

Summary
Complex interplay between T helper (Th) cells and macrophages contributes to the formation and progression of atherosclerotic plaques. While Th1 cytokines promote inflammatory activation of lesion macrophages, Th2 cytokines attenuate macrophage-mediated inflammation and enhance their repair functions. In spite of its biologic importance, the biochemical and molecular basis of how Th2 cytokines promote maturation of anti-inflammatory macrophages is not understood. We show here that in response to interleukin-4 (IL-4), signal transducer and activator of transcription 6 (STAT6) and PPARγ-coactivator-1β (PGC-1β ) induce macrophage programs for fatty acid oxidation and mitochondrial biogenesis. Transgenic expression of PGC-1β primes macrophages for alternative activation and strongly inhibits proinflammatory cytokine production, whereas inhibition of oxidative metabolism or RNAi-mediated knockdown of PGC-1β attenuates this immune response. These data elucidate a molecular pathway that directly links mitochondrial oxidative metabolism to the anti-inflammatory program of macrophage activation, suggesting a potential role for metabolic therapies in treating atherogenic inflammation.
Introduction
Atherosclerosis is the primary cause of mortality from coronary artery disease in the Western societies (Hansson, 2005). Although initially considered to be a passive disorder arising from deposition of lipids in the vessel wall, atherogenesis is now recognized as an active inflammatory condition, involving complex interactions between the endothelium, smooth muscle cells, and the immune system (Glass and Witztum, 2001; Hansson, 2005). In this contemporary view, endothelial dysfunction, resulting from hypercholesterolemia, hypertension, diabetes, or smoking, initiates a proinflammatory reaction in the vessel wall, resulting in recruitment of lymphocytes and macrophages. If persistent, this influx of leukocytes promotes a chronic inflammatory state, eventually leading to clinically significant stenosis or thrombosis.
Development and progression of atherosclerotic plaques is dictated by a balance between pro- and anti-inflammatory factors (Binder et al., 2002; Hansson, 2005). Interestingly, macrophages recruited to the injured endothelium orchestrate both the initiation and resolution of atherogenic inflammation (Goerdt and Orfanos, 1999; Gordon, 2003). For instance, early in atherogenesis, Th1 cytokine interferon γ (IFNγ) and the invading lipoproteins promote classical activation of lesional macrophages, resulting in the secretion of proinflammatory cytokines, metalloproteinases, and hydrolases. This proinflammatory program serves to protect the host by scavenging, degrading, and clearing offending lipoproteins. However, if excessive and unabated, this inflammatory response becomes detrimental and retards the healing process (Goerdt and Orfanos, 1999). In this context, recent studies suggest that the Th2 cytokines interleukin-4 (IL-4) and IL-13 activate STAT6 to promote maturation of alternatively activated macrophages to counteract excessive inflammation and enhance repair of tissues (Gordon, 2003; Herbert et al., 2004; Rauh et al., 2005; Stout and Suttles, 2004). These macrophages can be distinguished by their expression of arginase I, which metabolizes arginine to proline, leading to collagen production and fibrosis (Gordon, 2003; Mosser, 2003). Since the two arginine-metabolizing enzymes, inducible nitric oxide synthase (iNOS) and arginase I, are reciprocally regulated by Th1- and Th2-type cytokines, respectively, the magnitude and duration of macrophage respiratory burst is directly controlled by the Th1/Th2 balance (Rauh et al., 2005; Rutschman et al., 2001). In support of this notion, atherosclerotic lesions in IFNγ R/apoE double-knockout mice are markedly smaller and exhibit a substantial increase in their collagen content (Gupta et al., 1997), indicating that loss of Th1 signaling polarizes macrophages to the less-inflammatory alternative state. Furthermore, it has been postulated that the Th2-biasing effects of statins might account for a significant portion of their antiatherogenic actions (Arnaud et al., 2005; Schonbeck and Libby, 2004). However, despite the therapeutic potential of alternatively activated macrophages, little is known about the regulatory factors and pathways that control their maturation.
The PGC-1 family of transcriptional coactivators provide a potential molecular link between cellular programs of inflammation and lipid homeostasis (Lin et al., 2005a). Although initially identified as master regulators of oxidative metabolism (Puigserver et al., 1998), these coactivator proteins are highly versatile in their functions. By interacting with tissue-specific transcription factors, the PGC-1 proteins regulate various cellular processes, including thermogenesis, muscle fiber type specification, gluconeogenesis, lipogenesis, and cytokine signaling (Lin et al., 2002, 2005b; Puigserver et al., 1998, 2001; Yoon et al., 2001). For instance, the catabolic effects of proinflammatory cytokines are mediated by PGC-1α. In this case, stimulation of muscle cells by Th1-type cytokines leads to phosphorylation and activation of PGC-1α, which in turn increases uncoupled respiration and energy expenditure in myocytes. Given the ability of PGC-1 proteins to transcriptionally integrate cytokine and lipid signals, we investigated whether PGC-1 might also regulate programs of macrophage activation. We show here that stimulation of macrophages with IL-4 robustly induces PGC-1β and upregulates the genetic program for oxidative metabolism. Notably, PGC-1β coactivates the transcriptional functions of STAT6 to metabolically polarize macrophages to the alternative state, thereby attenuating their inflammatory burst. Taken together, these findings delineate a novel mechanism by which metabolic regulators can control macrophage activation, thereby providing an additional therapeutic strategy with which to combat atherogenic inflammation.
Results
Induction of oxidative metabolism and PGC-1β during alternative macrophage activation
To examine the crosstalk between immune programs of macrophage activation and lipid homeostasis, we used cDNA micro-arrays to interrogate gene expression patterns of classically (highly inflammatory) and alternatively (less inflammatory) activated macrophages. Clustering analysis revealed that genes involved in fatty acid oxidation were strongly induced in alternatively, but not classically, activated macrophages (Figure 1A). Notably, expression of enzymes important in mitochondrial oxidation of fatty acids, including acyl-CoA dehydrogenases and enoyl CoA hydratases, was significantly enhanced by IL-4. Northern blot analyses further revealed that mRNAs encoding proteins necessary for fatty acid uptake (LPL, lipoprotein lipase; PPARγ, peroxisome proliferator activated receptor-γ), transport (CD36, fatty acid transporter; CPT1, carnitine palmitoyltransferase I), and oxidation (MCAD, medium chain acyl-CoA dehydrogenase; LCAD, long chain acyl-CoA dehydrogenase) were concurrently induced in the less-inflammatory alternatively activated macrophages (Figure 1B and Figure S1A in the Supplemental Data available with this article online), thereby providing independent verification of the microarray data. Interestingly, induction of the macrophage oxidative program was accompanied by an increase in the expression of PGC-1β mRNA (Figure 1B). Immunoblot analyses further verified that stimulation of bone marrow-derived or thioglycolate-elicited macrophages with IL-4 robustly induced the expression of PGC-1β protein (Figure 1C). Since these changes in gene expression were absent in STAT6-deficient macrophages, they are likely a direct effect of IL-4. Lastly, a reciprocal pattern of regulation was observed between alternatively and classically activated macrophages for genes involved in β-oxidation of fatty acids and inflammation (Figures 1A and S1A), suggesting that oxidative metabolism might be intrinsically linked to the less-inflammatory alternative state.
Figure 1. Induction of oxidative metabolism and PGC-1β during alternative macrophage activation.
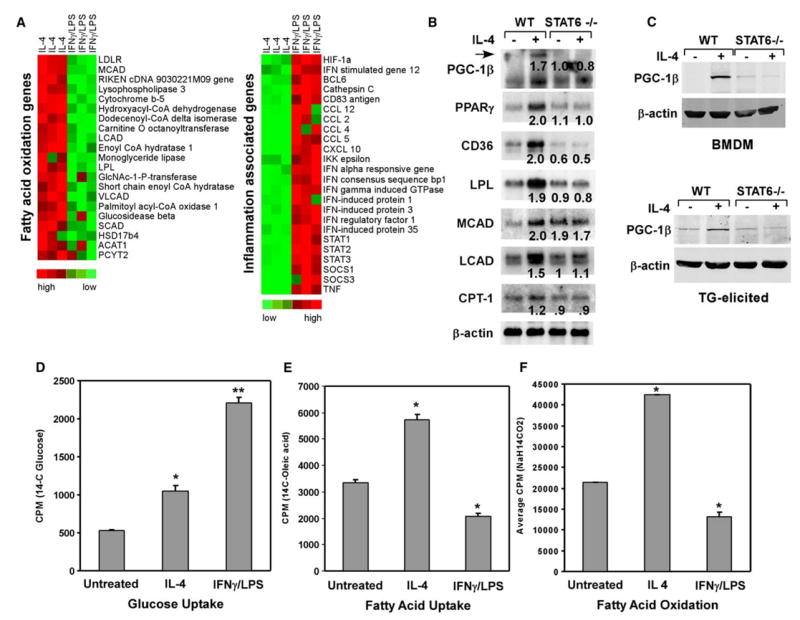
A) Cluster analysis of macrophage gene expression during alternative and classical activation. Note the reciprocal pattern of expression for inflammatory genes with those involved in fatty acid metabolism.
B) IL-4 induces genes required for fatty acid metabolism in a STAT6-dependent manner. Average fold induction relative to untreated wild-type sample is indicated.
C) Induction of endogenous PGC-1β protein by IL-4 in a STAT6-dependent manner. 75 μg of total cellular lysates from wild-type or STAT6 null macrophages were subjected to immunoblot analysis with a PGC-1β antibody. Top panel: bone marrow-derived macrophages (BMDM); bottom panel: thioglycolate (TG)-elicited macrophages.
D–F) Alternative activation increases reliance on fatty acid metabolism, as measured by uptake of glucose (C), uptake of fatty acids (D), and β-oxidation of fatty acids (E). *p < 0.05, **p < 0.005.
To determine the significance of increased expression of β-oxidation genes in IL-4-stimulated macrophages, we measured the rates of glucose uptake, fatty acid uptake, and fatty acid oxidation in macrophages stimulated to undergo classical or alternative activation. As expected, classical activation of macrophages by IFNγ and lipopolysaccharide (LPS) strongly induced uptake of glucose by ~450% (Figure 1D) and concomitantly suppressed the rates of fatty acid uptake and oxidation by ~40% (Figures 1E and 1F). In contrast, uptake of both glucose and fatty acids was increased in alternatively activated macrophages by ~200% (Figures 1D and 1E). Importantly, stimulation of macrophages with IL-4 increased the rate of β-oxidation by ~200% (Figure 1F), indicating that a switch in nutrient utilization accompanies alternative macrophage activation. Together, these data clearly show that while classically activated macrophages preferentially utilize glucose, the alternative program of macrophage activation switches over to fatty acid oxidation for energy homeostasis.
Oxidative metabolism is required for maturation of anti-inflammatory macrophages
Induction of oxidative metabolism in alternatively activated macrophages suggested that it might be integrally linked to the expression of the anti-inflammatory alternative phenotype. To investigate this relationship, we pharmacologically inhibited mitochondrial respiration in macrophages stimulated to undergo alternative activation. Inhibition of fatty acid oxidation by etomoxir (Djouadi et al., 1999) dramatically decreased the induction of arginase activity (Figure 2A), a hallmark of anti-inflammatory alternatively activated macrophages (Goerdt and Orfanos, 1999; Gordon, 2003; Mosser, 2003). Similarly, oligomycin and FCCP, which inhibit oxidative phosphorylation and uncouple mitochondrial respiration (Wu et al., 1999), respectively, strongly reduced arginase activity, as detected by conversion of arginine to urea (Figure 2A) (Rutschman et al., 2001). Consistent with this decrease in enzymatic activity, mRNAs encoding arginase I and two endocytic receptors, dectin-1 and mannose receptor, showed an absolute dependence on oxidative metabolism for their induction by IL-4 (Figure 2B) (Munder et al., 1999; Stein et al., 1992; Willment et al., 2003). Flow cytometric analyses of these and other alternative activation makers (Gordon, 2003; Loke and Allison, 2003) further supported this conclusion (Figures S2A–S2C). Moreover, since alternatively activated macrophages are known to counteract excessive production of proinflammatory cytokines (Goerdt and Orfanos, 1999; Gordon, 2003; Herbert et al., 2004), we examined whether inhibition of mitochondrial respiration could also abrogate this response. Indeed, as assayed by the secretion of IL-6, tumor necrosis factor α (TNF-α), and IL-12, pretreatment of macrophages with mitochondrial inhibitors completely abolished the anti-inflammatory effects of IL-4 (Figures 2C–2E). In contrast, inhibition of mitochondrial respiration did not have any significant effect on inflammatory activation of macrophages by IFNγ and lipopolysaccharide (Figures 2C–2E). It is important to note that incubation of macrophages with metabolic inhibitors did not significantly alter the cellular ATP, calcium, STAT6, or PGC-1β levels (Figures 2F, S2D, and S2E). In aggregate, these results provide evidence for direct involvement of mitochondrial oxidative metabolism, potentially independent of STAT6 and PGC-1β, in full expression of the anti-inflammatory alternative phenotype.
Figure 2. Requirement for oxidative metabolism in maturation of anti-inflammatory alternatively activated macrophages .
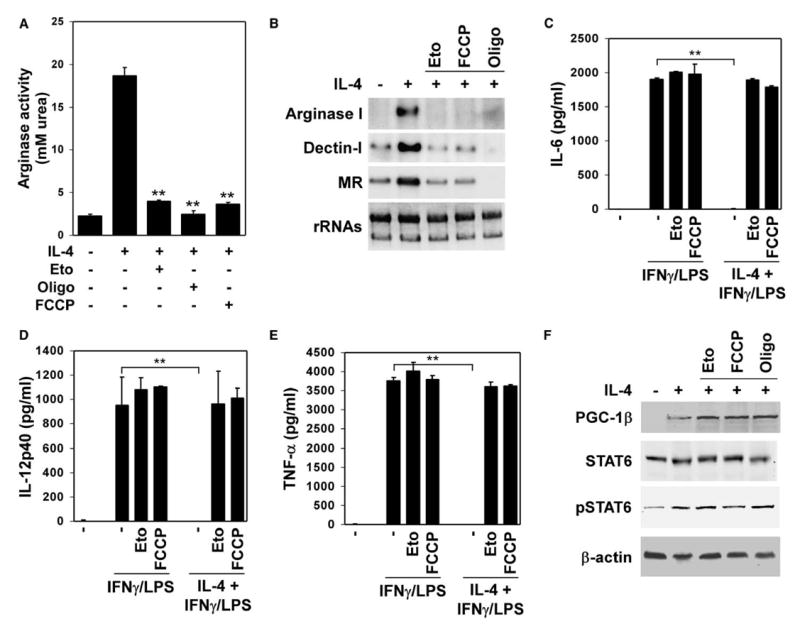
A) Inhibition or uncoupling of mitochondrial respiration prevents induction of arginase activity by IL-4.
B) Mitochondrial respiration is required for expression of the alternative phenotype, as quantified by induction of various mRNAs.
C–E) Mitochondrial inhibitors diminish the anti-inflammatory effects of IL-4. Control and treated macrophages were costimulated with IFNγ (1 u/ml) / LPS (2.5 ng/ml) in the presence or absence of IL-4 (5 ng/ml). ELISAs were used to quantify the secreted (C) IL-6, (D) IL-12p40 and (E) TNF-α by macrophages. Eto, etomoxir; Oligo, oligomycin. **p < 0.005.
F) IL-4 signaling is intact in macrophages treated with metabolic inhibitors, as measured by induction of PGC-1β and phosphorylation of STAT6.
STAT6 transcriptionally regulates oxidative metabolism in macrophages
Although IL-4 exerts a majority of its immunological effects on macrophages via STAT6 proteins (Ihle, 2001; Takeda and Akira, 2000; Wurster et al., 2000), the role of this transcription factor in orchestrating the macrophage metabolic response has not been investigated to date. Gene expression analysis of macrophages revealed that IL-4 failed to upregulate key metabolic genes, including CD36, LPL, MCAD, LCAD, and PGC-1β, in STAT6 null macrophages (Figures 1B and 1C). To investigate whether these metabolic genes might be targets for transcriptional regulation by STAT6 homodimers, we identified a potential STAT6 response element (STAT6RE) in the proximal 0.5kb of the MCAD promoter. Gel mobility shift assays with nuclear extracts from wild-type and STAT6 null macrophages confirmed binding to the MCAD-STAT6RE in an IL-4 dependent manner (Figure 3A). Moreover, competition with excess unlabeled probe and antibody supershift assays verified the specificity of STAT6 binding and its presence in the IL-4-induced retarded complexes, respectively (Figure 3A) (Ohmori et al., 1996). To examine whether the identified MCAD-STAT6RE could function in vivo, transient transfection experiments were performed with a reporter construct containing the 0.5 kb of the MCAD promoter. Treatment with IL-4 led to robust activation of the MCAD promoter in transiently transfected RAW264.7 cells (Figure 3B). Moreover, mutation of STAT6 binding site completely abolished the inductive effects of IL-4 on this promoter. Interestingly, consistent with increased basal expression of MCAD mRNA in STAT6-deficient macrophages (Figure 1B), mutation of STAT6RE increased basal activity of the MCAD promoter. These findings imply that in addition to transcriptionally activating gene expression, STAT6 might also have an important role in target gene repression (Schroder et al., 2002).
Figure 3. STAT6 regulates oxidative metabolism in alternatively activated macrophages.
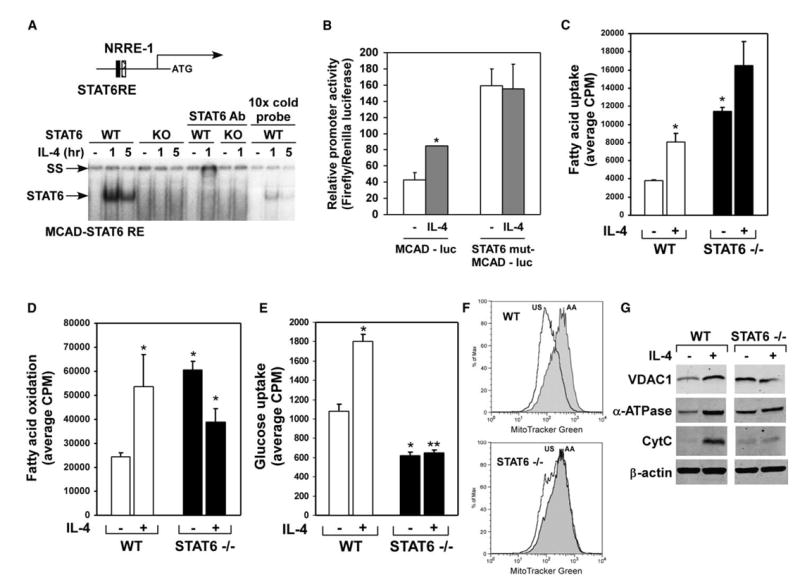
A) Identification of STAT6 binding sites in MCAD promoter. NRRE, nuclear receptor response element. Gel mobility shift assays were performed with nuclear extracts from wild-type and STAT6 null macrophages; SS-supershift of STAT6.
B) STAT6 homodimers transactivate the MCAD promoter. Note that mutation of STATRE in the MCAD promoter abolishes IL-4 induction, but raises basal promoter activity.
C–E) Loss of STAT6 affects both basal and IL-4 induced metabolic fluxes in macrophages; (C) fatty acid uptake, (D) fatty acid oxidation, and (E) glucose uptake. *p < 0.05, **p < 0.005.
F and G) Stimulation with IL-4 fails to increase cellular mitochondrial content in STAT6−/− macrophages, as measured by (F) MitoFluor Green and (G) VDAC1, α-ATPase, and CytC protein levels. Equivalent loading was confirmed by immunoblotting for β-actin.
To elucidate the regulatory role of STAT6 in oxidative metabolism, we measured the uptake and utilization of fatty acids in wild-type and STAT6 null macrophages. Consistent with the impaired induction of mRNAs for fatty acid catabolism, stimulation with IL-4 did not significantly change fatty acid uptake or oxidation in STAT6 null macrophages (Figures 3C and 3D). However, the basal rate for fatty acid oxidation was higher in STAT6 null macrophages, potentially reflecting a compensatory response, as evidenced by increased expression of MCAD mRNA in these cells (Figures 3C and 3D). Balanced, and often reciprocal, metabolism of glucose and fatty acids supports cellular energy homeostasis. Since STAT6-deficient macrophages exhibit higher reliance on fatty acid metabolism, we postulated that this might represent a compensatory response to decreased glycolytic flux. Indeed, as shown in Figure 3E, both basal and IL-4-stimulated rates of glucose uptake were reduced by ~45% and ~70%, respectively, in STAT6 null macrophages (Figure 3E). Together, these results demonstrate that STAT6 is required for both basal and IL-4-dependent changes in glucose and fatty acid homeostasis.
The ability of STAT6 to regulate macrophage oxidative metabolism suggested that this transcription factor might also play an important role in mitochondrial biogenesis. Alternative, but not classical, activation of wild-type macrophages led to a dramatic increase in cellular mitochondrial mass, as quantified by staining with MitoFluor Green (Figures 3F and S3A). Strikingly, IL-4 failed to induce PGC-1β expression (Figures 1B and 1C) and increase cellular mitochondrial content in STAT6-deficient macrophages (Figure 3F). As expected from their higher oxidative capacity, STAT6 null macrophages had more mitochondria and increased expression of some mitochondrial proteins, including voltage dependent anion channel 1 (VDAC1) and ATP synthase α (α-ATPase), in their basal state (Figure 3G). However, the ability of IL-4 to enhance the expression of all three mitochondrial proteins, VDAC1, α-ATPase, and cytochrome C (CytC), was completely abrogated in STAT6 null macrophages (Figure 3G), indicating that IL-4 is the physiologic trigger that couples macrophage oxidative metabolism to the larger program of immune activation via STAT6. Lastly, this compensatory increase in macrophage oxidative metabolism was not accompanied by any significant changes in the expression of transcription factors regulating mitochondrial biogenesis (Figure S3B) or an aberrant increase in the coactivator protein PGC-1α (data not shown).
Constitutive expression of PGC-1β downregulates macrophage inflammatory responses
The induction of PGC-1β by IL-4 suggested that this coactivator might in turn modulate the transcriptional activity of STAT6, thereby directly participating in the maturation of anti-inflammatory macrophages. To investigate this possibility, we examined the ability of PGC-1β to coactivate the STAT6-responsive arginase I promoter. As expected, activation of latent STAT6 proteins by IL-4 led to ~2-fold increase in the arginase I promoter activity in transiently transfected RAW264.7 cells (Figure 4A) (Pauleau et al., 2004). Notably, addition of PGC-1β strongly enhanced STAT6 activity on the arginase I promoter (~8-fold over basal levels, Figure 4A). Mutation of the STAT6 binding site, which is located in the enhancer region of arginase I promoter (Pauleau et al., 2004), completely abolished the ability of STAT6 and PGC-1β to activate this promoter, indicating that PGC-1β might directly interact with STAT6 on the arginase I promoter. Indeed, in coimmunoprecipitation assays, Flag-PGC-1β (f-PGC-1β) existed in a soluble complex with and efficiently precipitated STAT6 (Figure 4B). To determine whether PGC-1β is recruited to the STAT6RE in the distal enhancer of arginase I, we performed quantitative chromatin immunoprecipitation (ChIP) assays. As shown in Figure 4C, stimulation of macrophages with IL-4 led to increased corecruitment of STAT6 and PGC-1β to the arginase I enhancer, indicating that transcriptionally active STAT6 can efficiently recruit PGC-1β in close proximity to its binding site.
Figure 4. PGC-1β enhances maturation of anti-inflammatory alternatively activated macrophages.
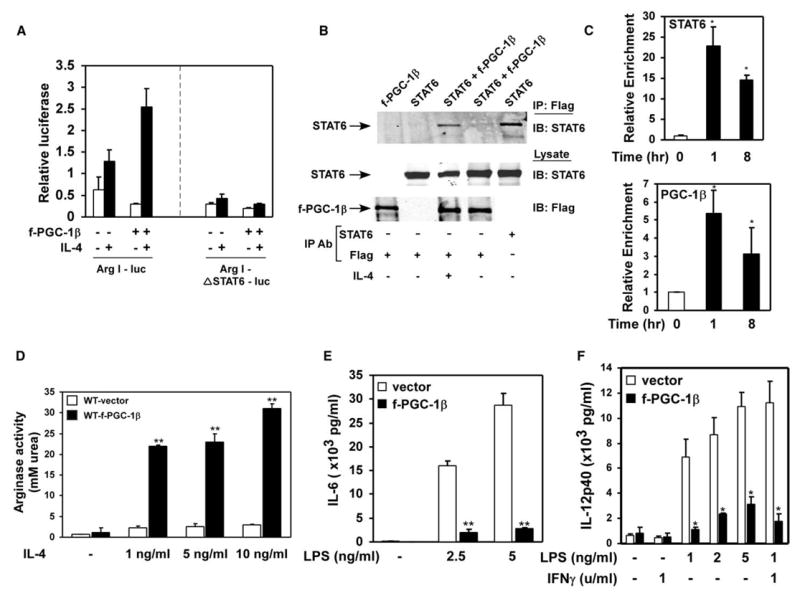
A) Coactivation of STAT6 by PGC-1β on arginase I promoter. RAW264.7 cells were transiently transfected with wild-type arginase I promoter (Arg I-luc) or STAT6RE mutant (Arg-ΔSTAT6-luc) reporter constructs, in the presence or absence of PGC-1β.
B) Coimmunoprecipitation of PGC-1β and STAT6.
C) Real-time ChIP analysis for the arginase promoter. Both STAT6 and PGC-1β are recruited to the arginase promoter after stimulation of macrophages with IL-4.
D) PGC-1β enhances arginase activity during alternative macrophage activation.
E and F) Expression of PGC-1β attenuates inflammatory activation of macrophages. ELISA measurements for secreted IL-6 (E) and IL-12 p40 subunit (F). *p < 0.02, **p < 0.005.
A unique feature of PGC-1 coactivators is their ability to couple mitochondrial biogenesis to biological processes that are associated with increased oxidative metabolism (Lin et al., 2002, 2005a). To investigate whether constitutive expression of PGC-1β could similarly polarize macrophages toward the more oxidative alternative state, we retrovirally introduced f-PGC-1β into wild-type macrophages (Figure S4A). As expected, fatty acid oxidation rates were ~160% higher in f-PGC-1β expressing cells, consistent with the known metabolic effects of this coactivator (Figure S4B). Strikingly, constitutive expression of f-PGC-1β increased IL-4 induced urea production via arginase I by ~800%–900% (Figure 4D), suggesting that PGC-1β can indeed potentiate macrophage activation along the less-inflammatory alternative axis. Notably, this enhancement of arginase I activity by f-PGC-1β was independent of changes in total or phosphorylated STAT6 protein levels (Figure S4C). To further examine whether priming by PGC-1β might attenuate inflammatory activation of macrophages, control and f-PGC-1β expressing cells were challenged with LPS, and cytokine secretion was quantified by ELISAs. f-PGC-1β expression strongly reduced the secretion of inflammatory cytokines, IL-6 and IL-12, in LPS-challenged macrophages (Figures 4E and 4F). Moreover, cell surface expression of dectin-1, a marker of alternatively activated macrophages, was robustly induced by PGC-1β (Figure S4D). Taken together, these results suggest that cooperation between the IL-4 signaling pathway and PGC-1β is critical in polarizing macrophages to the more oxidative, less-inflammatory alternative state.
PGC-1β knockdown impairs alternative macrophage activation
The induction of PGC-1β by IL-4 suggested that PGC-1β might be required for transcriptional activation of STAT6 functions in macrophages. To test this possibility, we used shRNAs that have previously been shown to be highly specific and effective in knocking down PGC-1β protein expression (Lin et al., 2005b). As shown in Figure 5A, retrovirally mediated stable expression of two distinct shRNAs led to greater than 80% knockdown of endogenous PGC-1β in macrophages. As expected, the ability of IL-4 to promote β-oxidation of fatty acids was reduced by 35%–52% in the PGC-1β knockdown cells (Figure 5B). Demonstrating a key role of PGC-1β in the maturation of less-inflammatory alternatively activated macrophages, knockdown of PGC-1β by RNAi reduced arginase activity by 61%–72% (Figure 5C). Furthermore, the anti-inflammatory actions of the Th2 cytokine IL-4 were greatly diminished in PGC-1β knockdown macrophages, as evidenced by the inability of IL-4 to completely suppress the release of IL-6 or IL-12 p40 subunit (Figures 5D and 5E). It is important to note that, in the absence of IL-4 stimulation, there was no significant difference between the inflammatory activation of control and PGC-1β knockdown macrophages (Figures S5A and S5B), indicating exquisite specificity for the anti-inflammatory functions of PGC-1β to the alternative state. Finally, in contrast to the potent inhibition of alternative macrophage activation by inhibitors of oxidative metabolism, macrophages with PGC-1β knockdown still retained some capacity to undergo alternative activation, perhaps reflecting the residual activity of PGC-1β protein or IL-4 induced increase in oxidative metabolism.
Figure 5. Requirement for PGC-1β in alternative macrophage activation.
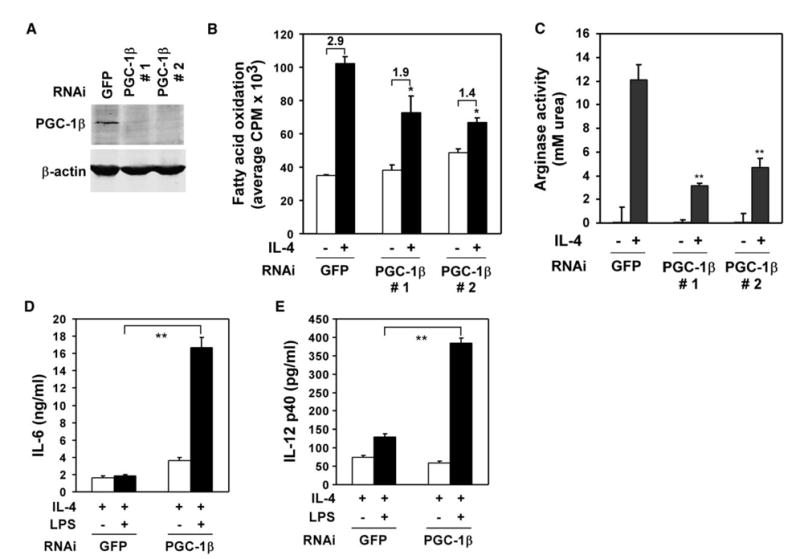
A) Knockdown of endogenous PGC-1β protein by retroviral expression of RNAi. Wild-type macrophages were stably infected with retroviral vectors expressing shRNAs against GFP or PGC-1β. Whole-cell lysates were analyzed by immunoblotting for endogenous PGC-1β.
B) Requirement for PGC-1β in IL-4-stimulated increase in β-oxidation of fatty acids.
C) Arginase activity in control and PGC-1β knockdown macrophages. Macrophages were stimulated IL-4 (5 ng/ml) for 24 hr prior to assaying for urea production.
D and E) Role of PGC-1β in inhibition of macrophage inflammatory activation. Macrophages were stimulated by LPS (1 ng/ml) in the presence of IL-4 (2 ng/ml), and secreted cytokine culture supernatants were quantified by ELISAs. Representative data are shown above (n = 3). *p < 0.05; **p < 0.004.
Decreased Th1-type macrophage inflammation in PGC-1β transgenic mice
To address whether PGC-1β can prime macrophages toward the less-inflammatory alternative state in vivo, we generated transgenic mice expressing f-PGC-1β under the control of human CD68 promoter, a promoter that preferentially directs transgene expression to macrophages (Figure 6A) (Gough et al., 2001; Lang et al., 2002). Two positive founder lines, which gave germline transmission, were expanded to establish the Mac-PGC-1β transgenic colonies. Importantly, macrophages derived from the transgenic animals showed robust expression of the PGC-1β protein (Figure 6A). Consistent with the tissue-specificity of the CD68 promoter, we failed to detect expression of Flag-PGC-1β in skeletal muscle or adipose tissue of lean mice (data not shown). Moreover, analysis of macrophage proteins revealed that founder #1 expresses PGC-1β to the same level as that seen in IL-4-stimulated wild-type macrophages (Figure 6A). To examine whether transgenic expression of PGC-1β potentiated the IL-4 driven macrophage program of alternative activation, we measured arginase I mRNA and activity levels in BMDM after IL-4 stimulation. As shown in Figures 6B and 6C, expression of arginase I mRNA and induction of arginase activity was dramatically higher in macrophages derived from PGC-1β transgenic mice. In addition to biasing arginine metabolism toward the less-inflammatory state, increased expression of PGC-1β protein led to the attenuation of macrophage-mediated inflammation, as evidenced by decreased secretion of IL-6 and IL-12p40 in the culture supernatants (Figures 6D and 6E). In aggregate, these results demonstrate that PGC-1β can potentiate the alternative program of macrophage activation in vivo, and that direct coactivation of STAT6 by PGC-1β likely accounts for a significant portion of this phenotypic switching in macrophage immune responses.
Figure 6. Transgenic expression of PGC-1β potentiates alternative activation and attenuates macrophage-mediated inflammation in vivo.
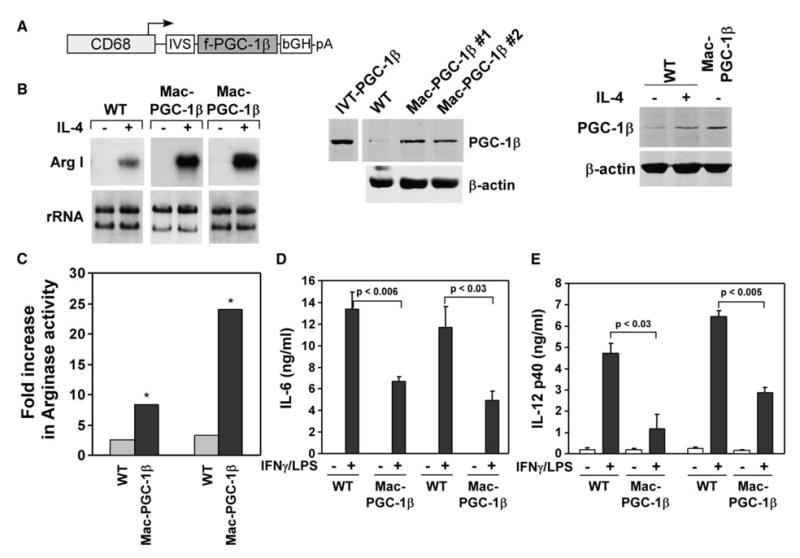
A) hCD68 promoter directs PGC-1β transgene expression to macrophages. Lysates from wild-type or Mac-PGC-1β macrophages were analyzed by immunoblotting for PGC-1β (middle panel). PGC-1β is expressed at a level equivalent to that found in IL-4-stimulated wild-type macrophages (right panel).
B) PGC-1β potentiates expression of arginase I mRNA, as assessed by Northern blots.
C) Fold increase in arginase activity in macrophages from wild-type and Mac-PGC-1β mice after stimulation with IL-4 (2 ng/ml) for 24 hr.
D and E) Transgenic expression of PGC-1β attenuates macrophage inflammatory burst. BMDM from wild-type and PGC-1β transgenic mice were stimulated with IFNγ (1 u/ml) for 20 hr prior to challenge with LPS (0.5 ng/ml). Cytokine secretion was assayed 6 hr later by ELISAs. *p < 0.04.
Discussion
Recent studies have shown a key role for glycolytic pathways controlled by hypoxia inducible factor-1α (HIF-1α) in promoting the effector responses of classically activated macrophages (Cramer et al., 2003). However, the metabolic and transcriptional bases for maturation of anti-inflammatory alternatively activated macrophages have remained largely unknown. The data presented here demonstrates that alternative activation of macrophages by IL-4 induces cellular pathways for fatty acid oxidation and mitochondrial biogenesis. In addition to directly regulating genes important in β-oxidation of fatty acids, STAT6, which mediates the transcriptional responses of IL-4, is also required for induction of the coactivator protein PGC-1β. Interestingly, a feedforward loop, in which PGC-1β is induced by and coactivates STAT6, amplifies and sustains the metabolic, biogenic and anti-inflammatory programs of alternatively activated macrophages. Lastly, the ability of PGC-1β to potentiate the macrophage program of alternative activation provides a unique example of how immune responses can be transcriptionally directed by metabolic coactivators.
Oxidative metabolism fuels alternative macrophage activation
Four independent lines of evidence support the importance of oxidative metabolism in priming and elaboration of the less-inflammatory alternative macrophage phenotype. First, stimulation of macrophages with IL-4 robustly induces the entire program for fatty acid metabolism, including uptake and oxidation of fatty acids, and mitochondrial biogenesis. Second, pharmacologic inhibition of oxidative metabolism potently suppresses the effector functions of alternatively, but not classically, activated macrophages. Third, STAT6, the transcriptional regulator of Th2 responses in lymphocytes and macrophages, is the principal factor that coordinates the macrophage metabolic programs in response to IL-4. And finally, transgenic expression of the metabolic coactivator protein PGC-1β potentiates alternative activation, whereas its knockdown by RNAi impairs both the metabolic and anti-inflammatory functions of alternatively activated macrophages.
Although our results provide unequivocal proof for the requirement of STAT6 in mediating the metabolic response of the Th2 cytokine IL-4, STAT6-deficient macrophages exhibit a paradoxical increase in oxidative metabolism. For instance, basal rates of fatty acid uptake and oxidation are higher in STAT6 null macrophages (Figures 3C and 3D), perhaps reflecting altered energy homeostasis in these cells. Consistent with this notion, glucose uptake was dramatically reduced in STAT6 null macrophages (Figure 3E). Since altered metabolism of glucose is likely to impact cellular energy homeostasis, we postulated that compensatory activation of the energy-sensing AMP-activated protein kinase (AMPK) pathway or induction of the coactivator protein PGC-1α might be the driving force for increased reliance on β-oxidation of fatty acids (Lin et al., 2005a; Ruderman et al., 2003). However, despite repeated attempts, we failed to detect any significant differences between wild-type and STAT6 null macrophages in the expression or phosphorylation of AMPK, and its substrate the acetyl-CoA carboxylase (ACC) (data not shown). Moreover, expression of PGC-1α mRNA was undetectable by quantitative RT-PCR in both wild-type and STAT6 null macrophages under various stimulatory conditions (data not shown), making it unlikely that these pathways are responsible for the observed metabolic differences between these macrophages. An alternative explanation might be that, in the absence of IL-4, STAT6 represses a subset of its target genes, including those involved in β-oxidation. Congruent with this idea, the deletion of STAT6 binding site in the MCAD promoter or a genetic deficiency of STAT6 protein in macrophages leads to activation of the MCAD promoter, as reflected in higher basal promoter activity and expression of MCAD mRNA (Figures 3B and 1B). Interestingly, similar findings were reported by Schroder et. al. in activated B cells (Schroder et al., 2002). In their study, greater than 50% of the differentially expressed genes were derepressed in STAT6 null cells, implying that STAT6 directly or indirectly represses a subset of its target genes. More detailed studies will be required to delineate the activation and repression functions of STAT6 in glucose and fatty acid homeostasis.
Our work raises an intriguing question as to why oxidative metabolism is required for expression of the biologic program driven by IL-4. One possibility is that metabolic skewing of macrophages, toward glycolytic or oxidative metabolism, is necessary for performance of their critical functions in host defense. For example, the Th1-type cytokine, IFNγ, primes macrophages to increase production of proinflammatory cytokines, reactive oxygen species and nitric oxide for safe clearance of intracellular pathogens (Gordon, 2003; Stout and Suttles, 2004). Energy required for this respiratory burst is rapidly provided by glycolytic pathways, which are transcriptionally controlled by hypoxia inducible transcription factor-1α (HIF-1α) (Cramer et al., 2003). Pharmacological inhibition of glycolysis or genetic deletion of HIF-1α in myeloid cells greatly reduces cellular ATP content and has the functional consequence of diminishing their antimicrobial activity (Cramer et al., 2003; Peyssonnaux et al., 2005). In contrast, the Th2-type cytokines IL-4 and IL-13 promote alternative macrophage activation to provide a defense against extracellular parasites (Goerdt and Orfanos, 1999; Gordon, 2003; Stout and Suttles, 2004). In this case, STAT6 controls the genetic program for long-term macrophage activation (Takeda and Akira, 2000; Wurster et al., 2000). By increasing the secretion of chitinases, chemokines, and collagen, these alternatively activated macrophages neutralize the invading parasites, recruit eosinophils, and promote granuloma formation (Gordon, 2003). This macrophage secretory program is energetically demanding, both in terms of its intensity and duration. Since oxidative metabolism is best suited to meet the bioenergetic demands for long-term macrophage activation, our data suggest that IL-4 mediated activation of STAT6 serves to transcriptionally couple the metabolic pathways to the immune functions performed by these cells (Figure 7).
Figure 7.
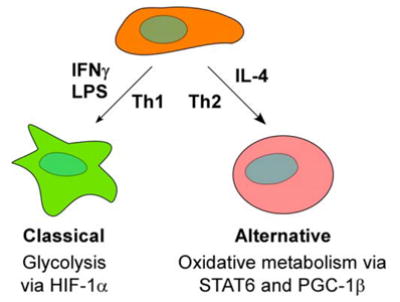
Model for regulation of macrophage activation by metabolic regulators HIF-1α and Th1-type stimuli increase glycolytic metabolism to support proinflammatory classical activation of macrophages. In contrast, in response to Th2 cytokine IL-4, STAT6, and PGC-1β enhance oxidative metabolism and promote expression of the less-inflammatory alternative phenotype.
Integration of macrophage metabolic and immune functions by PGC-1β
Previous studies and the data presented here indicate a critical role for STAT6 and oxidative metabolism in promoting maturation of anti-inflammatory alternatively activated macrophages. However, how these distinct cellular pathways are integrated into a coherent program of macrophage gene expression has not been described to date. We report here that the coactivator protein PGC-1β transcriptionally regulates the development of alternatively activated macrophages. Several key pieces of data support this claim. First, in a STAT6-dependent manner, IL-4 induces the expression of PGC-1β mRNA and protein in both quiescent and activated macrophages. Second, ectopic expression of PGC-1β leads to marked changes in macrophage metabolism and function. Transgenic expression of PGC-1β to levels equal to those induced by IL-4 increases macrophage oxidative metabolism and stimulates expression of proteins characteristic of the less-inflammatory alternative state. This likely represents a physiologic function of PGC-1β, because knockdown of PGC-1β by RNAi greatly diminishes these responses.
Taken together, the findings presented here greatly broaden our mechanistic understanding of how Th2 cytokines attenuate macrophage-mediated inflammation, and provide another example for metabolic integration of cellular processes by a family member of the PGC-1 coactivator proteins. In a manner analogous to muscle fiber conversion by metabolic regulators (Lin et al., 2002; Wang et al., 2004), our results suggest a model in which metabolic pathways and their transcriptional regulators serve as key nodal points for directing pro- and anti-inflammatory programs of macrophage activation (Figure 7). For instance, HIF-1α and glycolysis promote proinflammatory activation of macrophages, whereas oxidative metabolism regulated by STAT6 and PGC-1β primes macrophages for the less-inflammatory alternative state. In addition, these findings raise the possibility that metabolic priming of macrophages to the less-inflammatory alternative state might be a principal mechanism by which therapies that promote utilization of fatty acids, such as fibric acids and thiazolidinediones, attenuate macrophage-mediated inflammation in atherosclerosis (Duval et al., 2002; Zhang and Chawla, 2004).
Experimental procedures
Cell culture
For bone marrow-derived macrophages (BMDM), wild-type and STAT6−/− bone marrow was isolated from femurs and tibias, and cultured in IMDM supplemented with 10% fetal calf serum and 20% L929 conditioned media. Embryonic stem cell-derived macrophages (ESDM) were cultured as described previously (Chawla et al., 2001), except with the following modifications. ES-derived myeloid progenitors were conditionally immortalized with eco-tropic retroviruses encoding HOXA9-ER, a fusion protein that blocks myeloid differentiation in an estrogen-dependent manner (Sykes and Kamps, 2001). Wild-type HOXA9-ER lines were proliferated in the presence of SCF (25 ng/ml), IL-3 (1.25 ng/ml) and estradiol (1 μM). Macrophage differentiation was induced by withdrawing estradiol and culturing cells in IMDM supplemented with 10% bovine calf serum and 20% L929 conditioned media. Differentiated macrophages developed over 6–7 days. Proliferating WT-HOXA9-ER lines were infected with control or f-PGC-1β encoding retro-viruses, and subjected to selection with puromycin (4 μg/ml). Knockdown of PGC-1β was accomplished by cloning PGC-1β #1 and #2 shRNAs (Lin et al., 2005b) into pSIREN-Puro retroviral vector. WT-HOXA9-ER cells were infected with GFP, PGC-1β #1, or PGC-1β #2 shRNA-expressing retro-viruses, and selected with puromycin (4 μg/ml). For classical or alternative macrophage activation, macrophages were stimulated with murine IFNγ (100 u/ml) plus LPS (5 ng/ml) or murine IL-4 (10–20 ng/ml) for 24 hr, respectively.
Generation of PGC-1β transgenic mice
cDNA fragment encoding Flag-tagged mPGC-1β was subcloned into a vector containing the hCD68 promoter (Gough et al., 2001). DNA was excised from the cloning vector and injected into CBA 3 C57Bl6 mouse oocytes. Transgenic offspring were genotyped by PCR using the following primers: TY1: 5′-TTCTCGGCTCTGTGAATGACA-3′ and 5′ -CAGCCCTCTCTTGGAAAGGAGG-3′. Founder mice were backcrossed onto C57Bl6 background, and F1-F3 progeny were used for the macrophage experiments.
Gel mobility shift assays and transient transfections
Gel mobility shift assays were performed with nuclear extracts from wild-type or STAT6−/− macrophages, and 32P end-labeled oligonucleotides. RAW 264.7 cells were transiently transfected by electroporation using GenePulser II (BioRad) at 300V, 1000 μF, and subsequently stimulated with IL-4 (10 ng/ml) for 12–20 hr. Luciferase assays were performed with the Dual-Luciferase reporter assay system (Promega) as per manufacturer’s protocols. The 0.5 kb mouse MCAD promoter fragment was generated by PCR and cloned into the luciferase reporter construct pGL3-Basic (Promega). All transfections were done in triplicate and repeated at least three times.
Chromatin immunoprecipitation assays
Chromatin immunoprecipitation assays (ChIPs) were performed as described (Pauleau et al., 2004) with the following modifications. 2.5 × 106 RAW cells were plated in 10 cm plates, rested overnight, and stimulated with 10 ng/ml IL-4. Cells were crosslinked with formaldehyde and processed for ChIP. Each STAT6 immunoprecipitation was performed with 1 μg M200 anti-STAT6 polyclonal antibody (Santa Cruz Biotechnology) mixed with 1 μl polyclonal anti-STAT6 antibody supplied by James Ihle (St. Jude Children’s Research Hospital). PGC-1β immunoprecipitations were performed using affinity purified rabbit polyclonal antibody. Immunoprecipitates were processed as described (Pauleau et al., 2004) and subjected to qPCR analysis using primers specific for the Arginase I enhancer. Data is expressed as relative enrichment compared to a mouse genomic DNA standard curve. Each experiment was performed in duplicate, and repeated three independent times.
Western blot analysis, ELISAs, and flow cytometry
Western blot analyses were performed using standard procedures with BMDM. Equivalent amounts of cellular protein were analyzed by immunoblotting for cytochrome C (1:1000, BD Pharmingen), VDAC1 (1:2000, Sigma), α-ATPase (1:1000, Molecular Probes), Flag (1:1000, Sigma), PGC-1β (1:500), and β-actin (1:5000, Sigma). IR™Dye 800 conjugated secondary antibodies (1:30000, Rockland) were used for protein detection on Odyssey Infrared Detection System. For ELISAs, cells were plated in 48- or 96-well plates and stimulated with cytokines or LPS for 6–24 hr. TNF-α, IL-6, and IL-12 secretion was quantified by ELISA, per manufacturer’s protocols (BD Pharmingen). For flow cytometry, control and IL-4-stimulated macrophages were incubated with FITC-conjugated antibodies against mannose receptor, dectin-1 and PD-L2 prior to analysis on FACs Caliber. For measurement of intracellular calcium, treated macrophages were resuspended in medium containing 1 μM fluo-4AM (added 1:1 with pluronic, a nonionic detergent) for 45 min at 37ºC and subsequently analyzed by flow cytometry. All assays were repeated at least three independent times.
Gene expression analysis
Total RNA was prepared from three independent experimental replicates using Trizol reagent (Invitrogen) and analyzed by Northern blot analyses (Chawla et al., 2001). Microarray experiments were performed with 20–25 μg of total RNA, which was labeled with fluorescent nucleotides and hybridized to murine cDNA slides. Hybridized slides were interrogated via an Agilent scanner, data was subjected to background subtraction and normalization, and analyzed with statistical analysis of microarrays software package.
Protein interaction assays
Physical association of PGC-1β with STAT6 was assessed in coimmunoprecipitation assays. 293-T cells were transfected with plasmids encoding f-PGC-1β and murine STAT6. Twenty-four hours after transfection, cells were stimulated with hIL-4 (10 ng/ml) for 60 min. PGC-1β containing complexes were immunoprecipitated using the Flag antibody (Sigma), resolved on a denaturing gel and immunoblotted for STAT6 (1:1000, Santa Cruz).
Metabolic assays
For glucose and fatty acid uptake, macrophages were prestimulated with Th1 or Th2 cytokines and incubated with 14C-labeled 2-deoxy-glucose or fatty acid-free BSA conjugated 14C-oleic acid, respectively. Subsequently, cells were extensively washed and lysed, and incorporated radioactivity was quantified. Fatty acid oxidation assays were performed as described previously (Muoio et al., 2002). For arginase assays, 5 × 105 macrophages were plated in a 24-well plate and stimulated with IL-4 for 15 hr. Total cellular arginase activity was monitored via a colorimetric assay that detects the production of urea (Rutschman et al., 2001). Total cell number or protein content was used for normalization. For studies in which oxidative metabolism was pharmacologically inhibited or uncoupled, macrophages were pretreated with etomoxir (50 μM), oligomycin (2 μg/ml) or FCCP (50 nM) for 24 hr prior to stimulation with IL-4. All assays were performed in duplicate or triplicate and repeated at least three times.
Statistical analyses and array dataset
All statistical analyses were performed using paired Student’s t test, and results are expressed as mean ± SEM. A p value < 0.05 was considered to be significant. The data discussed in this publication have been deposited in NCBIs Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) and are accessible through GEO Series accession number GSE5003.
Supplemental data
Supplemental data include five figures and can be found with this article online at http://www.cellmetabolism.org/cgi/content/full/4/1/13/DC1/.
Supplementary Material
Acknowledgments
We thank members of the Chawla lab and C. Lee for valuable comments; A. Loh, R. Evans, R. Kraemer, and E. Butcher for critique on the manuscript; R. Wagner for assistance with microarray experiments. We also thank Y. Ohmori, J. Ihle, and A. Kakizuka for providing key reagents. This work was supported by grants made available: to A.C. by NIH (DK062386 and HL076746), AHA Western Affiliate, Rita Allen Foundation, Astellas USA Foundation, Rockefeller Brothers Fund, and by Goldman Philanthropic Partnerships; to P.J.M. by NIH grant AI062921, the Sandler Program for Asthma Research, Cancer Center CORE (P30 CA 21765), and by the American Leb-anese Syrian Associated Charities. A.C. is a Charles E. Culpeper Medical Scholar. A dean’s fellowship provided support to D.V. and L.M. All animal care was in accordance with Stanford University’s A-PLAC committee guidelines.
References
- Arnaud C, Braunersreuther V, Mach F. Toward immunomodulatory and anti-inflammatory properties of statins. Trends Cardiovasc Med. 2005;15:202–206. doi: 10.1016/j.tcm.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Binder CJ, Chang MK, Shaw PX, Miller YI, Hartvigsen K, Dewan A, Witztum JL. Innate and acquired immunity in atherogenesis. Nat Med. 2002;8:1218–1226. doi: 10.1038/nm1102-1218. [DOI] [PubMed] [Google Scholar]
- Chawla A, Barak Y, Nagy L, Liao D, Tontonoz P, Evans RM. PPAR-gamma dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat Med. 2001;7:48–52. doi: 10.1038/83336. [DOI] [PubMed] [Google Scholar]
- Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–657. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djouadi F, Brandt JM, Weinheimer CJ, Leone TC, Gonzalez FJ, Kelly DP. The role of the peroxisome proliferator-activated receptor alpha (PPAR alpha) in the control of cardiac lipid metabolism. Prostaglandins Leukot Essent Fatty Acids. 1999;60:339–343. doi: 10.1016/s0952-3278(99)80009-x. [DOI] [PubMed] [Google Scholar]
- Duval C, Chinetti G, Trottein F, Fruchart JC, Staels B. The role of PPARs in atherosclerosis. Trends Mol Med. 2002;8:422–430. doi: 10.1016/s1471-4914(02)02385-7. [DOI] [PubMed] [Google Scholar]
- Glass CK, Witztum JL. Atherosclerosis. the road ahead Cell. 2001;104:503–516. doi: 10.1016/s0092-8674(01)00238-0. [DOI] [PubMed] [Google Scholar]
- Goerdt S, Orfanos CE. Other functions, other genes: alternative activation of antigen- presenting cells. Immunity. 1999;10:137–142. doi: 10.1016/s1074-7613(00)80014-x. [DOI] [PubMed] [Google Scholar]
- Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- Gough PJ, Gordon S, Greaves DR. The use of human CD68 transcriptional regulatory sequences to direct high-level expression of class A scavenger receptor in macrophages in vitro and in vivo. Immunology. 2001;103:351–361. doi: 10.1046/j.1365-2567.2001.01256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Pablo AM, Jiang X, Wang N, Tall AR, Schindler C. IFN-gamma potentiates atherosclerosis in ApoE knock-out mice. J Clin Invest. 1997;99:2752–2761. doi: 10.1172/JCI119465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- Herbert DR, Holscher C, Mohrs M, Arendse B, Schwegmann A, Radwanska M, Leeto M, Kirsch R, Hall P, Mossmann H, et al. Alternative macrophage activation is essential for survival during schistosomiasis and downmodulates T helper 1 responses and immunopathology. Immunity. 2004;20:623–635. doi: 10.1016/s1074-7613(04)00107-4. [DOI] [PubMed] [Google Scholar]
- Ihle JN. The Stat family in cytokine signaling. Curr Opin Cell Biol. 2001;13:211–217. doi: 10.1016/s0955-0674(00)00199-x. [DOI] [PubMed] [Google Scholar]
- Lang R, Rutschman RL, Greaves DR, Murray PJ. Autocrine deactivation of macrophages in transgenic mice constitutively overexpressing IL-10 under control of the human CD68 promoter. J Immunol. 2002;168:3402–3411. doi: 10.4049/jimmunol.168.7.3402. [DOI] [PubMed] [Google Scholar]
- Lin J, Handschin C, Spiegelman BM. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005a;1:361–370. doi: 10.1016/j.cmet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, Michael LF, Puig-server P, Isotani E, Olson EN, et al. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797–801. doi: 10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- Lin J, Yang R, Tarr PT, Wu PH, Handschin C, Li S, Yang W, Pei L, Uldry M, Tontonoz P, et al. Hyperlipidemic effects of dietary saturated fats mediated through PGC-1beta coactivation of SREBP. Cell. 2005b;120:261–273. doi: 10.1016/j.cell.2004.11.043. [DOI] [PubMed] [Google Scholar]
- Loke P, Allison JP. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc Natl Acad Sci USA. 2003;100:5336–5341. doi: 10.1073/pnas.0931259100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosser DM. The many faces of macrophage activation. J Leukoc Biol. 2003;73:209–212. doi: 10.1189/jlb.0602325. [DOI] [PubMed] [Google Scholar]
- Munder M, Eichmann K, Moran JM, Centeno F, Soler G, Modolell M. Th1/Th2-regulated expression of arginase isoforms in murine macrophages and dendritic cells. J Immunol. 1999;163:3771–3777. [PubMed] [Google Scholar]
- Muoio DM, MacLean PS, Lang DB, Li S, Houmard JA, Way JM, Winegar DA, Corton JC, Dohm GL, Kraus WE. Fatty acid homeostasis and induction of lipid regulatory genes in skeletal muscles of peroxisome proliferator-activated receptor (PPAR) alpha knock-out mice. Evidence for compensatory regulation by PPAR delta. J Biol Chem. 2002;277:26089–26097. doi: 10.1074/jbc.M203997200. [DOI] [PubMed] [Google Scholar]
- Ohmori Y, Smith MF, Jr, Hamilton TA. IL-4-induced expression of the IL-1 receptor antagonist gene is mediated by STAT6. J Immunol. 1996;157:2058–2065. [PubMed] [Google Scholar]
- Pauleau AL, Rutschman R, Lang R, Pernis A, Watowich SS, Murray PJ. Enhancer-mediated control of macrophage-specific arginase I expression. J Immunol. 2004;172:7565–7573. doi: 10.4049/jimmunol.172.12.7565. [DOI] [PubMed] [Google Scholar]
- Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, Gallo RL, Hurtado-Ziola N, Nizet V, Johnson RS. HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J Clin Invest. 2005;115:1806–1815. doi: 10.1172/JCI23865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puigserver P, Rhee J, Lin J, Wu Z, Yoon JC, Zhang CY, Krauss S, Mootha VK, Lowell BB, Spiegelman BM. Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPAR-gamma coactivator-1. Mol Cell. 2001;8:971–982. doi: 10.1016/s1097-2765(01)00390-2. [DOI] [PubMed] [Google Scholar]
- Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- Rauh MJ, Ho V, Pereira C, Sham A, Sly LM, Lam V, Huxham L, Minchinton AI, Mui A, Krystal G. SHIP Represses the Generation of Alternatively Activated Macrophages. Immunity. 2005;23:361–374. doi: 10.1016/j.immuni.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Ruderman NB, Saha AK, Kraegen EW. Minireview: malonyl CoA, AMP-activated protein kinase, and adiposity. Endocrinology. 2003;144:5166–5171. doi: 10.1210/en.2003-0849. [DOI] [PubMed] [Google Scholar]
- Rutschman R, Lang R, Hesse M, Ihle JN, Wynn TA, Murray PJ. Cutting edge: Stat6-dependent substrate depletion regulates nitric oxide production. J Immunol. 2001;166:2173–2177. doi: 10.4049/jimmunol.166.4.2173. [DOI] [PubMed] [Google Scholar]
- Schonbeck U, Libby P. Inflammation, immunity, and HMG-CoA reductase inhibitors: statins as antiinflammatory agents? Circulation. 2004;109:II18–II26. doi: 10.1161/01.CIR.0000129505.34151.23. [DOI] [PubMed] [Google Scholar]
- Schroder AJ, Pavlidis P, Arimura A, Capece D, Rothman PB. Cutting edge: STAT6 serves as a positive and negative regulator of gene expression in IL-4-stimulated B lymphocytes. J Immunol. 2002;168:996–1000. doi: 10.4049/jimmunol.168.3.996. [DOI] [PubMed] [Google Scholar]
- Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med. 1992;176:287–292. doi: 10.1084/jem.176.1.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stout RD, Suttles J. Functional plasticity of macrophages: reversible adaptation to changing microenvironments. J Leukoc Biol. 2004;76:509–513. doi: 10.1189/jlb.0504272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykes DB, Kamps MP. Estrogen-dependent E2a/Pbx1 myeloid cell lines exhibit conditional differentiation that can be arrested by other leukemic oncoproteins. Blood. 2001;98:2308–2318. doi: 10.1182/blood.v98.8.2308. [DOI] [PubMed] [Google Scholar]
- Takeda K, Akira S. STAT family of transcription factors in cytokine-mediated biological responses. Cytokine Growth Factor Rev. 2000;11:199–207. doi: 10.1016/s1359-6101(00)00005-8. [DOI] [PubMed] [Google Scholar]
- Wang YX, Zhang CL, Yu RT, Cho HK, Nelson MC, Bayuga-Ocampo CR, Ham J, Kang H, Evans RM. Regulation of muscle fiber type and running endurance by PPARdelta. PLoS Biol. 2004;2:e294. doi: 10.1371/journal.pbio.0020294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willment JA, Lin HH, Reid DM, Taylor PR, Williams DL, Wong SY, Gordon S, Brown GD. Dectin-1 expression and function are enhanced on alternatively activated and GM-CSF-treated macrophages and are negatively regulated by IL-10, dexamethasone, and lipopolysaccharide. J Immunol. 2003;171:4569–4573. doi: 10.4049/jimmunol.171.9.4569. [DOI] [PubMed] [Google Scholar]
- Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- Wurster AL, Tanaka T, Grusby MJ. The biology of Stat4 and Stat6. Oncogene. 2000;19:2577–2584. doi: 10.1038/sj.onc.1203485. [DOI] [PubMed] [Google Scholar]
- Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, Rhee J, Adelmant G, Stafford J, Kahn CR, Granner DK, et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001;413:131–138. doi: 10.1038/35093050. [DOI] [PubMed] [Google Scholar]
- Zhang L, Chawla A. Role of PPARgamma in macrophage biology and atherosclerosis. Trends Endocrinol Metab. 2004;15:500–505. doi: 10.1016/j.tem.2004.10.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.