of d(T2AG3)4 site-specifically substituted with 2-aminopurine are consistent with HXDV-induced destacking of the adenines at positions 15 and 21, but not the adenine at position 9
While the UV melting studies described in the preceding section are indicative of an interaction between HXDV and d(T2AG3)4, they provide little information regarding the molecular nature of the ligand–quadruplex complex. Toward this goal, we substituted the adenine residue at either position 9, 15 or 21 of d(T2AG3)4 with AP (see Figure 1B). Control UV melting studies of the resulting AP-substituted oligomers (9AP, 15AP and 21AP) reveal that the AP substitutions induce little or no alteration in the thermal stability of d(T2AG3)4, nor do they alter the interaction of d(T2AG3)4 with HXDV (see Figure S1 of the Supplementary Data).
We monitored the impact of HXDV binding on the fluorescence of 9AP, 15AP and 21AP, with the resulting emission spectra being shown in Figure 3A–C, respectively. HXDV binding induces a substantive increase in the fluorescence of 15AP and 21AP, while minimally affecting the fluorescence of 9AP. We quantified these fluorescence changes by analyzing the fluorescence intensities of 9AP, 15AP and 21AP and their HXDV complexes to derive the quantum yield (Φ) values listed in Table 1. HXDV binding increases the Φ values of 15AP and 21AP by 0.013 and 0.007, respectively. In contrast, the Φ value of 9AP is essentially unchanged by HXDV. These fluorescence results are consistent with HXDV binding being coupled to the destacking of the AP bases at positions 15 and 21, but not the base at position 9.
Figure 3.

Steady-state fluorescence emission spectra at 25°C of 9AP (A), 15AP (B) and 21AP (C) in the absence (solid lines) and presence (dashed lines) of saturating concentrations of HXDV. The solution conditions were 10 mM EPPS (pH 7.5) and sufficient KCl to bring the total K+ concentration to 150 mM.
Table 1.
Fluorescence quantum yields (Φ) of 9AP, 15AP, 21AP and their HXDV complexes at 25°Ca
| Sample | Φ (±0.001) |
|---|---|
| 9AP | 0.034 |
| 9AP-HXDV | 0.035 |
| 15AP | 0.004 |
| 15AP-HXDV | 0.017 |
| 21AP | 0.014 |
| 21AP-HXDV | 0.021 |
a Solution conditions were 10 mM EPPS (pH 7.5) and sufficient KCl to bring the total K+ concentration to 150 mM.
HXDV binds d(T2AG3)4 via a ‘terminal capping’ rather than an intercalative mode of interaction
Previously reported NMR and computational studies (29–32) of d(T2AG3)4 and related oligomers in the presence of potassium ions indicate that the adenine residues in the edgewise loops (A15 and A21) are stacked on opposing terminal G-tetrads (schematically depicted in Figure 4). Unlike these two adenine residues, the adenine residue in the double-chain reversal loop (A9) adopts an unstacked conformation. Our fluorescence studies described above suggest that HXDV binding to d(T2AG3)4 induces the destacking of A15 and A21, but not A9. A potential mode of interaction that would be consistent with these results is one in which two HXDV molecules bind to the quadruplex, one at each end of the structure (schematically depicted in Figure 4). This ‘terminal capping’ model for the HXDV–d(T2AG3)4 interaction excludes the involvement of an intercalative binding mode. To test this hypothesis, we used a fluorescence resonance energy transfer (FRET) approach to monitor the impact of HXDV binding on the length of the d(T2AG3)4 quadruplex. Intercalation is typically associated with an increase in the length of the host nucleic acid structure. In contrast, a ‘capping’ type of interaction should not alter the length of the nucleic acid.
Figure 4.

Schematic representation of the ‘terminal capping’ model for the interaction between HXDV and d(T2AG3)4. In the absence of HXDV, the adenine residues at positions 15 and 21 (A15 and A21) are stacked on the terminal G-tetrads, while the adenine residue at position 9 (A9) does not engage in such stacking interactions. Upon the binding of an HXDV molecule to each end of the quadruplex (i.e. ‘terminal capping’ of the quadruplex), A15 and A21 become destacked, while A9 remains essentially unaffected. Adenine bases are depicted in red, guanine bases are depicted in green and thymine bases are depicted in orange. HXDV molecules are depicted in yellow.
We designed the AF-Dab and AF variants of d(T2AG3)4 shown in Figure 1B. AF-Dab has an Alexa Fluor 546 molecule conjugated to its 5′-end and a dabcyl molecule conjugated to its 3′-end. AF has only the 5′-conjugated Alexa Fluor 546 molecule. Through a FRET-mediated process, dabcyl (the acceptor) quenches the fluorescence of Alexa Fluor 546 (the donor) with an associated Förster distance (R0) of 29 Å (as reported in the 2005 Molecular Probes Handbook). Given this R0 value, the increase in quadruplex length of ∼3.5 Å that might be induced by the intercalation of a single ligand molecule would yield a detectable decrease in the extent of dabcyl-induced quenching of Alexa Fluor 546 fluorescence. Figure 5 shows the fluorescence emission spectra at 25°C of the quadruplex forms of AF (A) and AF-Dab (B) in the absence and presence of HXDV. In addition, the corresponding emission spectrum of the single-stranded form of AF-Dab is also presented (Figure 5B). Note the enhanced fluorescence intensity of the single-stranded relative to the quadruplex form of AF-Dab, an observation reflecting the increased distance between the Alexa Fluor 546 and dabcyl functionalities in the single-stranded versus the quadruplex form. The presence of HXDV induces a small decrease in the fluorescence intensity of quadruplex AF-Dab. This result argues against an intercalative mode of binding, since such a mode of interaction should induce an opposite effect (i.e. increase the fluorescence intensity of quadruplex AF-Dab). The HXDV-induced decrease in quadruplex AF-Dab fluorescence does not appear to reflect a decrease in the distance between the Alexa Fluor 546 and dabcyl moieties, as a similar decrease in the fluorescence of AF is also induced by the presence of HXDV (Figure 5A). Control fluorescence melting studies confirm the binding of HXDV to the AF-Dab and AF quadruplexes used in our FRET experiments (see Figure S2 of the Supplementary Data).
Figure 5.

(A) Fluorescence emission spectra at 25°C of the quadruplex form of AF in the absence (solid line) and presence (dashed line) of saturating concentrations of HXDV. (B) Fluorescence emission spectra at 25°C of the quadruplex form of AF-Dab in the absence (solid line) and presence (dashed line) of saturating concentrations of HXDV. This panel also shows the corresponding emission spectrum of the single-stranded form of AF-Dab in the absence of HXDV (dashed-dotted line). The 25°C spectrum of single-stranded AF-Dab reflects the spectrum of AF-Dab acquired at 90°C (a temperature at which AF-Dab is fully denatured) after correction for the temperature dependence of AF fluorescence. This correction was achieved by multiplying the 90°C spectrum of AF-Dab by the ratio of the 25°C AF spectrum relative to the 90°C AF spectrum. The solution conditions were as described in the legend to Figure 3.
The nonintercalative mode of HXDV-quadruplex binding suggested by the FRET results is not surprising if one considers the structural propensities of HXDV itself. We constructed and refined a structural model for HXDV using the Sybyl software suite (Tripos Inc., St. Louis, MO, USA). This model is depicted in Figure 6. Significantly, HXDV adopts a concave rather than a planar structure, with the plane formed by three of its oxazole moieties being angled ∼150° relative to the plane formed by its other three oxazole moieties (see side view of Figure 6). It is likely that such a nonplanar ligand molecule would have difficulty stably intercalating between neighboring G-tetrads. Taken together, the computational, FRET and AP fluorescence results described above, are fully consistent with a nonintercalative ‘terminal capping’ mode of interaction between HXDV and d(T2AG3)4.
Figure 6.

Top and side views of a structural model for HXDV, in which carbon atoms are depicted in gray, oxygen atoms are depicted in red, nitrogen atoms are depicted in blue and hydrogen atoms are depicted in white. Note the nonplanar nature of the hexaoxazole torand, as highlighted by the side view. The SYBYL 7.2 software suite (Tripos Inc.) was used to build a starting 3D structure of the ligand molecule. This structure was subsequently energy minimized using the Tripos force field with 1000 iterations of the conjugate gradient method. A nonbonded cutoff of 8 Å was used in these calculations, and atomic charges were assigned based on the Gasteiger–Hückel method (82).
HXDV binds d(T2AG3)4 with a stoichiometry of two ligand molecules per quadruplex
The ‘terminal capping’ model for the HXDV–d(T2AG3)4 interaction invokes the binding of one ligand molecule to each end of the quadruplex. We sought to determine experimentally the stoichiometry with which HXDV binds d(T2AG3)4. To this end, we designed the 15,21AP variant of d(T2AG3)4 shown in Figure 1B, in which the adenine residues at both positions 15 and 21 are substituted with AP residues. As was the case with a single AP substitution at position 9, 15 or 21, the double AP substitution at positions 15 and 21 induces little or no alteration in the thermal stability of d(T2AG3)4, nor does it alter the interaction of d(T2AG3)4 with HXDV (see Figure S1 of the Supplementary Data). We monitored the fluorescence of 15,21AP as a function of added HXDV. Figure 7A shows a representative fluorescence titration profile (depicted semilogarithmically) acquired at 25°C. This fluorescence titration profile was analyzed as described in the Materials and Methods to yield concentrations of free and bound HXDV ([HXDV]free and [HXDV]bound, respectively). These concentrations enabled us to construct the binding curves shown in Figure 7B and C, in which [HXDV]bound/[15,21AP]total (where [15,21AP]total denotes the total concentration of 15,21AP quadruplex) is plotted as a function of [HXDV]free. We fit the resulting binding curves with the following formalisms for one or two binding sites (83):
| 6 |
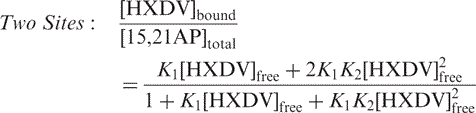 |
7 |
Figure 7.

(A) Semilogarithmic depiction of the fluorescence profile for the titration of 15,21AP with HXDV at 25°C. I370 denotes the buffer-corrected fluorescence emission intensity at 370 nm. The solid line reflects the fit of the experimental data points with Equation (1). (B, C) Binding curves of HXDV to 15,21AP derived from the fluorescence titration profile shown in panel A. The solid line in panel B reflects the fit of the experimental data using the formalism for one binding site [Equation (6)], while the solid line in panel C reflects the fit of the data using the formalism for two binding sites [Equation (7)]. The solution conditions were as described in the legend to Figure 3.
In these formalisms, Ki denotes the association constant for the binding of HXDV to a given site on 15,21AP. A comparison of Figure 7B and C reveals that the formalism for two binding sites affords a substantively better fit (χ2 = 0.004) of the binding curve than the corresponding formalism for one binding site (χ2 = 0.011). Note that applying a formalism for three binding sites (not shown) did not significantly improve the fit afforded by the two-site formalism (i.e. no further reduction in χ2 was observed). Collectively, these results indicate that HXDV binds to d(T2AG3)4 with a stoichiometry of two ligand molecules per quadruplex, a binding stoichiometry consistent with our proposed ‘terminal capping’ mode of interaction.
The binding of HXDV to d(T2AG3)4 is entropy driven
In addition to conducting fluorescence titration experiments at 25°C, we also conducted similar experiments at 15, 35 and 45°C. The binding curves ([HXDV]bound/[15,21AP]total versus [HXDV]free) resulting from these temperature-dependent studies are shown in Figure 8A. Fitting these binding curves with the formalism for two binding sites [Equation (7)] enabled us to derive values of K1 and K2 at each temperature, with these values being listed in Table 2. At all the temperatures examined, K1 (which ranges from 9.2 × 104 to 1.9 × 105 M−1) is greater than K2 (which ranges from 3.0 to 5.9 × 104 M−1). Thus, one HXDV molecule binds the host quadruplex with a greater affinity than the other.
Figure 8.

(A) Binding curves of HXDV to 15,21AP derived from fluorescence titration profiles acquired at 15, 25, 35 and 45°C. The solid lines reflect fits of the experimental data points with Equation (7). (B) van't Hoff plots {ln(K) versus 1/T} depicting the temperature dependence of the association constants for the binding of HXDV to its two target sites on 15,21AP. The solid lines reflect linear fits of the experimental data points with Equation (8).
Table 2.
Temperature dependence of the association constants (K) for the binding of HXDV to its two target sites on 15,21APa
We analyzed the temperature dependence of K1 and K2 using the following van't Hoff relationship:
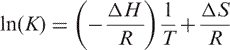 |
8 |
where ΔH and ΔS are the respective enthalpy and entropy changes for the binding of HXDV to the quadruplex. Equation (8) postulates that van't Hoff plots of ln(K) versus 1/T will be linear, with the slopes and y-intercepts of these plots yielding estimates for ΔH and ΔS, respectively.
Figure 8B shows the van't Hoff plots constructed using the temperature-dependent K data listed in Table 2. The values of ΔH and ΔS derived from linear regression analyses of these plots and subsequent application of Equation (8) are listed in Table 3. In addition, Table 3 also lists the free energy changes that accompany the binding reactions at 35°C (ΔG35). These ΔG35 values were derived using the corresponding values of K and the standard relationship
| 9 |
Table 3.
Thermodynamic parameters for the binding of HXDV to its two target sites on 15,21AP at 35°C
| Site | ΔG35a (kcal/mol) | ΔHb (kcal/mol) | ΔSb (cal/mol • K) |
|---|---|---|---|
| 1 | −7.4 ± 0.1 | +3.8 ± 2.1 | +36.0 ± 7.0 |
| 2 | −6.5 ± 0.1 | +4.0 ± 0.7 | +34.4 ± 2.1 |
Inspection of the thermodynamic data in Table 3 reveals positive ΔH and ΔS values for the binding of HXDV to both quadruplex sites. Thus, the binding of HXDV to both sites is enthalpically unfavorable, while being entropically favorable. Viewed as a whole, these results demonstrate that HXDV binding to the host d(T2AG3)4 quadruplex is entropically driven.
Defining the molecular origins of the entropic driving force for the HXDV–d(T2AG3)4 binding reactions
One commonly invoked explanation for entropically driven ligand-nucleic acid binding reactions is the coupled release of counterions. However, salt-dependent binding studies (not shown) suggest that the affinity of HXDV for d(T2AG3)4 is unaffected by changing ionic strength. This result is not surprising given the noncharged nature of HXDV. It is therefore unlikely that the entropic driving force for the HXDV–d(T2AG3)4 binding reactions reflects significant contributions from coupled counterion release.
A second commonly invoked explanation for entropically driven binding reactions is the coupled release of constrained water molecules. We sought to explore this possibility by using an osmotic stress approach to characterize the hydration changes, if any, that accompany the binding of HXDV to 15,21AP. In the osmotic stress approach, one monitors the impact of neutral solutes or cosolvents (osmolytes) on the ligand-DNA association constants (K). An osmolyte-induced increase in K reflects a net release of water molecules in conjunction with binding, while an osmolyte-induced decrease in K reflects a net uptake of water molecules. We employed ethylene glycol (MW = 62.1 g/mol) and glycerol (MW = 92.1 g/mol) as the osmolytes in our studies, since their molecular weights are sufficiently small relative to that of HXDV so as to minimize the potential for the introduction of volume exclusion (crowding) effects. Control CD studies reveal that the structure of d(T2AG3)4 is unaffected by the presence of ethylene glycol or glycerol at the osmolality (2.0) used in the osmotic stress studies (see Figure S3 of the Supplementary Data).
Table 4 lists the K values derived from fits of binding curves acquired at 35°C in the absence and presence of either ethylene glycol or glycerol. The presence of neither osmolyte significantly alters the value of K2, with any observed differences in K2 being almost within the experimental uncertainty. In contrast, the presence of both osmolytes induces a modest increase in the value of K1 from (1.9 ± 0.1) to (2.8–2.9 ± 0.2) × 105 M−1. These collective results suggest that HXDV binding to at least one of the two sites on d(T2AG3)4 may be accompanied by a net release of water molecules, a hydration change that could contribute, at least in part, to the entropic driving force for the binding of HXDV to d(T2AG3)4.
Table 4.
Osmolyte dependence of the association constants (K) for the binding of HXDV to 15,21AP at 35°Ca
Another potential explanation for the entropic nature of the driving force underlying the HXDV–d(T2AG3)4 binding reactions is a concomitant increase in the configurational entropy of the host DNA. We explored this possibility by using time-resolved fluorescence anisotropy to monitor the impact of HXDV binding on the conformational dynamics of d(T2AG3)4. Specifically, we evaluated how HXDV binding affects the mobilities of the AP residues in 9AP and 21AP. The low fluorescence quantum yield of unbound 15AP (see Table 1) precluded our use of this AP-labeled quadruplex in the fluorescence anisotropy studies.
Figure 9 shows a representative set of fluorescence intensity decays for 9AP, 21AP and their HXDV complexes with the emission polarizer oriented either parallel (IVV) or perpendicular (IVH) to the excitation polarization. These polarized fluorescence intensity decays were deconvolved as described in the Materials and Methods to yield the anisotropy decay parameters listed in Table 5. The anisotropy decays were best described by the sum of two exponential terms [Equation (4)], with associated r(0) values ranging from 0.308 to 0.417. The longer of the two rotational correlation times (ϕ1) reflects the overall tumbling of the quadruplex, while the shorter rotational correlation time (ϕ2) reflects the internal motion of the AP residue in the quadruplex. HXDV binding increases the mobility of the AP residue at position 21 (Δϕ2 = −0.33 ns), while exerting essentially no impact on the mobility of the AP residue at position 9 (Δϕ2 = −0.03 ns). This differential impact of HXDV on the mobilities of the AP residues at positions 9 and 21 is consistent with the steady-state fluorescence measurements described above revealing the binding-induced destacking of the AP residue at position 21, but not at position 9. Recall that these steady-state fluorescence measurements also revealed the binding-induced destacking of the AP residue at position 15. Thus, it is reasonable to suggest that HXDV binding would likely increase the mobility of this residue as well. In the aggregate, our time-resolved fluorescence anisotropy results suggest that favorable HXDV-induced alterations in the configurational entropies of the edgewise loops of d(T2AG3)4 contribute to the entropic driving force for the HXDV–d(T2AG3)4 binding reactions. In addition, the fluorescence anisotropy results also lend further support to the ‘terminal capping’ model for the mode of the HXDV–d(T2AG3)4 interaction.
Figure 9.

Polarized fluorescence decay profiles at 25°C for 9AP (A), 21AP (C) and their complexes with HXDV (B and D, respectively). The decay profile with the emission polarizer oriented parallel to the excitation polarization (IVV) is depicted in red, while the decay profile with the emission polarizer oriented perpendicular to the excitation polarization (IVH) is depicted in blue. In each panel, the hollow circles represent the experimental data points, while the solid lines reflect the nonlinear least squares fits of the data with Equation (2). The inset in each panel shows the autocorrelation functions of the weighted residuals for the fits of the corresponding decay profiles. The solution conditions were as described in the legend to Figure 3.
Table 5.
Fluorescence anisotropy decay parameters for 9AP, 21AP and their complexes with HXDV at 25°Ca
| Sample | β1 | ϕ1 (ns) | β2 | ϕ2 (ns) | r(0)b |
|---|---|---|---|---|---|
| 9AP | 0.144 ± 0.031 | 5.06 ± 1.00 | 0.164 ± 0.054 | 0.58 ± 0.10 | 0.308 ± 0.085 |
| 9AP-HXDV | 0.209 ± 0.022 | 3.47 ± 0.03 | 0.133 ± 0.041 | 0.55 ± 0.16 | 0.342 ± 0.064 |
| 21AP | 0.086 ± 0.009 | 10.09 ± 5.65 | 0.332 ± 0.003 | 0.74 ± 0.03 | 0.417 ± 0.011 |
| 21AP-HXDV | 0.215 ± 0.039 | 5.02 ± 1.99 | 0.189 ± 0.060 | 0.41 ± 0.05 | 0.404 ± 0.099 |
a For each sample, the values of βi and ϕi represent averages of two independent experiments, with the indicated uncertainties reflecting the standard deviations from the mean. Solution conditions were as described in the footnote to Table 1.
b Values of r(0) were calculated from the sum of β1 and β2, with the indicated uncertainties reflecting the maximum possible errors in βi as propagated through this summation.
SUMMARY AND CONCLUSIONS
The studies described here demonstrate that the macrocyclic hexaoxazole HXDV binds solely to the G-quadruplex form of nucleic acids under physiological conditions, with no detectable binding to duplex or triplex forms. This behavior may prove to be a general feature of macrocyclic oxazole-containing ligands, as the natural product telomestatin (a heptaoxazole-containing macrocycle) appears to exhibit a similar degree of binding specificity (38,39,61,81). The bistriazoles are the only other class of compounds reported to date that approach this degree of quadruplex binding specificity (61). Significantly, this degree of target specificity is likely to be an important determinant of the therapeutic utility of quadruplex-directed agents.
A broad range of spectroscopic and computational evidence is also presented indicating that HXDV binds the G-quadruplex formed by the human telomeric d(T2AG3)4 sequence in the presence of K+ ions via a nonintercalative ‘terminal capping’ mode in which one ligand molecule binds to each end of the quadruplex. Furthermore, the binding of HXDV to d(T2AG3)4 is entropy driven, with this thermodynamic driving force reflecting contributions from favorable binding-induced alterations in the configurational entropies of the edgewise loops of the host quadruplex as well as in net hydration. Hurley and coworkers used docking and molecular dynamics simulations to propose a ‘terminal capping’ binding model for the interaction of telomestatin with d(T2AG3)4 (37). Our studies described here provide the first experimental demonstration for the G-quadruplex DNA binding of an oxazole-containing macrocycle via a ‘terminal capping’ mode. This mode of interaction with G-quadruplex DNA may be a common feature of the oxazole-containing macrocyclic class of compounds.
The recently reported high-resolution structures of G-quadruplexes adopted by human telomeric DNA model oligonucleotides (25,27,32) form a tempting database for structure-based drug design efforts. However, such efforts should be undertaken with caution, since drug binding can be coupled to conformational changes in the host quadruplex that are difficult to mimic in computational studies, particularly docking methodologies. An intercalative mode of interaction is typically associated with a drug-induced lengthening of the host DNA. It is important to note that drug-induced conformational changes in the host DNA need not be restricted to an intercalative mode of interaction, since our studies described here suggest that they can also occur in conjunction with a ‘terminal capping’ mode of interaction.
Yang and coworkers have proposed a model for human telomeric DNA in which the quadruplex structure formed by d(T2AG3)4 is multiply repeated along the length of the 3′-terminal overhang region of the telomeric DNA (30). This overhang region of human telomeric DNA consists of ∼ 16–35 repeats of the 5′-T2AG3-3′ sequence (23,24). Thus, the 3′-terminal overhang region of human telomeric DNA could potentially contain as many as eight repeats of the quadruplex structure formed by d(T2AG3)4. Our present results imply that oxazole-containing macrocyclic ligands like HXDV and telomestatin could conceivably bind to these repeating telomeric quadruplex structures with a stoichiometry of at least one and perhaps two ligand molecules per quadruplex structure.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
This work was supported by NIH grants CA097123 (D.S.P.) and CA098127 (E.J.L.) and American Cancer Society grant RSG-99-153-04-CDD (D.S.P.). C.M.B. was supported in part by an NIH training grant (T32 CA108455) in Cancer Pharmacology. Funding to pay the Open Access publication charges for this article was provided by the National Cancer Institute.
Conflict of interest statement. None declared.
REFERENCES
- 1.Blackburn EH. Switching and signaling at the telomere. Cell. 2001;106:661–673. doi: 10.1016/s0092-8674(01)00492-5. [DOI] [PubMed] [Google Scholar]
- 2.Autexier C, Lue NF. The structure and function of telomerase reverse transcriptase. Annu. Rev. Biochem. 2006;75:493–517. doi: 10.1146/annurev.biochem.75.103004.142412. [DOI] [PubMed] [Google Scholar]
- 3.Bertuch AA, Lundblad V. The maintenance and masking of chromosome termini. Curr. Opin. Cell Biol. 2006;18:247–253. doi: 10.1016/j.ceb.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 4.Hastie ND, Dempster M, Dunlop MG, Thompson AM, Green DK, Allshire RC. Telomere reduction in human colorectal carcinoma and with ageing. Nature. 1990;346:866–868. doi: 10.1038/346866a0. [DOI] [PubMed] [Google Scholar]
- 5.Counter CM, Avilion AA, LeFeuvre CE, Stewart NG, Greider CW, Harley CB, Bacchetti S. Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J. 1992;11:1921–1929. doi: 10.1002/j.1460-2075.1992.tb05245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, Shay JW. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011–2015. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- 7.Counter CM, Botelho FM, Wang P, Harley CB, Bacchetti S. Stabilization of short telomeres and telomerase activity accompany immortalization of epstein-barr virus-transformed human B lymphocytes. J. Virol. 1994;68:3410–3414. doi: 10.1128/jvi.68.5.3410-3414.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karlseder J, Broccoli D, Dai Y, Hardy S, de Lange T. p53- and ATM-dependent apoptosis induced by telomeres lacking TRF2. Science. 1999;283:1321–1325. doi: 10.1126/science.283.5406.1321. [DOI] [PubMed] [Google Scholar]
- 9.Karlseder J, Smogorzewska A, de Lange T. Senescence induced by altered telomere state, not telomere loss. Science. 2002;295:2446–2449. doi: 10.1126/science.1069523. [DOI] [PubMed] [Google Scholar]
- 10.Blackburn EH, Greider CW, Szostak JW. Telomeres and telomerase: the path from maize, tetrahymena and yeast to human cancer and aging. Nat. Med. 2006;12:1133–1138. doi: 10.1038/nm1006-1133. [DOI] [PubMed] [Google Scholar]
- 11.Han H, Hurley LH. G-Quadruplex DNA: a potential target for anti-cancer drug design. Trends Pharmacol. Sci. 2000;21:136–142. doi: 10.1016/s0165-6147(00)01457-7. [DOI] [PubMed] [Google Scholar]
- 12.Hurley LH, Wheelhouse RT, Sun D, Kerwin SM, Salazar M, Fedoroff OY, Han FX, Han H, Izbicka E, et al. G-Quadruplexes As targets for drug design. Pharmacol. Ther. 2000;85:141–158. doi: 10.1016/s0163-7258(99)00068-6. [DOI] [PubMed] [Google Scholar]
- 13.Neidle S, Read MA. G-Quadruplexes as therapeutic targets. Biopolymers. 2000;56:195–208. doi: 10.1002/1097-0282(2000)56:3<195::AID-BIP10009>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 14.Hurley LH. Secondary DNA structures as molecular targets for cancer therapeutics. Biochem. Soc. Trans. 2001;29:692–696. doi: 10.1042/0300-5127:0290692. [DOI] [PubMed] [Google Scholar]
- 15.Perry PJ, Jenkins TC. DNA Tetraplex-binding drugs: structure-selective targeting is critical for antitumour telomerase inhibition. Mini Rev. Med. Chem. 2001;1:31–41. doi: 10.2174/1389557013407304. [DOI] [PubMed] [Google Scholar]
- 16.Cech TR. Beginning to understand the end of the chromosome. Cell. 2004;116:273–279. doi: 10.1016/s0092-8674(04)00038-8. [DOI] [PubMed] [Google Scholar]
- 17.Komata T, Kanzawa T, Kondo Y, Kondo S. Telomerase as a therapeutic target for malignant gliomas. Oncogene. 2002;21:656–663. doi: 10.1038/sj.onc.1205072. [DOI] [PubMed] [Google Scholar]
- 18.Garvik B, Carson M, Hartwell L. Single-stranded DNA arising at telomeres in cdc13 mutants may constitute a specific signal for the RAD9 checkpoint. Mol. Cell. Biol. 1995;15:6128–6138. doi: 10.1128/mcb.15.11.6128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qi H, Li TK, Kuo D, Nur EKA, Liu LF. Inactivation of Cdc13p triggers MEC1-dependent apoptotic signals in yeast. J. Biol. Chem. 2003;278:15136–15141. doi: 10.1074/jbc.M212808200. [DOI] [PubMed] [Google Scholar]
- 20.Tahara H, Shin-Ya K, Seimiya H, Yamada H, Tsuruo T, Ide T. G-quadruplex stabilization by telomestatin induces TRF2 protein dissociation from telomeres and anaphase bridge formation accompanied by loss of the 3′ telomeric overhang in cancer cells. Oncogene. 2006;25:1955–1966. doi: 10.1038/sj.onc.1209217. [DOI] [PubMed] [Google Scholar]
- 21.Gomez D, O’Donohue MF, Wenner T, Douarre C, Macadre J, Koebel P, Giraud-Panis M-J, Kaplan H, Kolkes A, Shinya K, et al. The G-quadruplex ligand telomestatin inhibits pot1 binding to telomeric sequences in vitro and induces GFP-POT1 dissociation from telomeres in human cells. Cancer Res. 2006;66:6908–6912. doi: 10.1158/0008-5472.CAN-06-1581. [DOI] [PubMed] [Google Scholar]
- 22.Gomez D, Wenner T, Brassart B, Douarre C, O’Donohue MF, El Khoury V, Shin-Ya K, Morjani H, Trentesaux C, Riou J-F. Telomestatin-induced telomere uncapping is modulated by POT1 through G-overhang extension in HT1080 human tumor cells. J. Biol. Chem. 2006;281:38721–38729. doi: 10.1074/jbc.M605828200. [DOI] [PubMed] [Google Scholar]
- 23.Makarov VL, Hirose Y, Langmore JP. Long G tails at both ends of human chromosomes suggest a C strand degradation mechanism for telomere shortening. Cell. 1997;88:657–666. doi: 10.1016/s0092-8674(00)81908-x. [DOI] [PubMed] [Google Scholar]
- 24.Wright WE, Tesmer VM, Huffman KE, Levene SD, Shay JW. Normal human chromosomes have long G-rich telomeric overhangs at one end. Genes Dev. 1997;11:2801–2809. doi: 10.1101/gad.11.21.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y, Patel DJ. Solution structure of the human telomeric repeat d[AG3(T2AG3)3] G-Tetraplex. Structure. 1993;1:263–282. doi: 10.1016/0969-2126(93)90015-9. [DOI] [PubMed] [Google Scholar]
- 26.Mergny JL, Phan AT, Lacroix L. Following G-quartet formation by UV-spectroscopy. FEBS Lett. 1998;435:74–78. doi: 10.1016/s0014-5793(98)01043-6. [DOI] [PubMed] [Google Scholar]
- 27.Parkinson GN, Lee MP, Neidle S. Crystal structure of parallel quadruplexes from human telomeric DNA. Nature. 2002;417:876–880. doi: 10.1038/nature755. [DOI] [PubMed] [Google Scholar]
- 28.Ren J, Qu X, Trent JO, Chaires JB. Tiny telomere DNA. Nucleic Acids Res. 2002;30:2307–2315. doi: 10.1093/nar/30.11.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li J, Correia JJ, Wang L, Trent JO, Chaires JB. Not so crystal clear: the structure of the human telomere G-quadruplex in solution differs from that present in a crystal. Nucleic Acids Res. 2005;33:4649–4659. doi: 10.1093/nar/gki782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ambrus A, Chen D, Dai J, Bialis T, Jones RA, Yang D. Human telomeric sequence forms a hybrid-type intramolecular G-quadruplex structure with mixed parallel/antiparallel strands in potassium solution. Nucleic Acids Res. 2006;34:2723–2735. doi: 10.1093/nar/gkl348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu Y, Noguchi Y, Sugiyama H. The new models of the human telomere d[AGGG(TTAGGG)3] in K+ solution. Bioorg. Med. Chem. 2006;14:5584–5591. doi: 10.1016/j.bmc.2006.04.033. [DOI] [PubMed] [Google Scholar]
- 32.Luu KN, Phan AT, Kuryavyi V, Lacroix L, Patel DJ. Structure of the human telomere in K+ solution: an intramolecular (3 + 1) G-quadruplex scaffold. J. Am. Chem. Soc. 2006;128:9963–9970. doi: 10.1021/ja062791w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paeschke K, Simonsson T, Postberg J, Rhodes D, Lipps HJ. Telomere end-binding proteins control the formation of G-quadruplex DNA structures in vivo. Nat. Struct. Biol. 2005;12:847–854. doi: 10.1038/nsmb982. [DOI] [PubMed] [Google Scholar]
- 34.Chang C-C, Chu J-F, Kao F-J, Chiu Y-C, Lou P-J, Chen H-C, Chang T-C. Verification of antiparallel G-quadruplex structure in human telomeres by using two-photon excitation fluorescence lifetime imaging microscopy of the 3,6-Bis(1-methyl-4-vinylpyridinium)carbazole diiodide molecule. Anal. Chem. 2006;78:2810–2815. doi: 10.1021/ac052218f. [DOI] [PubMed] [Google Scholar]
- 35.Helder MN, Wisman GB, van der Zee GJ. Telomerase and telomeres: from basic biology to cancer treatment. Cancer Invest. 2002;20:82–101. doi: 10.1081/cnv-120000370. [DOI] [PubMed] [Google Scholar]
- 36.Shin-ya K, Wierzba K, Matsuo K, Ohtani T, Yamada Y, Furihata K, Hayakawa Y, Seto H. Telomestatin, a novel telomerase inhibitor from Streptomyces anulatus. J. Am. Chem. Soc. 2001;123:1262–1263. doi: 10.1021/ja005780q. [DOI] [PubMed] [Google Scholar]
- 37.Kim MY, Vankayalapati H, Shin-Ya K, Wierzba K, Hurley LH. Telomestatin, a potent telomerase inhibitor that interacts quite specifically with the human telomeric intramolecular G-quadruplex. J. Am. Chem. Soc. 2002;124:2098–2099. doi: 10.1021/ja017308q. [DOI] [PubMed] [Google Scholar]
- 38.Kim M-Y, Gleason-Guzman M, Izbicka E, Nishioka D, Hurley LH. The different biological effects of telomestatin and TMPyP4 can be attributed to their selectivity for interaction with intramolecular or intermolecular G-quadruplex structures. Cancer Res. 2003;63:3247–3256. [PubMed] [Google Scholar]
- 39.Rosu F, Gabelica V, Shin-ya K, De Pauw E. Telomestatin-induced stabilization of the human telomeric DNA quadruplex monitored by electrospray mass spectrometry. Chem. Comm. 2003;21:2702–2703. doi: 10.1039/b309394h. [DOI] [PubMed] [Google Scholar]
- 40.Tauchi T, Shin-Ya K, Sashida G, Sumi M, Nakajima A, Shimamoto T, Ohyashiki JH, Ohyashiki K. Activity of a novel G-quadruplex-interactive telomerase inhibitor, telomestatin (SOT-095), against human leukemia cells: involvement of ATM-dependent DNA damage response pathways. Oncogene. 2003;22:5338–5347. doi: 10.1038/sj.onc.1206833. [DOI] [PubMed] [Google Scholar]
- 41.Gomez D, Paterski R, Lemarteleur T, Shin-Ya K, Mergny J-L, Riou JF. Interaction of telomestatin with the telomeric single-strand overhang. J. Biol. Chem. 2004;279:41487–41494. doi: 10.1074/jbc.M406123200. [DOI] [PubMed] [Google Scholar]
- 42.Sumi M, Tauchi T, Sashida G, Nakajima A, Gotoh A, Shin-Ya K, Ohyashiki JH, Ohyashiki K. A G-quadruplex-interactive agent, telomestatin (SOT-095), induces telomere shortening with apoptosis and enhances chemosensitivity in acute myeloid leukemia. Int. J. Oncol. 2004;24:1481–1487. [PubMed] [Google Scholar]
- 43.Minhas GS, Pilch DS, Kerrigan JE, Lavoie EJ, Rice JE, Neidle S. Synthesis and G-quadruplex stabilizing properties of a series of oxazole-containing macrocycles. Bioorg. Med. Chem. Lett. 2006;16:3891–3895. doi: 10.1016/j.bmcl.2006.05.038. [DOI] [PubMed] [Google Scholar]
- 44.Perry PJ, Reszka AP, Wood AA, Read MA, Gowan SM, Dosanjh HS, Trent JO, Jenkins TC, Kelland LR, Neidle S. Human telomerase inhibition by regioisomeric disubstituted amidoanthracene-9,10-diones. J. Med. Chem. 1998;41:4873–4884. doi: 10.1021/jm981067o. [DOI] [PubMed] [Google Scholar]
- 45.Sun D, Thompson B, Cathers BE, Salazar M, Kerwin SM, Trent JO, Jenkins TC, Neidle S, Hurley LH. Inhibition of human telomerase by a G-quadruplex-interactive compound. J. Med. Chem. 1997;40:2113–2116. doi: 10.1021/jm970199z. [DOI] [PubMed] [Google Scholar]
- 46.Gowan SM, Heald R, Stevens MF, Kelland LR. Potent inhibition of telomerase by small-molecule pentacyclic acridines capable of interacting with G-quadruplexes. Mol. Pharmacol. 2001;60:981–988. doi: 10.1124/mol.60.5.981. [DOI] [PubMed] [Google Scholar]
- 47.Gowan SM, Harrison JR, Patterson L, Valenti M, Read MA, Neidle S, Kelland LR. A G-quadruplex-interactive potent small-molecule inhibitor of telomerase exhibiting in vitro and in vivo antitumor activity. Mol. Pharmacol. 2002;61:1154–1162. doi: 10.1124/mol.61.5.1154. [DOI] [PubMed] [Google Scholar]
- 48.Harrison RJ, Cuesta J, Chessari G, Read MA, Basra SK, Reszka AP, Morrell J, Gowan SM, Incles CM, Tanious FA, et al. Trisubstituted acridine derivatives as potent and selective telomerase inhibitors. J. Med. Chem. 2003;46:4463–4476. doi: 10.1021/jm0308693. [DOI] [PubMed] [Google Scholar]
- 49.Harrison RJ, Reszka AP, Haider SM, Romagnoli B, Morrell J, Read MA, Gowan SM, Incles CM, Kelland LR, Neidel S. Evaluation of disubstituted acridone derivatives as telomerase inhibitors: the importance of G-quadruplex binding. Bioorg. Med. Chem. Lett. 2004;14:5845–5849. doi: 10.1016/j.bmcl.2004.09.037. [DOI] [PubMed] [Google Scholar]
- 50.Moore MJ, Schultes CM, Cuesta J, Cuenca F, Gunaratnam M, Tanious FA, Wilson WD, Neidle S. Trisubstituted acridines as G-quadruplex telomere targeting agents. Effects of extensions of the 3,6- and 9-side chains on quadruplex binding, telomerase activity, and cell proliferation. J. Med. Chem. 2006;49:582–599. doi: 10.1021/jm050555a. [DOI] [PubMed] [Google Scholar]
- 51.Anantha NV, Azam M, Sheardy RD. Porphyrin binding to quadruplexed T4G4. Biochemistry. 1998;37:2709–2714. doi: 10.1021/bi973009v. [DOI] [PubMed] [Google Scholar]
- 52.Arthanari H, Basu S, Kawano TL, Bolton PH. Fluorescent dyes specific for quadruplex DNA. Nucleic Acids Res. 1998;26:3724–3728. doi: 10.1093/nar/26.16.3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Arthanari H, Bolton PH. Porphyrins can catalyze the interconversion of DNA quadruplex structural types. Anticancer Drug Des. 1999;14:317–326. [PubMed] [Google Scholar]
- 54.Han FX, Wheelhouse RT, Hurley LH. Interactions of TMPyP4 and TMPyP2 with quadruplex DNA. Structural basis for the differential effects on telomerase inhibition. J. Am. Chem. Soc. 1999;121:3561–3570. [Google Scholar]
- 55.Haq I, Trent JO, Chowdhry BZ, Jenkins TC. Intercalative G-tetraplex stabilization of telomeric DNA by a cationic porphyrin. J. Am. Chem. Soc. 1999;121:1768–1779. [Google Scholar]
- 56.Izbicka E, Wheelhouse RT, Raymond E, Davidson KK, Lawrence RA, Sun D, Windle BE, Hurley LH, Von Hoff DD. Effects of cationic porphyrins as g-quadruplex interactive agents in human tumor cells. Cancer Res. 1999;59:639–644. [PubMed] [Google Scholar]
- 57.Han H, Langley DR, Rangan A, Hurley LH. Selective interactions of cationic porphyrins with G-quadruplex structures. J. Am. Chem. Soc. 2001;123:8902–8913. doi: 10.1021/ja002179j. [DOI] [PubMed] [Google Scholar]
- 58.Shi DF, Wheelhouse RT, Sun D, Hurley LH. Quadruplex-interactive agents as telomerase inhibitors: synthesis of porphyrins and structure-activity relationship for the inhibition of telomerase. J. Med. Chem. 2001;44:4509–4523. doi: 10.1021/jm010246u. [DOI] [PubMed] [Google Scholar]
- 59.Grand CL, Han H, Munoz RM, Weitman S, Von Hoff DD, Hurley LH, Bearss DJ. The cationic porphyrin TMPyP4 down-regulates c-MYC and human telomerase reverse transcriptase expression and inhibits tumor growth in vivo. Mol. Cancer Ther. 2002;1:565–573. [PubMed] [Google Scholar]
- 60.Yamashita T, Uno T, Ishikawa Y. Stabilization of guanine quadruplex DNA by the binding of porphyrins with cationic side arms. Bioorg. Med. Chem. 2005;13:2423–2430. doi: 10.1016/j.bmc.2005.01.041. [DOI] [PubMed] [Google Scholar]
- 61.Moorhouse AD, Santos AM, Gunaratnam M, Moore M, Neidle S, Moses JE. Stabilization of G-quadruplex DNA by highly selective ligands via click chemistry. J. Am. Chem. Soc. 2006;128:15972–15973. doi: 10.1021/ja0661919. [DOI] [PubMed] [Google Scholar]
- 62.Fedoroff OY, Salazar M, Han H, Chemeris VV, Kerwin SM, Hurley LH. NMR-based model of a telomerase-inhibiting compound bound to G-quadruplex DNA. Biochemistry. 1998;37:12367–12374. doi: 10.1021/bi981330n. [DOI] [PubMed] [Google Scholar]
- 63.Han H, Cliff CL, Hurley LH. Accelerated assembly of G-quadruplex structures by a small molecule. Biochemistry. 1999;38:6981–6986. doi: 10.1021/bi9905922. [DOI] [PubMed] [Google Scholar]
- 64.Mazzitelli CL, Brodbelt JS, Kern JT, Rodriguez M, Kerwin SM. Evaluation of binding of perylene diimide and benzannulated perylene diimide ligands to DNA by electrospray ionization mass spectrometry. J. Am. Soc. Mass Spectrom. 2006;17:593–604. doi: 10.1016/j.jasms.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 65.Guo Q, Lu M, Marky LA, Kallenbach NR. Interaction of the dye ethidium bromide with DNA containing guanine repeats. Biochemistry. 1992;31:2451–2455. doi: 10.1021/bi00124a002. [DOI] [PubMed] [Google Scholar]
- 66.Koeppel F, Riou JF, Laoui A, Mailliet P, Arimondo PB, Labit D, Petitgenet O, Helene C, Mergny JL. Ethidium derivatives bind to G-quartets, inhibit telomerase and act as fluorescent probes for quadruplexes. Nucleic Acids Res. 2001;29:1087–1096. doi: 10.1093/nar/29.5.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Perry PJ, Read MA, Davies RT, Gowan SM, Reszka AP, Wood AA, Kelland LR, Neidle S. 2,7-Disubstituted amidofluorenone derivatives as inhibitors of human telomerase. J. Med. Chem. 1999;42:2679–2684. doi: 10.1021/jm990084q. [DOI] [PubMed] [Google Scholar]
- 68.Mergny J-L, Lacroix L, Teulade-Fichou M-P, Hounsou C, Guittat L, Hoarau M, Arimondo PB, Vigneron J-P, Lehn J-M, Riou JE, et al. Telomerase inhibitors based on quadruplex ligands selected by a fluorescence assay. Proc. Natl Acad. Sci. USA. 2001;98:3062–3067. doi: 10.1073/pnas.051620698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Leonetti C, Amodei S, D’Angelo C, Rizzo A, Benassi B, Antonelli A, Elli R, Stevens MF, D’Incalci M, Zupi G, et al. Biological activity of the G-quadruplex ligand RHPS4 (3,11-Difluoro-6,8,13-trimethyl-8H-quino[4,3,2-kl]acridinium Methosulfate) is associated with telomere capping alteration. Mol. Pharmacol. 2004;66:1138–1146. doi: 10.1124/mol.104.001537. [DOI] [PubMed] [Google Scholar]
- 70.Cookson JC, Dai F, Smith V, Heald RA, Laughton CA, Stevens MF, Burger AM. Pharmacodynamics of the G-quadruplex-stabilizing telomerase inhibitor 3,11-difluoro-6,8,13-trimethyl-8H-quino[4,3,2-kl]acridinium methosulfate (RHPS4) in vitro: activity in human tumor cells correlates with telomere length and can be enhanced, or antagonized, with cytotoxic agents. Mol. Pharmacol. 2005;68:1551–1558. doi: 10.1124/mol.105.013300. [DOI] [PubMed] [Google Scholar]
- 71.Duan W, Rangan A, Vankayalapati H, Kim MY, Zeng Q, Sun D, Han H, Fedoroff OY, Nishioka D, Rha SY, et al. Design and synthesis of fluoroquinophenoxazines that interact with human telomeric G-quadruplexes and their biological effects. Mol. Cancer Ther. 2001;1:103–120. [PubMed] [Google Scholar]
- 72.Kim MY, Duan W, Gleason-Guzman M, Hurley LH. Design, synthesis, and biological evaluation of a series of fluoroquinoanthroxazines with contrasting dual mechanisms of action against topoisomerase II and G-quadruplexes. J. Med. Chem. 2003;46:571–583. doi: 10.1021/jm0203377. [DOI] [PubMed] [Google Scholar]
- 73.Binz N, Shalaby T, Rivera P, Shin-ya K, Grotzer MA. Telomerase inhibition, telomere shortening, cell growth suppression and induction of apoptosis by telomestatin in childhood neuroblastoma cells. Eur. J. Cancer. 2005;41:2873–2881. doi: 10.1016/j.ejca.2005.08.025. [DOI] [PubMed] [Google Scholar]
- 74.Plum GE. In: Current Protocols in Nucleic Acid Chemistry. Beaucage SL, Bergstrom DE, Glick GD, Jones RA, editors. Vol. 1. New York: John Wiley & Sons; 2000. pp. 7.3.1–7.3.17. [Google Scholar]
- 75.Szabo AG. In: Spectrophotometry and Spectrofluorimetry. Gore MG, editor. New York: Oxford University Press; 2000. pp. 33–67. [Google Scholar]
- 76.Chamberlin MJ. Comparative properties of DNA, RNA, and hybrid homopolymer pairs. Federation Proceedings. 1965;24:1446–1457. [PubMed] [Google Scholar]
- 77.Riley M, Maling B, Chamberlin MJ. Physical and chemical characterization of two- and three-stranded adenine-thymine and adenine-uracil homopolymer complexes. J. Mol. Biol. 1966;20:359–389. doi: 10.1016/0022-2836(66)90069-6. [DOI] [PubMed] [Google Scholar]
- 78.Shafer RH. Stability and structure of model DNA triplexes and quadruplexes and their interactions with small ligands. Prog. Nucleic Acid Res. Mol. Biol. 1998;59:55–94. doi: 10.1016/s0079-6603(08)61029-6. [DOI] [PubMed] [Google Scholar]
- 79.Cheong C, Moore PB. Solution structure of an unusually stable RNA tetraplex containing G- and U-quartet structures. Biochemistry. 1992;31:8406–8414. doi: 10.1021/bi00151a003. [DOI] [PubMed] [Google Scholar]
- 80.Deng J, Xiong Y, Sundaralingam M. X-Ray analysis of an RNA tetraplex (UGGGGU)4 with divalent Sr2+ ions at subatomic resolution (0.61 Å) Proc. Natl Acad. Sci. USA. 2001;98:13665–13670. doi: 10.1073/pnas.241374798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.De Cian A, Guittat L, Shin-Ya K, Riou J-F, Mergny J-L. Affinity and Selectivity of G4 Ligands Measured by FRET. Nucleic Acids Symp. Ser. 2005;49:235–236. doi: 10.1093/nass/49.1.235. [DOI] [PubMed] [Google Scholar]
- 82.Gasteiger J, Marsili M. Iterative partial equalization of orbital electronegativity—a rapid access to atomic charges. Tetrahedron. 1980;36:3219–3228. [Google Scholar]
- 83.Wyman J, Gill SJ. Binding and Linkage: Functional Chemistry of Biological Macromolecules. Mill Valley: University Science Books; 1990. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.