

Abstract
GAP1IP4BP is a member of the GAP1 family of Ras GTPase-activating proteins (Ras GAPs) that includes GAP1m, CAPRI, and RASAL. Composed of a central Ras GAP domain, surrounded by amino-terminal C2 domains and a carboxyl-terminal pleckstrin homology/Bruton’s tyrosine kinase domain, GAP1IP4BP has previously been shown to possess an unexpected GAP activity on the Ras-related protein Rap, besides the predicted Ras GAP activity (Cullen, P. J., Hsuan, J. J., Truong, O., Letcher, A. J., Jackson, T. R., Dawson, A. P., and Irvine, R. F. (1995) Nature 376, 527–530). Here we have shown that GAP1IP4BP is indeed an efficient Ras/Rap GAP, having Kms of 213 and 42 μM and estimated kcats of 48 and 16 s−1 for Ras and Rap, respectively. For this dual activity, regions outside the Ras GAP domain are required, as the isolated domain (residues 291–569) retains a pronounced Ras GAP activity yet has very low activity toward Rap. Interestingly, mutagenesis of the Ras GAP argi-nine finger, and surrounding residues important in Ras binding, inhibit both Ras and Rap GAP activity of GAP1IP4BP. Although the precise details by which GAP1IP4BP can function as a Rap GAP remain to be determined, these data are consistent with Rap associating with GAP1IP4BP through the Ras-binding site within the Ras GAP domain. Finally, we have established that such dual Ras/Rap GAP activity is not restricted to GAP1IP4BP. Although GAP1m appears to constitute a specific Ras GAP, CAPRI and RASAL display dual activity. For CAPRI, its Rap GAP activity is modulated upon its Ca2+-induced association with the plasma membrane.
The Ras-like family of small GTPases includes, in addition to the “classic” Ras proteins H-Ras, N-Ras, and K-Ras4A and 4B, the Rap proteins Rap1A, 1B, 2A, and 2B (1–3). These ubiquitously expressed, evolutionarily conserved proteins couple extracellular signals to various cellular responses through an ability to undergo conformational changes in response to the alternate binding of GDP and GTP. The GDP-bound “off” state and the GTP-bound “on” state recognize distinct effector proteins, thereby allowing these proteins to function as binary molecular switches (1–3). Although Ras is the best known and best studied member of the family, Rap1A and 1B have recently attracted considerable attention (3–6).
Rap1 was identified as a protein that could suppress the transformed phenotype of fibroblasts oncogenically transformed by one of the mutated Ras genes, K-ras (7). This coupled with Rap1 having an effector domain virtually identical to that of Ras led to a model in which Rap1 was considered to function as an antagonist of Ras by trapping Ras effectors in an inactive complex (4). However, recent analysis has suggested that Rap1 may possess more complex biological functions. Active Rap1 has been implicated in several cellular processes, including super-oxide formation, cyclic adenosine monophosphate (cAMP)-induced neurite outgrowth, cell proliferation, integrin-mediated cell adhesion, and secretion (3–6).
As with other small GTPases, the extent and duration of Ras and Rap1 signaling is controlled by the interplay between guanine-nucleotide exchange factors (GEFs),3 which induce activation by stimulating the exchange of GDP for GTP, and GTPase-activating proteins (GAPs), which modulate inactivation by enhancing the intrinsic GTPase activity. A wide range of GEFs and GAPs for these GTPases have been identified, and in a few instances the mechanisms by which signals originating from activated cell surface receptors converge on these proteins have been mapped. However, for most Ras-like GEFs and GAPs we have a poor understanding of their regulation (8–10).
In mammalian cells, Ras-specific GAPs include p120GAP, NF1, GAP1 proteins, and the SynGAPs (DAB2IP, nGAP, and Syn GAP). Rap1-specific GAPs comprise Rap GAPs I and II, the SPA-1 family (SPA-1, SPAR, SPAL, E6TP1), tuberin, and DOCK4 (Ref. 11, reviewed in Ref. 8). Although Rap1 is a close homologue of Ras it does not possess the catalytic glutamine residue that is critical for GTP hydrolysis in all other Ras-like GTPases. The mechanism by which Ras and Rap1 GAPs enhance the GTPase activity of their respective GTPase is therefore distinct. For Ras GAPs a catalytic arginine residue, the arginine finger, is supplied by the GAP molecule into the active site of Ras, thereby stabilizing the transition state of the GTPase reaction and increasing the reaction rate by >1,000-fold (12–14). Rap1 GAPs are not related to other GAPs and do not employ a catalytic arginine residue (15, 16); instead, Rap1 GAPs provide a catalytic asparagine, the asparagine thumb, to stimulate GTP hydrolysis (17). It has been proposed that the carboxamide side chain of the asparagine residue has a function similar to the glutamine residue in Ras-like GTPases in stabilizing the relative position of the nucleophilic water and γ-phosphate in the transition complex (17).
The GAP1 family, which comprises GAP1m, GAP1IP4BP, CAPRI, and RASAL (18–22), is characterized by a conserved domain structure comprising amino-terminal tandem C2 domains, a highly conserved central Ras GAP domain, and a carboxyl-terminal pleckstrin homology domain that is associated with a Bruton’s tyrosine kinase motif (23). Consistent with the presence of the Ras GAP domain all of these proteins have been shown to function as Ras GAP, although each is differentially regulated following receptor stimulation (19, 21, 22, 24, 25). Surprisingly, however, given that these proteins contain no detectable sequence homology with any known Rap GAP, GAP1IP4BP is also capable of enhancing the GTPase activity of Rap (19). This protein therefore has dual Ras and Rap GAP activity. In the current study we describe a kinetic characterization of this dual activity and have begun the process of dissecting out the mechanistic details of these activities. In addition, we address whether dual Ras/Rap GAP activity is restricted solely to GAP1IP4BP or is a function conserved within other members of the GAP1 family.
MATERIALS AND METHODS
Purification of GTP-binding Proteins
C-terminal-truncated Rap1B (Rap1B C′, residue 1–167) was purified from Escherichia coli strain CK600K using the ptac-expression system as described for Ras by Tucker et al. (26). After lysis in 32 mM Tris (pH 7.5), 100 μM phenyl-methylsulfonyl fluoride, 2 mM EDTA, the supernatant was applied to a Q-Sepharose column equilibrated with 32 mM Tris (pH 7.5), 10 mM MgCl2, 5 mM DTE. After washing with the same buffer, proteins were eluted using a salt gradient (0–300 mM NaCl) in 32 mM Tris (pH 7.5), 10 mM MgCl2, 5 mM DTE. Fractions containing Rap1B C′ were precipitated with ammonium sulfate (3 M final concentration). The pellet was resuspended in 64 mM Tris (pH 7.5), 10 mM MgCl2, 5 mM DTE, 200 μM GDP, 0.4 M NaCl and further purified on a Sephadex 75 gel filtration column. Fractions containing Rap1B C′ were pooled and concentrated using an Amicon concentrator (10-kDa cut-off). Truncated H-Ras-(1–166) was purified as described before (26, 27).
Purification of GAP1IP4BP
For stability reasons, full-length wild-type GAP1IP4BP was purified as GST fusion protein GST-GAP1IP4BP, while the mutant full-length GST-GAP1IP4BP-(R371A) and the GAP domain construct GST-GAP1IP4BP-(291–569) was cleaved from GST. Freshly transformed E. coli Rosetta strain was grown in 5 liters of TB medium containing 50 μg/ml ampicillin and 50 μg/ml chloramphenicol at 37 °C. At A600 ~0.8, expression was induced by addition of 100 μM isopropyl 1-thio-β-D-galactopyranoside overnight at 18 °C. After lysis by micro-fluidizer (Microfluidics Corp.) in 50 mM Hepes (pH 7.5), 5 mM DTE, 50 mM NaCl, 2.5 mM EDTA, 100 μM phenylmethylsulfonyl fluoride, the extract was applied to glutathione-Sepharose 4-B (Amersham Biosciences) in 50 mM Hepes (pH 7.5), 5 mM DTE, 50 mM NaCl. Following extensive washes, the protein was eluted with buffer containing 30 mM glutathione (pH 7.5). Protein was concentrated using an Amicon concentrator (30-kDa cut-off), and aliquots were snap frozen in liquid nitrogen and stored at −80 °C. Cleavage of GAP1IP4BP-(R371A) and GAP1IP4BP-(291–569) from GST was performed on the glutathione-Sepharose column with 300–600 units of thrombin (Serva) overnight at 4 °C under continuous circulation (0.5 ml/min). Proteins were eluted with 50 mM Hepes (pH 7.5), 5 mM DTE, 50 mM NaCl. GAP1IP4BP-(291–569) was further purified on a Superdex 200 gel filtration column in 50 mM Hepes (pH 7.5), 5 mM DTE, 50 mM NaCl. Proteins were concentrated using an Amicon concentrator (15 and 30-kDa cut-off), and aliquots were snap frozen in liquid nitrogen and stored at −80 °C. Full-length constructs cannot be fully purified and contain additional lower molecular mass bands, most probably derived from proteolytic degradation.
Nucleotide Exchange
200 μM Rap or Ras in 25 mM Tris (pH 7.5), 100 mM NaCl, 5 mM DTE were incubated with 15 mM EDTA, 100 mM ammonium sulfate, and 10 mM GTP (stock 100 mM nucleotide in 1 M Tris, pH 7.5) for 60 min at room temperature. Exchange was terminated by the addition of 25 mM MgCl2. The separation of unbound nucleotides and EDTA from Rap-GTP and Ras-GTP was carried out by washing the proteins several times with buffer using an Amicon concentrator (10-kDa cut-off) at 4 °C. Nucleotide exchange was controlled by reversed-phase high performance liquid chromatography. Radioactive [γ-32P]GTP-bound Rap1 and Ras were prepared by incubating 1.5 mM GTP-bound protein with 20 μCi of [γ-32P]GTP (800 Ci/mmol; Amer-sham Biosciences) in the presence of 12 mM EDTA for 30 min on ice. The exchange reaction was stopped by the addition of 25 mM MgCl2.
GAP Assay
The GAP-stimulated GTP hydrolysis of Rap1 and H-Ras was assayed by measuring Pi release using the charcoal method. Briefly, increasing amounts of radioactively labeled Rap-GTP and Ras-GTP were added to fixed concentrations of GAP1IP4BP proteins at 25 °C in standard buffer (25 mM Tris (pH 7.5), 5 mM MgCl2, 5 mM DTE, 50 mM NaCl) as described before (15). To determine end points of GTP hydrolysis, all Rap-GTP and Ras-GTP were hydrolyzed by addition of a highly concentrated GAP1IP4BP solution, and a last aliquot was taken. Initial GTP hydrolysis rates were evaluated by linear regression, and Km and kcat were determined by Michaelis-Menten equation using the program Grafit (Erithacus software).
GAP Assays of GAP1IP4BP Site-directed Mutants
Full-length GAP1IP4BP mutants L484A, R485Q, K517E, and K534A were isolated as GST fusion proteins, and analysis of the GAP activities of these mutants was performed under first order kinetics as described by Bottomley et al. (28). The particular GTP-binding protein was loaded with [γ-32P]GTP (3000 Ci mmol−1; Amersham Biosciences) for 5 min at 25 °C. GTPase activity was assayed at 25 °C by addition of the various GAP proteins to the loaded GTP-binding protein. After 10 min, activity was stopped by addition of 5 mM silicotungstate, 1 mM H2SO4. The liberated [32P]Pi was extracted with isobutanol/toluene (1/1 v/v), 5% (w/v) ammonium molybdate, 2 M H2SO4, and the upper phase was removed for scintillation counting.
Ras Pulldown Assays
Glutathione S-transferase fusion of the Ras-GTP-binding domain from Raf-1 (GST-RBD) was purified from BL21(DE3) E. coli cells harboring the plasmid pGEX KG containing the Raf Ras-binding domain (amino acids 1–149). After induction of a bacterial culture (A600 between 0.4 and 0.6) for 3 h at 37 °C with 1 mM isopropyl-1-thio-β-D-galactopyranoside, cells were lysed by sonication in phosphate-buffered saline containing 1 mM EDTA, 1% Triton X-100, 10 μg/ml of aprotinin, and 10 μg/ml of leupeptin. The lysate was clarified by centrifugation, and the resultant supernatant was stored in aliquots at −80 °C. On the required day, aliquots were thawed prior to incubation with glutathione-Sepharose (Amersham Biosciences) for 1 h at room temperature. The Sepharose beads were washed twice with phosphate-buffered saline, 1 mM EDTA, 1% (v/v) Triton X-100 before finally being suspended as a 1:1 slurry. This was used immediately in pulldown assays. Here, dishes (100 mm) of CHO-T cells (8 × 105 cells) were transiently transfected by lipofection (GeneJuice; Novagen) with 2.5 μg of H-Ras cDNA and 1 μg of vector control or vector encoding the particular GAP1 protein. The cells were serum starved for 2 h at 37 °C in serum-free F-12 (HAM; Invitrogen) prior to the experimental procedures. On completion of the manipulation, cells were lysed in 1 ml of ice-cold extraction buffer (50 mM Hepes, pH 7.5, 100 mM NaCl, 1 mM EGTA, 5 μg/ml of benzamidine, 5 μg/ml of aprotinin, 5 μg/ml of leupeptin, 5 μg/ml of pepstatin A, 5 μg/ml of trypsin inhibitor, 0.5 mM phenylmethylsulfonyl fluoride, and 1 mM dithiothreitol containing 1% Triton X-100 and 10 mM MgCl2). Nuclear-free supernatants were incubated with GST-RBD on glutathione-Sepharose beads at 4 °C for 30 min. The beads were then collected by centrifugation and washed three times with ice-cold phosphate-buffered saline, 0.1% Triton X-100, and 10 mM MgCl2. Ras proteins were separated by SDS-PAGE and visualized by immunoblotting on nitrocellulose filters using pan-Ras antibodies (F132; Santa Cruz Biotechnology) and enhanced chemiluminescence (Amersham Biosciences). Blots were analyzed by volume integration using ImageQuant software (Amersham Biosciences) as previously described (22, 24).
Rap1 Pulldown Assays
For the analysis of active Rap, a glutathione S-transferase fusion of the Rap-GTP-binding domain from RalGDS (GST-RalGDS) was used (29). Dishes (100 mm) of CHO-T cells (8 × 105 cells) were transiently co-transfected by lipofection (GeneJuice; Novagen) with 2.5 μg of HA-tagged Rap1A cDNA and 1 μg of vector control or vector encoding the particular GAP1 protein. The cells were serum starved for 2 h at 37 °C in serum-free F-12 (HAM) medium 24 h post-transfection prior to stimulation. Cell lysis and Rap1A-GTP pulldown were carried out as described above. Immobilized Rap1A was detected using the HA probe (Y-11; Santa Cruz Biotechnology). Blots were analyzed by volume integration using ImageQuant software (Amersham Biosciences) (22, 24).
RESULTS
Efficient Ras and Rap GAP Activity Requires Structural Elements beyond the Ras GAP Domain of GAP1IP4BP
To extend the published characterization of the Ras and Rap GAP activity of GAP1IP4BP (19, 28), we initially performed a detailed kinetic analysis (Fig. 1). Here, the GAP activity was monitored by following the production of Pi using the char-coal method (15). The isolated GAP domain of GAP1IP4BP, residue 291–569, corresponding to the GAP constructs GAP334 and NF1–333 from p120GAP and neurofibromin, respectively, has pronounced Ras GAP activity that, in contrast to those other Ras GAPs, cannot be saturated under the conditions used (Fig. 1, A and B). Using 100 nM GAP1IP4BP-(291–569) and 800 μM Ras-GTP, the kcat of the reaction reaches a value of 8 s−1, which is in the range observed for other Ras GAPs (30). Under these conditions, no stimulation of the Rap GTPase was observed. In the presence of 20 μM GAP domain, however, Rap GAP activity was detected (Fig. 1, C and D) but reached a kcat of only 0.02 s−1 with 800 μM Rap-GTP, indicating that the apparent second order rate constant kcat/Km is at least 2 orders of magnitude lower for the Rap versus the Ras GTPase activation. Inefficient hydrolysis is at least partially due to the absence of a glutamine residue in Rap, because with the Rap-(T61Q) mutant, kcat reached 0.5 s−1, which is only 10-fold lower than that observed with Ras (Fig. 1, A and B).
FIGURE 1. Michaelis-Menten kinetics of the GAP domain of GAP1IP4BP.


A, 100 nM GAP1IP4BP-(291–569) were incubated in standard buffer at 25 °C with increasing concentrations of H-Ras[γ-32P]GTP (•) and Rap1A[γ-32P]GTP (▪). The insert shows the reaction with mutant Rap-(T61Q) (▴) in comparison with wild-type Rap (▪). B, the data from panel A are plotted as kcat in panel B. C and D, 20 μM GAP1IP4BP-(291–569) were used to assay GTP hydrolysis with increasing concentrations of Rap1A[γ-32P]GTP. GTPase activity was monitored by measuring Pi release, and data were evaluated as described under “Materials and Methods.” Data are plotted as rates (C) or rate constants (D).
Because robust Rap GAP activity has been observed before (19) and Ras GAP activity is apparently similar to values observed for other Ras GAPs, it became obvious that GAP1IP4BP requires additional domains for efficient Rap GAP activity. Indeed, when we used full-length GST-GAP1IP4BP enriched via GSH-Sepharose, we observed efficient Ras and Rap GAP activity (Fig. 2). Using 5 nM GAP1IP4BP we observed similar maximal rates of 0.24 μmol/s for Ras and only a 3-fold lower rate of 0.08 μmol/s for Rap (Fig. 2A). Although these rates cannot be converted to kcat values due to the degradation products present in the preparation of the full-length protein, kcat was estimated to be 48 s−1 for Ras and 16 s−1 for Rap, indicating very efficient catalysis for both GTPase reactions. Furthermore, full-length protein had a 5-fold lower Km for Rap (42 μM) versus Ras (213 μM). Thus the apparent second order rate constant kcat/Km for the GAP-mediated catalysis, estimated to be between 2.2 and 3.8 ×105 M−1 s−1, is, independent of the exact value, very similar for Ras and Rap. This indicates that the additional domain(s) not only affects the catalytic efficiency of the Rap GAP reaction but also dramatically enhances the affinity toward Rap-GTP. Thus, whereas the isolated Ras GAP domain of GAP1IP4BP is, like other Ras GAPs, sufficient to enhance the GTPase activity of Ras, other regions of the molecule such as the amino-terminal C2 domains and/or the carboxyl-terminal region including the pleckstrin homology/Bruton’s tyrosine kinase domain are required for Rap GAP activity.
FIGURE 2. Michaelis-Menten kinetics of full-length GAP1IP4BP.

5 nM full-length enriched GST-GAP1IP4BP were incubated in standard buffer at 25 °C with increasing concentrations of radioactively labeled [γ-32P]GTP Ras (•) (A) or Rap-GTP (▪) (A, B) as indicated, and GTP hydrolysis was measured as described in Fig. 1. The GTPase reaction of Rap was also measured with the arginine finger mutant GAP1IP4BP-(R371A) (♦) in panel A or the T61Q mutant (▴) of Rap in panel B.
For Ras GAPs it has been shown that the glutamine 61 residue of Ras and the arginine finger of Ras GAPs are the two crucial residues required for efficient GAP-mediated reaction (13, 14). Rap does not have a glutamine in position 61 and Rap GAP does not act via a catalytic arginine (17), so the question arises as to how GAP1IP4BP-mediated Rap GAP activity is achieved. When we analyze the GTPase reaction using the arginine finger mutant of GAP1IP4BP, GAP1IP4BP-(R371A), the RapGAP (and RasGAP, not shown) activity was severely affected (Fig. 2A), arguing that the basic machinery for GAP activation seems to reside in the GAP domain. As with the GAP domain, catalysis is more efficient for the Rap mutant Rap-(T61Q), but the affinity is reduced because the reaction is no longer saturated with 800 μM Rap-GTP (Fig. 2B). Thus, the mechanism for GAP-mediated GTPase activity of GAP1IP4BP seems to be different from both the Ras GAP and the Rap GAP mechanisms.
Further Mutations in the GAP1IP4BP GAP Domain
To extend this analysis we generated a series of site-directed mutants targeting residues that lie within the Ras-binding site of the Ras GAP domain and are known to be required for the Ras GAP activity of p120GAP and NF1. Arginine at position 485 in GAP1IP4BP is equivalent to arginine 903 and 1391 in p120GAP and NF-1, respectively, residues that by stabilizing the catalytic position of the arginine finger are vital for GTP hydrolysis on Ras (13). The arginine finger is also stabilized by leucine 902 and 1390 in p120GAP and NF-1, respectively, equivalent to leucine 484 in GAP1IP4BP. Furthermore, lysine 949 in p120GAP resides within the variable loop that has been proposed to stabilize the α6c helix within the effector loop region of Ras-GTP. This residue is equivalent to lysine 534 of GAP1IP4BP. Finally, lysine 517 in GAP1IP4BP is equivalent to lysine 935, which has been proposed to stabilize the p120GAP/Ras-GTP complex. To examine the role of these residues in the Ras and Rap GAP activity, we introduced the following individual mutations into full-length GAP1IP4BP: GAP1IP4BP-(L484A), GAP1IP4BP-(R485Q), GAP1IP4BP-(K517E), and GAP1IP4BP-(K534A). As predicted from their conservation with residues critical for the activity of p120GAP and NF-1, all of these mutations lead to a significant decrease in the Ras GAP activity of GAP1IP4BP (Fig. 3). Interestingly, however, each mutation also resulted in a significantly reduced ability of GAP1IP4BP to function as a Rap GAP (Fig. 3). These data are consistent with a model in which Rap associates with GAP1IP4BP through the Ras-binding domain present in the Ras GAP domain.
FIGURE 3. Ras and Rap1 associate with GAP1IP4BP through the Ras GAP domain in vitro.

The GAP activities of the four GAP1IP4BP Ras GAP domain point mutants were assayed using H-Ras or Rap1A. GAP activity is expressed as a percentage of wild-type GAP1IP4BP.
GAP1IP4BP Also Displays Dual Ras and Rap1 GAP Activity in Vivo
To confirm the activity described in vitro we next examined the GAP activity of GAP1IP4BP in vivo using well established pulldown assays for Ras and Rap1 (29). For assays examining Ras GAP activity, CHO-T cells were transiently co-transfected with plasmids encoding for H-Ras and either control plasmid or constructs encoding for GAP1IP4BP. After 24 h cells were serum starved for 2 h prior to quantification of the levels of Ras-GTP following pulldown, using glutathione-Sepharose coupled to a GST fusion of the Ras-GTP-binding domain from Raf-1. Compared with control cells, in those expressing wild-type GAP1IP4BP a significant decrease in the level of Ras-GTP was observed (Fig. 4A). This decrease, indicative of an increase in the GTPase activity of this protein, was dependent on the catalytic activity of GAP1IP4BP as GAP1IP4BP-(L484A) was unable to elicit the decrease in Ras-GTP (Fig. 4A). Moreover, a site-directed mutant targeting the conserved catalytic arginine finger GAP1IP4BP-(R371Q) was also unable to elicit the decrease in Ras-GTP (Fig. 4A). Such data are entirely consistent with GAP1IP4BP functioning as a Ras GAP in vivo.
FIGURE 4. In vivo GAP1IP4BP also functions as a dual Ras and Rap1 GAP.

i, CHO-T cells were transiently co-transfected with 2.5 μg of H-Ras (A) or 2.5 μg of HA-tagged Rap1A (B) and 1 μg of the relevant GAP1IP4BP expression vector. Compared with control cells, the quantity of Ras- and Rap1-GTP is significantly decreased in cells expressing wild-type GAP1IP4BP. This decrease in Ras- and Rap1-GTP required a functional GAP-related domain (compare GAP1IP4BP with GAP1IP4BP-(L484A) and GAP1IP4BP-(R371Q)). ii, quantification of Ras- (A) and Rap1-GTP (B) levels from CHO-T cells expressed as a percentage of the pulldown in control cells (average of six separate experiments ± S.E.).
To analyze the potential in vivo Rap1 GAP activity, CHO-T cells were transiently co-transfected with plasmids encoding for Rap1 and either control plasmid or constructs encoding for GAP1IP4BP (Fig. 4B). After 24 h cells were serum starved for 2 h prior to quantification of the levels of Rap1-GTP following pulldown, using glutathione-Sepharose coupled to a GST fusion of the RBD domain from RalGDS (29). Consistent with the in vitro data, in cells expressing GAP1IP4BP a significant decrease in the level of Rap1-GTP was observed. This decrease in recoverable Rap1-GTP was not observed in those cells expressing GAP1IP4BP-(L484A) or the arginine finger mutant GAP1IP4BP-(R371Q) (Fig. 4B).
GAP1m Appears to Function as a Specific Ras GAP in Vivo
An outstanding issue concerns whether this ability to function as a dual Ras and Rap GAP is restricted to GAP1IP4BP or whether it is a property of other GAP1 family members. Previous work has demonstrated that in vitro GAP1m displays GAP activity against Ras but not Rap1 (28). However, given the subsequent demonstration that GAP1m undergoes a receptor-mediated association with the plasma membrane as a result of its pleckstrin homology/Bruton’s tyrosine kinase domain associating with phosphatidylinositol 3,4,5-trisphosphate (21), we examined whether in vivo GAP1m was capable of functioning as a Rap1 GAP following such a receptor-mediated plasma membrane association. In CHO-T cells transiently co-transfected with plasmids encoding Rap1 and either control plasmid or constructs encoding GAP1m, no detectable drop in the level of Rap1-GTP was observed in those cells expressing GAP1m (Fig. 5A). In parallel experiments examining the GAP activity against Ras, a clear decrease in the level of Ras-GTP was observed (Fig. 5B); these data confirm the previous in vitro study of GAP1m (28). To examine whether the plasma membrane association of GAP1m stimulated its ability to function as a Rap1 GAP, serum-starved cells were stimulated with epidermal growth factor (EGF), an agonist that induces the plasma membrane association of GAP1m (21). Under these conditions, still no detectable decrease in the level of Rap1 GTP was observed (Fig. 5A). These data suggest that unlike GAP1IP4BP, GAP1m does not display detectable Rap1 GAP activity in vivo when assayed under these conditions.
FIGURE 5. In contrast to GAP1IP4BP, GAP1m appears to function as a specific Ras GAP in vivo.
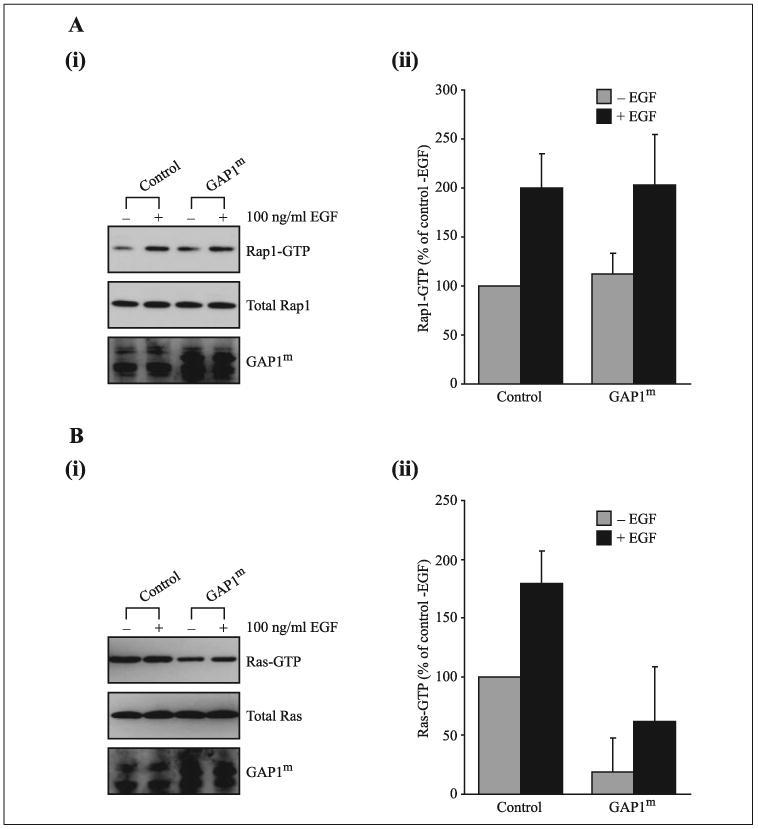
A, panel i, CHO-T cells were transiently co-transfected with 2.5 μg of HA-tagged Rap1A and 1 μg of either a control plasmid or the GAP1m expression vector. In control cells, after 60 s of stimulation with 100 ng/ml EGF the quantity of Rap1-GTP increases 2-fold. In cells expressing GAP1m no decrease in the quantity of Rap1-GTP was observed irrespective of EGF stimulation (compare GAP1m −EGF with GAP1m +EGF) (panel ii). B, panel i, CHO-T cells were transiently co-transfected with 2.5 μg of H-Ras and 1 μg of either a control plasmid or the GAP1m expression vector. In control cells, after 60 s of stimulation with 100 ng/ml EGF the quantity of Ras-GTP increases ~2-fold. In cells expressing Gap1m a significant decrease in the quantity of Ras-GTP was observed irrespective of EGF stimulation (compare GAP1m − EGF with GAP1m + EGF (panel ii)). This decrease in Ras-GTP indicates that the ability of GAP1m to undergo membrane association is not required for it to function as a Ras GAP. (panel ii). Quantification of Rap1- (A) and Ras-GTP (B) levels from CHO-T cells before and 60 s after stimulation with 100 ng/ml EGF is expressed as a percentage of the pull-down in control, serum-starved cells prior to EGF stimulation (average of six separate experiments ± S.E.).
In Contrast to GAP1m, RASAL Displays Dual Ras and Rap1 GAP Activity in Vivo
The remaining GAP1 family members, CAPRI and RASAL, are cytosolic proteins that undergo a rapid Ca2+-induced association with the plasma membrane following receptor activation (22, 24, 25). This recruitment, which results from their C2 domains binding phospholipids in a Ca2+-dependent manner, increases their Ras GAP activity (22, 24, 25). To examine whether these proteins display Rap GAP activity, we initially transiently co-transfected CHO-T cells with Rap1 and RASAL. In those cells expressing RASAL a significant decrease in the amount of Rap1-GTP was observed compared with controls (Fig. 6). This basal Rap GAP activity was dependent on the Ras GAP domain, as expression of RASAL-(Q507N) had no effect on Rap1-GTP levels, but was independent of the Ca2+/phospholipid-binding C2 domains; RASAL-(D202A) is a C2 domain mutant unable to undergo Ca2+-induced membrane association (24) (Fig. 6). These data suggest that in vivo when localized to the cytosol RASAL has a basal Rap GAP activity.
FIGURE 6. RASAL appears to function as a constitutively active Rap GAP.

A, CHO-T cells were transiently co-transfected with 2.5 μg of HA-tagged Rap1A and 1 μg of the relevant RASAL expression vector. In control cells, after 60 s of stimulation with 50 μM ATP the quantity of Rap1-GTP increased 2-fold. In cells expressing Rap1A and wild-type RASAL a significant decrease in the quantity of Rap1-GTP was observed before and after stimulation with ATP (compare RASAL − ATP with RASAL + ATP). This decrease in Rap-GTP required a functional GAP-related domain but does not appear to require the ability of RASAL to undergo Ca2+-induced membrane association (compare RASAL with RASAL-(Q507N) and RASAL-(D202A)). B, quantification of Rap1-GTP levels from CHO-T cells before and 60 s after stimulation with 50 μM ATP expressed as a percentage of the pulldown in control, serum-starved cells prior to ATP stimulation (average of ten separate experiments ± S.E.).
To establish the role of membrane association in the control of the Rap GAP activity, we examined the ability of RASAL to function as a Rap GAP following stimulation with ATP, an agonist that elevates intra-cellular free Ca2+ and induces the plasma membrane association of RASAL (24). In control serum-starved cells, addition of ATP induced an increase in Rap1-GTP levels (Fig. 6). In cells expressing RASAL the ATP-induced elevation in Rap1-GTP was significantly reduced (Fig. 6). Interestingly, the ability of RASAL to decrease the ATP-induced elevation in Rap1-GTP, although clearly dependent on the Ras GAP domain (see RASAL-(Q507N)), was independent of the Ca2+-induced membrane translocation as RASAL-(D202A) reduced the ATP-stimulated increase in Rap1-GTP to a similar level as wild-type protein (Fig. 6). Taken with previous data (24) these results establish that in vivo RASAL acts as a dual Ras and Rap GAP. However, whereas its Ras GAP activity is dependent on its Ca2+-induced plasma membrane translocation (24), the Rap GAP activity appears to be irrespective of such a membrane association.
CAPRI Also Has Dual Ras and Rap GAP Activity
Given the sequence similarity between RASAL and the Ca2+-regulated Ras GAP CAPRI (22, 24, 25), we also examined the ability of the latter to function as a Rap GAP (Fig. 7). Here, the basal level of Rap1-GTP was not significantly affected by overexpression of CAPRI. However, in parallel experiments to those described above, the ATP-induced elevation in Rap1-GTP was significantly reduced in CAPRI-expressing CHO-T cells (Fig. 7). This increase in Rap1 GAP activity required the ability of CAPRI to associate with the plasma membrane as CAPRI-(D202A), a C2 domain mutant unable to undergo Ca2+-induced plasma membrane association (data not shown), failed to decrease the ATP-induced elevation in Rap1-GTP (Fig. 7). Thus, CAPRI can function as a dual Ras and Rap GAP, but in contrast to RASAL the latter activity is only detectable upon the Ca2+-induced plasma membrane association.
FIGURE 7. CAPRI displays an ability to function as a Ca2+-regulated Rap GAP.

A, CHO-T cells were transiently co-transfected with HA-tagged Rap1A and 1 μg of expression vector containing either wild-type CAPRI or CAPRI(D202A) (22). In control cells, after 60 s of stimulation with 50 μM ATP the quantity of Rap1-GTP was increased. In cells expressing Rap1A and CAPRI a significant decrease in the quantity of Rap1-GTP was observed after 60 s of stimulation with ATP (compare CAPRI −ATP with CAPRI +ATP). This decrease in Rap1-GTP was not observed in ATP-stimulated cells expressing the CAPRI-(D202A) mutant (compare CAPRI + ATP with CAPRI-(D202A) + ATP). B, quantification of Rap1-GTP levels from CHO-T cells before and 60 s after stimulation with 50 μM ATP expressed as a percentage of the pulldown in control, serum-starved cells prior to ATP stimulation (average of five separate experiments ± S.E.).
DISCUSSION
In this study, detailed kinetic analysis has demonstrated that full-length, recombinant GAP1IP4BP possesses a robust Ras and Rap GAP activity, with estimated kcats of 16 and 48 s−1 and Kms of 42 and 213 μM for Rap and Ras, respectively. These values are very similar for the two GTPase substrates and are in the range of values for known specific Ras and Rap GAPs. The Ras-specific GAPs, NF1 and p120GAP, have Km values of 0.23 and 5.0 μM and kcats of 5.4 and 8.0 s−1 on H-Ras, respectively (30), whereas Rap1 GAP, a Rap-specific GAP, has a kcat of 5.6 s−1 and a Km of 52 μM for Rap (15). Thus, GAP1IP4BP has similar activity toward the individual small GTPases, but, unlike NF1, p120GAP, and Rap1 GAP, it has retained both of these activities within a singular molecular entity.
How is such a dual activity achieved? At present we cannot define the details of the actual mechanism. This will require further work. However, it is clear that although the isolated Ras GAP domain retains Ras GAP activity (this is consistent with the same domain having Ras GAP activity in NF1 and p120GAP) the isolated GAP1IP4BP Ras GAP domain loses its efficiency as a Rap GAP. These data point to other regions outside the GAP domain being required to support the catalytic activity toward Rap. Indeed, for other GAP molecules there is evidence that additional domains are required to regulate the catalytic activity (31–33). Although the absence in Rap of the Gln-61 residue of Ras is partially responsible for the lower catalytic activity of both the GAP domain and full-length protein, its presence in the T61Q mutant of Rap also influences the affinity toward GAP in opposite ways, by increasing the affinity in the GAP domain and decreasing it in full-length protein. Structural studies will be required to understand these observations in mechanistic terms.
Our analysis of various site-directed mutants, some of which target residues within the predicted Ras binding region of the GAP domain, is consistent with Rap associating with GAP1IP4BP through the conserved Ras-binding site. This conclusion, not unexpected given that Rap-GTP associates with p120GAP through the Ras-binding site (although obviously in this case the GTPase activity of Rap1 is not enhanced), argues that GAP1IP4BP cannot function simultaneously as a Ras and Rap GAP. In other words, at any one time GAP1IP4BP will only stimulate the GTPase activity of either Ras or Rap. At present we do not know whether additional factors, such as phosphorylation, association with particular phospholipids, or other protein-protein interactions, modulate the preference for Ras versus Rap GAP. However, given the precedent that all of these modes of modulation have been proposed to regulate the catalytic activity of a wide variety of GAP proteins (reviewed in Ref. 10), it is tempting to speculate that one or more of these mechanisms may differentially regulate the activity of GAP1IP4BP. Clearly, such modulatory mechanisms will have an important bearing on the physiological function of GAP1IP4BP.
Besides GAP1IP4BP, our data establish that, with the possible exception of GAP1m, all members of the GAP1 family act as dual Ras and Rap GAPs. For these proteins the ability to display their particular GAP activity is controlled by distinct second messenger pathways, GAP1IP4BP being controlled by phosphoinositides (19, 34, 35) and CAPRI and RASAL modulated to differing degrees by receptor-mediated increases in [Ca2+]i (Refs. 22,24,25, and the present study). This argues for a complex interplay between these second messenger pathways and the modulation of Ras and Rap signaling. This is particularly true for increases in [Ca2+]i (36).
It has long been recognized that increases in [Ca2+]i can regulate Ras and Rap1 activation. Only recently, however, have molecular entities been described through which this regulation may occur (reviewed in Ref. 35). In the context of the present study, the CalDAG-GEF/GRP family of exchange factors is of particular interest. Members of this family have Ca2+- and diacylglycerol-binding sites and are regulated by one or both of these messenger molecules (4–6). For CalDAG-GEFI, and also GRP2 and GRP3, there is evidence that receptor-mediated increases in [Ca2+]i and diacylglycerol stimulate the ability of these proteins to function as dual exchange factors for Ras and Rap (4, 36, 37). These data, taken with the ability of Ca2+ to regulate the Ras and Rap GAP activity of CAPRI and RASAL, suggest that receptor-mediated increases in [Ca2+]i may constitute a complex means by which cells can modulate the relative activation profiles of Ras and Rap during receptor activation (38). In this regard it is interesting to note that Ras and Rap have been proposed to function independently, but in a coordinated manner, in delivering and removing α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) receptors from the post-synaptic membrane during long-term potentiation and long-term depression (39). An important modulator of such synaptic plasticity is SynGAP (40, 41), a Ras GAP whose activity appears to be modulated through Ca2+/cal-modulin-dependent protein kinase II (CaMKII) (42, 43). Interestingly, this protein has also been shown to have Rap GAP activity (43), although it remains to be established what, if any, effect CaMKII-mediated phosphorylation has on the relative Ras versus Rap GAP activity. Like the GAP1 proteins discussed in this study, SynGAP may well constitute a molecular machine that can coordinate changes in the activation profiles of these two small GTPases.
Finally, the identification of RASAL and CAPRI as candidate tumor suppressors (44, 45) and the proposed role of CAPRI in innate immunity (46) have increased interest in the biology of these proteins. The data presented here argue that when considering the function of RASAL and CAPRI one needs to consider not only their Ras GAP activity but also their ability to inactivate Rap1 signaling.
Acknowledgments
We thank Hans Bos for provision of constructs for expression of GST-RalGDS and Rap1.
Footnotes
This work was supported in part by The Wellcome Trust and by Component and Career Establishment grants from the Medical Research Council.
The abbreviations used are: GEF, guanine-nucleotide exchange factor; GST, glutathione S-transferase; CHO, Chinese hamster ovary; DTE, dithioerythritol; HA, hemagglutinin; EGF, epidermal growth factor.
References
- 1.Shields JM, Pruitt K, McFall A, Shaub A, Der CJ. Trends Cell Biol. 2000;10:147–153. doi: 10.1016/s0962-8924(00)01740-2. [DOI] [PubMed] [Google Scholar]
- 2.Hancock JF. Nat Rev Mol Cell Biol. 2003;4:373–384. doi: 10.1038/nrm1105. [DOI] [PubMed] [Google Scholar]
- 3.Bos JL. Curr Opin Cell Biol. 2005;17:123–128. doi: 10.1016/j.ceb.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 4.Stork PJS. Trends Biochem Sci. 2003;28:267–275. doi: 10.1016/S0968-0004(03)00087-2. [DOI] [PubMed] [Google Scholar]
- 5.Caron E. J Cell Sci. 2003;116:435–440. doi: 10.1242/jcs.00238. [DOI] [PubMed] [Google Scholar]
- 6.Hattori M, Minato N. J Biochem. 2003;134:479–484. doi: 10.1093/jb/mvg180. [DOI] [PubMed] [Google Scholar]
- 7.Kitayama H, Sugimoto Y, Matsuzaki T, Ikawa Y, Noda M. Cell. 1989;56:77–84. doi: 10.1016/0092-8674(89)90985-9. [DOI] [PubMed] [Google Scholar]
- 8.Donovan S, Shannon KM, Bollag G. Biochim Biophys Acta. 2002;1602:23–45. doi: 10.1016/s0304-419x(01)00041-5. [DOI] [PubMed] [Google Scholar]
- 9.Bernards A. Biochim Biophys Acta. 2003;1603:47–82. doi: 10.1016/s0304-419x(02)00082-3. [DOI] [PubMed] [Google Scholar]
- 10.Bernards A, Settleman J. Trends Cell Biol. 2004;14:377–385. doi: 10.1016/j.tcb.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 11.Yajnik V, Paulding C, Sordella R, McClatchey AI, Saito M, Wahrer DCR, Reynolds P, Bell DW, Lake R, van den Heuvel S, Settleman J, Haber DA. Cell. 2003;112:673–684. doi: 10.1016/s0092-8674(03)00155-7. [DOI] [PubMed] [Google Scholar]
- 12.Scheffzek K, Lautwein A, Kabsch W, Ahmadian MR, Wittinghofer A. Nature. 1996;384:591–596. doi: 10.1038/384591a0. [DOI] [PubMed] [Google Scholar]
- 13.Scheffzek K, Ahmadian MR, Kabsch W, Wiesmuller L, Lautwein A, Schmitz F, Wittinghofer A. Science. 1997;277:333–338. doi: 10.1126/science.277.5324.333. [DOI] [PubMed] [Google Scholar]
- 14.Ahmadian MR, Stege P, Scheffzek K, Wittinghofer A. Nat Struct Biol. 1997;4:686–689. doi: 10.1038/nsb0997-686. [DOI] [PubMed] [Google Scholar]
- 15.Brinkmann T, Daumke O, Herbrand U, Kuhlmann D, Stege P, Ahmadian MR, Wittinghofer A. J Biol Chem. 2002;277:12525–12531. doi: 10.1074/jbc.M109176200. [DOI] [PubMed] [Google Scholar]
- 16.Chakrabarti PP, Suveyzdis Y, Wittinghofer A, Gerwert K. J Biol Chem. 2004;279:46226–46233. doi: 10.1074/jbc.M405603200. [DOI] [PubMed] [Google Scholar]
- 17.Daumke O, Weyand M, Chakrabarti PP, Vetter IR, Wittinghofer A. Nature. 2004;429:197–201. doi: 10.1038/nature02505. [DOI] [PubMed] [Google Scholar]
- 18.Maekawa M, Iwamatsu SA, Morishita T, Yokota K, Imai Y, Kohsaka S, Nakamura S, Hattori S. Mol Cell Biol. 1994;14:6879–6885. doi: 10.1128/mcb.14.10.6879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cullen PJ, Hsuan JJ, Truong O, Letcher AJ, Jackson TR, Dawson AP, Irvine RF. Nature. 1995;376:527–530. doi: 10.1038/376527a0. [DOI] [PubMed] [Google Scholar]
- 20.Allen M, Chu S, Brill S, Stotler C, Buckler A. Gene. 1998;218:17–25. doi: 10.1016/s0378-1119(98)00394-1. [DOI] [PubMed] [Google Scholar]
- 21.Lockyer PJ, Wennstrom S, Kupzig S, Venkateswarlu K, Downward J, Cullen PJ. Curr Biol. 1999;9:265–268. doi: 10.1016/s0960-9822(99)80116-x. [DOI] [PubMed] [Google Scholar]
- 22.Lockyer PJ, Kupzig S, Cullen PJ. Curr Biol. 2001;11:981–986. doi: 10.1016/s0960-9822(01)00261-5. [DOI] [PubMed] [Google Scholar]
- 23.Cullen PJ. Biochim Biophys Acta. 1998;1436:35–47. doi: 10.1016/s0005-2760(98)00149-0. [DOI] [PubMed] [Google Scholar]
- 24.Walker SA, Kupzig S, Bouyoucef D, Davies LC, Tsuboi T, Bivona T, Cozier GE, Lockyer PJ, Buckler A, Rutter GA, Allen MJ, Philips MR, Cullen PJ. EMBO J. 2004;23:1749–1760. doi: 10.1038/sj.emboj.7600197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu Q, Walker SA, Gao D, Taylor JA, Dai YF, Arkell RS, Bootman MD, Roderick HL, Cullen PJ, Lockyer PJ. J Cell Biol. 2005;170:183–190. doi: 10.1083/jcb.200504167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tucker J, Sczakiel G, Feuerstein J, John J, Goody RS, Wittinghofer A. EMBO J. 1986;5:1351–1358. doi: 10.1002/j.1460-2075.1986.tb04366.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.John J, Frech M, Wittinghofer A. J Biol Chem. 1988;263:11792–11799. [PubMed] [Google Scholar]
- 28.Bottomley JR, Reynolds JS, Lockyer PJ, Cullen PJ. Biochem Biophys Res Commun. 1998;250:143–149. doi: 10.1006/bbrc.1998.9179. [DOI] [PubMed] [Google Scholar]
- 29.Franke B, Akkerman JW, Bos JL. EMBO J. 1997;16:252–259. doi: 10.1093/emboj/16.2.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ahmadian MR, Hoffmann U, Goody RS, Wittinghofer A. Biochemistry. 1997;36:4535–4541. doi: 10.1021/bi962556y. [DOI] [PubMed] [Google Scholar]
- 31.Drugan JK, Rogers-Graham K, Gilmer T, Campbell S, Clark GJ. J Biol Chem. 2000;275:35021–35027. doi: 10.1074/jbc.M004386200. [DOI] [PubMed] [Google Scholar]
- 32.Fauchereau F, Herbrand U, Chafey P, Eberth A, Koulakoff A, Vinet MC, Ahmadian MR, Chelly J, Billuart P. Mol Cell Neurosci. 2003;23:574–586. doi: 10.1016/s1044-7431(03)00078-2. [DOI] [PubMed] [Google Scholar]
- 33.Moskwa P, Paclet MH, Dagher MC, Ligeti E. J Biol Chem. 2005;280:6716–6720. doi: 10.1074/jbc.M412563200. [DOI] [PubMed] [Google Scholar]
- 34.Lockyer PJ, Bottomley JR, Reynolds JS, McNulty TJ, Venkateswarlu K, Potter BVL, Dempsey CE, Cullen PJ. Curr Biol. 1997;7:1007–1010. doi: 10.1016/s0960-9822(06)00423-4. [DOI] [PubMed] [Google Scholar]
- 35.Cozier GE, Lockyer PJ, Reynolds JS, Kupzig S, Bottomley JR, Millard TM, Banting G, Cullen PJ. J Biol Chem. 2000;275:28261–28268. doi: 10.1074/jbc.M000469200. [DOI] [PubMed] [Google Scholar]
- 36.Cullen PJ, Lockyer PJ. Nat Rev Mol Cell Biol. 2002;3:339–348. doi: 10.1038/nrm808. [DOI] [PubMed] [Google Scholar]
- 37.Bos JL, de Rooij J, Reedquist KA. Nat Rev Mol Cell Biol. 2001;2:369–377. doi: 10.1038/35073073. [DOI] [PubMed] [Google Scholar]
- 38.Kupzig S, Walker SA, Cullen PJ. Proc Natl Acad Sci U S A. 2005;102:7577–7582. doi: 10.1073/pnas.0409611102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu JJ, Qin Y, Zhao M, Van Aelst L, Malinow R. Cell. 2002;110:443–455. doi: 10.1016/s0092-8674(02)00897-8. [DOI] [PubMed] [Google Scholar]
- 40.Chen HJ, Rojas-Soto M, Oguni A, Kennedy MB. Neuron. 1998;20:895–904. doi: 10.1016/s0896-6273(00)80471-7. [DOI] [PubMed] [Google Scholar]
- 41.Kim JH, Liao DZ, Lau LF, Huganir RL. Neuron. 1998;20:683–691. doi: 10.1016/s0896-6273(00)81008-9. [DOI] [PubMed] [Google Scholar]
- 42.Oh JS, Manzerra P, Kennedy MB. J Biol Chem. 2004;279:17980–17988. doi: 10.1074/jbc.M314109200. [DOI] [PubMed] [Google Scholar]
- 43.Krapivinsky G, Medina I, Krapivinsky I, Gapon S, Clapham DE. Neuron. 2004;43:563–574. doi: 10.1016/j.neuron.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 44.Kolfschoten IGM, van Leeuwen B, Berns K, Mullenders J, Beijersbergen RL, Bernards R, Voorhoeve PM, Agami R. Cell. 2005;121:849–858. doi: 10.1016/j.cell.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 45.Westbrook TF, Martin ES, Schlabach MR, Leng YM, Liang AC, Feng B, Zhao JJ, Roberts TM, Mandel G, Hannon GJ, DePinho RA, Chin L, Elledge SJ. Cell. 2005;121:837–848. doi: 10.1016/j.cell.2005.03.033. [DOI] [PubMed] [Google Scholar]
- 46.Zhang J, Guo J, Dzhagalov I, He YW. Nat Immunol. 2005;6:911–919. doi: 10.1038/ni1232. [DOI] [PMC free article] [PubMed] [Google Scholar]