


Abstract
Phage integrases are required for recombination of the phage genome with the host chromosome either to establish or exit from the lysogenic state. ϕC31 integrase is a member of the serine recombinase family of site-specific recombinases. In the absence of any accessory factors integrase is unidirectional, catalysing the integration reaction between the phage and host attachment sites, attP × attB to generate the hybrid sites, attL and attR. The basis for this directionality is due to selective synapsis of attP and attB sites. Here we show that mutations in attB can block the integration reaction at different stages. Mutations at positions distal to the crossover site inhibit recombination by destabilizing the synapse with attP without significantly affecting DNA-binding affinity. These data are consistent with the proposal that integrase adopts a specific conformation on binding to attB that permits synapsis with attP. Other attB mutants with changes close to the crossover site are able to form a stable synapse but cleavage of the substrates is prevented. These mutants indicate that there is a post-synaptic DNA recognition event that results in activation of DNA cleavage.
INTRODUCTION
ϕC31 integrase and several of its relatives are being widely used for precise engineering of complex genomes (1–8) and are emerging as promising new tools for gene therapy (9–15). In addition to being highly portable ϕC31 integrase is, unlike other recombinases used for genome manipulation such as Cre and Flp, unidirectional (7,16,17). In nature phage integrases are required for recombination of the phage genome with the host chromosome either to establish or exit from the lysogenic state. For integration the host-encoded attB site undergoes a conservative and reciprocal recombination with the phage attP site to form the hybrid product sites, attL and attR. During induction into the lytic cycle, the phage genome excises and this reaction normally requires integrase and an accessory protein Xis (18,19). Phage-encoded integrases can belong to the tyrosine or the serine recombinase families (20). Both families of proteins act by binding to their cognate substrates and bringing the DNAs together in a synapse. Recombination is initiated by cleaving DNA strands, which undergo strand exchange to form recombinant products and these are then released (21). While the mechanism of phage λ integrase, a tyrosine recombinase, is well understood (18,22,23), the mechanism of action of integrases such as ϕC31 integrase that belong to the serine recombinase family, is less clear.
All serine recombinases have a conserved catalytic domain required for DNA cleavage and rejoining (20,24). The resolvase/invertases also have a small (∼60 amino acids; aa) C-terminal DNA-binding domain (25). The serine integrases, some transposases and the staphylococcal cassette recombinases (Ccr proteins; required for the movement of methicillin resistance gene in MRSA) are so-called large serine recombinases as they have extensive C-terminal domains (∼300–500 aa in length; 20). Sequence alignments of these large serine recombinases indicate that they are an extremely diverse family. Experiments with ϕC31 integrase, mycobacteriophage Bxb1 integrase and TnpX transposase suggest that the recombination mechanism used by the large serine recombinases resembles that of the well-studied resolvase/invertases (17,24,26–33). DNA cleavage occurs at a 2 bp crossover sequence to form a staggered break and a transient covalent phosphoserine bond between the recessed 5′ ends and the recombinase is formed (17,27). Strand exchange most likely occurs by rotation of two recombinase subunits bound to half sites relative to the other two subunits (34–37). Rejoining of the products is dependent on the complementarity of the DNA sequence at the staggered breaks; if there is a mismatch at this sequence, joining of the products is severely inhibited but iteration of strand exchange results in changes in the topology of the substrates (17,28,38).
Divergence from the resolvase paradigm by the serine integrases occurs in the nature of substrate recognition and the formation of the synapse. The pairs of recombination sites used by the serine integrases have different sequences; for example, the ϕC31 attP and attB sites share 39% sequence identity [Figure 1, (39)]. The recombination sites are generally short, ∼50 bp (10,17,29,40). The minimal sites for ϕC31 integrase have been defined as a 39 bp attP site and a 34 bp attB site (10). Under in vitro conditions ϕC31 integrase converts ∼80% of attB and attP to products in the absence of accessory proteins and there are no restrictions on the topology of the substrates (16,27,28). Moreover, in these in vitro reactions, ϕC31 integrase is catalytically inert on all other combinations of substrates including attL and attR (28). Hatfull and colleagues have shown that Bxb1 integrase has similar properties and they have gone on to show that Bxb1 integrase binds to its substrates as a dimer (17,26). The synapse is therefore likely to contain a tetramer of integrase subunits (26).
Figure 1.

ϕC31 attB and attP sites. (A) The double-stranded DNA sequences of the S. coelicolor attB site (green) and the attP site (blue) are shown. The crossover dinucleotides are shown in black. The colons connecting the two sequences indicate the positions of sequence identity between the aligned attB and attP sites. The grey shading indicates the positions where sequence conservation can be detected between the attB or attP sites and their pseudo-sites from Streptomyces or Mycobacteria (pseudo-attB sites) or from human or mouse cell lines (pseudo-attP sites) (9,41–43). (B) Summary of mutation scanning in attB. The attB site is shown as a single-strand sequence where each base acts as point on the x-axis of a histogram. The y-axis shows the fold reduction in product made when mutations are introduced in attB. The positions are annotated according to the numbering shown. The activities of attB sites with double mutations at symmetrical positions (eg −/+1, −/+2, etc.) are shown in pink and the activities of mutants with single mutations are shown in black. The data for the summary graph were calculated from the estimated absolute activities shown in Table 1, Figure 2 and Supplementary Data, Figure S1. Beneath the attB sequence, three of the S. coelicolor pseudo-attB sites [pseB1, pseB2 and pseB3 (41)] are shown for comparison with the wild-type attB. The four sites have been aligned and are shaded according to whether there is 100% identity (black background and white text) or 75% identity (grey background) between the sites.
A major focus in our lab has been to understand why ϕC31 integrase can only recombine attB and attP in vitro. We have shown previously that integrase cannot synapse pairs of recombination sites other than attP with attB indicating that the formation of the synapse is the major block to excision in vitro (27). We and others have proposed that integrase adopts specific conformations when bound to attP and attB sites that enable the formation of a synapse, but when bound to attL and attR disable or destabilize the synapse (26,27,29). In this model, the interactions between integrase and attP and attB are central to the formation of the synaptic interface. Some clues as to the preferred sequences of attP and attB have been obtained previously through studies that have characterized the substrates used by integrase when one of the cognate sites is not present (9,41,42). Pseudo-attB sites in the bacterial host, Streptomyces coelicolor and other actinomycetes show a strong preference for certain bases [Figure 1, (41,43)]. Similarly, pseudo-attP sites have been characterized in mammalian genomes and these also show base specific preferences (Figure 1). Many of the bases that are conserved in the pseudo-attP and pseudo-attB sites are also conserved between attP and attB (Figure 1).
To examine the integrase–attB interaction in more detail, the minimal attB site was subjected to mutagenesis and the activities of the mutants assayed in recombination and binding assays. Recombination defective attB mutants that could still bind to integrase with affinities not dissimilar to the wild-type attB site were found to be blocked either at synapsis or at DNA cleavage. The most likely explanation is that there are two separate recognition events that occur between integrase and the attB site. The first event results in a protein–protein interface that enables synapsis and the second post-synapsis event results in activation of DNA cleavage.
MATERIALS AND METHODS
Bacterial strains and plasmids
Escherichia coli strains DH5α and DS941 were used as general cloning hosts and were grown in LB or 2xYT (44). E. coli transformation, plasmid preparations and DNA manipulation were performed as described previously (44).
Plasmids pRT600 and pRT700 were constructed previously by insertion of annealed oligonucleotides RM1/RM2 containing attB (51 bp) and RM3/RM4 attP (50 bp) sites inserted into pGEM7 cut with EcoRI and Csp45I (29). For this work, the attP site from pRT700 was excised with BamHI and EcoRI and inserted into BamHI and EcoRI cut pSP72 to form pRT702. Plasmids containing mutant attB sites at all positions except for −/+3, −/+8 and −/+12 were constructed as for pRT600; annealed oligonucleotides (see Supplementary Data, Table S1) were inserted into pGEM7 cut with EcoRI and Csp45I. Plasmids containing mutations at −/+3, −/+8 and −/+12 were constructed differently; PCR amplification using primers containing a randomized base at positions 3, 8 or 12 (Supplementary Data, Table S2) resulted in fragments that could be inserted into pGEM7 and these were then sequenced to determine the nature of the mutations. To create the double mutants with mutations at symmetrical positions, fragments containing the two single mutations were spliced together using the unique StyI site in the centre of the attB site. All the plasmids containing the mutant attB sites were subjected to confirmation by sequencing.
Recombination assays
Standard recombination assays between two attachment sites located on two separate plasmids were performed as described previously. Plasmids (100 ng each) containing attB (or the mutant attBs) and attP were mixed with 18 μl of recombination buffer (10 mM Tris pH 7.5, 1 mM EDTA pH 8, 100 mM NaCl, 5 mM DTT, 5 mM spermidine, 4.5% glycerol and 0.5 mg/ml bovine serum albumin) and ϕC31 integrase was added to the recombination reaction to final concentrations 0, 441, 110, 55, 27 or 14 nM unless otherwise stated. Reactions were incubated at 30°C for 1 h unless otherwise stated and terminated by incubation at 65°C for 10 min. After addition of an equal volume of 2× restriction buffer (20 mM Tris-HCl pH 7.9, 100 mM NaCl, 20 mM MgCl2, 2 mM DTT) the plasmids were treated with HindIII restriction endonuclease (37°C for 2 h) and the fragments were separated by electrophoresis through 0.8% agarose gels in 1× TBE buffer (100 V). HindIII linearizes the substrates containing attB (or mutant attBs) and attP to give DNA molecules of 3035 and 2491 bp, respectively. The recombination product is a cointegrate of the two substrate plasmids and is cut by HindIII into two fragments; 5435 bp containing attL and 91 bp containing attR. Only the attL fragment is detected routinely after electrophoresis.
Recombination reactions were also performed using a plasmid encoding the attP site, pRT702, and annealed oligonucleotides containing the attB sequence or its mutant derivatives (the ‘oligo-plasmid’ assay; see Table S2 in the Supplementary Data for the sequences of the oligos) (17). pRT702 (100 ng) was mixed with 4.5 ng of annealed oligonucleotides encoding a mutant attB site and 18 μl of recombination buffer. ϕC31 integrase (1 μl) was added to give final concentrations as described above (i.e. 0, 441, 110, 55, 27 and 14 nM) and the reactions were incubated at 30°C for 1 h. The recombination reactions were terminated by heat inactivating the samples at 65°C for 10 min and the products of recombination were analysed on 0.8% agarose gels in 1× TBE buffer (100 V). The products of recombination were identified as linear DNAs (2546 bp).
DNA binding and radioactive recombination assays
DNA affinity and synapse assays were performed as described previously (29). DNA fragments for radioactive labelling were prepared by digestion of pRT600 (encoding wt attB), pRT700 (encoding attP), or plasmids containing cloned annealed oligonucleotide pairs encoding mutant attB sites with HindIII and XhoI restriction enzymes. The 72 bp fragments containing the att sites were separated on 4% agarose gel (Nusieve agarose) and then purified using gel extraction columns (QIAGEN) as per the manufacturer's protocol. The concentration of the purified fragment was determined on 4% agarose gels following which the fragments were end-labelled using DNA polymerase I large (Klenow) fragment in the presence of [α−32P]dCTP (as described previously in Sambrook et al. (44)). Unless otherwise stated, binding affinity assays were performed with 1.0 ng labelled probe in binding buffer (20 mM Tris-HCl pH 8.0, 0.1 mM EDTA, 50 mM KCl, 5% glycerol), 1 μg sonicated salmon sperm DNA and integrase added to final concentrations of 0, 351, 87, 43, 21 and 10 nM. Reactions with no integrase contained 1 μg BSA. Reactions were incubated at 30°C for 30 min prior to electrophoresis following which the reaction mix were separated on 5% non denaturing 0.5× TBE polyacrylamide gels in 0.5× TBE running buffer (200 V, 5 W for 2 h).
For radioactive recombination assays, unlabelled ‘partner’ fragments prepared by PCR amplification of pRT600 and pRT700 with SP6 and T7 primers containing either attP (193 bp) or wt or mutant attBs (194 bp) were added to the radiolabelled attachment site in the presence of integrase. Complexes containing either the uncleaved synapse, the cleaved intermediates with integrase bound covalently to the att sites, and integrase bound to the labelled substrate and products were observed by non-denaturing PAGE as described previously (27). These assays were performed using 1.5 ng of labelled probe (72 bp), 20 ng of the unlabelled fragment containing a ‘partner’ attachment site and 66 nM integrase in binding buffer. Unless otherwise stated, all reactions were incubated for a period of 2 h at 30°C prior to electrophoresis on 5% non-denaturing polyacrylamide gels (200V, 5W for 2 h).
To detect the cleavage of the DNA fragments by integrase, reactions were set up as for the radioactive recombination assays but after incubation at 30°C, reactions were heat inactivated (72°C for 10 min) then incubated with 1μl of subtilisin A (Sigma 0.1 mg/ml in 1× binding buffer) for 15 min at 30°C. Subtilisin was inactivated (72°C for 10 min) and the reactions were loaded onto a 0.5× TBE, 5% non-denaturing polyacrylamide gel.
After electrophoresis gels were dried and exposed to a phosphorimager screen (Fuji) for 16 h and then scanned (Fuji FLA3200 phosphorimager). Quantification of radioactivity was performed using the AIDA software (Raytest, Straubenhardt, Germany).
Purification of integrase
Wild-type ϕC31 and S12A integrase were purified as described previously (27). Integrase concentration was assayed using a method based on the dye-binding procedure of Bradford (45) employing the BioRad protein assay solution, and bovine serum albumin as a standard.
RESULTS
Identification of defective mutations in attB
The minimal attB site, according to Groth et al., (10) is 34 bp with the crossover 5′TT (abbreviated to XO) at the centre (Figure 1). Footprinting confirmed that integrase binds either side of the crossover site in all the attachment sites and, as integrase is a dimer in solution it probably binds as a dimer (29). Moreover, we have shown that integrase binds to attB and attP in a functionally symmetrical manner. Thus in order to maximize any phenotype arising from mutations in attB we generated a set of doubly mutated sites with base pair changes at symmetrical positions with respect to the crossover sequence. To aid in the description of the positions of mutations, the base pairs in the minimal attB and attP sites were annotated with either a negative number when they lie to the left of the crossover dinucleotide sequence (5′TT) i.e. B or P arm to use the λ terminology) or positive when it lies to the right of the crossover (B′ or P′ arm); the numbers count upwards as the position extends away from the crossover (Figure 1). Thus mutations in a double mutant involving the two base pairs adjacent to the crossover is at −/+1 and mutations at the next position moving outwards are at −/+2, etc. Mutations were chosen that would introduce sequence symmetry at the desired position. Thus each double mutant was designed to contain one of the four bases, A, T, C or G, at position –x on the B arm and at +x on the B′ arm its complement, T, A, G or C, respectively, was inserted. The choice of mutation was made on the basis that the introduced bases had to be different from those present in both arms of attB and preferably also different to those seen in the pseudo-attB sites (Figure 1). For example, position 15 is a T in the B arm and a C in the B′ arm and the pseudo-attB sites have a G in the B arm and a C or A in the B′ arm. T-15C:C+15G and the T-15A:C+15T contain changes at −/+15 on the B and the B′ arm to base pairs that are different from both the wt and the pseudo-site sequences and should be functionally the same mutation in both arms. For most positions at least two mutant forms were made but for some sites (positions 4, 7, 16) only one option was available. Other positions where only a single mutant form was made are at 14, 17 and 18.
Except for mutations at positions −/+3, −/+8 and −/+12 the activities of the double substituted attB sites was first assayed using annealed oligonucleotides. Oligonucleotides containing the double substitutions were purified by PAGE, annealed and used in an oligo-plasmid recombination assay (17). In this assay, a supercoiled plasmid containing attP was mixed with the oligonucleotide containing attB or one of the mutant forms and various concentrations of integrase. The extent of linearization of the attP plasmid indicated the extent of recombination and this was assayed after separation of the DNA in an agarose gel. A control reaction using the wild-type attB site was performed in every assay so that the activities could be compared under identical conditions. The lowest integrase concentration at which recombination could be observed was scored (Figure S1 and Table 1).
Table 1.
List of mutant attB sites and their activity compared to the wild-type attB
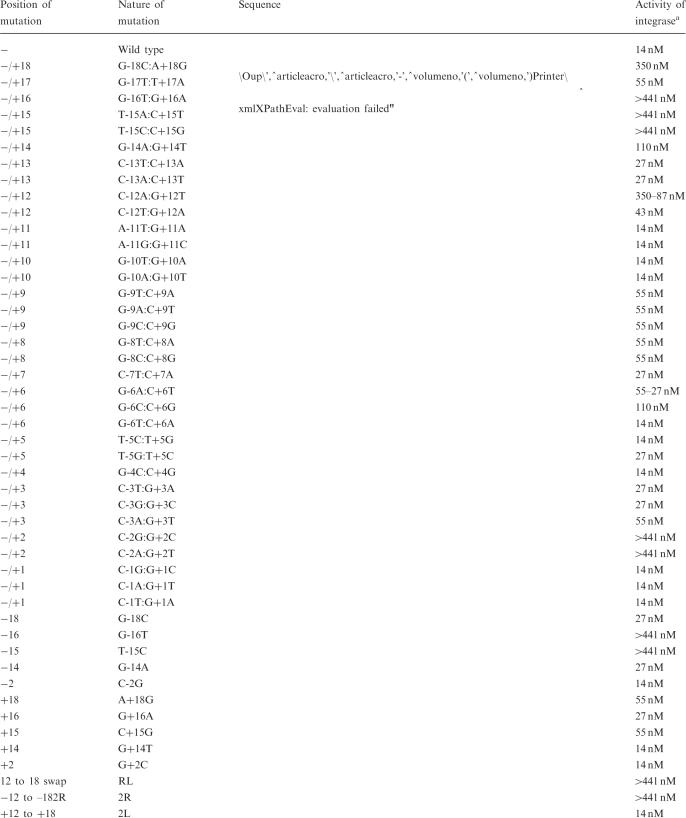 |
aConcentration of integrase required to observe recombinants in a 0.8% TBE gel stained with ethidium bromide. Two concentrations (350–87 or 55–27 nM) are given when the level of recombination obtained at the lower concentration is much less than that obtained with 14 nM integrase with wild-type attB.
Many of the mutant attB sites showed little or only 2-fold change in activity compared to the wild-type site. These sites were changed at −/+1, −/+4, −/+5, −/+7, −/+10, −/+11 and −/+13 (Table 1, Figure 1). The remaining mutants showed defective or partially defective activity ranging from 4-fold less active than wild type to apparently inactive. Oligos encoding sites C-2G:G+2C, C-2A:G+2T, G-6A:C+6T, (G-6T:C+6A, G-9T:C+9A, G-9A:C-9T, G-9C:C-9G, T-15C:C+15G, G-16T:G+15A and G-18C:A+18G were cloned into pGEM7 (Promega) so that the activities of the mutant attB sites could be verified by a standard recombination assay using both att sites residing on plasmids. Only one of the mutant attB sites that was partially defective (at position −/+14) was not represented in the cloned mutant attB site collection; this site was instead subjected to single site substitutions (see later). T-15A:C+15T was not cloned as a plasmid containing another mutant at −/+15 (T-15C:C+15G) with the same activity was quickly obtained. A plasmid encoding G-6C:C+6G was not obtained due to technical difficulties. Plasmids containing mutations in C-3T:G+3A, C-3G:G+3C, C-3A:G+3T, G-8T:C+8A, G-8C:G+8C, C-12A:G+12T and C-12T:G+12A were obtained by PCR mutagenesis as described in the Material and Methods section. The relative activities of the double substitution mutants were estimated compared to a standard reaction with wild-type attB (Table 1 and Figures 1 and 2). As for the oligo-plasmid assay the activity of each mutant site was scored as the concentration of integrase required to observe recombinants in an agarose gel stained with ethidium bromide (Table 1). The relative activities compared to the wild-type site are summarized graphically (Figure 1).
Figure 2.

Recombination activities of attB mutant sites. Recombination activities are shown for the wild-type attB site (A), mutant sites at position 2 (B), 6 (C), 12 (D), 15 (E), 16 (F) 18 (G). Panel H shows the activities of partially symmetrized attB sites that contain the right sequence between +12 and +18 changed to the same sequence as on the left (−12 to −18), 2L (+12 to +18) or vice versa, 2R (−12 to −18). Recombination assays were performed using the standard plasmid assay containing the plasmid indicated in each panel and pRT702 encoding attP. The concentrations of integrase used for each set of six reactions in panels A to C and E, F and H was 0, 441, 110, 55, 27 and 14 nM. The concentrations of integrase used for each set of six reactions in panels D and G was 0, 351, 87, 43, 21 and 10 nM.
Three of the mutant attB sites were very defective for recombination and these contained substitutions at −/+2, −/+15 and −/+16. In all cases no recombination was observed in either the oligo-plasmid or the standard assay using these double substituted attB sites (Figure 2 and Figure S1). Recombination was just detectable with attB containing substituted −/+18 in the plasmid assay with 351 nM integrase (Figure 2). The low activity of the −/+18 double mutant was surprising given that this position is outside the minimal attB site defined previously by Groth et al. (10). The nature of the mutations made small differences to activity in only a few mutants. The −/+12 mutant containing the double transversion C-12A:G+12T was only just active with 87 nM integrase while the −/+12 mutant containing the transitions C-12T:G+12A was active with 43 nM integrase (Figure 2). The G-6T:C+6A transversions had similar activity to wild-type attB but another −/+6 mutant, containing transitions (G-6A:C+6T) was 2-to 4-fold less active than attB (Figure 2).
All of the mutant attB sites described in this section that were cloned into plasmids were used to test whether they would recombine with attL, attR or attB but no activity was detected in any case. Thus none of these mutant sites had any detectable gain-of-function.
The sequence on the left side of attB has a greater role in attB function than the right side
The effects of mutations at positions −/+2, −/+14, −/+15, −/+16 and −/+18 were studied further. Oligonucleotides were synthesized that had single mutations at either the −x position in the B arm or in the +x position in the B′ arm. Recombination was performed with the oligo-plasmid assay and with the standard recombination assay using the sites cloned into pGEM7. The attB sites containing the single mutations C-2G and G+2C regained much of the activity of the wild-type attB site suggesting that a correct interaction on one or other side of the crossover at this position is sufficient for recombination (Figure 2). Similarly the single mutation at –18 or +18 also regained some activity compared to wild-type attB (Figure S1). Single mutations at the 15 and 16 positions behaved differently. Mutants at –15 or –16 had much greater effects on recombination than the mutants at +15 or +16. The single mutations C+15G and G+16A regained some activity compared to the double mutants T-15C:C+15G and G-16T:G+16A whereas the single mutants at T-15C and G-16T did not (Figure 2). A similar difference, but less so, was also observed at position 14 where the left B arm was more sensitive to mutation than the right B′ arm (Table 1). To test this further we experimented with partially symmetrical sites. The B arm of attB that included the region from –12 to –18 was replaced with the +12 to +18 sequence from the B′ side [2R (−12 to −18)] and vice versa, [2L (+12 to +18)]. The 2L (+12 to +18) attB site was as active as the wild-type attB site whereas the 2R (−12 to −18) site was inactive (Figure 2). These data indicate that the sequence in the left arm of attB plays a major role in attB function and its loss removes all activity. A mutant attB site RL, with the straight swap of the B arm sequence between −12 and −18 with the B′ arm sequence at +12 to +18 was inactive (Figure S1) indicating that whatever positive role the −12 to −18 sequence plays in attB function, it is not acting independently of other sequences in the attB site.
Mutant attB sites have little or no reduction in affinity for integrase
This mutational analysis of attB showed that double mutations at three positions −/+2, −/+15 −/+16 and the single mutants at –15 and –16 were particularly defective for recombination.
We have shown previously that it is possible to assay several intermediate steps in recombination i.e. DNA binding, formation of the synapse and cleavage of the DNA to form the covalent intermediate in which integrase is covalently bound to its cleaved substrate (27). The mutant attB sites were used first in affinity assays with integrase. As seen previously integrase bound to the wild-type attB site with an affinity of ∼60 nM (27,29). Most of the mutant attB sites bound with a similar affinity to the wild-type attB site including the severely recombination defective sites C-2A:G+2T and G-16T:G+16A (Figure 3, Table 2). The mutant T-15C:C+15G had a slightly lower affinity for integrase (∼128 nM) but this loss of affinity was abolished in the single mutant at −15 (T-15C) which was still defective in recombination (Table 2, Figures 1 and 2). Differences in binding affinities by integrase for mutant attB sites C-2A:G+2T, G-16T:G+16A, T-15C and G-16T cannot therefore account for the defectiveness of these sites in recombination. Mutations involving position 18 from the crossover dinucleotide showed an ∼3-fold lower affinity for integrase than wild-type attB which could contribute to the observed decrease in recombination activity (Figure 2). It seems likely that attB sites with mutations at −/+2, −/+15, −/+16 were blocked elsewhere in the recombination pathway.
Figure 3.

Binding affinities by integrase for the wild-type and mutant attB sites. Integrase was incubated with radiolabelled wild type (panel A) and mutant attB sites (panels B–F). In each panel, the phosphorimage shows the complexes obtained with increasing integrase concentrations and, below, the quantitative analysis of the% bound versus the concentration of integrase. Only the −/+15 mutant (T-15C:C+15G) and the −/+18 mutant (G-18C:A+18G) sites showed reduced binding affinities for integrase under the conditions used. A summary of the integrase concentrations required for 50% binding of the different attB mutants is shown in Table 2.
Table 2.
Apparent binding affinities by integrase for mutant attB sites
| attB Site; position of mutation | Nature of mutation | Mean integrase concentration for 50% binding ± SDa (nM) |
|---|---|---|
| Wt | – | 61.3 ± 12.7 |
| −/+2 | C-2A:G+2T | 56.7 ± 16.1 |
| −2 | C-2G | 55.7 ± 3.5 |
| +2 | G+2C | 58.3 ± 6.7 |
| −/+15 | T-15C:C+15G | 127.7 ± 24.2 |
| −15 | T-15C | 68.3 ± 5.8 |
| +15 | C+15G | 72.7 ± 5.5 |
| −/+16 | G-16T:G+16A | 61.3 ± 12.1 |
| −16 | G-16T | 66.0 ± 5.3 |
| +16 | G+16A | 53.3 ± 6.1 |
| −/+18 | G-18C:A+18G | 189.7 ± 30.7 |
| −18 | G-18C | 194.7 ± 17.1 |
| +18 | A+18G | 186.3 ± 19.8 |
| Partially symmetrical site | 2L(+12 - +18) | 61.7 ± 5.7 |
aMeans and SDs were calculated from the integrase concentrations required for 50% binding from three separate binding experiments.
Cleavage by integrase of attB sites with mutations at −/+2 is severely inhibited
The formation of both the cleaved covalent intermediate and the synapse can be observed in a recombination assay using a radiolabelled attB or attP site, a cold partner att site and integrase (27). These assays are performed in a buffer that is sub-optimal for recombination (binding buffer) that enriches for synaptic complexes and the cleaved covalent complex compared with standard recombination conditions (27). attP was labelled with [α−32P] dCTP, mixed with cold wild-type or mutant attB sites and integrase and run in a non-denaturing PAGE gel (Figure 4A, left panel). Compared to wild-type attB, sites with −/+2 changes (C-2G:G+2C and C-2A:G+2T) showed an accumulation of synapse with almost undetectable cleaved covalent complex or product formed (Figure 4A, left panel). Treatment of the recombination intermediates with the protease, subtilisin showed a small amount of cleaved probe with C-2G:G+2C but this was undetectable with C-2A:G+2T (Figure 4B). Subtilisin treatment of reactions containing wild-type attB clearly revealed the two recombination products attL and attR but these were not visible with C-2A:G+2T and barely visible with C-2G:G+2C (Figure 4B). As seen in the recombination assay reverting one of the two mutations in C-2G:G+2C back to the wild-type sequence was sufficient to regain activity similar to the wild type attB site (C-2G or G+2C in Figure 4A, left panel). The catalytically inactive integrase mutant (S12A) was able to bind to C-2G:G+2C, C-2A:G+2T, C-2G and G+2C normally to form a synaptic complex indistinguishable from the wild type attB site (Figure 4A, right panel). Experiments in which the labelled probes were attB or the −/+2 mutant derivatives and unlabelled attP was used to supershift the complexes showed similar results, i.e. very little cleavage of the −/+2 mutant attB was observed (Supplementary Data—Figure S2). These data indicate that attB sites with a double substitution at −/+ 2 are able to generate a stable synapse but are severely defective in cleavage of the substrates.
Figure 4.

Synapse assays with wild-type and mutant attB sites. Panel (A) Radiolabelled attP was incubated with wild-type (left panel) or the catalytically inactive integrase, S12A, (right panel) and a cold partner fragment containing wild-type attB (wt) or the indicated mutant attB sites. The arrows show the positions of the synapse containing the radiolabelled substrate, the cold partner fragment and integrase (Int:synapse attP/B), the covalently linked cleaved substrate (Int:cleaved attP/B), the shifted and free products (Int:attL/R and attL/R, respectively) and the positions of the attP bound only to integrase (two complexes labelled Int:attP) or free (attP). Panel (B) The protease subtilisin was used to reveal the extent of cleavage of attB sites and the products formed during the synapse assay. Arrows show the positions of the products, attL and attR, the radiolabelled substrate and attB. The smear of radioactivity migrating faster than the free probe results from subtilisin treated cleaved covalently linked complexes.
The stability of the synapse is reduced in attB sites with mutations at −/+15, −/+16, −15 and −16
The attB sites with mutations at −/+15 and −/+16 that were defective in the standard recombination assay did not appear to be defective in the radioactive assay to detect intermediates (Figure 4A, left panel). The amounts of cleaved covalent intermediate and shifted attL/attR products were indistinguishable from the reaction with the wild-type attB site (Figure 4A, left panel). The only observable difference was in the amount of synapse, which was reduced in the reactions with the most defective sites i.e. T-15C:C+15G, T-15G, G-16T:G+16A and G-16T and the appearance of some free product. These differences were also observed when the attB sites were labelled and incubated with integrase and cold attP (Supplementary Data—Figure S2). When the S12A catalytically inactive integrase mutant was used, there was a small reduction in accumulation of the synaptic complex with T-15C:C+15G, T-15G, G-16T:G+16A and G-16T compared with wild-type attB (Figure 4A, right panel).
The inconsistency whereby mutants T-15C:C+15G and G-16T:G+16A were inactive in the recombination assay but active in the assay for intermediates was addressed. As the standard recombination assay is performed over 1 h and the synapse assay is over 2 h, time courses were performed for each assay. In the standard recombination assay, products were observed in 2 and 3 h with mutants T-15C:C+15G and G-16T:G+16A (Figure 5A). Using the more sensitive radioactive assay, cleaved substrates, T-15C:C+15G and G-16T:G+16A, and their recombinant products started to appear at 30 min of incubation and accumulated further over the next 30 min whereas with the wild-type attB, most of the substrate had been converted to intermediates or products at 15 min. Even after 60 min, less attP in the presence of T-15C:C+15G or G-16T:G+16A was converted to product compared to attP in the presence of wild-type attB (Figure 5B). Thus both the mutants T-15C:C+15G and G-16T:G+16A could undergo recombination but the reaction is considerably slower than that for the wild-type attB site. As there is a consistently reduced level of synaptic complex observed with these mutant attB sites, it is likely that changes in attB at −/+15 and −/+16 both result in an unstable synapse that explains the slow rate of recombination.
Figure 5.

The rate of recombination with mutant attB sites T-15C:C+15G and G-16T:G+16A is greatly reduced. Panel (A) shows the appearance of products from recombination assays using T-15C:C+15G and G-16T:G+16A as substrates after prolonged incubation. Plasmids encoding the wild-type attB (wt) or the indicated attB mutants were incubated with pRT702 (attP) for 1, 2 or 3 h at 30°C and then the products analysed by restriction and agarose gel electrophoresis. After 2 and 3 h some product (attL) is visible in the lanes containing the −/+15 and −/+16 mutations. Panel (B) shows the time-dependent appearance of recombination intermediates when wild-type attB (wt) was used compared to C-2A:G+2T, T-15C:C+15G or G-16T:G+16A. The −/+2 mutant site rapidly forms a synapse (Int:synapse attP/B) and thereafter the reaction is blocked. The −/+15 and −/+16 attB sites slowly accumulated the cleaved intermediate (Int:cleaved attP/B) and some shifted product complexes (Int:attL/R). The remaining complexes on the gel are as described in Figure 4.
We reasoned that altered recombination conditions might partially suppress the defect in T-15C:C+15G and G-16T:G+16A by stabilizing the putative protein–protein interface. Recombination was observed when the NaCl concentration was increased to 500 mM or 1 M in recombination buffer (Figure 6A). However, increasing the concentration of NaCl did not increase the amount of synaptic complex observed with these mutant attB sites (Figure 6B). Indeed at 1 M NaCl there was a reduction in the level of synapse observed with the mutants at −/+15 and −/+16 and a slight reduction in the affinity for the attB site by integrase (Figure 6B and C).
Figure 6.

High NaCl concentrations can partially suppress the recombination defective phenoytpe of the mutations. Panel (A) shows the appearance of products from recombination assays using T-15C:C+15G, G-16T:G+16A, G-16T and T-15C as substrates after incubation in either 500 mM or 1 M NaCl. Plasmids encoding the wild-type attB (wt), or the above mutants were incubated with pRT702 (attP) in recombination buffer adjusted to 100 mM, 500 mM or 1 M NaCl. After digesting with HindIII the DNA was separated in an agarose gel. The appearance of the 5435 bp fragment encoding attL is indicative of recombination. Panel (B) shows the synapse assays using the wild-type attB (wt), C-2A:G+2T, T-15C:C+15G and G-16T:G+16A under different NaCl conditions with wild type (left panel) or S12A integrase (right panel) with labelled attP. The complexes are annotated as described in Figure 4. −/+15 and −/+16 mutant attB sites accumulated both the cleaved complex (Int:cleaved attP/B), the shifted products (Int:attL/R) and released some free product (attL/R) with 500 mM and 1 M NaCl with the wild-type integrase. The synapse however as indicated using the S12A integrase did not become more abundant in high NaCl buffer, if anything it reduced. Panel (C) shows that the binding affinity of attB sites for integrase in the presence of different NaCl concentrations. Radiolabelled attB sites were incubated in binding buffer containing 50 mM, 500 mM or 1 M NaCl. Integrase was added at 66 nM. The shifted attB complexes are indicated by arrows. Only at 1 M NaCl, there was a slight increase in the free DNA for all four attB sites.
DISCUSSION
The interactions between ϕC31 integrase and its attachment sites are critical in determining the directionality of recombination. In vitro integrase only recombines attB and attP to form the hybrid products, attL and attR. We have shown previously that, in vitro, integrase selectively brings attP and attB together to form the synapse and no other combination of sites forms a stable synapse under these conditions (27). These observations have led to the proposal that integrase adopts specific conformations when bound to attP or attB that permit formation of the protein:protein interface required for stable synapsis (27,29). Here we showed that mutations in attB can significantly affect the ability of integrase to form a stable synapse or to cleave the substrates. These perturbations in the reaction are likely to be due to the absence of important interactions between integrase and attB and could be indicative of ‘non-permissive’ conformations of integrase that block recombination at these different stages.
The mutations at −/+2 in attB showed a failure to cleave the DNA but these substrates could still form a stable synapse (Figures 4–6). These data show clearly that there is a post-synaptic activation step required for recombination by ϕC31 integrase. This activation step depends on an interaction that has been disrupted in the attB −/+2 mutants, C-2G:G+2C or C-2A:G+2T. The nature of the interaction is not known but could be a specific base-pair contact or a DNA conformation that is recognized. The block in DNA cleavage occurred in both the mutant attB sites themselves and in the wild-type attP sites (Figures 4–6 and Figure S2). This behaviour is consistent with concerted DNA cleavage in the reaction with wild-type recombination sites. Possibly the block in cleavage in reactions containing the −/+2 mutant sites could be due to failure to undergo a conformational change in the whole synaptic complex which would normally lead to cleavage. Alternatively the DNA conformation of the mutant sites prevents the catalytic sites gaining access to the scissile phosphate. As reversion of just one of the bases from the double mutant back to the wild type was sufficient to regain most of the attB activity it would seem that activation only requires a ‘correct’ interaction at one half-site of attB.
The mutants T-15C:C+15G and G-16T:G+16A were able to recombine but at a slow rate compared to wild-type attB (Figure 5). There was a consistent reduction in the amount of synapse observed during recombination with these mutants suggesting that the synaptic complex was unstable (Figures 4 and 5). Raising the concentration of NaCl partially suppressed the defect in recombination with the −/+15 and −/+16 mutants but it is not clear which step was affected by NaCl (Figure 6). The stability of the synapse did not increase in the presence of a higher concentration of NaCl, if anything the binding affinity and the level of synapse was reduced at 1 M NaCl (Figure 6). Despite this, suppression was still observed suggesting that high NaCl activates or stabilizes an event later in the recombination pathway. The single point mutants at –15 and –16 were sufficient to severely affect recombination while mutations at +15 or +16 had a lesser effect (Table 1, Figures 2, 4–6). Thus the single mutations at positions –15 and –16 accounted for most of the defect in the −/+15 and −/+16 double mutants. These data argue that there could be a specific interaction between the B arm and integrase that contributes significantly to the activity of the attB site. The partial symmetrization of the attB sites (with either the sequence from the B arm [2L (+12 to +18)] or with that from the B′ arm [2R (−12 to −18)]; Figure 2, panel H) showed that the B arm was indeed more active than the B′ arm. However, it is known from previous work that the attB and attP sites act with integrase in a functionally symmetrical manner as integrase does not control the relative orientation of the sites when they come together at synapsis (28). Thus the interactions by each subunit of integrase bound to each arm of attB are not independent of each other and we propose that a specific integrase conformation that results from the –15, −16 interactions in the B arm is communicated through both subunits.
These conclusions can be combined with information from other large serine recombinases and the resolvases to generate a model that focuses on substrate recognition and formation of the synapse by integrase (adapted from that published previously for Bxb1 integrase, 26; Figure 7). In the resolvases, the DNA is contacted in the minor groove in the centre of each binding site and through specific contacts in the major groove towards the outer flank of the site via the C-terminal DNA binding domain (24,25). The geometry of DNA-binding is such that the C-terminal domain of resolvase extends around the DNA and contacts on the opposite side of the DNA to the catalytic serine (25). As in Bxb1 and TnpX, ϕC31 integrase has a proteolytically sensitive site between the N and C terminal domains (K152, unpublished data)(26,30,46). Moreover, the C-terminal domains of Bxb1 and TnpX have been shown previously to be capable of binding specifically to DNA (26,30,46). Thus we propose that the C-terminal domains interact with the outer flanks of the att sites, that these interactions determine the conformations of integrase bound to each site and therefore whether they are compatible for synapsis. In attB this information is ‘read’ at least in part from –15 and –16 where disruption of this interaction disables the ability of integrase to form a stable synapse (Figures 4–6). The model predicts that there is communication between the putative DNA-binding motifs in the C-terminal domain and the regions of integrase that generate the protein-protein interface for synapsis. We currently envisage this communication as an allosteric switch mediated by conformational changes. In γδ resolvase, the synaptic interface is located at the DNA distal surface of the catalytic domain and it is likely that the serine integrases use the equivalent of this interface for synapsis, although it is possible that the C-terminal domain may also have a role in synapsis. After synapsis an activation step is required for DNA cleavage and in attB this depends on the base pairs at position −/+2. In attB only one of the −/+2 bases needs to be wild type for activity and this can be either on the B or B′ arm. Given the proximity of −/+2 to the scissile phosphate, position 2 is more likely to interact with the catalytic domain than with the C-terminal domain. As in resolvase there may be significant conformation changes that occur with activation of recombination (34,37).
Figure 7.

Model for the mechanism of integrase. The integrase subunits are shown with a small N-terminal (catalytic) domain through which the subunits may dimerize (26) and a large C-terminal domain that we propose recognizes the sequences in the outer flanks of the recombination sites. These recognition events, specifically −15 and −16 (annotated as blue bars) in attB, lead to an ‘induced fit’ or stabilization of a specific conformation of integrase that enables synapsis with integrase bound to attP. Different conformations of integrase bound to either attB or attP are shown as different colours. The synaptic interface via the N-terminal catalytic domains is indicated based on the resolvase precedent; there is no evidence to indicate that the C-terminal domains could also participate in a synaptic interface. Mutations at positions −15 or −16 (such as in T-15C:C+15G, G-16T:G+16A), T-15C or G-16T do not induce the conformation of integrase that can form a stable synapse with attP so the rate of reaction decreases (thin arrow). Mutations at −/+2 in attB (red bars) are severely inhibited in cleavage but are capable of forming a stable synapse. These mutants indicate that the formation of the synaptic complex is followed by a well-defined activation step that results in concerted DNA cleavage. After strand exchange integrase is bound to the hybrid sites and adopts a conformation that cannot synapse attL and attR. The putative tetrameric complex rapidly dissociates to binary complexes containing integrase and either attL or attR. See text for more details.
The data presented here provides a source of information that could be used for the design of alternative attB sites for genome engineering using ϕC31 integrase. Indeed we have already used this information to create a non-methylatable attB site for use in vertebrate cell lines (4). In this site, attBm, the CpG steps have been replaced with bases that we have shown here were neutral with respect to recombination activity. It is noteworthy that the positions in attB that are critical for recombination other than position 2 i.e. positions 15, 16 were not highlighted as being particularly preferred in the pseudo-attB sites (Figure 1). However all of the pseudo-attB sequences have either the wild-type C at −2 or a wild-type G at +2 (Figure 1) (41,43). It is also noticeable that the regions where there are most identities between attB and attP are not particularly sensitive to mutation (Figure 1). A plausible explanation for both these observations is that the 24 bp of att site DNA from about position 11 to the crossover site is a core sequence that integrase binds to specifically. We propose that the role of positions 15 and 16 in attB revealed by this study is to greatly enhance the efficiency of the reaction and effectively discriminate between the pseudo-sites and the cognate attB site. It is envisaged that this data and a similar analysis with attP will enable an understanding of the optimal sequences to target for continued application of ϕC31 integrase.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We are grateful for comments on this manuscript from Paul Rowley and Paul Hoskisson. M.G was funded by a scholarship from the University of Aberdeen. This work was funded by the Biotechnology and Biological Sciences Research Council, UK. Funding to pay the open access publication charges for this article was provided by the Biotechnology and Biological Sciences Research Council, UK.
Conflict of interest statement. None declared.
REFERENCES
- 1.Bateman JR, Lee AM, Wu CT. Site-specific transformation of Drosophila via ϕC31 integrase-mediated cassette exchange. Genetics. 2006;173:769–777. doi: 10.1534/genetics.106.056945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andreas S, Schwenk F, Kuter-Luks B, Faust N, Kuhn R. Enhanced efficiency through nuclear localization signal fusion on phage ϕC31-integrase: activity comparison with Cre and FLPe recombinase in mammalian cells. Nucleic Acids Res. 2002;30:2299–2306. doi: 10.1093/nar/30.11.2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Belteki G, Gertsenstein M, Ow DW, Nagy A. Site-specific cassette exchange and germline transmission with mouse ES cells expressing ϕC31 integrase. Nat Biotechnol. 2003;21:321–324. doi: 10.1038/nbt787. [DOI] [PubMed] [Google Scholar]
- 4.Dafhnis-Calas F, Xu Z, Haines S, Malla SK, Smith MCM, Brown WR. Iterative in vivo assembly of large and complex transgenes by combining the activities of ϕC31 integrase and Cre recombinase. Nucleic Acids Res. 2005;33:e189. doi: 10.1093/nar/gni192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khan MS, Khalid AM, KMalik KA. Phage ϕC31 integrase: a new tool in plastid genome engineering. Trends in Plant Science. 2005;10:1–3. doi: 10.1016/j.tplants.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 6.Malla S, Dafhnis-Calas F, Brookfield JF, Smith MCM, Brown WR. Rearranging the centromere of the human Y chromosome with ϕC31 integrase. Nucleic Acids Res. 2005;33:6101–6113. doi: 10.1093/nar/gki922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thomason LC, Calendar R, Ow DW. Gene insertion and replacement in Schizosaccharomyces pombe mediated by the Streptomyces bacteriophage ϕC31 site-specific recombination system. Mol. Gen. Genom. 2001;265:1031–1038. doi: 10.1007/s004380100498. [DOI] [PubMed] [Google Scholar]
- 8.Nkrumah LJ, Muhle RA, Moura PA, Ghosh P, Hatfull GF, Jacobs WR, Jr, Fidock DA. Efficient site-specific integration in Plasmodium falciparum chromosomes mediated by mycobacteriophage Bxb1 integrase. Nat. Methods. 2006;3:615–621. doi: 10.1038/nmeth904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thyagarajan B, Olivares EC, Hollis RP, Ginsburg DS, Calos MP. Site-specific genomic integration in mammalian cells mediated by phage ϕC31 integrase. Mol Cell Biol. 2001;21:3926–3934. doi: 10.1128/MCB.21.12.3926-3934.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Groth AC, Olivares EC, Thyagarajan B, Calos MP. A phage integrase directs efficient site-specific integration in human cells. Proc. Natl Acad. Sci. USA. 2000;97:5995–6000. doi: 10.1073/pnas.090527097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Olivares EC, Hollis RP, Chalberg TW, Meuse L, Kay MA, Calos MP. Site-specific genomic integration produces therapeutic Factor IX levels in mice. Nat. Biotechnol. 2002;20:1124–1128. doi: 10.1038/nbt753. [DOI] [PubMed] [Google Scholar]
- 12.Held PK, Olivares EC, Aguilar CP, Finegold M, Calos MP, Grompe M. In vivo correction of murine hereditary tyrosinemia type I by ϕC31 integrase-mediated gene delivery. Mol. Ther. 2005;11:399–408. doi: 10.1016/j.ymthe.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 13.Portlock JL, Keravala A, Bertoni C, Lee S, Rando TA, Calos MP. Long-term increase in mVEGF164 in mouse hindlimb muscle mediated by phage ϕC31 integrase after nonviral DNA delivery. Hum. Gene Ther. 2006;17:871–876. doi: 10.1089/hum.2006.17.871. [DOI] [PubMed] [Google Scholar]
- 14.Keravala A, Portlock JL, Nash JA, Vitrant DG, Robbins PD, Calos MP. ϕC31 integrase mediates integration in cultured synovial cells and enhances gene expression in rabbit joints. J. Gene Med. 2006;8:1008–1017. doi: 10.1002/jgm.928. [DOI] [PubMed] [Google Scholar]
- 15.Bertoni C, Jarrahian S, Wheeler TM, Li Y, Olivares EC, Calos MP, Rando TA. Enhancement of plasmid-mediated gene therapy for muscular dystrophy by directed plasmid integration. Proc. Natl. Acad. Sci. USA. 2006;103:419–424. doi: 10.1073/pnas.0504505102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thorpe HM, Smith MCM. In vitro site-specific integration of bacteriophage DNA catalyzed by a recombinase of the resolvase/invertase family. Proc. Natl Acad. Sci. USA. 1998;95:5505–5510. doi: 10.1073/pnas.95.10.5505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghosh P, Kim AI, Hatfull GF. The orientation of mycobacteriophage Bxb1 integration is solely dependent on the central dinucleotide of attP and attB. Mol. Cell. 2003;12:1101–1111. doi: 10.1016/s1097-2765(03)00444-1. [DOI] [PubMed] [Google Scholar]
- 18.Azaro MA, Landy A. In: Mobile DNA II. Craig NL, Craigie R, Gellert M, Lambowitz AM, editors. Washington D.C.: ASM Press; 2002. pp. 118–148. [Google Scholar]
- 19.Ghosh P, Wasil LR, Hatfull GF. Control of phage Bxb1 excision by a novel recombination directionality factor. PLoS Biol. 2006;4:e186. doi: 10.1371/journal.pbio.0040186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith MCM, Thorpe HM. Diversity in the serine recombinases. Mol. Microbiol. 2002;44:299–307. doi: 10.1046/j.1365-2958.2002.02891.x. [DOI] [PubMed] [Google Scholar]
- 21.Craig NL. In: Mobile DNA II. Craig NL, Craigie R, Gellert M, Lambowitz AM, editors. Washington DC: American Society for Microbiology; 2002. pp. 3–11. [Google Scholar]
- 22.Biswas T, Aihara H, Radman-Livaja M, Filman D, Landy A, Ellenberger T. A structural basis for allosteric control of DNA recombination by lambda integrase. Nature. 2005;435:1059–1066. doi: 10.1038/nature03657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Radman-Livaja M, Biswas T, Ellenberger T, Landy A, Aihara H. DNA arms do the legwork to ensure the directionality of lambda site-specific recombination. Curr. Opin. Struct. Biol. 2006;16:42–50. doi: 10.1016/j.sbi.2005.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grindley NDF. In: Mobile DNA II. Craig NL, Craigie R, Gellert M, Lambowitz AM, editors. Washington: ASM Press; 2002. pp. 272–302. [Google Scholar]
- 25.Yang W, Steitz TA. Crystal-structure of the site-specific recombinase gamma-delta resolvase complexed with a 34bp cleavage site. Cell. 1995;82:193–207. doi: 10.1016/0092-8674(95)90307-0. [DOI] [PubMed] [Google Scholar]
- 26.Ghosh P, Pannunzio NR, Hatfull GF. Synapsis in phage Bxb1 integration: selection mechanism for the correct pair of recombination sites. J. Mol. Biol. 2005;349:331–348. doi: 10.1016/j.jmb.2005.03.043. [DOI] [PubMed] [Google Scholar]
- 27.Smith MCA, Till R, Brady K, Soultanas P, Thorpe H, Smith MCM. Synapsis and DNA cleavage in ϕC31 integrase-mediated site-specific recombination. Nucleic Acids Res. 2004;32:2607–2617. doi: 10.1093/nar/gkh538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith MCA, Till R, Smith MCM. Switching the polarity of a bacteriophage integration system. Mol. Microbiol. 2004;51:1719–1728. doi: 10.1111/j.1365-2958.2003.03942.x. [DOI] [PubMed] [Google Scholar]
- 29.Thorpe HM, Wilson SE, Smith MCM. Control of directionality in the site-specific recombination system of the Streptomyces phage ϕC31. Mol. Microbiol. 2000;38:232–241. doi: 10.1046/j.1365-2958.2000.02142.x. [DOI] [PubMed] [Google Scholar]
- 30.Adams V, Lucet IS, Tynan FE, Chiarezza M, Howarth PM, Kim J, Rossjohn J, Lyras D, Rood JI. Two distinct regions of the large serine recombinase TnpX are required for DNA binding and biological function. Mol. Microbiol. 2006;60:591–601. doi: 10.1111/j.1365-2958.2006.05120.x. [DOI] [PubMed] [Google Scholar]
- 31.Crellin PK, Rood JI. The resolvase/invertase domain of the site-specific recombinase TnpX is functional and recognizes a target sequence that resembles the junction of the circular form of the Clostridium perfringens transposon Tn4451. J. Bacteriol. 1997;179:5148–5156. doi: 10.1128/jb.179.16.5148-5156.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang H, Mullany P. The large resolvase TndX is required and sufficient for integration and excision of derivatives of the novel conjugative transposon Tn5397. J. Bacteriol. 2000;182:6577–6583. doi: 10.1128/jb.182.23.6577-6583.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson RC. In: Mobile DNA II. Craig NL, Craigie R, Gellert M, Lambowitz AM, editors. Washington DC: ASM Press; 2002. pp. 230–271. [Google Scholar]
- 34.Li W, Kamtekar S, Xiong Y, Sarkis GJ, Grindley NDF, Steitz TA. Structure of a synaptic gammadelta resolvase tetramer covalently linked to two cleaved DNAs. Science. 2005;309:1210–1215. doi: 10.1126/science.1112064. [DOI] [PubMed] [Google Scholar]
- 35.Nollmann M, He J, Byron O, Stark WM. Solution structure of the Tn3 resolvase-crossover site synaptic complex. Mol. Cell. 2004;16:127–137. doi: 10.1016/j.molcel.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 36.Stark WM, Boocock MR, Sherratt DJ. Catalysis by site-specific recombinases. Trends Genet. 1992;8:432–439. [PubMed] [Google Scholar]
- 37.Kamtekar S, Ho RS, Cocco MJ, Li W, Wenwieser SV, Boocock MR, Grindley NDF, Steitz TA. Implications of structures of synaptic tetramers of gamma delta resolvase for the mechanism of recombination. Proc. Natl. Acad. Sci. USA. 2006;103:10642–10647. doi: 10.1073/pnas.0604062103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stark WM, Grindley NDF, Hatfull GF, Boocock MR. Resolvase-catalyzed reactions between sites differing in the central dinucleotide of subsite-I. EMBO J. 1991;10:3541–3548. doi: 10.1002/j.1460-2075.1991.tb04918.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuhstoss S, Rao RN. Analysis of the integration function of the streptomycete bacteriophage ϕc31. J. Mol. Biol. 1991;222:897–908. doi: 10.1016/0022-2836(91)90584-s. [DOI] [PubMed] [Google Scholar]
- 40.Breuner A, Brondsted L, Hammer K. Resolvase-like recombination performed by the TP901-1 integrase. Microbiology. 2001;147:2051–2063. doi: 10.1099/00221287-147-8-2051. [DOI] [PubMed] [Google Scholar]
- 41.Combes P, Till R, Bee S, Smith MCM. The streptomyces genome contains multiple pseudo-attB sites for the (ϕ)C31-encoded site-specific recombination system. J. Bacteriol. 2002;184:5746–5752. doi: 10.1128/JB.184.20.5746-5752.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chalberg TW, Portlock JL, Olivares EC, Thyagarajan B, Kirby PJ, Hillman RT, Hoelters J, Calos MP. Integration specificity of phage ϕC31 integrase in the human genome. J. Mol. Biol. 2006;357:28–48. doi: 10.1016/j.jmb.2005.11.098. [DOI] [PubMed] [Google Scholar]
- 43.Murry J, Sassetti CM, Moreira J, Lane J, Rubin EJ. A new site-specific integration system for mycobacteria. Tuberculosis (Edinb) 2005;85:317–323. doi: 10.1016/j.tube.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 44.Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. 3rd edn. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2001. edn. [Google Scholar]
- 45.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 46.Lucet IS, Tynan FE, Adams V, Rossjohn J, Lyras D, Rood JI. Identification of the structural and functional domains of the large serine recombinase TnpX from Clostridium perfringens. J. Biol. Chem. 2005;280:2503–2511. doi: 10.1074/jbc.M409702200. [DOI] [PubMed] [Google Scholar]