


Abstract
Posttranslational histone modifications associated with actively expressed genes are generally believed to be introduced primarily by histone-modifying enzymes that are recruited by transcription factors or their associated co-activators. We have performed a comprehensive spatial and temporal analyses of the histone modifications that are deposited upon activation of the MHC class II gene HLA-DRA by the co-activator CIITA. We find that transcription-associated histone modifications are introduced during two sequential phases. The first phase precedes transcription initiation and is characterized exclusively by a rapid increase in histone H4 acetylation over a large upstream domain. All other modifications examined, including the acetylation and methylation of several residues in histone H3, are restricted to short regions situated at or within the 5′ end of the gene and are established during a second phase that is concomitant with ongoing transcription. This second phase is completely abrogated when elongation by RNA polymerase II is blocked. These results provide strong evidence that transcription elongation can play a decisive role in the deposition of histone modification patterns associated with inducible gene activation.
INTRODUCTION
The basic building block of eukaryotic chromatin is the nucleosome, which consists of 147 bp of DNA wrapped around an octamer of the four core histone proteins H3, H4, H2A and H2B. The packaging of DNA into nucleosomes creates a restrictive environment that reduces the accessibility of DNA to factors that mediate chromatin-templated processes such as transcription. Dynamic structural rearrangements that render the chromatin permissive for transcription are therefore intimately associated with the regulation of gene expression. These structural rearrangements are believed to be facilitated by various posttranslational modifications of nucleosomal histones that affect chromatin structure either directly or by creating docking sites for the recruitment of effector proteins [for recent reviews see (1–3)].
Among the most well-documented modifications associated with actively transcribed genes are the acetylation of various lysine residues in the N-terminal tails of H3 and H4 (H3Ac and H4Ac) (1), the di- and tri-methylation of lysine 4 in the N-terminal tail of H3 (H3K4Me2 and H3K4Me3) (2) and the methylation of arginine 17 in the N-terminal tail of H3 (H3R17Me2) (3). More recently, it has been shown that the methylation of lysine 9 in the N-terminal tail of H3 (H3K9Me3)—a modification that was until then thought to be a hallmark of silent heterochromatin (2)—is actually also found within the 5′ ends of several actively transcribed mammalian genes (4). It is now well established from studies in multiple model systems and species that there is a tight correlation between the presence of these histone modifications and active gene expression (1–3). A widespread view is that histone-modifying activities are recruited by transcription factors and/or associated co-activators, and that they impact primarily on assembly of the general transcription machinery and transcription initiation (1–3,5). However, there is also growing evidence suggesting that factors associated with elongating RNA polymerase can play a key role in establishing histone modifications associated with actively expressed genes (2,6–11).
A full understanding of the mechanisms that mediate the deposition and functional consequences of specific histone modification patterns requires the analysis and integration of at least three related parameters: where in the gene the various different histone modifications are made, in what temporal order they are introduced and what steps in the transcription process they are implicated in (12–16). Yet in the majority of higher eukaryotic systems these three parameters have not been integrated into a single comprehensive and dynamic description of the spatial distribution and temporal order of chromatin modification events that occur during the activation of transcription. We have performed such an analysis in one of the most well-defined human model systems, the activation of major histocompatibility complex class II (MHC-II) genes (17–19).
Two modes of MHC-II expression, constitutive and inducible, are recognized (17–19). Constitutive expression is restricted mainly to specialized cells of the immune system (thymic epithelial cells, dendritic cells, macrophages and B cells). Most other cell types do not express MHC-II genes unless they are exposed to interferon-γ (IFN-γ). The molecular machinery that regulates MHC-II expression has been exceptionally well defined thanks to the elucidation of the genetic defects that are responsible for the Bare Lymphocyte Syndrome, a rare hereditary immunodeficiency disease resulting from mutations in genes encoding transcription factors that are essential for MHC-II expression (17,20–24). One of these factors, the class II transactivator (CIITA) (20), is a transcriptional co-activator that is exquisitely specific for the activation of MHC-II genes (Figure 1A). CIITA serves as the master regulator of MHC-II genes and is expressed in a cell-type-specific and IFN-γ-inducible manner that dictates both the constitutive and inducible patterns of MHC-II expression (19). It is recruited to the regulatory regions of MHC-II genes by protein–protein interactions with a multi-protein ‘enhanceosome’ complex that assembles on a characteristic enhancer known as the MHC-II S-Y module (Figure 1A) (17,18,25–29). A heterotrimeric DNA-binding factor called regulatory factor X (RFX) is a central component of the enhanceosome complex (Figure 1A) (17,21–24).
Figure 1.
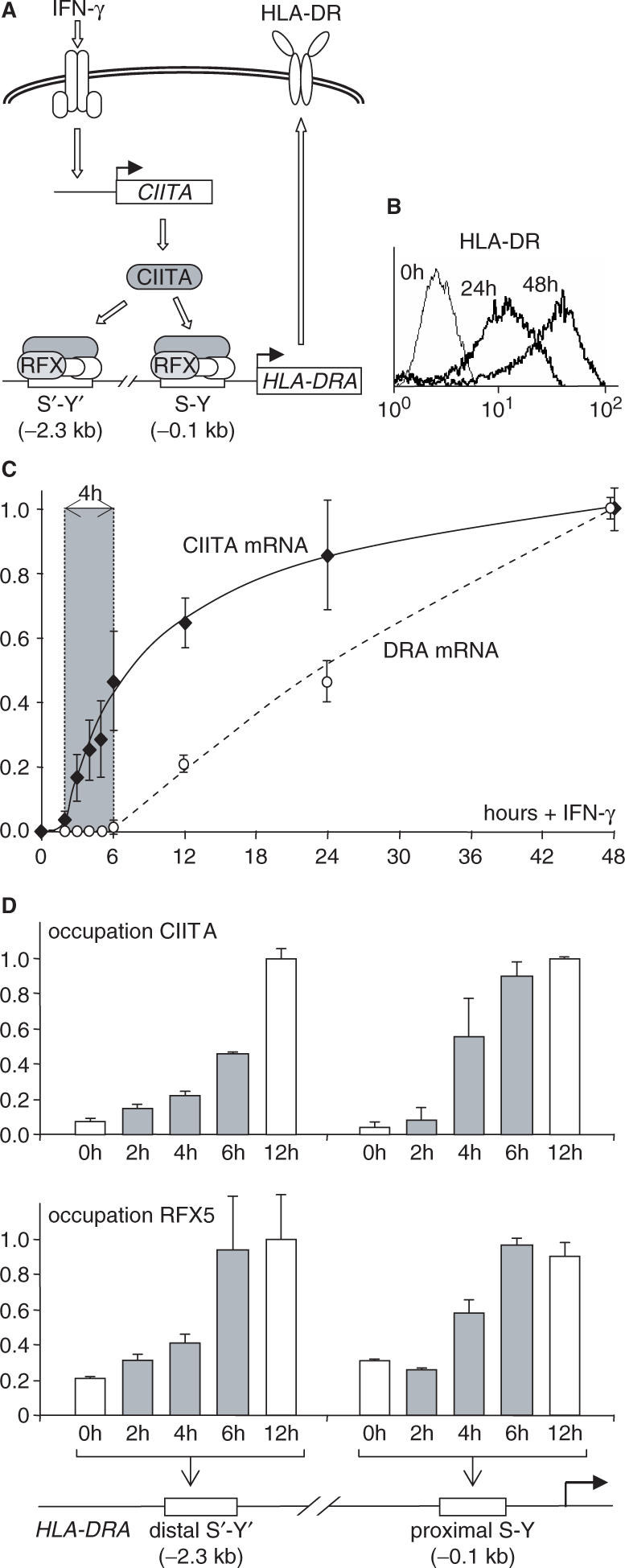
Precise timing of IFN-γ-induced HLA-DRA expression. (A) Molecular mechanism mediating IFN-γ-induced HLA-DRA expression. IFN-γ induces rapid expression of the gene coding for the transcriptional co-activator CIITA, which activates transcription of the HLA-DRA gene by associating with an enhanceosome complex that assembles at its proximal (S-Y) and distal (S′-Y′) regulatory modules. RFX is a key component of the enhanceosome complex. (B) Cell surface HLA-DR expression was measured by FACS in Me67.8 cells stimulated with IFN-γ for 0, 24 and 48 h. (C) The accumulation of CIITA and HLA-DRA mRNAs were quantified by real-time RT-PCR in IFN-γ-stimulated Me67.8 cells over a 48-h time period. Shading highlights a 4-h lag period between the induction of CIITA and HLA-DRA mRNA accumulation. Results are expressed relative to the values measured after 48 h. The mean and SD are shown for four experiments. (D) Occupation of the HLA-DRA S-Y and S′-Y′ modules by CIITA and RFX was measured by quantitative chromatin immunoprecipitation (ChIP) at the indicated times following stimulation with IFN-γ. Shaded bars highlight the occupation that is induced during the lag period preceding HLA-DRA gene activation. Results are expressed relative to the plateau values reached after 12 h. The mean and SD are shown for two experiments.
The activation of MHC-II genes by CIITA is associated with an increase in many of the major histone modifications known to correlate with active transcription, including H3Ac, H4Ac, H3K4Me2, H3K4Me3 and H3R17Me2 (26–32). Most studies examining these modifications have restricted themselves to the promoters of MHC-II genes (28,30–32), despite the fact that several reports have shown that they may affect distal regions as well (26,27,29). Moreover, only limited information is available on when and in what order these modifications are introduced during the activation process (28,29,32). We have therefore performed a comprehensive analysis of the spatial distribution, order and timing of the histone modifications that are introduced during IFN-γ-induced activation of the prototypical MHC-II gene HLA-DRA. We demonstrate that there are two spatially and temporally distinct chromatin-modification phases. The first phase precedes the initiation of transcription and is characterized by a global increase in H4 acetylation throughout a large upstream domain. All other modifications are restricted to short regions situated within or near the 5′ end of the gene and are introduced in a second phase that coincides with, and is a consequence of, ongoing transcription. With the exception of H4Ac, most modifications observed at the HLA-DRA gene are thus established by transcription-coupled mechanisms.
MATERIALS AND METHODS
Cells
Me67.8 cells were grown in RPMI+Glutamax medium (Invitrogen) supplemented with 10% fetal calf serum and antibiotics, and were induced with 200 U/ml of human IFN-γ (Invitrogen) as described (33). Cell surface HLA-DR expression was analyzed by FACS as described (34). DRB (Sigma) was added to culture medium at a concentration of 100 μM.
Chromatin immunoprecipitation (ChIP)
Chromatin preparation and ChIP experiments were performed as described using antibodies specific for RFX, CIITA and modified histones (26,27,31). Results for modified histones were normalized using the TATA-binding protein (TBP) promoter as internal control and were compensated for fluctuations in nucleosome density by performing ChIP experiments in parallel with an antibody against unmodified H3. Histone antibodies were obtained from Abcam, Cambridge, UK (H3, Ab1791; H4K5Ac, Ab1758; H3K4Me3, Ab8580; H3K4Me2, Ab7766; H3K9Me3, Ab8898; H3K36me3, Ab9050) or Upstate, Lake Placid, NY (H4Ac, 06-866; H4K8Ac, 07-328; H4K12Ac, 07-595; H4K16Ac, 07-329; H3Ac, 06-599; H3K9Ac, 07-352; H3K14Ac, 07-353; H3K23Ac, 07-355; H3K27Ac, 07-360; H3R17Me3, 07-214). The RNA Pol II antibody was from Abcam (Ab817). Results were generated by real-time PCR using the primers listed in Supplementary Table 1. PCR was performed using the iCycler iQ Real-Time PCR Detection System (BioRad) and a SYBR-Green-based kit for quantitative PCR (iQ Supermix—BioRad). Amplification specificity was controlled by gel electrophoresis and dissociation curve analysis. Results were quantified using a standard curve generated with serial dilutions of input DNA. PCR amplifications were performed in triplicate. Experiments were repeated between two and five times with different inductions.
RNA quantification
CIITA and DRA mRNAs were quantified by real-time RT-PCR and normalized with respect to TBP mRNA as described (31,34). Chromatin-bound nascent and intergenic transcripts were isolated and quantified by real-time RT-PCR as described (26). Primers used for nascent and intergenic transcripts are listed in Supplementary Table 1. PCR measurements were made in triplicate. Experiments were repeated between two and four times with different inductions.
Immunoblotting
Cells were lysed for 1 h on ice in 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 10 mM EDTA, 1% NP40, 1 mM DTT containing 1× complete™ protease inhibitors (Roche) and a phosphatase inhibitor cocktail (Sigma). Lysates were clarified by centrifugation and protein concentrations were determined. Proteins were dissolved in 2× sample buffer and equal amounts were fractionated by SDS-PAGE. Proteins were transferred to Immobilon-P membranes (Millipore), which were incubated with rabbit polyclonal CARM1 antibodies (Upstate 07-080, used at 1/1000 dilution), rabbit polyclonal CBP antibodies (SantaCruz sc-369, used at 1/500 dilution), rabbit polyclonal pCAF antibodies (Abcam ab12188, gift from W. Herr, used at 1/600 dilution), rabbit polyclonal Set1c antibodies (gift from W. Herr, used at 1/500 dilution), rabbit polyclonal WDR5 antibodies (gift from W. Herr, used at 1/500 dilution) and mouse monoclonal S2-phosphorylated-Pol II antibodies (Covance, MMS-129R). Detection was performed using peroxidase conjugated anti-mouse or anti-rabbit IgG antibodies (Promega) and chemiluminescence visualization (ECL, Amersham) according to the manufacturer's instructions. Blots were quantified using Quantity One software.
RESULTS
Precise timing of IFN-γ-induced HLA-DRA gene transcription
Histone modifications introduced during IFN-γ-induced activation of the HLA-DRA gene were studied in the melanoma cell line Me67.8, which was chosen because it exhibits a robust and reproducible induction of CIITA and MHC-II expression in response to INF-γ (Figure 1A) (33). Me67.8 cells respond to IFN-γ in a synchronized manner, such that the entire population exhibits a homogeneous shift over time in the level of cell surface MHC-II expression (Figure 1B).
Time course experiments were performed to document the activation of CIITA and HLA-DRA mRNA expression (Figure 1C), as well as occupation of the HLA-DRA S-Y modules by CIITA and RFX (Figure 1D). The time courses obtained for these parameters are highly reproducible from one experiment to another (see error bars in Figure 1). CIITA mRNA accumulation starts 2 h after induction, whereas the increase in HLA-DRA mRNA only becomes evident after 6 h (Figure 1C). During this 4-h lag period, occupation of the S-Y modules by CIITA and RFX increases to reach a plateau by 6 h (Figure 1D). These results define a precise 4-h time window during which key events implicated in CIITA-mediated transcriptional initiation of the HLA-DRA gene are most likely to occur.
Two distinct phases of histone acetylation
We performed chromatin immunoprecipitation (ChIP) experiments to examine changes in the levels of H3Ac and H4Ac that are induced over time within a 7-kb domain that spans 5 kb of the upstream regulatory region and 2 kb of the 5′ end of the HLA-DRA gene. Two spatially and temporally distinct histone acetylation phases were evident (Figure 2). During the first phase, there was a strong increase in H4Ac over the entire region examined. This global increase in H4Ac was first evident after 3 h of induction and reached nearly maximal levels by 6 h. During the second phase, there was an increase in H3Ac that was restricted to a short 1-kb region situated within the 5′ end of the gene. This local increase in H3Ac occurred mainly between 6 and 12 h after the start of induction.
Figure 2.

Spatial and temporal patterns of the induction of histone acetylation at the HLA-DRA gene. (A) The levels of H4 (top, H4Ac) and H3 (bottom, H3Ac) acetylation were measured by quantitative ChIP at different positions within the HLA-DRA gene in non-induced cells and cells treated with IFN-γ for 12 h. A schematic map of the HLA-DRA gene is shown below. (B) The levels of H4 (left) and H3 (right) acetylation were measured by quantitative ChIP at the indicated positions after induction with IFN-γ for the indicated times. For H4Ac, similar time courses were also observed at the other positions examined in (A) (data not shown). Asterisk denotes not measured in all experiments. All results are expressed relative to the value observed at the promoter in non-induced cells. The mean and SD are shown for four (H4Ac) and five (H3Ac) experiments.
To define the precise lysine (K) residues that are implicated in the two phases, we performed ChIP experiments with antibodies directed against specific acetylated K residues of histones H4 and H3. During the first phase, H4 was acetylated mainly at K5 and K8 (Figure 3A). No obvious increases in the acetylation of H4K12 and H4K16 were evident (Figure 3A). As observed for H4Ac, the increases in H4K5Ac and H4K8Ac were early events that were essentially complete by 6 h of induction (Figure 3A) and concerned the entire 7-kb region (Figure 3B). During the second phase, the strongest increase in acetylation was observed at K9 of H3 (Figure 4A). There was a more modest increase for acetylation at H3K14, whereas no obvious changes were evident for acetylation at H3K23 and H3K27 (Figure 4A). As for H3Ac, the increase in H3K9Ac was a late event occurring mainly after 6 h of induction (Figure 4A) and was restricted to the 5′ end of the gene (Figure 4B). The acetylation specificity that we observed is consistent with previous reports showing that H4K8 and H3K9 are among the major residues that are acetylated at the HLA-DRA promoter in B cells and IFN-γ-induced cells (29,30).
Figure 3.

Specificity of H4 acetylation induced at the HLA-DRA gene. (A) The levels of acetylation at lysines K5, K8, K12 and K16 of H4 were measured by quantitative ChIP at the HLA-DRA promoter after induction with IFN-γ for the indicated times. Similar results were obtained at the other positions in the region analyzed in (B) (data not shown). (B) Acetylation at H4K5 and H4K8 was measured by quantitative ChIP at the indicated positions in the HLA-DRA gene after 0 and 12 h of induction. All results are expressed relative to the value observed at the promoter in non-induced cells. The mean and SD are shown for two experiments.
Figure 4.

Specificity of H3 acetylation induced at the HLA-DRA gene. (A) The levels of acetylation at lysines K9, K14, K23 and K27 of H3 were measured by quantitative ChIP at position +300 of the HLA-DRA gene after induction with IFN-γ for the indicated times. Similar results were obtained at the promoter and at +600 (data not shown). (B) H3 acetylation at K9 was measured by quantitative ChIP at the indicated positions in the HLA-DRA gene after 0 and 12 h of induction. All results are expressed relative to the value observed at the promoter in non-induced cells. The mean and SD are shown for three experiments.
To situate the two phases of histone acetylation with respect to the initiation of transcription, we performed ChIP experiments to determine the timing of Pol II recruitment at the HLA-DRA promoter and the appearance of nascent chromatin-bound HLA-DRA transcripts (Figure 5A). Pol II recruitment coincided with the increase in H4 acetylation and reached nearly maximal levels by 6 h. In contrast, nascent transcripts appeared mainly after 6 h, in parallel with the increase in H3 acetylation. The first phase of H4 acetylation is thus associated with Pol II recruitment but precedes the initiation of transcription. In contrast, the subsequent H3 acetylation phase appeared to coincide with active transcription. To confirm the latter, we performed a finer time course between 6 and 12 h (Figure 5B). The results indicate that the increase in H3 acetylation and the appearance of nascent transcripts are simultaneous events.
Figure 5.

Temporal relationship between the induction of histone acetylation, the recruitment of Pol II and the initiation of transcription. (A) H4 acetylation, H3 acetylation, recruitment of Pol II and the appearance of nascent chromatin-bound transcripts were quantified after the indicated times in IFN-γ-induced cells. For H4Ac, the results represent the mean and SD for measurements made in three to four experiments at four positions (−1.3, −0.1, +0.3 and +0.6 kb) in the HLA-DRA gene. For H3Ac, the results represent the mean and SD for measurements made in three to five experiments at three positions (−0.1, +0.3 and +0.6 kb) in the HLA-DRA gene. For nascent transcripts, the results represent the mean and SD for measurements made in two experiments at three positions (+0.3, +0.6 and +1.7 kb) within the HLA-DRA gene. For Pol II, results represent the mean and SD for measurements made at the promoter (−0.04 kb) in four experiments. Asterisk denotes not measured in all experiments. (B) Nascent chromatin-bound transcripts (open bars) and H3 acetylation (gray bars) were quantified in IFN-γ-induced cells after the indicated times. Results represent the mean and SD from two experiments.
H4 acetylation is associated with intergenic transcription
Constitutive expression of the HLA-DRA gene in B cells is associated with intergenic transcription of its upstream regulatory region (26). We therefore determined whether these intergenic transcripts are also observed in IFN-γ-induced cells (Figure 6). Intergenic transcripts were induced during the early phase of H4 acetylation, prior to transcription of the HLA-DRA gene itself. The abundance of these intergenic transcripts is highest just upstream of the promoter and in regions that flank the distal S′-Y′ module (Figure 6). This IFN-γ-induced pattern of intergenic transcription is similar to the one observed previously in B cells (26).
Figure 6.

Intergenic transcription induced by IFN-γ in the upstream region of the HLA-DRA gene. Intergenic transcripts were quantified by real-time RT-PCR at the indicated times and positions in the upstream region of the HLA-DRA gene. In the time course experiment (top), the results represent the mean and SD of measurements made in three experiments at six positions (−4.9, −3.5, −2.3, −1.8, −1.3 and −0.1 kb) in the upstream region. The spatial distribution (bottom) shows the mean and SD derived from three experiments in cells induced for 0 and 12 h. Asterisk denotes not measured in all experiments.
H3 acetylation coincides with increased H3 methylation
To examine where and when changes in H3 methylation occur during IFN-γ-induced expression of the HLA-DRA gene, we performed ChIP experiments with antibodies directed against H3K4Me3, H3K4Me2, H3R17Me2 and H3K9Me3 (Figure 7). Increases restricted to short regions situated near the 5′ end of the HLA-DRA gene were observed for all four modifications. The increases in H3K4Me3, H3K4Me2 and H3K9Me3 peaked at a position situated after the transcription initiation site. The strongest increase in H3R17Me2 was situated at the transcription initiation site. These methylation events occurred mainly after 6 h, although the increase in H3K4Me2 appeared to precede the others somewhat. Taken together, these results show that introduction of the H3 methylation marks overlaps temporally and spatially with increased H3 acetylation.
Figure 7.

Temporal and spatial pattern of IFN-γ-induced H3 methylation. The induction of (A) H3K4Me2, (B) H3K4Me3, (C) H3R17Me2 and (D) H3K9Me3 modifications were assessed at the indicated times and positions in the HLA-DRA gene. In the left panels, results are presented for cells induced with IFN-γ for 0 and 12 h. In the right panels, time courses are shown for positions at which the modifications attain a peak. The mean and SD are shown for two experiments. Asterisk denotes not measured in all experiments.
All H3 modifications are transcription dependent
With respect to both their timing and position, the increases in H3 acetylation and methylation coincide with active transcription of the HLA-DRA gene, raising the possibility that they might actually be a consequence of transcription. To address this possibility, we induced cells with IFN-γ for 6 h to permit the induction of CIITA expression and completion of the early H4 acetylation phase, and then continued the induction in the presence of the drug 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole (DRB) (Figure 8A), which blocks transcription elongation by inhibiting phosphorylation of the C-terminal domain (CTD) of Pol II (Figure 8B) (35,36). This protocol completely blocked the induction of HLA-DRA mRNA accumulation (Figure 8C) and reduced Pol II density within the body of the gene (Supplementary Figure 1). As expected, DRB did not significantly affect HLA-DRA promoter occupation by RFX and CIITA, the acetylation of H4 or recruitment of Pol II to the promoter (Figure 8B and C). Events that precede the initiation of HLA-DRA transcription had thus been completed and were not reversed by the addition of DRB. In contrast, the increases in H3Ac, H3K4Me3, H3K4Me2, H3R17Me2 and H3K9Me3, which occur mainly after 6 h, were all completely eliminated by DRB. The introduction of these five modifications thus behaves in a manner similar to that of the H3K36Me3 modification (Figure 8C), which is known to be strictly dependent on active Pol II elongation (2).
Figure 8.

Most IFN-γ-induced histone modifications deposited at the HLA-DRA gene are a consequence of active transcription. (A) The diagram depicts the protocol used to document the effect of DRB on IFN-γ-induced HLA-DRA gene activation. (B) The effect of DRB on the phosphorylation of Pol II was assessed by western blotting (left), whereas its effect on HLA-DRA promoter occupation by RFX (right) and CIITA (middle) was measured by ChIP. (C) HLA-DRA mRNA accumulation, the recruitment of Pol II at the promoter, and the levels of the indicated modifications were measured after 0, 6 and 12 h of induction with IFN-γ, and after 12 h of induction in the presence of DRB for the last 6 h. Results are shown for measurements made at the promoter (Pol II and R17Me2), at +300 (H4Ac, H3Ac, H3K4Me3, H3K4Me2 and H3K9Me3) and in exon 5 (H3K36Me3). Similar results were obtained at other positions in the modified regions (data not shown). The mean and SD are shown for two experiments. (D) Protein levels of the indicated histone modifying factors were measured by western blotting in cells treated as in panel C.
To ascertain that the DRB sensitivity of the H3Ac, H3K4Me3, H3K4Me2, H3R17Me2 and H3K9Me3 modifications is not a non-specific consequence of a general block in the transcription of genes encoding histone-modifying factors, we examined the levels of several key proteins by western blotting experiments. These included the histone acetyltransferase (HAT) factors [cyclic AMP responsive-element binding-protein (CBP) and p300/CBP-associated factor (pCAF)], the histone methyltransferase (HMT) factors (Set1 and WDR5), and the H3R17-specific HMT CARM1 (Figure 8D). We also examined the HAT GCN5 (data not shown). No drop in the abundance of any of these proteins was induced by the 6-h DRB treatment.
Taken together, these results are consistent with the model that the H3Ac, H3K4Me3, H3K4Me2, H3R17Me2 and H3K9Me3 modifications are introduced in a transcription-coupled manner during IFN-γ-induced activation of the HLA-DRA gene.
DISCUSSION
Our results demonstrate that histone modifications associated with IFN-γ-induced HLA-DRA gene activation are introduced during two sequential phases that differ with respect to their timing, the regions of the gene that are concerned, the precise modifications that are made and their dependence on ongoing transcription. The first phase precedes the initiation of transcription and is characterized by a rapid increase in H4K5 and H4K8 acetylation over a large upstream domain. The second phase is concomitant with active transcription and is characterized by increases in H3K9Ac, H3K4Me2, H3K4Me3, H3K9Me3 and H3R17Me2 in short regions situated at or within the 5′ end of the gene. This second phase is completely blocked by the transcription elongation inhibitor DRB, indicating that it is a consequence of active elongation by Pol II.
The timing, localization and transcription-dependence of the increases in H3Ac, H3K4Me2, H3K4Me3, H3K9Me3 and H3R17Me2 suggest that these marks are introduced by HATs and HMTs that are recruited by actively transcribing Pol II, associated elongation factors and/or prior histone modifications that were introduced in a transcription-dependent manner. Moreover, they imply that most histone modifications introduced during HLA-DRA gene activation do not play a role in facilitating assembly of the general transcription machinery and transcription initiation, but are instead likely to be implicated in subsequent processes such as promoter clearance, transcription elongation and/or the establishment of transcriptional memory. These conclusions are consistent with previous reports suggesting that phosphorylation of the CTD of Pol II by the CDK7 subunit of the general transcription factor TFIIH and the CDK9 subunit of the transcription elongation factor pTEFb are key events in CIITA-induced MHC-II gene expression (28,37). In contrast, our results challenge current models proposing that CIITA serves as a scaffolding protein that recruits and coordinates all key chromatin-modifying activities at MHC-II promoters, and that the modifications introduced by these activities are primarily required for transcription initiation at MHC-II genes (5).
Among the histone modifications we have examined, only the rapid and long-range increase in H4 acetylation coincides temporally with the recruitment of CIITA and occurs prior to and independently of active transcription of the HLA-DRA gene. This suggests that only the early H4 acetylation phase is likely to be mediated by HATs that are recruited directly by CIITA. HATs that are believed to be able to cooperate with CIITA include CBP, pCAF, GCN5 and steroid receptor co-activator (SRC)-1. These HATs have been reported to be recruited to MHC-II promoters in B cells and IFN-γ-induced cells, can interact with CIITA in vitro and upon over expression in transfected cells, and can activate MHC-II reporter genes in synergy with CIITA in transient transfection experiments (38–42). However, we have so far been unable to document a robust and reproducible recruitment of any of these HATs in a pattern that coincides temporally and spatially with the binding of CIITA and the early increase in H4 acetylation. One explanation that could account for this discrepancy is that these HATs associate only transiently with the HLA-DRA upstream region. Alternatively, other HATs could be implicated. Since CIITA has been reported to have intrinsic HAT activity (43), one possibility is that CIITA itself might be responsible for the increase in H4 acetylation. A second intriguing possibility is suggested by the finding that intergenic transcription is induced in the HLA-DRA upstream region according to a pattern that is similar to that observed for H4 acetylation with respect to both timing and spatial distribution. Intergenic transcription has been attributed a regulatory function in the establishment of open chromatin domains in several systems (44–49). It has notably been suggested to contribute to the function of locus control regions (LCRs) (44,45,49). Among other models, it has been postulated that the regulatory role of intergenic transcription could be due to chromatin remodeling activities—such as HATs—that track along the chromatin with Pol II (50–55). Since the HLA-DRA upstream regions exhibit properties typical of LCRs (26,56–58), it is tempting to speculate that such a tracking mechanism is operating during the early phase of H4 acetylation observed during HLA-DRA gene activation. We can however not exclude the possibility that the intergenic transcription is a consequence rather than a cause of increased H4 acetylation.
Our results highlight a dominant role of transcription elongation in the recruitment of histone-modifying activities during the induction of MHC-II gene expression. This is at odds with widespread models of gene regulation postulating that HATs and HMTs are primarily recruited by DNA-bound transcription factors and their associated co-activators (1–3,5). However, there is growing evidence that a decisive role of transcription elongation in the establishment of histone modifications is not just a peculiarity of the MHC-II system.
There is strong evidence for a link between deposition of the H3K4Me3 mark and transcription elongation in Saccharomyces cerevisiae. Set1—the HMT responsible for methylating H3K4 in yeast—is recruited to the elongating CTD-phosphorylated form of Pol II by its interaction with the elongation factor Paf1 (2,6–8). This mode of recruitment is consistent with the results of genome-wide mapping studies, which have shown that the density of H3K4 methylation generally peaks within the 5′ transcribed portion of active yeast genes (59,60). The link between H3K4 methylation and transcription elongation is less well established in higher eukaryotes. Set1 homolog in higher eukaryotes—of which there are several—have in fact been reported to be recruited by interactions with specific transcription factors (61–65). However, large-scale mapping studies in Drosophila melanogaster and humans, as well as a more limited study in the chicken, have demonstrated that H3K4 methylation is, like in yeast, frequently concentrated within the 5′ transcribed regions of active genes (66–70). Moreover, a protein fraction enriched in H3K4-specific HMT activity was recently reported to introduce the H3K4Me3 modification in a transcription-coupled manner in a reconstituted human in vitro transcription system (71). These findings suggest that a transcription-dependent mode of recruitment of H3K4-specific HMTs is conserved in higher eukaryotes.
There are also indications that H3 acetylation may be coupled to transcription elongation at numerous genes in diverse species. Large-scale mapping studies performed in S. cerevisiae, Schizosaccharomyces pombe, D. melanogaster and humans have established that the density of H3 acetylation tends to peak, as observed here at the HLA-DRA gene, within the 5′ transcribed region of expressed genes (59,60,67,68,70,72,73). This intragenic localization would be consistent with a widespread role of active transcription in increasing H3 acetylation. Transcription-dependent H3 acetylation could be mediated by HATs, such as the elongator complex, that travel along chromatin in association with elongating Pol II (9,51–54). Alternatively, several findings suggest that H3 acetylation could be coupled to H3K4 methylation. Large-scale mapping studies have shown that the patterns of H3Ac and H3K4Me3 often coincide (59,60,67,68,70,72,73). Moreover, methylation at K4 makes H3 a preferential target for dynamic changes in acetylation (74). Finally, histone-modifying complexes containing both HAT and H3K4-specific HMT subunits have been identified in yeast, D. melanogaster and humans (11,65,75).
The recruitment of CARM1, the HMT responsible for making the H3R17Me2 modification, is believed to be mediated by interactions with specific transcription factors or co-activators (3,14,15,32,76). However, in two systems CARM1 recruitment has been shown to be dependent on the prior acetylation of H3 (14,15). Tethering of CARM1 to chromatin modifications established during active transcription, such as H3 acetylation, might thus constitute an alternative pathway for CARM1 recruitment.
Finally, our results extend the recent discovery that the H3K9Me3 mark is introduced into the 5′ transcribed region of mouse and human genes by a transcription-elongation-dependent process (4). As observed here at the HLA-DRA gene, this previous report also demonstrated co-localization between the H3K9Me3 and H3K4Me3 marks, suggesting that their introduction might be coupled (4).
Taken together, the findings outlined above suggest that transcription elongation may play a critical role in establishing histone-modification patterns associated with many actively expressed genes in species ranging from yeast to humans. The results reported here for activation of the HLA-DRA gene are thus likely to be of widespread relevance to numerous gene regulatory systems in diverse species.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We are grateful to Michel Strubin, Iannis Talianidis, and current and past members of the laboratory for valuable discussions and critical reading of the manuscript. This work was supported by the Swiss National Science Foundation. Funding to pay the Open Access publication charges for this article was provided by the Swiss National Science Foundation.
Conflict of interest statement. None declared.
REFERENCES
- 1.Kurdistani SK, Grunstein M. Histone acetylation and deacetylation in yeast. Nat. Rev. Mol. Cell Biol. 2003;4:276–284. doi: 10.1038/nrm1075. [DOI] [PubMed] [Google Scholar]
- 2.Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 2005;6:838–849. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- 3.Wysocka J, Allis CD, Coonrod S. Histone arginine methylation and its dynamic regulation. Front Biosci. 2006;11:344–355. doi: 10.2741/1802. [DOI] [PubMed] [Google Scholar]
- 4.Vakoc CR, Mandat SA, Olenchock BA, Blobel GA. Histone H3 lysine 9 methylation and HP1gamma are associated with transcription elongation through mammalian chromatin. Mol. Cell. 2005;19:381–391. doi: 10.1016/j.molcel.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 5.Wright KL, Ting JP. Epigenetic regulation of MHC-II and CIITA genes. Trends Immunol. 2006;27:405–412. doi: 10.1016/j.it.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 6.Krogan NJ, Dover J, Wood A, Schneider J, Heidt J, Boateng MA, Dean K, Ryan OW, Golshani A, et al. The Paf1 complex is required for histone H3 methylation by COMPASS and Dot1p: linking transcriptional elongation to histone methylation. Mol. Cell. 2003;11:721–729. doi: 10.1016/s1097-2765(03)00091-1. [DOI] [PubMed] [Google Scholar]
- 7.Ng HH, Robert F, Young RA, Struhl K. Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol. Cell. 2003;11:709–719. doi: 10.1016/s1097-2765(03)00092-3. [DOI] [PubMed] [Google Scholar]
- 8.Eissenberg JC, Shilatifard A. Leaving a mark: the many footprints of the elongating RNA polymerase II. Curr. Opin. Genet. Dev. 2006;16:184–190. doi: 10.1016/j.gde.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 9.Close P, Hawkes N, Cornez I, Creppe C, Lambert CA, Rogister B, Siebenlist U, Merville MP, Slaugenhaupt SA, et al. Transcription impairment and cell migration defects in elongator-depleted cells: implication for familial dysautonomia. Mol. Cell. 2006;22:521–531. doi: 10.1016/j.molcel.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 10.Saunders A, Core LJ, Lis JT. Breaking barriers to transcription elongation. Nat. Rev. Mol. Cell Biol. 2006;7:557–567. doi: 10.1038/nrm1981. [DOI] [PubMed] [Google Scholar]
- 11.Smith ST, Petruk S, Sedkov Y, Cho E, Tillib S, Canaani E, Mazo A. Modulation of heat shock gene expression by the TAC1 chromatin-modifying complex. Nat. Cell Biol. 2004;6:162–167. doi: 10.1038/ncb1088. [DOI] [PubMed] [Google Scholar]
- 12.Metivier R, Penot G, Hubner MR, Reid G, Brand H, Kos M, Gannon F. Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell. 2003;115:751–763. doi: 10.1016/s0092-8674(03)00934-6. [DOI] [PubMed] [Google Scholar]
- 13.Kouskouti A, Talianidis I. Histone modifications defining active genes persist after transcriptional and mitotic inactivation. EMBO J. 2005;24:347–357. doi: 10.1038/sj.emboj.7600516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.An W, Kim J, Roeder RG. Ordered cooperative functions of PRMT1, p300, and CARM1 in transcriptional activation by p53. Cell. 2004;117:735–748. doi: 10.1016/j.cell.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 15.Daujat S, Bauer UM, Shah V, Turner B, Berger S, Kouzarides T. Crosstalk between CARM1 methylation and CBP acetylation on histone H3. Curr. Biol. 2002;12:2090–2097. doi: 10.1016/s0960-9822(02)01387-8. [DOI] [PubMed] [Google Scholar]
- 16.Agalioti T, Chen G, Thanos D. Deciphering the transcriptional histone acetylation code for a human gene. Cell. 2002;111:381–392. doi: 10.1016/s0092-8674(02)01077-2. [DOI] [PubMed] [Google Scholar]
- 17.Reith W, Mach B. The bare lymphocyte syndrome and the regulation of MHC expression. Ann. Rev. Immunol. 2001;19:331–373. doi: 10.1146/annurev.immunol.19.1.331. [DOI] [PubMed] [Google Scholar]
- 18.Ting JP, Trowsdale J. Genetic control of MHC class II expression. Cell. 2002;109:S21–S33. doi: 10.1016/s0092-8674(02)00696-7. [DOI] [PubMed] [Google Scholar]
- 19.Reith W, LeibundGut-Landmann S, Waldburger JM. Regulation of MHC class II gene expression by the class II transactivator. Nat. Rev. Immunol. 2005;5:793–806. doi: 10.1038/nri1708. [DOI] [PubMed] [Google Scholar]
- 20.Steimle V, Otten LA, Zufferey M, Mach B. Complementation cloning of an MHC class II transactivator mutated in hereditary MHC class II deficiency. Cell. 1993;75:135–146. [PubMed] [Google Scholar]
- 21.Steimle V, Durand B, Barras E, Zufferey M, Hadam MR, Mach B, Reith W. A novel DNA binding regulatory factor is mutated in primary MHC class II deficiency (bare lymphocyte syndrome) Genes Dev. 1995;9:1021–1032. doi: 10.1101/gad.9.9.1021. [DOI] [PubMed] [Google Scholar]
- 22.Durand B, Sperisen P, Emery P, Barras E, Zufferey M, Mach B, Reith W. RFXAP, a novel subunit of the RFX DNA binding complex is mutated in MHC class II deficiency. EMBO J. 1997;16:1045–1055. doi: 10.1093/emboj/16.5.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Masternak K, Barras E, Zufferey M, Conrad B, Corthals G, Aebersold R, Sanchez JC, Hochstrasser DF, Mach B, et al. A gene encoding a novel RFX-associated transactivator is mutated in the majority of MHC class II deficiency patients. Nat. Genet. 1998;20:273–277. doi: 10.1038/3081. [DOI] [PubMed] [Google Scholar]
- 24.Nagarajan UM, Louis-Plence P, DeSandro A, Nilsen R, Bushey A, Boss JM. RFX-B is the gene responsible for the most common cause of the bare lymphocyte syndrome, an MHC class II immunodeficiency. Immunity. 1999;10:153–162. doi: 10.1016/s1074-7613(00)80016-3. [DOI] [PubMed] [Google Scholar]
- 25.Masternak K, Muhlethaler-Mottet A, Villard J, Zufferey M, Steimle V, Reith W. CIITA is a transcriptional coactivator that is recruited to MHC class II promoters by multiple synergistic interactions with an enhanceosome complex. Genes Dev. 2000;14:1156–1166. [PMC free article] [PubMed] [Google Scholar]
- 26.Masternak K, Peyraud N, Krawczyk M, Barras E, Reith W. Chromatin remodeling and extragenic transcription at the MHC class II locus control region. Nature Immunol. 2003;4:132–137. doi: 10.1038/ni883. [DOI] [PubMed] [Google Scholar]
- 27.Krawczyk M, Peyraud N, Rybtsova N, Masternak K, Bucher P, Barras E, Reith W. Long distance control of MHC class II expression by multiple distal enhancers regulated by regulatory factor X complex and CIITA. J. Immunol. 2004;173:6200–6210. doi: 10.4049/jimmunol.173.10.6200. [DOI] [PubMed] [Google Scholar]
- 28.Spilianakis C, Kretsovali A, Agalioti T, Makatounakis T, Thanos D, Papamatheakis J. CIITA regulates transcription onset viaSer5-phosphorylation of RNA Pol II. EMBO J. 2003;22:5125–5136. doi: 10.1093/emboj/cdg496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gomez JA, Majumder P, Nagarajan UM, Boss JM. X box-like sequences in the MHC class II region maintain regulatory function. J. Immunol. 2005;175:1030–1040. doi: 10.4049/jimmunol.175.2.1030. [DOI] [PubMed] [Google Scholar]
- 30.Beresford GW, Boss JM. CIITA coordinates multiple histone acetylation modifications at the HLA- DRA promoter. Nat. Immunol. 2001;2:652–657. doi: 10.1038/89810. [DOI] [PubMed] [Google Scholar]
- 31.Masternak K, Reith W. Promoter-specific functions of CIITA and the MHC class II enhanceosome in transcriptional activation. EMBO J. 2002;21:1379–1388. doi: 10.1093/emboj/21.6.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zika E, Fauquier L, Vandel L, Ting JP. Interplay among coactivator-associated arginine methyltransferase 1, CBP, and CIITA in IFN-gamma-inducible MHC-II gene expression. Proc. Natl Acad. Sci. USA. 2005;102:16321–16326. doi: 10.1073/pnas.0505045102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muhlethaler-Mottet A, Di Berardino W, Otten LA, Mach B. Activation of the MHC class II transactivator CIITA by interferon-g requires cooperative interaction between Stat1 and USF-1. Immunity. 1998;8:157–166. doi: 10.1016/s1074-7613(00)80468-9. [DOI] [PubMed] [Google Scholar]
- 34.Landmann S, Muhlethaler-Mottet A, Bernasconi L, Suter T, Waldburger JM, Masternak K, Arrighi JF, Hauser C, Fontana A, et al. Maturation of dendritic cells is accompanied by rapid transcriptional silencing of class II transactivator (CIITA) expression. J. Exp. Med. 2001;194:379–391. doi: 10.1084/jem.194.4.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chodosh LA, Fire A, Samuels M, Sharp PA. 5,6-Dichloro-1-beta-D-ribofuranosylbenzimidazole inhibits transcription elongation by RNA polymerase II in vitro. J. Biol. Chem. 1989;264:2250–2257. [PubMed] [Google Scholar]
- 36.Yankulov K, Yamashita K, Roy R, Egly JM, Bentley DL. The transcriptional elongation inhibitor 5,6-dichloro-1-beta-D-ribofuranosylbenzimidazole inhibits transcription factor IIH-associated protein kinase. J. Biol. Chem. 1995;270:23922–23925. doi: 10.1074/jbc.270.41.23922. [DOI] [PubMed] [Google Scholar]
- 37.Kanazawa S, Okamoto T, Peterlin BM. Tat competes with CIITA for the binding to P-TEFb and blocks the expression of MHC class II genes in HIV infection. Immunity. 2000;12:61–70. doi: 10.1016/s1074-7613(00)80159-4. [DOI] [PubMed] [Google Scholar]
- 38.Kretsovali A, Agalioti T, Spilianakis C, Tzortzakaki E, Merika M, Papamatheakis J. Involvement of CREB binding protein in expression of major histocompatibility complex class II genes via interaction with the class II transactivator. Mol. Cell. Biol. 1998;18:6777–6783. doi: 10.1128/mcb.18.11.6777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fontes JD, Kanazawa S, Jean D, Peterlin BM. Interactions between the class II transactivator and CREB binding protein increase transcription of major histocompatibility complex class II genes. Mol. Cell. Biol. 1999;19:941–947. doi: 10.1128/mcb.19.1.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu XS, Ting JP. A 36-amino-acid region of CIITA is an effective inhibitor of CBP: novel mechanism of gamma interferon-mediated suppression of collagen alpha(2)(I) and other promoters. Mol. Cell. Biol. 2001;21:7078–7088. doi: 10.1128/MCB.21.20.7078-7088.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spilianakis C, Papamatheakis J, Kretsovali A. Acetylation by PCAF enhances CIITA nuclear accumulation and transactivation of major histocompatibility complex class II genes. Mol. Cell. Biol. 2000;20:8489–8498. doi: 10.1128/mcb.20.22.8489-8498.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tzortzakaki E, Spilianakis C, Zika E, Kretsovali A, Papamatheakis J. Steroid receptor coactivator 1 links the steroid and interferon gamma response pathways. Mol. Endocrinol. 2003;17:2509–2518. doi: 10.1210/me.2002-0439. [DOI] [PubMed] [Google Scholar]
- 43.Raval A, Howcroft TK, Weissman JD, Kirshner S, Zhu XS, Yokoyama K, Ting J, Singer DS. Transcriptional coactivator, CIITA, is an acetyltransferase that bypasses a promoter requirement for TAF(II)250. Mol. Cell. 2001;7:105–115. doi: 10.1016/s1097-2765(01)00159-9. [DOI] [PubMed] [Google Scholar]
- 44.Plant KE, Routledge SJ, Proudfoot NJ. Intergenic transcription in the human beta-globin gene cluster. Mol. Cell. Biol. 2001;21:6507–6514. doi: 10.1128/MCB.21.19.6507-6514.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Routledge SJ, Proudfoot NJ. Definition of transcriptional promoters in the human beta globin locus control region. J. Mol. Biol. 2002;323:601–611. doi: 10.1016/s0022-2836(02)01011-2. [DOI] [PubMed] [Google Scholar]
- 46.Rogan DF, Cousins DJ, Santangelo S, Ioannou PA, Antoniou M, Lee TH, Staynov DZ. Analysis of intergenic transcription in the human IL-4/IL-13 gene cluster. Proc. Natl Acad. Sci. USA. 2004;101:2446–2451. doi: 10.1073/pnas.0308327100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bolland DJ, Wood AL, Johnston CM, Bunting SF, Morgan G, Chakalova L, Fraser PJ, Corcoran AE. Antisense intergenic transcription in V(D)J recombination. Nat. Immunol. 2004;5:630–637. doi: 10.1038/ni1068. [DOI] [PubMed] [Google Scholar]
- 48.Schmitt S, Prestel M, Paro R. Intergenic transcription through a polycomb group response element counteracts silencing. Genes Dev. 2005;19:697–708. doi: 10.1101/gad.326205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gribnau J, Diderich K, Pruzina S, Calzolari R, Fraser P. Intergenic transcription and developmental remodeling of chromatin subdomains in the human beta-globin locus. Mol. Cell. 2000;5:377–386. doi: 10.1016/s1097-2765(00)80432-3. [DOI] [PubMed] [Google Scholar]
- 50.Engel JD, Tanimoto K. Looping, linking, and chromatin activity: new insights into beta-globin locus regulation. Cell. 2000;100:499–502. doi: 10.1016/s0092-8674(00)80686-8. [DOI] [PubMed] [Google Scholar]
- 51.Svejstrup JQ. The RNA polymerase II transcription cycle: cycling through chromatin. Biochim. Biophys. Acta. 2004;1677:64–73. doi: 10.1016/j.bbaexp.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 52.Wittschieben BO, Otero G, de Bizemont T, Fellows J, Erdjument-Bromage H, Ohba R, Li Y, Allis CD, Tempst P, et al. A novel histone acetyltransferase is an integral subunit of elongating RNA polymerase II holoenzyme. Mol. Cell. 1999;4:123–128. doi: 10.1016/s1097-2765(00)80194-x. [DOI] [PubMed] [Google Scholar]
- 53.Hawkes NA, Otero G, Winkler GS, Marshall N, Dahmus ME, Krappmann D, Scheidereit C, Thomas CL, Schiavo G, et al. Purification and characterization of the human elongator complex. J. Biol. Chem. 2002;277:3047–3052. doi: 10.1074/jbc.M110445200. [DOI] [PubMed] [Google Scholar]
- 54.Kim JH, Lane WS, Reinberg D. Human elongator facilitates RNA polymerase II transcription through chromatin. Proc. Natl Acad. Sci. USA. 2002;99:1241–1246. doi: 10.1073/pnas.251672198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Travers A. Chromatin modification by DNA tracking. Proc. Natl Acad. Sci. USA. 1999;96:13634–13637. doi: 10.1073/pnas.96.24.13634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carson S, Wiles MV. Far upstream regions of class II MHC Ea are necessary for position-independent, copy-dependent expression of Ea transgene. Nucleic Acids Res. 1993;21:2065–2072. doi: 10.1093/nar/21.9.2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Carson S. DNase I hypersensitive sites flank the mouse class II major histocompatibility complex during B cell development. Nucleic Acids Res. 1991;19:5007–5014. doi: 10.1093/nar/19.18.5007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dorn A, Fehling HJ, Koch W, Lemeur M, Gerlinger P, Benoist C, Mathis D. B cell control region at the 5′ end of a major histocompatibility complex class II gene: sequences and factors. Mol. Cell. Biol. 1988;8:3975–3987. doi: 10.1128/mcb.8.10.3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pokholok DK, Harbison CT, Levine S, Cole M, Hannett NM, Lee TI, Bell GW, Walker K, Rolfe PA, et al. Genome-wide map of nucleosome acetylation and methylation in yeast. Cell. 2005;122:517–527. doi: 10.1016/j.cell.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 60.Liu CL, Kaplan T, Kim M, Buratowski S, Schreiber SL, Friedman N, Rando OJ. Single-nucleosome mapping of histone modifications in S. cerevisiae. PLoS Biol. 2005;3:e328. doi: 10.1371/journal.pbio.0030328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sedkov Y, Cho E, Petruk S, Cherbas L, Smith ST, Jones RS, Cherbas P, Canaani E, Jaynes JB, et al. Methylation at lysine 4 of histone H3 in ecdysone-dependent development of Drosophila. Nature. 2003;426:78–83. doi: 10.1038/nature02080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Milne TA, Briggs SD, Brock HW, Martin ME, Gibbs D, Allis CD, Hess JL. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol. Cell. 2002;10:1107–1117. doi: 10.1016/s1097-2765(02)00741-4. [DOI] [PubMed] [Google Scholar]
- 63.Nakamura T, Mori T, Tada S, Krajewski W, Rozovskaia T, Wassell R, Dubois G, Mazo A, Croce CM, et al. ALL-1 is a histone methyltransferase that assembles a supercomplex of proteins involved in transcriptional regulation. Mol. Cell. 2002;10:1119–1128. doi: 10.1016/s1097-2765(02)00740-2. [DOI] [PubMed] [Google Scholar]
- 64.Wysocka J, Myers MP, Laherty CD, Eisenman RN, Herr W. Human Sin3 deacetylase and trithorax-related Set1/Ash2 histone H3-K4 methyltransferase are tethered together selectively by the cell-proliferation factor HCF-1. Genes Dev. 2003;17:896–911. doi: 10.1101/gad.252103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dou Y, Milne TA, Tackett AJ, Smith ER, Fukuda A, Wysocka J, Allis CD, Chait BT, Hess JL, et al. Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell. 2005;121:873–885. doi: 10.1016/j.cell.2005.04.031. [DOI] [PubMed] [Google Scholar]
- 66.Miao F, Natarajan R. Mapping global histone methylation patterns in the coding regions of human genes. Mol. Cell. Biol. 2005;25:4650–4661. doi: 10.1128/MCB.25.11.4650-4661.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schubeler D, MacAlpine DM, Scalzo D, Wirbelauer C, Kooperberg C, van LF, Gottschling DE, O'Neill LP, Turner BM, et al. The histone modification pattern of active genes revealed through genome-wide chromatin analysis of a higher eukaryote. Genes Dev. 2004;18:1263–1271. doi: 10.1101/gad.1198204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liang G, Lin JC, Wei V, Yoo C, Cheng JC, Nguyen CT, Weisenberger DJ, Egger G, Takai D, et al. Distinct localization of histone H3 acetylation and H3-K4 methylation to the transcription start sites in the human genome. Proc. Natl Acad. Sci. USA. 2004;101:7357–7362. doi: 10.1073/pnas.0401866101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schneider R, Bannister AJ, Myers FA, Thorne AW, Crane-Robinson C, Kouzarides T. Histone H3 lysine 4 methylation patterns in higher eukaryotic genes. Nat. Cell Biol. 2004;6:73–77. doi: 10.1038/ncb1076. [DOI] [PubMed] [Google Scholar]
- 70.Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, McMahon S, Karlsson EK, Kulbokas EJ, III, et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005;120:169–181. doi: 10.1016/j.cell.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 71.Pavri R, Zhu B, Li G, Trojer P, Mandal S, Shilatifard A, Reinberg D. Histone H2B monoubiquitination functions cooperatively with FACT to regulate elongation by RNA polymerase II. Cell. 2006;125:703–717. doi: 10.1016/j.cell.2006.04.029. [DOI] [PubMed] [Google Scholar]
- 72.Sinha I, Wiren M, Ekwall K. Genome-wide patterns of histone modifications in fission yeast. Chromosome Res. 2006;14:95–105. doi: 10.1007/s10577-005-1023-4. [DOI] [PubMed] [Google Scholar]
- 73.Roh TY, Cuddapah S, Zhao K. Active chromatin domains are defined by acetylation islands revealed by genome-wide mapping. Genes Dev. 2005;19:542–552. doi: 10.1101/gad.1272505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hazzalin CA, Mahadevan LC. Dynamic acetylation of all lysine 4-methylated histone H3 in the mouse nucleus: analysis at c-fos and c-jun. PLoS Biol. 2005;3:e393. doi: 10.1371/journal.pbio.0030393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pray-Grant MG, Daniel JA, Schieltz D, Yates JR, III, Grant PA. Chd1 chromodomain links histone H3 methylation with. Nature. 2005;433:434–438. doi: 10.1038/nature03242. [DOI] [PubMed] [Google Scholar]
- 76.Covic M, Hassa PO, Saccani S, Buerki C, Meier NI, Lombardi C, Imhof R, Bedford MT, Natoli G, et al. Arginine methyltransferase CARM1 is a promoter-specific regulator of NF-kappaB-dependent gene expression. EMBO J. 2005;24:85–96. doi: 10.1038/sj.emboj.7600500. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.