

Abstract
A 192-member library of N,N′-disubstituted urea inhibitors was synthesized by a solid-phase method. The ureas were tested for their inhibitory activities against recombinant human soluble epoxide hydrolase. Simple carbocyclic or para/meta-substituted phenyl groups showed inhibition potencies that were equal to or greater than adamantane-based sEH inhibitors, while the presence of bulky or ionizable groups close to the urea group dramatically decreased their activities.
Keywords: Soluble epoxide hydrolase, Urea, Hypertension, Anti-inflammation
Soluble epoxide hydrolase (sEH, EC 3.3.2.3) is involved in the metabolism of endogenously derived fatty acid epoxides, such as arachidonic acid, linoleic acid, and other lipid epoxides.1 Epoxyeicosatrienoic acids (EETs), the cytochrome P450 epoxygenase products of arachidonic acid, act at vascular, renal, and cardiac levels of blood pressure regulation. Recent studies have showed that EETs are involved in antihypertensive e3ects as an endothelium-derived hyperpolarizing factor (EDHF) that mediates vasodilation by activating Ca2+-activated K+ channels in smooth muscle cells.1,2 TRPV4, a Ca2+ entry channel belonging to the vanilloid subfamily of the transient receptor potential (TRP) channels, acts as an extracellular receptor for EETs.3 The sEH enzyme catalytically hydrolyzes EETs into dihydroxyeicosatrienoic acids (DHETs) which show reduced biological activity.1 We have demonstrated that sEH inhibition significantly reduces the blood pressure of the spontaneous hypertensive rats (SHRs) and angiotensin II-induced hypertensive rats.4,5 EETs also possess anti-inflammatory properties in endothelial cells by inhibiting the expression of tumor necrosis factor-α (TNF-α)-induced vascular cell adhesion molecule-1 (VCAM-1) which is a pro-atherogenic mediator or by activating PPARγ which suppresses the NF-κB-mediated expression of molecules, that is, VCAM-1, intercellular adhesion molecule-1 (ICAM-1), and endothelin-1.6 In addition, diols derived from epoxy-linoleate (leukotoxin) via sEH hydrolysis perturb membrane permeability and calcium homeostasis, which results in inflammation.7 These results suggest that sEH inhibition may represent a novel approach for the treatment of hypertension and inflammatory diseases.
Earlier inhibitors developed for sEH were substrate-like compounds, such as chalcone oxides.8 More recently, we have explored urea, amide, and carbamate-based transition state analog inhibitors of sEH.9 Several recent studies have emphasized adamantane-based urea compounds due to their ease of synthesis, high potency, and ease of analysis. The recent development of fluorescent substrates10,11 for sEH which both discriminate among low nanomolar and picomolar inhibitors and facilitate high-throughput analysis makes the development of inhibitors through a combinatorial approach attractive. In this report we illustrate this combinatorial approach in the further exploration of the role of R1 (see compound D, Scheme 1) as it influences the potency of sEH inhibitors.
Scheme 1.
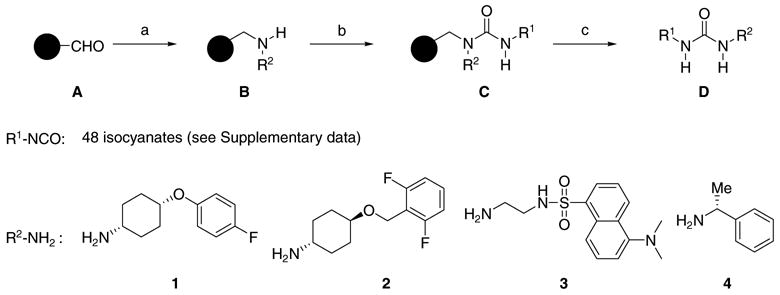
Reagents and conditions: (a) i—R2-NH2, TEOF, THF, rt, 8 h; ii—1 M NaBH3CN in THF, AcOH, THF, rt, 4 h; (b) R1-NCO, Et3N, CH2Cl2, rt, 1 d; (c) 1% TFA in CH2Cl2, rt, 4 h.
A focused library of 192-ureas was designed in order to optimize R1 group as well as to avoid the tedious purification step caused by the main contaminant during urea formation, a symmetrical urea (R1NHC(=O)NHR1, by-product from the step b in Scheme 1), which also has significant inhibitory activity against the human sEH enzyme.
The solid-phase procedure was set up using a previously reported urea formation method starting from the commercially available acid-labile formylindole resin A.12
The reactions were performed on an IRORI AccuTag™ Combinatorial Chemistry System13 according to the general reaction pathway outlined in Scheme 1. Four amines 1–4 and commercially available 48 isocyanates were used to construct the 192-member urea library.
The four amines 1–4 were selected based on the following criteria. First, selection was based on inhibitory activities of the corresponding adamantane-based ureas (see IC50 values in Table 1). Second, three amines are relatively large (amine 1, 2, and 3), thus driving their orientation in the catalytic tunnel, while one amine is relatively small (amine 4).14 Finally, amines containing a benzene ring were chosen in order to evaluate the purity of the products by UV detection on HPLC.
Table 1.
Inhibitory activities of the adamantane-based ureas 1{1}, 2{1}, 3{1}, and 4{1} derived from the amines 1 to 4
| Compounda | R2-NH2 | IC50b(nM) | % inhibitionc |
|---|---|---|---|
| 1{1} | 1 | 0.5 | 86 ± 9 |
| 2{1} | 2 | 0.5 | 75 ± 12 |
| 3{1} | 3 | 30 | 19 ± 7 |
| 4{1} | 4 | 100 | 10 ± 3 |
The notation for the compound number: amine number {isocyanate number}, for example, 2{1} indicates a compound made by the combination of amine 2 with isocyanate 1.
As determined via a kinetic fluorescent assay.10
Determined via an end-point fluorescent assay, results are means ± SD of three separate experiments.11
Based on the previous SAR analysis of the chalcone oxide derivatives8 and other urea inhibitors,15 R1 groups in the isocyanates were selected as follows: (1) an adamantane as a control, (2) simple carbocyclic rings, (3) para-, meta-, and ortho-mono-substituted phenyl rings, (4) sterically hindered groups, such as tert-butyl or ortho-di-substituted phenyl rings, (5) 1- and 2-naphthyl group, and (6) phenylalkyl groups having various lengths of alkyl spacers.16 The purity was assessed by HPLC and all mass spectra were consistent with the anticipated product structure. Out of the total 192-member library, 181 compounds having over 90% purity were tested for their inhibitory activity at a 100 nM concentration using recombinant human sEH by an endpoint assay.11,17
Overall, compounds derived from amines 1 and 2 showed good inhibitory activity compared to those made from amines 3 and 4 (see Table 1 in Supplementary data). These results strongly suggest that the tested compounds orient in the same direction as the adamantane-based inhibitors do when they bind at the active site, which can be predicted from the recent X-ray crystal structure of human sEH with urea-based inhibitors.14
In order to determine the influence on the inhibitory activity by R1 group itself, compounds having similar activity compared to the corresponding adamantane-based ureas (1{1}, 2{1}, 3{1}, and 4{1}) are highlighted in black squares in Figure 1.
Figure 1.

Relative inhibition of the 192-member urea library at 100 nM concentration compared to the corresponding adamantane-based ureas 1{1}, 2{1}, 3{1}, and 4{1}, respectively. Black square: equal to or greater than 80% of inhibition obtained when R1 is an adamantyl group; grey square: less than 80% of the inhibition obtained when R1 is an adamantyl group; white square: compounds were not tested due to the poor purity (less than 90%).
From the illustration in Figure 1, it is apparent that carbocyclic groups ({2–6}) and para-substituted phenyl groups ({8–17}) showed the best results, regardless of R2 group. There was no strong correlation between the inhibition and the σ values. It is likely that the binding site in which the R1 group resides is a narrow, hydrophobic tunnel. This is further supported by the fact that overall para-substituted phenyl groups are better than meta-substituted phenyl groups ({18–26}), the 2-naphthyl group ({45}) is better than the 1-naphthyl group ({44}), and the phenyl groups having longer alkyl spacers ({47–48}) gave the better activities. Meanwhile, sterically hindered derivatives, such as inhibitors from the isocyanates ({27–44 and 46}), showed poor inhibition. Based on these results, poor activities most likely came from steric e3ects of groups on R1. In other words, the adamantyl group, which is generally considered as a bulky group, might be the marginal biggest group as the R1.
In addition, as seen in Figure 2, ureas derived from amines 1 and 2 generally show similar inhibition; one can expect such results because of the similar IC50 values obtained for their corresponding adamantane-based ureas (Table 1). However, for ureas having sterically hindered groups on R1 (R1 = {27–44}), ureas derived from amine 1 showed overall much better inhibition. It is likely that a repulsion caused by sterically hindered groups on R1 influences the positioning of the R2 group in the active site tunnel. Such altered positioning seems to result in the further destabilization e3ect for ureas derived from amine 2 than for those from amine 1.
Figure 2.
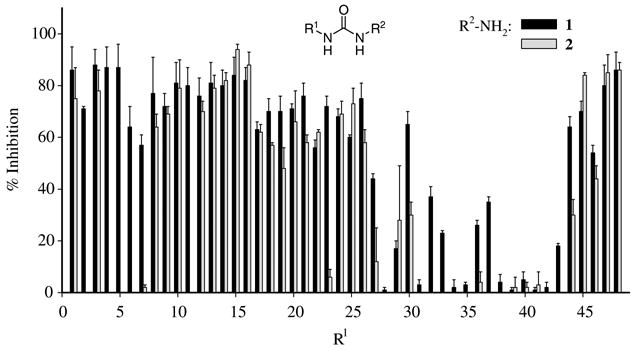
Percent inhibition of ureas derived from amines 1 and 2 at 100 nM concentration. Note, ureas 2{2}, 2{4}, 2{5}, 2{6}, 2{11}, and 2{43} were not tested for inhibition because of their low purity.
To validate our high-throughput screening results, a subset of compounds having high percent inhibition, that is, 1{4}, 1{12}, 1{13}, 1{14}, 1{15}, 1{16} and having low percent inhibition, that is, 1{34}, 1{38}, 1{39}, 1{40}, were resynthesized. Their IC50 values were then determined using a continuous fluorescent assay.10 In general, data from both methods showed good correlation as shown in Table 2. These results confirm that sterically less hindered para- or meta-substituted phenyl groups, simple non-rigid carbocyclic groups, 2-naphthyl, and phenyl alkyl groups having at least an ethyl spacer are good alternatives to the adamantyl group. These results are also consistent with our previous SAR of chalcone oxide derivatives and other urea-based inhibitors.8,15
Table 2.
| Compound | IC50a(nM) | Inhibitionb(%) | R1 | Mp (°C) |
|---|---|---|---|---|
| 1{1} | 0.5 ± 0.1 | 86 ± 9 | Adamantyl | 242–245 |
| 1{4} | 0.5 ± 0.1 | 87 ± 8 | c-Hep | 158–167 |
| 1{12} | 1.0 ± 0.1 | 76 ± 7 | 4-I–Ph– | 198–201 |
| 1{13} | 0.7 ± 0.1 | 81 ± 8 | 4-Cl–Ph | 177–182 |
| 1{14} | 0.6 ± 0.1 | 80 ± 6 | 4-Br–Ph | 192–194 |
| 1{15} | 0.9 ± 0.1 | 84 ± 7 | 4-OCF3–Ph | 157–158 |
| 1{16} | 1.2 ± 0.2 | 82 ± 5 | 4-CF3–Ph | 171–174 |
| 1{34} | 100 ± 5 | 2 ± 3 | 2-OCF3–Ph | 158–160 |
| 1{38} | 1200 ± 100 | 4 ± 3 | 2,6-Di-Me–Ph | 196–199 |
| 1{39} | 940 ± 60 | 1 ± 1 | 2,6-Di-Cl–Ph | 181–187 |
| 1{40} | 50500 ± 500 | 5 ± 3 | 2,6-Di-i-Pr–Ph | 218–222 |
| 7 | 2.9 ± 0.2 | 62 ± 13 | 4-CO2Me–Ph | 162–163 |
| 8 | 220 ± 5 | nd | 4-CO2H–Ph | 269–285 |
| 9 | 2.0 ± 0.2 | 72 ± 6 | 3-CO2Me–Ph | 116–123 |
| 10 | 590 ± 60 | nd | 3-CO2H–Ph | 240–254 |
nd denotes not determined.
Determined via a kinetic fluorescent assay, results are means ± SD of three separate experiments.
Determined via an end-point fluorescent assay, results are means ± SD of three separate experiments.
The coincidence between the current SAR with that of the chalcone oxide derivatives encouraged us to explore the carboxylated analogs to investigate the e3ect in the presence of the ionizable group on the R1. The required acid compounds 8 and 10 were synthesized using amine 1 as outlined in Scheme 2.
Scheme 2.
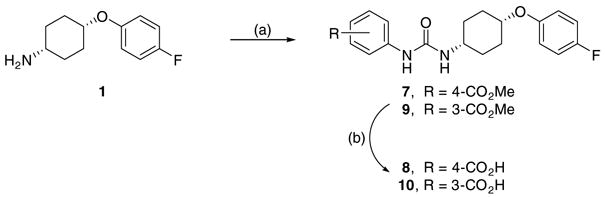
Reagents and conditions: (a) Ar-NCO, DMF, rt, 12 h; (b) LiOH, acetonitrile, water, 90 °C, 6 h.
In a similar manner as observed for chalcone oxide derivatives,8 the introduction of the free carboxylic acid at the para-position of the phenyl ring dramatically decreased its activity about 440-fold compared to compound 1{1}. Having a free carboxylic acid at the meta-position decreased inhibition potency even more dramatically. Corresponding ester compounds 7 and 9 are, however, only 2- to 6-fold less active compared to compound 1{1}. The reason for the poor inhibitory activities of inhibitors having the free carboxylic acid on R1 is at present unclear. Possible explanations include: (1) water solvation of the carboxylate might either prevent access of the inhibitor into the active site or cause the repulsion with the residues at the active site, (2) ionic interactions between the carboxylate anion and protonated imidazole on His523 preventing the optimal binding of the inhibitor at the active site. Recent X-ray crystal structure data of human sEH complexed with di3erent dialkylurea inhibitors bearing pendant carboxylate of varying length supported the latter explanation.14
Due to the fact that some of these compounds are as potent as adamantane-based inhibitors in vitro on the recombinant human sEH enzyme, we propose that sterically less hindered lipophilic groups, such as the para-trifluoromethoxy phenyl group, are good replacements for adamantyl group. Such compounds are UV dense, have increased water solubility, and should lead to altered routes of metabolism and distribution.
In summary, we have demonstrated that several groups, such as simple non-rigid carbocylic rings or para/meta-substituted phenyl rings, can replace the adamantane ring found currently in the most potent urea-based sEH inhibitors. Compounds with sterically hindered groups as R1, however, had significantly decreased potency. In addition, having ionizable free carboxylic acid on R1 dramatically decreased the inhibitory activity. These observations strongly suggest that the side of the active site tunnel where R1 of the N,N′-disubstituted urea binds favors sterically unhindered lipophilic groups.9c
Acknowledgments
The authors thank Dr. Mark J. Kurth for many helpful discussions. We also thank Dr. Jozsef Lango and Dr. Paul Whetstone for assistance with mass spectral determinations. This work was supported in part by NIEHS Grant ES02710, NIEHS Superfund Grant P42 ES04699, NIEHS Center Grant P30 ES05707, and NHLBI STTR Grant R41 HL078016.
Footnotes
Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bmcl.2006.08.078.
References and notes
- 1.(a) Capdevila JH, Falck JR, Harris RC. J Lipid Res. 2000;41:163. [PubMed] [Google Scholar]; (b) Fretland AJ, Omiecinski CJ. Chem Biol Interact. 2000;129:41. doi: 10.1016/s0009-2797(00)00197-6. [DOI] [PubMed] [Google Scholar]; (c) Meijer J, Depierre JW. Chem Biol Interact. 1988;64:207. doi: 10.1016/0009-2797(88)90100-7. [DOI] [PubMed] [Google Scholar]; (d) Morisseau C, Hammock BD. Annu Rev Pharmacol Toxicol. 2005;45:311. doi: 10.1146/annurev.pharmtox.45.120403.095920. [DOI] [PubMed] [Google Scholar]
- 2.(a) Fisslthaler B, Popp R, Kiss L, Potente M, Harder DR, Fleming I, Busse R. Nature. 1999;401:493. doi: 10.1038/46816. [DOI] [PubMed] [Google Scholar]; (b) Pagliaro P, Rastaldo R, Paolocci N, Gattullo D, Losano G. Ital Heart J. 2000;1:264. [PubMed] [Google Scholar]
- 3.(a) Watanabe H, Vriens J, Prenen J, Droogmans G, Voets T, Nilius B. Nature. 2003;424:434. doi: 10.1038/nature01807. [DOI] [PubMed] [Google Scholar]; (b) Earley S, Heppner TJ, Nelson MT, Brayden JE. Circ Res. 2005;97:1270. doi: 10.1161/01.RES.0000194321.60300.d6. [DOI] [PubMed] [Google Scholar]; (c) Vriens J, Owsianik G, Fisslthaler B, Suzuki M, Janssens A, Voets T, Morisseau C, Hammock B, Fleming I, Busse R, Nilius B. Circ Res. 2005;97:908. doi: 10.1161/01.RES.0000187474.47805.30. [DOI] [PubMed] [Google Scholar]
- 4.Yu Z, Xu F, Huse LM, Morisseau C, Draper AJ, Newman JW, Parker C, Graham L, Engler MM, Hammock BD, Zeldin DC, Kroetz DL. Circ Res. 2000;87:992. doi: 10.1161/01.res.87.11.992. [DOI] [PubMed] [Google Scholar]
- 5.Imig JD, Zhao X, Capdevila JH, Morisseau C, Hammock BD. Hypertension. 2002;39:690. doi: 10.1161/hy0202.103788. [DOI] [PubMed] [Google Scholar]
- 6.(a) Spiecker M, Liao J. Arch Biochem Biophys. 2005;433:413. doi: 10.1016/j.abb.2004.10.009. [DOI] [PubMed] [Google Scholar]; (b) Node K, Huo YQ, Ruan XL, Yang BC, Spiecker M, Ley K, Zeldin DC, Liao JK. Science. 1999;285:1276. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Liu Y, Zhang Y, Schmelzer K, Lee TS, Fang X, Zhu Y, Spector AA, Gill S, Morisseau C, Hammock BD, Shyy JY. Proc Natl Acad Sci USA. 2005;102:16747. doi: 10.1073/pnas.0508081102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zheng J, Plopper CG, Lakritz J, Storms DH, Hammock BD. Am J Respir Cell Mol Biol. 2001;25:434. doi: 10.1165/ajrcmb.25.4.4104. [DOI] [PubMed] [Google Scholar]
- 8.Morisseau D, Du G, Newman JW, Hammock BD. Arch Biochem Biophys. 1998;356:214. doi: 10.1006/abbi.1998.0756. [DOI] [PubMed] [Google Scholar]
- 9.(a) Kim IH, Morisseau C, Watanabe T, Hammock BD. J Med Chem. 2004;47:2110. doi: 10.1021/jm030514j. [DOI] [PubMed] [Google Scholar]; (b) Kim IH, Heirtzler FR, Morisseau C, Nishi K, Tsai HJ, Hammock BD. J Med Chem. 2005;48:3621. doi: 10.1021/jm0500929. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Jones PD, Tsai HJ, Do Z, Morisseau C, Hammock BD. Bioorg Med Chem Lett. 2006;16:5212. doi: 10.1016/j.bmcl.2006.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones PD, Wolf NM, Morisseau C, Whetstone P, Hock B, Hammock BD. Anal Biochem. 2005;343:66. doi: 10.1016/j.ab.2005.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wolf NM, Morisseau C, Jones PD, Hock B, Hammock BD. Anal Biochem. 2006;355:71. doi: 10.1016/j.ab.2006.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhattacharyya S, Gooding OW, Labadie J. Tetrahedron Lett. 2003;44:6099. [Google Scholar]
- 13.http://www.irori.com.
- 14.Gomez GA, Morisseau C, Hammock BD, Christianson DW. Protein Sci. 2006;15:58. doi: 10.1110/ps.051720206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morisseau C, Goodrow MH, Newman JW, Wheelock CE, Dowdy DL, Hammock BD. Biochem Pharmacol. 2002;63:1599. doi: 10.1016/s0006-2952(02)00952-8. [DOI] [PubMed] [Google Scholar]
- 16.See Table 1 in Supplementary data for a complete list of these diversity reagents.
- 17.See Tables 1 and 2 in Supplementary data for the mass data, the purities, the yields, and the percent inhibition values at 100 nM concentration of the library.