

Abstract
Macrophages participate in physiologic and pathologic processes through elaboration of distinct activation programs. Studies with macrophage cell systems have revealed much concerning the importance of this pleiotropic cell; however, these studies are inherently limited by three factors: heterogeneity of the target cell population, poor capacity to elaborate various activation programs, and lack of a genetically tractable model system for loss- and gain-of-function studies. Although definitive, hematopoietic lineages can be isolated from embryonic stem (ES) cells, these isolation procedures are inefficient and time-consuming and require elaborate cell-sorting protocols. We, therefore, examined whether myeloid precursors, capable of differentiating into macrophages, could be conditionally expanded in vitro. Here, we report methods for selective isolation and immortalization of ES cell-derived myeloid precursors by estrogen-regulated HoxA9 protein. Using this new macrophage differentiation system, an unlimited number of custom-designed macrophages with defined functional characteristics can be generated from any targeted ES cell. In combination with knockout or small interfering RNA knockdown technologies, this macrophage differentiation system provides a powerful tool for high throughput analysis of regulatory mechanisms controlling macrophage activation in health and disease.
Keywords: activation
INTRODUCTION
Macrophages, sentinels of innate immunity, comprise a heterogeneous group of cells that perform critical functions in host defense, immune surveillance, and tissue remodeling and regeneration [1]. These mononuclear phagocytes, which are activated by inflammatory, metabolic, or immune stimuli, are extremely efficient at clearing invading pathogens, cellular debris, apoptotic cells, and modified host proteins [2, 3]. In addition to their phagocytic functions, macrophages play a key role in the initiation and resolution of inflammatory responses via secretion of pro- and anti-inflammatory cytokines [3, 4].
It is interesting that recent studies indicate that immune and innate activation of macrophages contributes to the pathogenesis of various chronic inflammatory diseases. For instance, macrophages are critical effector cells that promote tissue inflammation and damage in autoimmune (e.g., multiple sclerosis), inflammatory (e.g., psoriasis), and metabolic (e.g., atherosclerosis and type 2 diabetes mellitus) diseases [5–7]. Despite a greater appreciation for the pathogenic activities of these cells in chronic inflammatory states, molecular studies of macrophage activation programs have been hampered by the lack of good, tractable genetic systems. Currently, experimental macrophages are derived from two sources: tumor-derived cell lines (e.g., RAW 267.4, J774, and THP-1 cells) and primary cells {e.g., bone marrow-derived macrophages (BMDM) and elicited peritoneal macrophages [8]}. The former source is endowed with several propitious characteristics, including an unlimited, replicative potential, genetic tractability, and population homogeneity. However, the use of macrophage-like cell lines is limited by their relative phenotypic immaturity and their inherent distance from normal biology as a result of their derivation from tumors. In contrast, BMDM and elicited-peritoneal macrophages are more reflective of quiescent and activated tissue macrophages, respectively. As such, however, the replicative potential, genetic tractability, and population homogeneity are all quite poor for these cells.
Embryonic stem (ES) cells, which can be differentiated into macrophages, provide a potential genetic system to study macrophage biology. However, conventional methods of generating ES cell-derived macrophages (ESDM) are tedious and give low yields, and the target population exhibits tremendous functional heterogeneity [9–11]. To develop a robust macrophage differentiation system, we investigated whether ES cell-derived myeloid progenitors could be isolated and expanded in vitro. Specifically, we asked whether oncoproteins, which arrest myeloid differentiation, could promote growth factor-dependent self-renewal of ES cell-derived myeloid progenitors. By testing various protocols for progenitor isolation, cytokine cocktails for progenitor renewal and differentiation, and different oncogenic proteins, we developed a simple, highly reproducible method for infinite expansion of ES-cell derived myeloid progenitors. In this report, we demonstrate that ectopic expression of estrogen-regulated HoxA9 protein (HoxA9-ER) conditionally immortalizes ES cell-derived myeloid progenitors, which are capable of differentiating synchronously into macrophages. It is important that immunologic and expression profiling of HoxA9-ER ESDM revealed a high degree of maturational and functional coherence. In contrast to other established culture systems, HoxA9-ER ESDM are cytokine-responsive and can express the macrophage programs of classical activation, alternative activation, and deactivation. Thus, these new methods provide a dramatic improvement in efficiency, purity, and homogeneity of cultured ESDM, making it an ideal system for high-throughput genetic analysis of macrophage functions.
MATERIALS AND METHODS
Derivation of ESDM cell lines
Wild-type 129/J1 ES cells or peroxisome proliferator-activated receptor (PPAR)δ, null ES cells were cultured on feeder cells as described [12, 13]. Embryoid bodies (EBs), generated as described previously [14], were harvested on Days 6–8, trypsinized, and replated in IMDM, supplemented with murine stem cell factor (mSCF; 25 ng/ml) and mIL-3 (5 ng/ml). Two days later, nonadherent, hematopoietic precursors were collected, counted, and infected with ecotropic retroviruses encoding the conditional myeloid oncoprotein, HoxA9-ER [15]. The immortalized progenitors, J1-HoxA9-ER or PPARδ, null HoxA9-ER, were expanded and maintained in proliferation medium: IMDM supplemented with 10% FCS, SCF (25 ng/ml), mIL-3 (1.25 ng/ml), and β-estradiol (1 μM). Macrophage differentiation was induced by withdrawing β-estradiol and SCF and adding 20% L929 conditioned medium, a source of M-CSF. Media were subsequently changed every 2 days, and differentiated macrophages developed over 6–7 days. Genetic rescue experiments were carried out by infecting proliferating PPARδ, null HoxA9-ER cells with ecotropic retroviruses encoding full-length Flag-PPARδ or empty vector and subjecting the selection with puromycin (4 μg/ml).
Gene expression studies
Total RNA was prepared using TRIzol reagent and used in Northern blot analysis as described previously [16]. For quantitative PCR (QPCR), total RNA was treated with DNase (1 U/ml) and reverse-transcribed using a first-strand cDNA synthesis kit, and QPCR reactions were performed on an Opticon 2 DNA engine. Primers used in these studies are: CD68 (f: GCGGTGGAATA-CAATGTGTC, r: TGGAGCTCTCGAAGAGATGA), F4/80 (f: GCATCATG-GCATACCTGTTC, r: AGTCTGGGAATGGGAGCTAA), M-CSF receptor (M-CSFR; f: ATCATGAGTCACCTGGGACA, r: CCTTCCTTCGGAGAAAGTTG), mannose receptor (MR; f: TGATTACGAGCAGTGGAAGC, r: GTTCACCGTA-AGCCCAATTT), PU.1 (f: TCTGATGGAGAAGCTGATGG, r: TTTCTTGCT-GCCTGTCTCC), and L32 (f: TGCTGATGTGCAACAAATCTT, r: GTTGG-GATTGGTGACTCTGA). QPCR data were normalized to L32 levels using the ΔΔC(t) method. Fold change was calculated relative to the expression levels of proliferating HoxA9 cells.
Protein analyses
Western blot analyses were performed using standard procedures. Cells were lysed in radioimmunoprecipitation assay buffer containing 1 mM Na2VO4, 1 protease inhibitor cocktail. Total cell lysate mM NaF, 1 mM Na4P2O8, and 1× (30 μg) was analyzed by immunoblotting for Flag-PPARδ with anti-Flag primary antibody (1:1000, Sigma Chemical Co., St. Louis, MO, USA) and β-actin (1:5000, Sigma Chemical Co.) and detected on an Odyssey infrared detection system with IRDye 800-conjugated secondary antibody (1:30,000, Rockland Immunochemicals, Gilbertsville, PA, USA). Arginase assays were preformed by well-established methodologies [16, 17]. For ELISAs, cells were plated in 48- or 96-well plates and stimulated as indicated for 6–24 h. TNF-α, IL-6, and IL-12 in culture supernatants were quantified per the manufacturer’s protocols (BD PharMingen, San Diego, CA, USA). All assays were done in triplicate, and results are indicative of at least three independent experiments. For flow cytometry, cells were washed with PBS, harvested, and resuspended in PBS containing 2% FCS and 0.2 mM EDTA. Samples were blocked with 200 μg/ml normal mIgG and stained on ice with fluro-conjugated antibodies against CD34, c-kit, stem cell antigen 1 (Sca-1), CD16/32, CD11b, F4/80, CD80, CD3, B220, Ter-119, GR-1, MR, and PD-L2 and analyzed on a Becton Dickinson LSR. For nitrite production, macrophages were stimulated with IFN-γ (100 U/ml) and LPS (5 ng/ml), and nitrite production was quantified by a Greiss reagent in culture supernatants.
Uptake studies
Macrophages were incubated with 1 mg/ml dextran-FITC (m.w. 70,000), 1 mg/ml Lucifer yellow, or 2 μm FITC-labeled latex beads in differentiation medium for 15 min at 37°C and 4°C. Cells were washed thrice with PBS, harvested, and analyzed immediately by FACS. Microbial uptake studies were performed with DH5α Escherichia coli, Salmonella typhimurium strain SLI344 (Stanley Falkow, Stanford University, CA, USA), or Candida albicans clinical isolate SC5314 [Alexander “Sandy” Johnson, University of California San Francisco (UCSF), USA]. Early log-phase cultures were incubated in light-brown (E. coli and S. typhimurium) or yeast-peptone-dextrose (C. albicans) for 15 min with 5 μM 5-chloromethylfluorescein diacetate (CMFDA; Molecular Probes, Eugene, OR, USA), washed thrice with PBS, and added directly to macrophage cultures at a microbe:macrophage ratio of 10:1 for 15 min at 37°C.
Allogeneic lymphocyte stimulation
HoxA9 macrophages were cultured in triplicate in a 96-well flat-bottom plate at the indicated numbers overnight in medium containing IFN-γ (10 U/ml). Wells were washed thrice with PBS, and 105 BALB/c allogeneic or SV129 syngeneic splenocytes in DMEM containing 10% FCS were added to each well. After 72 h, the cultures were pulsed with 0.5 μCi/well [3H]methyl-thymidine for 16 h, harvested onto glass fiber filters using the TomTec MachIII plate harvester, and counted in the Wallac 1450 Microbeta/TriLux detector. Proliferation is reported as 3H CPMwell – CPMbackground.
RESULTS
Conditional immortalization of ES-derived myeloid precursors by HoxA9-ER
Lineage-specific hematopoietic precursors can be isolated from EBs that arise from spontaneously differentiating ES cells [9]. Although populations of definitive hematopoietic lineages, including macrophages and neutrophils, can be obtained from EBs, erratic yields and impurity in cell populations plague these methodologies. One potential way to overcome these hurdles is to immortalize the ES-derived myeloid precursors with a conditional oncoprotein, thereby permitting controllable expansion and synchronous differentiation of myeloid precursors. To test this concept, EBs were disrupted at various stages of differentiation and cultured in the presence of SCF (25 ng/ml) and IL-3 (5 ng/ml), cytokines known to promote the growth of myeloid precursors [18]. We focused on isolating myeloid precursors from Days 6–8 EBs, as previous work had established that definitive hematopoiesis begins at this time. As schematically outlined in Figure 1, nonadherent hematopoietic precursors were collected 2 days later and were infected with ecotropic retroviruses encoding the conditional myeloid oncoprotein HoxA9-ER or E2a-ER-Pbx1 [15]. It is notable that although infection with E2a-ER-Pbx1 failed to establish a stable macrophage differentiation system (data not shown), transduction of Day 8 hematopoietic precursors with HoxA9-ER resulted in factor-dependent renewal of myeloid progenitors. These progenitors, termed J1-HoxA9-ER, exhibited an absolute dependence on growth factors (SCF and IL-3) and β-estradiol for proliferation and M-CSF for differentiation into macrophages.
Fig. 1.
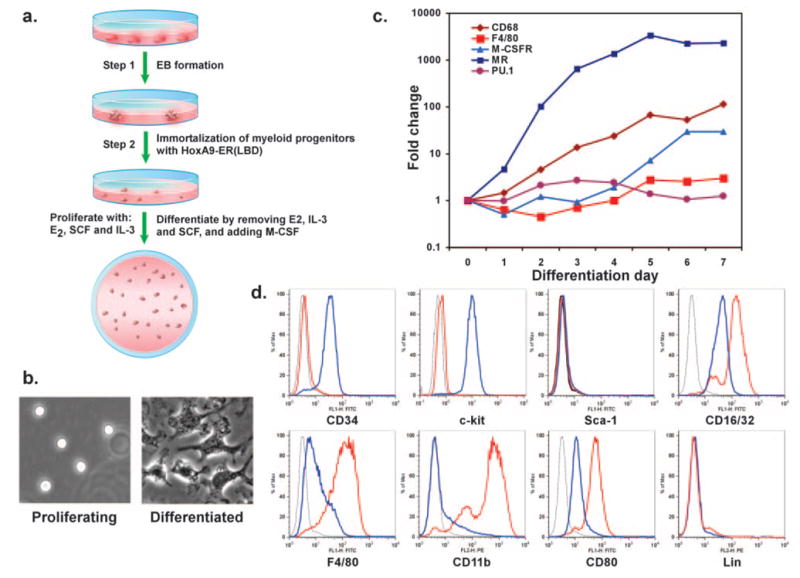
Generation of macrophage differentiating system by conditional immortalization of ES-derived myeloid progenitors. (a) Schematic representation for isolating and immortalizing ES-derived myeloid progenitors. (b–d) Differentiation of ES-derived myeloid progenitors yields mature macrophages, as assessed by morphology (b), progressive changes in gene expression (c), and expression of cell surface markers (d). For flow cytometry histograms, isotype-stained cells are represented by the thin, black line; proliferating cells by the blue line; and differentiated cells by the red line. LBD, Ligand-binding domain; E2, estradiol; Lin, lineage.
HoxA9-ER-immortalized ES cell-derived myeloid precursors faithfully express the macrophage differentiation program
To investigate whether the J1-HoxA9-ER myeloid progenitors faithfully executed macrophage differentiation, we performed experiments to delineate the morphologic characteristics and gene expression profiles of these cells. In contrast to proliferating myeloid precursors, which were highly refractile and nonadherent, M-CSF induced morphologic maturation into macrophages, as evidenced by increased adherence to tissue-culture plates, fibroblastoid appearance, and process extension (Fig. 1b). Terminal differentiation into macrophages was accompanied by cessation of cellular division, as assessed by serial cell counts and [3H]-methyl-thymidine incorporation (data not shown). Moreover, QPCR analyses revealed a pattern characteristic of ongoing macrophage differentiation. For instance, mRNAs, encoding macrophage-specific genes CD68 and F4/80 and the M-CSFR, increased by 114-, three-, and 29-fold, respectively, during the 7 days of differentiation (Fig. 1c) [19]. In contrast, expression of the myeloid-related transcription factor PU.1 did not change significantly, consistent with the published findings in BMDM (Fig. 1c). To further discern the differences between proliferating and differentiated J1-HoxA9-ER cells, we performed flow cytometric analyses for markers known to be present on myeloid progenitors or macrophages. Proliferating J1-HoxA9-ER cells expressed the myeloid progenitor markers (CD34, c-kit, and FcRγ) but were negative for the hematopoietic stem cell marker Sca-1 and other lineage markers (Gr1, CD3, B220, and Ter-119; Fig. 1d) [20, 21]. It is notable that maturation of J1-HoxA9-ER cells into macrophages was accompanied by down-regulation of progenitor markers (CD34: 7.4-fold; c-kit: 16-fold) and up-regulation of markers of mature macrophages (F4/80: 7.4-fold; CD11b: 28.4-fold; CD16/32: 3.5-fold; CD80: threefold), as shown in Figure 1d. Although the cell surface phenotype of J1-HoxA9-ER progenitors is similar to the one ascribed to common myeloid progenitors, these immortalized progenitors only gave rise to macrophage-containing colonies in the in vitro colony-forming assays (data not shown), in agreement with the reported oncogenic activity of HoxA9 in acute myeloid leukemia and adult bone marrow progenitors [15, 22].
Intact IFN-γ signaling and classical activation in HoxA9-immortalized ESDM
Many extant macrophage-like cell lines lack the ability to activate appropriately when stimulated with IFN-γ [23], and as such, their use in studies of macrophage biology is limited. To determine whether the J1-HoxA9-ER macrophages can enact the program of classical activation, these cells were stimulated with Th1 cytokine IFN-γ and assayed for expression of the proinflammatory phenotype. Immunoblots in Figure 2a showed that treatment of cells with IFN-γ led to robust and rapid phosphorylation of STAT1 and strong up-regulation of key proinflammatory mRNAs, including TNF-γ, IL-6, and inducible NO synthase (iNOS; Fig. 2b). Consistent with the known inflammatory priming effects of IFN-γ, J1-HoxA9-ER macrophages, costimulated with IFN-γ and LPS, had a marked increase in nitrite production (Fig. 2c), phlogistic cytokine response (Fig. 2d), and cell surface expression of costimulatory molecule PD-L1 (Fig. 2e) [3].
Fig. 2.
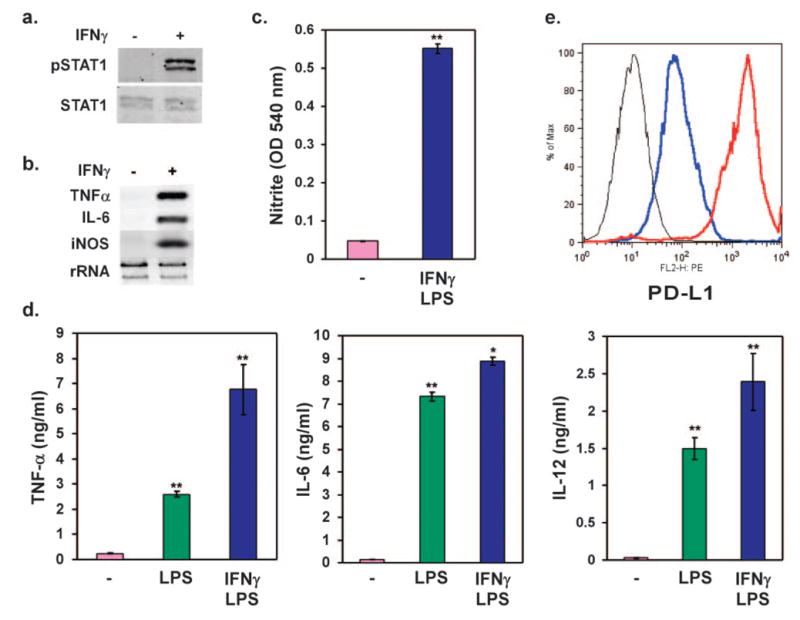
J1-HoxA9-ER macrophages undergo classical activation in response to IFN-γ. (a) Stimulation with IFN-γ leads to robust phosphorylation of STAT1. Mature HoxA9 ESDM were stimulated for 30 min with IFN-γ (100 u/ml), and total cellular protein was probed for pY701 STAT1 (pSTAT1) and total STAT1. (b) IFN-γ induces inflammatory gene expression in HoxA9 ESDM. Cells were stimulated with IFN-γ (100 U/ml) for 24 h, and gene expression analyses were performed by Northern blotting. (c, d) Combination of IFN-γ (100 u/ml) and LPS (5 ng/ml) promotes classical activation of HoxA9 ESDM. Nitrite production in culture supernatants (c) or inflammatory cytokine secretion (d) was monitored by Griess reagent and ELISAs, respectively. (e) Induction of cell surface expression of PD-L1 by IFN-γ in HoxA9-immortalized ESDM. Isotype, Black; vehicle, blue; IFN-γ, red. Results are representative of at least three different independent experiments.*, P = 0.04; **, P < 0.005.
Th2 cytokines promote alternative activation and deactivation of HoxA9-immortalized macrophages
Although a Th1 cytokine-driven program of classical activation provides a defense against intracellular bacteria and viruses, Th2 cytokines IL-4 and IL-13 promote alternative macrophage activation during parasitic infections. Furthermore, as polarization of macrophages toward the alternative state can attenuate Th1 inflammation, this program of macrophage activation holds great therapeutic promise for treating chronic inflammatory diseases. However, many of the commonly used macrophage model systems do not fully express the alternative phenotype. For instance, IL-4 fails to up-regulate the costimulatory protein PD-L2 or enhance arginase activity dramatically in RAW264.7 cells, two cardinal features of alternative activation [24]. To determine whether J1-HoxA9-ER macrophages can undergo alternative activation, we examined the IL-4 signaling pathways in these cells. It is remarkable that stimulation with IL-4 led to rapid phosphorylation of STAT6 (Fig. 3a) and marked induction of mRNAs for MR, arginase I, and dectin-1 (Fig. 3b), gene products specifically expressed by alternatively activated macrophages. Mirroring these changes in mRNA expression, stimulation of J1-HoxA9-ER macrophages with IL-4 led to a strong increase in arginase activity by ~30-fold (Fig. 3c) and dramatically enhanced cell surface expression of MR and dectin-1 by 2.2- and threefold, respectively (Fig. 3d). PD-L2, a high-stringency marker of macrophage alternative activation, is not expressed by any extant macrophage cell line in response to IL-4. It is remarkable that the Th2 cytokine IL-4 enhanced cell surface PD-L2 expression by threefold in J1-HoxA9-ER macrophages (Fig. 3d) [24]. Thus, unlike the existing macrophage cell lines, such as RAW264.7 or J774, the conditionally immortalized, ES-derived macrophages can faithfully execute the cellular programs of classical and alternative activation.
Fig. 3.
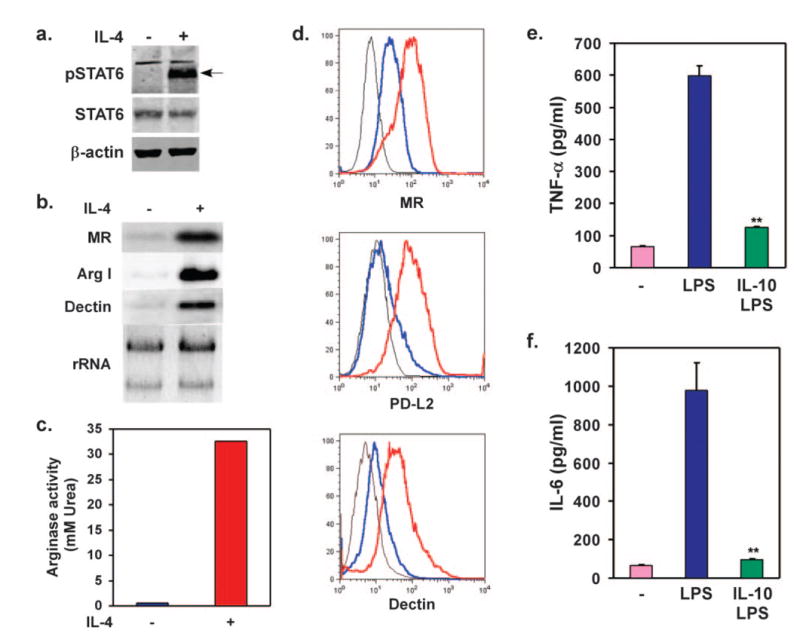
J1-HoxA9-ER macrophages undergo alternative activation in response to IL-4. (a) Treatment with IL-4 (10 ng/ml for 30 min) induces phosphorylation of STAT6 in HoxA9 ESDM. (b) IL-4 stimulates expression of mRNAs associated with the alternative activation phenotype. (c) HoxA9-immortalized ESDM induces arginase activity upon stimulation with IL-4 (10 ng/ml) for 24 h. (d) Cell surface expression of alternative activation markers. Flow cytometric analyses were performed on macrophages treated with vehicle or IL-4 (10 ng/ml) for 24 h. Isotype control, Black; vehicle, blue; IL-4, red. (e, f) IL-10 potently inhibits inflammatory activation of J1-HoxA9-ER macrophages. Cells were treated with LPS (5 ng/ml) in the presence or absence of IL-10 (10 ng/ml), and cytokine release was monitored by ELISAs. Results are representative of at least three different independent experiments. **, P < 0.005.
In contrast to IL-4, the Th2 cytokine IL-10 potently inhibits antigen presentation and inflammatory activation of macrophages [25]. STAT3, a member of the STAT family of transcription factors, mediates the biological effects of IL-10 in myeloid cells [26]. Congruent with these findings, tissue-specific deletion of STAT3 in myeloid cells or global deletion of IL-10 in mice results in inflammatory activation of macrophages, severe enterocolitis, and early postnatal mortality [27, 28]. To investigate whether ESDM can respond appropriately to IL-10, J1-HoxA9-ER macrophages were challenged with LPS in the presence or absence of IL-10, and secretion of proinflammatory cytokines was monitored by ELISAs. It is striking that IL-10 potently suppressed the release of TNF-α and IL-6 from J1-HoxA9-ER macrophages, indicating intact IL-10 signaling in these cells.
HoxA9-immortalized ESDM can perform essential macrophage functions
Resident and elicited macrophages continually sample their local environment. Large volumes of fluid phase can be ingested by pinocytosis, whereas endocytosis and phagocytosis mediate engulfment of larger particles, cellular debris, and invading microorganisms [29]. To determine whether the J1-HoxA9-ER macrophages were capable of performing these essential macrophage functions, cells were incubated with labeled substrates at 4°C or 37°C, and the increase in fluorescence was monitored by flow cytometry. As shown in Figure 4a, incubation of J1-HoxA9-ER macrophages at 37°C with Lucifer yellow or dextran-FITC (fluorescent molecules used to monitor pinocytosis and endocytosis, respectively) led to a strong increase in cellular fluorescence. Similarly, although J1-HoxA9-ER macrophages phagocytosed FITC-labeled latex beads rapidly, physiologically relevant phagocytosis was demonstrated by uptake of CMFDA-labeled Candida, E. coli, and S. typhimurium (Fig. 4a and data not shown).
Fig. 4.
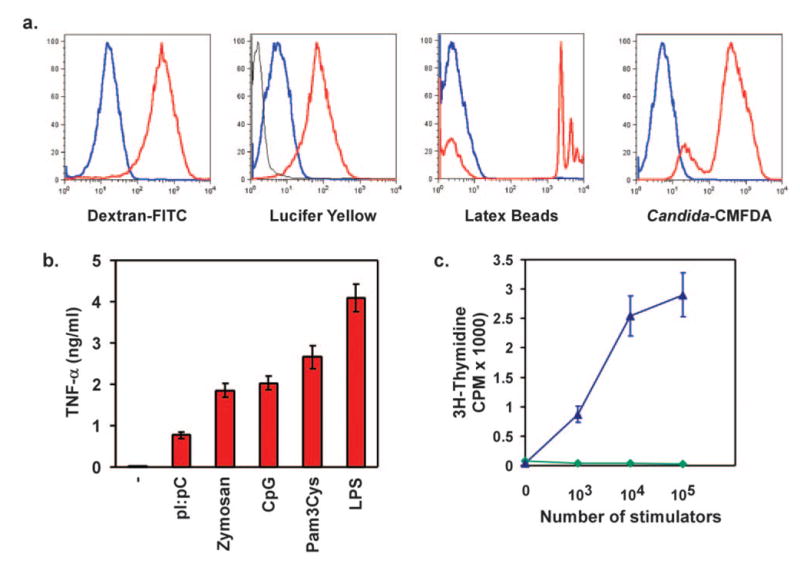
J1-HoxA9-ER macrophages are functionally patent for antigen uptake, identification, and presentation. (a) Cells were incubated with labeled endocytic, pinocytic, and phagocytic substrates for 15 min, and specific uptake (red) was followed by fluorescence gain over-reactions carried out at 4°C (blue). (b) TLR ligation induces TNF-α secretion in HoxA9-immortalized ESDM. Cells were stimulated with tripalmitoyl-S-glycero-Cys-(Lys)4 (Pam3Cys; 100 ng/ml), polyinosinic: polycytidylic (pI:pC; 25 μg/ml), CpG DNA (1 μM), zymosan (1 μg/ml), or LPS (10 ng/ml) for 6 h, and TNF-α secretion was monitored in supernatants by ELISA. (c) MLR between HoxA9-immortalized ESDM and allogenic or syngeneic splenocytes. Allogeneic (blue, ▴) or syngeneic (green,♦) splenocytes (105) were added to HoxA9 macrophages activated overnight with IFN-γ (10 U/ml). Cellular proliferation was monitored 72 h later with [3H]methyl-thymidine.
TLRs, which recognize pathogen-associated products, initiate signaling events that culminate in activation of innate antimicrobial defenses [30, 31]. For instance, ligation of TLR2, TLR3, and TLR4 by zymosan, dsRNA (pI:pC), and LPS, respectively, initiates not only phagocytosis of the microbe [32] but also the immune response by promoting the release of proinflammatory cytokines and chemokines. Thus, we next examined if J1-HoxA9-ER macrophages were capable of recognizing and responding to pathogen-associated products. Indeed, in a manner analogous to primary BMDM, stimulation of immortalized ESDM resulted in inflammatory activation of macrophages, as assessed by secretion of TNF-α and IL-6 (Fig. 4b and data not shown).
Last, in addition to orchestrating the cytokine milieu of the inflammatory response, macrophages bridge the innate and adaptive immune systems by presenting antigen for adaptive activation. The ability of J1-HoxA9-ER macrophages to activate adaptive immune responses was assessed by monitoring alloreactivity in a MLR. Data presented in Figure 4c show that IFN-γ-activated J1-HoxA9-ER macrophages supported robust proliferation of allogeneic, but not syngeneic, lymphocytes (Fig. 4c). In aggregate, these results suggest that the HoxA9-immortalized ESDM are not only adept at performing the critical functions of innate immunity but can also activate the adaptive immune response efficiently.
Generation of genetically engineered macrophages from targeted ES cells
As homologous recombination can be used to introduce deletions or mutations in ES cells, our procedures for isolating and immortalizing myeloid progenitors from EBs can potentially be used to generate large quantities of genetically modified macrophages. To test this hypothesis, we took advantage of genetically targeted ES cells to generate a line of PPARδ, null ESDM. Macrophages differentiated from these lines were indistinguishable from their wild-type counterparts in surface marker expression and in transcriptional programs of differentiation (data not shown). These cells demonstrated a lack of sensitivity to the synthetic PPARδ ligand GW501516, as assessed by induction of adipose differentiation-related protein (ADRP; Fig. 5b), a well-characterized PPARδ target gene [13]. To demonstrate that functional transgenes can be expressed ectopically in PPARδ, null HoxA9-ER macrophages, epitope-tagged PPARδ or empty vector was introduced retro-virally into proliferating PPARδ , null HoxA9-ER progenitors, and infected cells were subjected to selection by puromycin. Immunoblot analysis of selected cells with Flag antibody revealed robust expression of Flag-tagged PPARδ in the PPARδ , null HoxA9-ER macrophages. It is important that responsiveness to GW501516 was restored in PPARδ , null ESDM expressing Flag-PPARδ , as evidenced by ligand inducibility of ADRP mRNA expression. Collectively, these studies indicate that immortalized ESDM can be generated from genetically modified ES cells and used for transgene expression, thus making this model system ideal for biological pathway discovery by genetic manipulation.
Fig. 5.
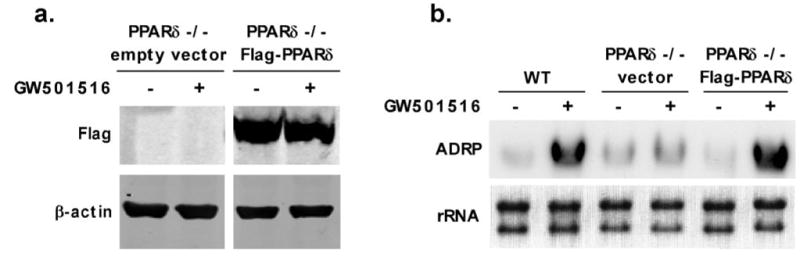
Genetic tractability of HoxA9-immortalized ESDM. (a) Rescue of PPAR−/− ESDM by ectopic expression of the Flag-PPARδ. Immunoblots showing expression of Flag-PPARδ in PPARδ, null ESDM. (b) Ectopic expression of PPARδ confers ligand responsiveness to PPARδ, null ESDM. Mature ESDM were stimulated with synthetic PPARδ ligand GW501516 (100 nM) for 24 h, and ADRP mRNA expression was monitored by Northern blot analysis. WT, Wild-type.
DISCUSSION
In this report, we describe a method for generating a macrophage differentiation system, which provides a convenient means of producing an unlimited number of genetically engineered macrophages, from mouse ES cells. This system capitalizes on the selective isolation and conditional immortalization of myeloid precursors from differentiating EBs, which can give rise to quiescent, terminally differentiated macrophages. Unlike many tumor-derived macrophage cell lines, macrophages obtained in this manner retain their full capacity to respond to the Th1 and Th2 cytokines. Last, as gene targeting by homologous recombination can be performed in murine ES cells, our novel macrophage differentiation system permits the generation of genetically modified macrophages without the need for whole animal-genetic manipulation.
Previous reports have described methodologies for isolating various definitive hematopoietic lineages, including macrophages, from spontaneously differentiating EBs [10, 14, 33]. However, lack of cellular homogeneity, poor yields, and the elaborate protocols for culturing EBs make these procedures unsuitable for generating large quantities of macrophages for biochemical and molecular studies. Thus, to fully exploit the genetic tractability of murine ES cells, methodologies are needed that facilitate the conditional expansion of myeloid precursor cells in vitro. As deregulated expression of the Hox family of transcription factors can lead to a block in hematopoiesis or expansion of hematopoietic stem cells [15, 22, 34], we investigated whether the conditional oncoprotein HoxA9-ER could arrest myeloid differentiation in the ES cell-derived hematopoietic precursors. As reported here, ectopic expression of HoxA9-ER leads to self-renewal of ES cell-derived myeloid precursors, which when stimulated with M-CSF, can terminally differentiate into macrophages. This new methodology for generating ESDM is superior to the conventional protocols in three different ways. First, rather than having to isolate myeloid progenitor cells repeatedly from EBs, these new methods allow for indefinite expansion of the myeloid progenitor cell population. In fact, the J1-HoxA9-ER-proliferating cells have been maintained stably in culture for over 3 years (>400 passages). Second, although the conventional protocols yield 10–20 million macrophages/week [14], we can routinely obtain 200–300 million macrophages/week, reflecting a 10- to 15-fold increase in the efficiency of the differentiation process. Third, our new methodology yields a homogenous population of cells that displays much better activation coherence, a point of great importance in dissecting the regulatory mechanisms controlling macrophage activation.
Unlike the tumor-derived or virally transformed macrophage cell lines [8, 35], HoxA9-immortalized ES cell progenitors retain many of the cellular and functional characteristics of primary monocytes and macrophages. For instance, although IL-3 is required for proliferation of ES cell-derived myeloid progenitors, M-CSF is essential for terminal differentiation and survival of ESDM. Furthermore, analyses of gene expression by QPCR and flow cytometry revealed a high degree of phenotypic and maturational coherence between ESDM and BMDM (Fig. 1). The ability of ESDM to faithfully recapitulate the classical, alternative, and deactivation phenotypes of BMDM further affirms the superiority of this macrophage differentiation system over other commonly used tumor-derived lines (Figs. 2 and 3). For example, treatment of HoxA9-immortalized ESDM with IL-4 led to robust expression of the complete, alternatively activated phenotype (Fig. 3), an activation program that is only partially expressed by tumor-derived macrophage cell lines. Furthermore, microarray analyses of classical or alternatively activated ESDM demonstrated the canonical cytokine responses [16]. Taken together, these results suggest that our novel ES cell-derived macrophage differentiation system provides a means to generate large numbers of genetically defined macrophages, which retain the salient features of quiescent and activated BMDM.
Introduction of heterologous genes or small interfering (si)RNA-mediated knockdown of endogenous proteins is a powerful tool in understanding the function of a particular gene in macrophage biology. As HoxA9-ER progenitors have an infinite replicative capacity, infection of proliferating cells with transgene-encoding retroviruses can be performed easily (Fig. 5). Indeed, we recently reported gain- and loss-of-function studies with the transcriptional proliferator-activated receptor-γ coactivator (PGC)-1β in J1-HoxA9-ER macrophages [16]. It is notable that transgenic expression of PGC-1β in mice, via the CD68 promoter, yielded results that were similar to those obtained with J1-HoxA9-ER macrophages, thus validating the use of our novel macrophage differentiation system for rapid analysis of gene function in macrophage biology.
Finally, testing macrophage-specific promoters and transgenic constructs is an important area where this novel macrophage differentiation system can be exploited. The standard methods used to generate transgenic mice, including pronuclear injection, founder screening, and transgene expression analysis, are costly and time-consuming [12]. As transgenic constructs can be introduced easily into ES cells, the methods outlined in this manuscript provide an excellent means of identifying genetically engineered ES cells, which express the desired transgene in macrophages. By initially screening for transgene expression in HoxA9-immortalized ESDM, one can be assured that ES cells injected into blastocysts will give rise to chimeric mice fully capable of transgene expression. This adaptation of the transgenic technology not only provides a reliable means of generating transgenic mice but also a genetically identical macrophage differentiation system that can be manipulated further by siRNA or overexpression to dissect regulatory cascades governing macrophage activation.
Acknowledgments
This work was supported by grants made available to A. C. by National Institutes of Health (DK062386 and HL076746), AHA Western Affiliate, Rita Allen Foundation, Rockefeller Brothers Fund, and Goldman Philanthropic Partnerships. A. C. is a Charles E. Culpeper Medical Scholar. J. I. O. was supported by Stanford Medical Scientist Training Program and R. R-G. by National Research Service Award fellowship (AI066402); a dean’s fellowship provided support to D. V. All animal care was in accordance with Stanford University’s Administrative Panels on Laboratory Animal Care committee guidelines. We thank members of the Chawla lab for valuable comments and A. Loh for critique of the manuscript. We also thank Stanley Falkow (Stanford University) for S. typhimurium strain SLI344 and Alexander “Sandy” Johnson (UCSF) for the clinical isolate of C. albicans SC5314.
References
- 1.Gordon S, Fraser I, Nath D, Hughes D, Clarke S. Macrophages in tissues and in vitro. Curr Opin Immunol. 1992;4:25–32. doi: 10.1016/0952-7915(92)90119-y. [DOI] [PubMed] [Google Scholar]
- 2.Stout RD, Suttles J. Functional plasticity of macrophages: reversible adaptation to changing microenvironments. J Leukoc Biol. 2004;76:509–513. doi: 10.1189/jlb.0504272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 4.Goerdt S, Orfanos CE. Other functions, other genes: alternative activation of antigen- presenting cells. Immunity. 1999;10:137–142. doi: 10.1016/s1074-7613(00)80014-x. [DOI] [PubMed] [Google Scholar]
- 5.Izikson L, Klein RS, Charo IF, Weiner HL, Luster AD. Resistance to experimental autoimmune encephalomyelitis in mice lacking the CC chemokine receptor (CCR)2. J Exp Med. 2000;192:1075–1080. doi: 10.1084/jem.192.7.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Glass CK, Witztum JL. Atherosclerosis. The road ahead. Cell. 2001;104:503–516. doi: 10.1016/s0092-8674(01)00238-0. [DOI] [PubMed] [Google Scholar]
- 7.Wang H, Peters T, Kess D, Sindrilaru A, Oreshkova T, Van Rooijen N, Stratis A, Renkl AC, Sunderkotter C, Wlaschek M, Haase I, Scharffetter-Kochanek K. Activated macrophages are essential in a murine model for T cell-mediated chronic psoriasiform skin inflammation. J Clin Invest. 2006;116:2105–2114. doi: 10.1172/JCI27180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walker WS. Establishment of mononuclear phagocyte cell lines. J Immunol Methods. 1994;174:25–31. doi: 10.1016/0022-1759(94)90007-8. [DOI] [PubMed] [Google Scholar]
- 9.Keller G, Kennedy M, Papayannopoulou T, Wiles MV. Hematopoietic commitment during embryonic stem cell differentiation in culture. Mol Cell Biol. 1993;13:473–486. doi: 10.1128/mcb.13.1.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moore KJ, Fabunmi RP, Andersson LP, Freeman MW. In vitro-differentiated embryonic stem cell macrophages: a model system for studying atherosclerosis-associated macrophage functions. Arterioscler Thromb Vasc Biol. 1998;18:1647–1654. doi: 10.1161/01.atv.18.10.1647. [DOI] [PubMed] [Google Scholar]
- 11.Anderson JS, Bandi S, Kaufman DS, Akkina R. Derivation of normal macrophages from human embryonic stem (hES) cells for applications in HIV gene therapy. Retrovirology. 2006;3:24. doi: 10.1186/1742-4690-3-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagy A, Gerstsenstein M, Vintersten K, Behringer R. Manipulating the Mouse Embryo: A Laboratory Manual. Cold Spring Harbor, NY, USA: Cold Spring Harbor; 2003. [Google Scholar]
- 13.Chawla A, Lee C, Barak Y, He W, Rosenfeld J, Liao D, Han J, Kang H, Evans R. PPAR δ is a very low-density lipoprotein sensor in macrophages. Proc Natl Acad Sci USA. 2003;100:1268–1273. doi: 10.1073/pnas.0337331100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chawla A, Barak Y, Nagy L, Liao D, Tontonoz P, Evans RM. PPAR-γ dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat Med. 2001;7:48–52. doi: 10.1038/83336. [DOI] [PubMed] [Google Scholar]
- 15.Calvo KR, Sykes DB, Pasillas M, Kamps MP. Hoxa9 immortalizes a granulocyte-macrophage colony-stimulating factor-dependent promyelocyte capable of biphenotypic differentiation to neutrophils or macrophages, independent of enforced meis expression. Mol Cell Biol. 2000;20:3274–3285. doi: 10.1128/mcb.20.9.3274-3285.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, Greaves DR, Murray PJ, Chawla A. Oxidative metabolism and PGC-1β attenuate macrophage-mediated inflammation. Cell Metab. 2006;4:13–24. doi: 10.1016/j.cmet.2006.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rutschman R, Lang R, Hesse M, Ihle JN, Wynn TA, Murray PJ. Cutting edge: Stat6-dependent substrate depletion regulates nitric oxide production. J Immunol. 2001;166:2173–2177. doi: 10.4049/jimmunol.166.4.2173. [DOI] [PubMed] [Google Scholar]
- 18.Sykes DB, Kamps MP. Estrogen-dependent E2a/Pbx1 myeloid cell lines exhibit conditional differentiation that can be arrested by other leukemic oncoproteins. Blood. 2001;98:2308–2318. doi: 10.1182/blood.v98.8.2308. [DOI] [PubMed] [Google Scholar]
- 19.McKnight AJ, Gordon S. Membrane molecules as differentiation antigens of murine macrophages. Adv Immunol. 1998;68:271–314. doi: 10.1016/s0065-2776(08)60562-3. [DOI] [PubMed] [Google Scholar]
- 20.Akashi K, Traver D, Miyamoto T, Weissman IL. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 2000;404:193–197. doi: 10.1038/35004599. [DOI] [PubMed] [Google Scholar]
- 21.Traver D, Miyamoto T, Christensen J, Iwasaki-Arai J, Akashi K, Weissman IL. Fetal liver myelopoiesis occurs through distinct, prospectively isolatable progenitor subsets. Blood. 2001;98:627–635. doi: 10.1182/blood.v98.3.627. [DOI] [PubMed] [Google Scholar]
- 22.Thorsteinsdottir U, Mamo A, Kroon E, Jerome L, Bijl J, Lawrence HJ, Humphries K, Sauvageau G. Overexpression of the myeloid leukemia-associated Hoxa9 gene in bone marrow cells induces stem cell expansion. Blood. 2002;99:121–129. doi: 10.1182/blood.v99.1.121. [DOI] [PubMed] [Google Scholar]
- 23.Stojdl DF, Lichty B, Knowles S, Marius R, Atkins H, Sonenberg N, Bell JC. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat Med. 2000;6:821–825. doi: 10.1038/77558. [DOI] [PubMed] [Google Scholar]
- 24.Loke P, Allison JP. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc Natl Acad Sci USA. 2003;100:5336–5341. doi: 10.1073/pnas.0931259100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moore KW, O’Garra A, de Waal Malefyt R, Vieira P, Mosmann TR. Interleukin-10. Annu Rev Immunol. 1993;11:165–190. doi: 10.1146/annurev.iy.11.040193.001121. [DOI] [PubMed] [Google Scholar]
- 26.Takeda K, Akira S. STAT family of transcription factors in cytokine-mediated biological responses. Cytokine Growth Factor Rev. 2000;11:199–207. doi: 10.1016/s1359-6101(00)00005-8. [DOI] [PubMed] [Google Scholar]
- 27.Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 28.Takeda K, Clausen BE, Kaisho T, Tsujimura T, Terada N, Forster I, Akira S. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. 1999;10:39–49. doi: 10.1016/s1074-7613(00)80005-9. [DOI] [PubMed] [Google Scholar]
- 29.Aderem A, Underhill DM. Mechanisms of phagocytosis in macrophages. Annu Rev Immunol. 1999;17:593–623. doi: 10.1146/annurev.immunol.17.1.593. [DOI] [PubMed] [Google Scholar]
- 30.O’Neill LA. The interleukin-1 receptor/Toll-like receptor superfamily: signal transduction during inflammation and host defense. Sci STKE 2000. 2000:RE1. doi: 10.1126/stke.442000re1. [DOI] [PubMed] [Google Scholar]
- 31.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 32.Blander JM, Medzhitov R. Regulation of phagosome maturation by signals from Toll-like receptors. Science. 2004;304:1014–1018. doi: 10.1126/science.1096158. [DOI] [PubMed] [Google Scholar]
- 33.Moore KJ, Rosen ED, Fitzgerald ML, Randow F, Andersson LP, Altshuler D, Milstone DS, Mortensen RM, Spiegelman BM, Freeman MW. The role of PPAR-γ in macrophage differentiation and cholesterol uptake. Nat Med. 2001;7:41–47. doi: 10.1038/83328. [DOI] [PubMed] [Google Scholar]
- 34.Antonchuk J, Sauvageau G, Humphries RK. HOXB4-induced expansion of adult hematopoietic stem cells ex vivo. Cell. 2002;109:39–45. doi: 10.1016/s0092-8674(02)00697-9. [DOI] [PubMed] [Google Scholar]
- 35.Blasi E, Radzioch D, Merletti L, Varesio L. Generation of macrophage cell line from fresh bone marrow cells with a myc/raf recombinant retrovirus. Cancer Biochem Biophys. 1989;10:303–317. [PubMed] [Google Scholar]