

Abstract
PURPOSE
Mutations in RP1are a common cause of dominant retinitis pigmentosa (RP), but the mechanism by which the identified mutations lead to photoreceptor cell death and blindness has not been determined. To investigate the function of the RP1 protein in photoreceptors and gain insight into the mechanism of disease, gene-targeting techniques were used to produce mice with a mutant Rp1allele that mimics the truncation alleles found to cause disease.
METHODS
RT-PCR was used to amplify illegitimate RP1transcripts from lymphoblasts. Gene targeting was used to create mice with a mutant Rp1-mycallele. Confocal immunofluorescence microscopy was used to identify the location of the mutant Rp1-myc protein in photoreceptors. The structure of the photoreceptors in the resultant Rp1-mycmice was studied by light and electron microscopy. The retinal function of the mutant mice was investigated using analysis of full-field ERGs.
RESULTS
Wild-type and mutant RP1mRNA were both detected in lymphoblasts from patients with RP1 disease. Rp1-mycmice produced a truncated version of the Rp1 protein, containing the N-terminal 662 amino acids, which localized correctly to the axoneme of the photoreceptor outer segments. Mice homozygous for the mutant Rp1-mycallele underwent a rapid-onset retinal degeneration characterized by incorrectly oriented outer segment discs that failed to stack properly into outer segments. In contrast, the photoreceptors of heterozygous mice remained relatively healthy.
CONCLUSIONS
The presence of mutant RP1mRNA in lympho-blasts from patients with RP1 disease implies that the mutant message can escape nonsense-mediated mRNA decay and that a truncated RP1 protein may be produced in the retina. The truncated Rp1-myc protein appears to be nonfunctional, and not to exert a dominant negative effect in the photoreceptors of heterozygous mice. Results from homozygous Rp1-mycmice indicate that RP1 is required for the correct orientation and higher order stacking of outer segment discs.
Mutations in RP1are a common cause of retinitis pigmentosa (RP).1-3Like many retinal degeneration genes, however, the mechanism by which mutations in RP1lead to photoreceptor cell death is not known. The location of the RP1 protein in the region of the axoneme of rod and cone photo-receptors provides some clues about its function.4The photo-receptor axoneme begins at the basal body in the distal inner segment, passes through the connecting cilium, and continues into the outer segment up to half its length.5Because the outer segment is continually renewed by shedding older discs at the tip and adding new discs at the base, there is a constant demand for polarized transport of proteins from the inner to the outer segment.6,7As the only connection between the inner and outer segments of rods and cones, the connecting cilium is considered to be the primary conduit for the proteins and membrane needed in outer segments.8Protein transport through the cilium is thought to occur by conserved axonemal transport processes, mediated by the motor proteins kinesin and dynein, along with intraflagellar transport (IFT) proteins and myosin VIIa.9-12
The junction between the connecting cilium and the outer segment is also the location of disc morphogenesis. Thus, proteins found in this region may also participate in this process. New outer segment discs are formed by a process of membrane evagination and fusion at the base of the outer segment, adjacent to the axoneme of the outer segment.7,13-15Actin is concentrated at the base of the outer segment and has been implicated in disc formation, based on microscopy studies and use of inhibitors of actin polymerization.16,17RPGRIP is also a component of connecting cilia, and RPGRIP knockout mice demonstrate defects in outer segment disc size and orientation, leading to the hypothesis that RPGRIP is required for disc morphogenesis.18The roles of other proteins shown to be located in or around the cilium, such as α-actinin, cadherin, centrin, and myosin II are not as clearly defined.19-22
Data from mice with targeted removal of exons 2 and 3 of the Rp1gene suggest roles for Rp1 in both protein transport and disc formation.23These mice produce an Rp1mRNA resulting from abnormal splicing of exons 1 and 4 of the Rp1gene. This abnormal mRNA is translated, and an abnormal Rp1 protein that is lacking the N-terminal 269 amino acids is produced in the retinas of heterozygous and homozygous Rp1-exon 2/3 deletion mice.23It is thus not clear whether the defect in outer segment formation observed in these mice is due to the lack of the Rp1 protein or to a dominant effect of the abnormal protein. In addition, the mislocalization of opsin is minimal compared with that observed with other mutations that alter protein trafficking.10,11
All the 20 pathologic mutations in RP1found to date are nonsense or frameshift mutations that are located between codons 500 and 1053; 18 are found between codons 658 and 872.3,24These mutations all cluster at the beginning of exon 4 (codons 263-2156) and are thought to generate a truncated RP1 protein that lacks the C-terminal one half to two thirds of the protein. Because these premature termination mutations are all located after the final intron–exon boundary of the RP1gene, it is predicted that the mutant mRNAs could escape nonsense mediated decay.25To study the function of the RP1 protein and gain insight into disease pathogenesis, we investigated the expression of mutant RP1mRNA in lymphoblasts from patients with RP1 disease. We then used gene-targeting techniques to produce mice with a mutant Rp1allele that mimics the truncation alleles found to cause disease. Our data show that RP1 has a role in controlling the orientation of outer segment discs and in the organization of discs into outer segments.
MATERIALS AND METHODS
Preparation of DNA and Mutation Screening
This research adhered to the tenets of the Declaration of Helsinki and was approved by the institutional review board of the University of Pennsylvania. Samples from the Foundation Fighting Blindness (FFB) Histopathology Collection (Scheie Eye Institute, Philadelphia, PA) were selected for genetic screening based on history and histology. Only those specimens with a history of autosomal dominant RP and retinal tissue containing some residual photoreceptors were chosen to be screened. Genomic DNA was isolated from frozen eye tissue or blood samples (QIAamp DNA Mini Kit; Qiagen, Valencia, CA), and the region of RP1(codon 603-891) that contains 18 of 20 known mutations was amplified by PCR. The primers used were forward 5′-AGGTTCAGTC-CTATTTCAGCAGATG-3′ and reverse 5′-CCCTGGTTGTAGCATGTT-GTTTA-3′. The PCR products (865 bp) were electrophoresed, eluted from the gel and purified by extraction with phenol-chloroform followed by precipitation with ethanol. Automated sequencing using both the forward and reverse primers was performed using 100 ng of purified DNA as a template.
Detection of RP1mRNA in Lymphoblasts
Lymphoblasts from members of the RP01 pedigree with the Arg677Ter mutation in the RP1gene were obtained from the Coriell Cell Repository (http://locus.umdnj.edu). The cells were grown in RPMI 1640 with 2 mM L-glutamine plus 15% fetal bovine serum at 37°C with 5% CO2. Total RNA (50 –100 μg) was extracted from approximately 10million cells (TRIzol; Invitrogen, Carlsbad, CA), and mRNA was prepared from this total RNA (Oligotex mRNA Minikit; Qiagen). Poly(A)+RNA (1– 4 μg) was treated with DNase I (Ambion, Austin, TX), and reverse transcribed (Superscript II; Invitrogen). The first PCR reaction was then performed using 1 μL of first-strand cDNA to amplify the region of RP1transcript containing codon 677. Two microliters of the first PCR reaction were used as the template for the second nested PCR reaction. Thirty cycles of amplification were used for both PCR reactions. The primers used for the first PCR reaction were: forward, 5′-CAATAACAATGATCAAATGGAGGAGTC-3′; and reverse, (5′-CCTT-GAACCTTGGAATTTTGAGTA-3′. The internal primers for the second PCR reaction were: forward, 5′-AGGTTCAGTCCTATTTCAGCAGATG-3′; and reverse, 5′-ATTCTACCTTTTGTGTTTATTCTCTCA-3′. The final PCR products were electrophoresed on agarose gels, extracted from the gel, and purified by extraction with phenol-chloroform, followed by precipitation with ethanol. The purified PCR products were then digested with Taqα1 to detect the Arg667Ter mutation, or sequenced. To control for genomic DNA contamination, parallel reactions without reverse transcriptase were included for each sample. These no-reverse-transcriptase control experiments were carried through the nested PCR steps with the experimental samples.
Generation of Rp1-mycMice
This research followed the tenets of the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research, and the guidelines of the University of Pennsylvania for Animal Care and Use. The Rp1-myctargeting vector was constructed using segments of genomic DNA amplified by high-fidelity PCR from a mouse bacterial artificial chromosome (BAC) clone that contains the entire mouse Rp1gene. This BAC clone, 314A, was identified by screening high-density filters from the RPCI-22 (129S6/SvEvTac) Mouse BAC Library (BACPAC web site: http://www.chori.org/bacpac/libraryres.htm/provided in the public domain by the National Center of Excellence in Genomics at Children’s Hospital Oakland Research Center, Oakland, CA). The 3′ arm of the targeting vector, including bases 16,110 to 16,879 of the Rp1gene (based on GenBank F291754; http://www.ncbi.nlm.nih.gov/Genbank; provided in the public domain by the National Center for Biotechnology Information, Bethesda, MD), plus an additional 1,174 bases 3′ to the gene, was amplified with PCR primers containing XhoI and KpnI restriction sites. The purified, digested PCR product was then cloned into loxpneoBS, which contains a floxed pgk-neo cassette,26creating plasmid pSP2. The 5′ arm of the targeting vector including bases 6,822 to 12,003 of the Rp1gene was amplified and cloned into the NotI and SalI sites of the pCMV-tag5 vector (Stratagene) adjacent to the myctag to create plasmid pSP6. The 5′-mycarm, including the myctag, stop codon and polyadenylation signal from pCMV-tag5, was then excised from pSP6, using the enzymes NotI and MluI, and cloned into the NotI-MluI sites of pSP2, upstream of the floxed pgk-neo cassette, creating the final Rp1-myctargeting vector (see Fig. 2).
FIGURE 2.
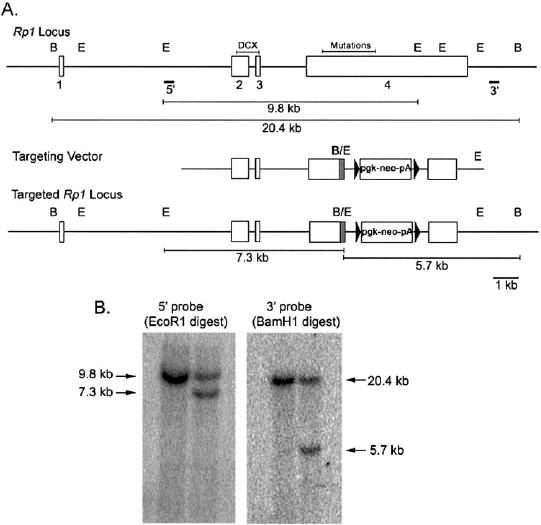
Targeted disruption of the Rp1gene in mice. (A)Maps of the wild-type Rp1locus, the targeting vector, and the targeted Rp1locus are shown. The exons of the Rp1gene are shown as open boxes. The DCX domain encoded by exons 2 and 3 is indicated, as is the region that contains all pathologic mutations at the beginning of exon 4. The map of the targeting vector shows the portion of exon 4 after Arg662 of the mouse Rp1coding sequence that is replaced with a neomycin resistance gene cassette (pgk-Neo-pA). A 10-amino-acid myctag coding sequence (shaded area)was placed at the C-terminal end of the truncated Rp1gene. The 5′ and 3′ probes used for Southern blot analyses are shown. The locations of EcoRI (E) and BamHI (B) are indicated, as are the expected sizes of the restriction fragments for the wild-type and targeted loci. (B)Southern blot analysis of EcoRI and BamHI digested genomic DNA isolated from control (left)and targeted (right)ES cells. The 7.3-kb band detected with the 5′ probe, and the 5.7-kb band detected with the 3′ probe confirm correct recombination at the Rp1locus.
The Rp1-myctargeting vector was linearized with SacII, and used to transfect 7.5 × 106TL-1 mouse embryonic stem (ES) cells.27Transfection was performed with 20 μg of a linearized targeting vector using a transfection reagent (Lipofectamine; Life Technologies, Gaithers-burg, MD) plus the nuclear localization signal peptide M9.28Approximately 200 G418 resistant (275 μg/mL; Invitrogen) colonies were selected and expanded according to established techniques.29DNA prepared from the isolated clones was digested with EcoRI and analyzed by Southern blot using the 5′ probe to identify correctly targeted ES cells. DNA from the single positive recombinant was also digested with BamHI, and analyzed by Southern blot using the 3′ probe. Probes for Southern blot analysis were amplified from BAC 314A DNA, and cloned into the pCRII-TOPO vector (Invitrogen). The 5′ probe was amplified with primers 5′-GGCAGGACCCCAGATTTTACAAC-3′ and 5′-AGCCTATCTTCTCAGCCCACCCTACA-3′, and the 3′ probe with primers 5′-CGGCGGCGGTCACGAACTC-3′ and 5′-CCTCGCCGGA-CACGCTGAACT-3′.
The single ES cell clone with correct recombination was microin-jected into C57BL/6 blastocysts to generate chimeric mice by the Chimeric and Transgenic Mouse Core Facility at the University of Pennsylvania School of Medicine (Philadelphia, PA). Highly chimeric founders were crossed with C57BL/6 mice to generate lines of heterozygous (Rp1+/myc) Rp1-mycmutant mice. Mice homozygous (Rp1myc/myc) for the targeted allele were generated by sibling crosses. Genotyping was performed by PCR analysis of tail DNA using two pairs of primers: 5′-GTGCCACAACAATGCTTTACCGTCAAC-3′ and 5′-GGA-CTGAGGGGCCTGAAATGA-3′ to amplify the mutant allele, and 5′-CTCAGCAGCGAATGATAACTTC-3′ and 5′-CACTTGGAACGGTGGTC-TGT-3′ to amplify the wild-type allele. The sizes of the PCR products amplified from wild-type and mutant Rp1alleles are 488 and 612 bp, respectively. Sequence analysis of the mutant PCR product verified correct gene targeting.
Western Blot Analysis
Retinas from animals of each genotype (Rp1+/myc, Rp1myc/myc, and Rp1+/+) at the desired ages were collected and homogenized in sample buffer (NuPAGE; Invitrogen). Solubilized retinal protein samples were then processed for Western blot analysis as described.4Briefly, equivalent amounts of protein from each genotype were separated on 4% to 12% Tris-acetate gel (NuPage; Invitrogen) and transferred to polyvinylidene difluoride (PVDF) membrane (Millipore, Bedford, MA).30,31The membranes were blocked with 10% nonfat dry milk in TBS-T (50 mM Tris-C1 [pH 8.0], 150 mM NaCl, 0.1% Tween-20) and incubated with primary antibodies. Chicken polyclonal anti-C ′-Rp1 antibody (1:2000) raised against codons 1708 to 1928 from the C-terminal region of mouse Rp1was used to detect the normal Rp1 protein.4Monoclonal anti-myc antibody (1:1000, Cell Signaling Technology; Beverly, MA) was used to detect the mutant Rp1-myc protein. Antibody binding was detected with alkaline phosphatase conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA) and enhanced chemifluorescence (ECF) substrate (Amer-sham Biosciences, Piscataway, NJ). Positive signals were visualized and quantified by fluorometry using a Storm 860 PhosphorImager (Amer-sham Biosciences-Molecular Dynamics, Sunnyvale, CA).
Immunofluorescence Microscopy
Eyes from postnatal day (P)14, P30, and 10-month-old mice were enucleated after cardiac perfusion with 4% PFA in 0.1 M phosphate buffer and fixed in 4% PFA for 3 hours. Fixed eyecups were infiltrated with 30% sucrose, frozen in OCT freezing medium, and cryosectioned at 10 μm. For immunostaining, retinal sections were blocked in PBS containing 1% normal goat serum, 1% bovine serum albumin, and 0.5% Triton X-100 for 1 hour and then incubated overnight with primary antibodies at 4°C. After they were rinsed with PBS, the sections were treated with fluorochrome-conjugated secondary antibodies for 1 hour, washed in PBS, and mounted (Fluoromount-G; Southern Biotechnology Associates, Birmingham, AL). The primary antibodies used were antiC′-Rp1,4anti-myc monoclonal antibody, mouse monoclonal anti-rhodopsin 4D2, anti-rds/peripherin (Per 5H2), anti-Rom1 (Rom 1D5), and anti-rod cyclic nucleotide gated channel α-subunit (PMc 1D1), anti-ABCR (rim 3F4) (Per 5H2, Rom 1D5, PMc 1D1 and rim 3F4 were obtained from Robert Molday, University of British Columbia, Vancouver, Canada) anti-arrestin,32anti-blue cone (JH455), and red/green cone (JH492).33Cy2- and Cy3-conjugated secondary antibodies were obtained from Jackson ImmunoResearch Laboratories. Alexa 468 – and Alexa 584 – conjugated secondary antibodies were from Molecular Probes (Eugene, OR). Control sections were treated with preimmune anti-C′-Rp1 or without primary antibodies. Stained sections were viewed with a confocal microscope (LSM 510; Carl Zeiss MicroImag-ing, Thornwood, NY). Frozen retinal sections from the same animals were also dried and stained with Richardson stain to evaluate the thickness of the outer nuclear layer. Bright-field digital images were captured using a microscope (TE300; Nikon USA, Melville, NY) equipped with a digital camera (Spot RT; Diagnostic Instruments, Sterling Heights, MI).
Light and Electron Microscopy
Anesthetized mice were perfused through the heart with freshly prepared 2% PFA+2% glutaraldehyde in 0.2 M sodium cacodylate buffer (pH 7.4). Eyes were removed and eyecups were fixed in the same buffer for 4 hours at room temperature. After they were rinsed with 0.2 M cold sodium cacodylate buffer (pH 7.4), eyecups were trimmed into 1 × 2-mm pieces and post fixed in 1% osmium tetroxide. Specimens were then stained with 1% uranyl acetate, dehydrated and embedded in Epon resin (EMbed812; Electron Microscopy Sciences) according to the manufacturer’s instructions. Sections of 1-μm thickness were cut and stained with alkaline toluidine blue for light microscopy. Ultrathin sections of 60- to 80-nm thickness were cut, stained with lead citrate and uranyl acetate, and examined using a transmission electron microscope (CX100; JEOL, Tokyo, Japan). Electron micrographs were captured using film or a digital-image capture system.
ERG Measurements
The methods for electrical recording of light stimulation have been described in detail previously.34In brief, full-field ERGs were recorded from both eyes of anesthetized mice with differential amplifiers having a bandwidth of 0.1 Hz to 1 kHz. The filtered traces were digitized at 5 kHz and stored on a computer for further analysis. The corneal electrodes were platinum wires embedded in the contact lenses, placed on the eye on a layer of hydroxypropyl methylcellulose (Goniosol; Ciba Vision, Duluth, GA). The reference electrode was a tungsten needle inserted subcutaneously in the forehead. The recording chamber served dually as a Faraday cage and a Ganzfeld, with appropriate ports and baffles to ensure uniform illumination. Intensities were calibrated as previously described.34Mice were dark adapted for a minimum of 12 hours before the ERG experiments. Preparations of the animals for recordings were made under dim red light. The mice were anesthetized with an intraperitoneal injection containing (in micrograms per gram body weight) 25 ketamine, 10 xylazine, and 1000 urethane and their pupils dilated with 1% tropicamide solution (Alconox, New York, NY). Before recording commenced, animals were maintained in complete darkness for 15 minutes.
RESULTS
Expression of Mutant RP1mRNA in Human Lymphoblasts
The 20 pathologic mutations found in RP1to date all cluster toward the beginning of exon 4 and are all truncation mutations.3,24Because all the pathologic mutations in RP1occur within the final exon, it is possible that the mutant RP1mRNA escapes nonsense-mediated mRNA decay to produce a truncated RP1 protein in the retina.25To ask if the mutant RP1mRNA escapes nonsense-mediated mRNA decay, we attempted to find retinal tissue samples of patients with RP1 disease in the FFB Histopathology Collection, which was then housed at the Scheie Eye Institute. One sample in this collection was from a patient known to have the Arg677Ter mutation in RP1. However, histopathologic evaluation of this sample showed end-stage retinal degeneration, with no photoreceptors remaining in the retina. Thus, this sample could not be used for detection of mutant RP1mRNA or protein in the retina. Tissue samples from 40 patients with less than end-stage retinal degeneration were identified by review of the pathologic descriptions for all retinal degeneration samples in the FFB collection. Genomic DNA was isolated from ocular or blood samples for each of these 40 patients, and the region of RP1that contains the known cluster of mutations was amplified and sequenced. Unfortunately, no mutations in the RP1gene were detected in these patients with early-stage RP (data not shown).
As a proxy for retinal tissue, we screened lymphoblasts isolated from patients with the Arg677Ter mutation, the most common RP1mutation, and related control subjects, for illegitimate RP1transcripts. Lymphoblasts from patients of the RP01 pedigree were obtained from the Coriell Cell Repository (http://locus.umdnj.edu/Coriell Institute for Medical Research, Camden, NJ).35mRNA was extracted from cultured lymphoblasts and nested RT-PCR was performed to amplify the region of the RP1transcript containing codon 677. This codon, and the primers used to amplify it, are within exon 4 of the RP1gene. As shown in Figure 1, RT-PCR successfully amplified RP1mRNA of patients homozygous and heterozygous for the Arg677Ter mutation, as well as unaffected control subject. Control PCR reactions using RNA that was not reverse transcribed (no RT) showed that the mRNA samples were not contaminated with genomic DNA (Fig. 1A). Restriction digests and DNA sequencing confirmed that the mutant mRNA was amplified in both heterozygous and homozygous patients (Figs. 1B, 1C).
FIGURE 1.
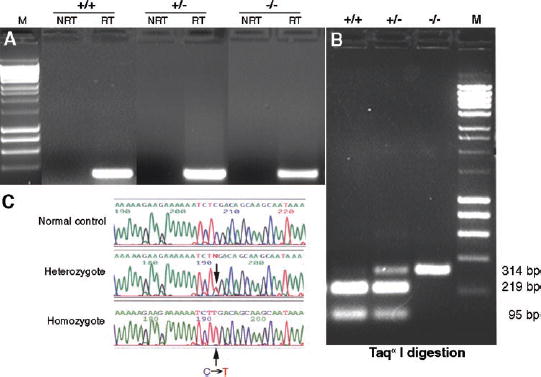
RP1mRNA is produced as an illegitimate transcript in lymphoblasts. (A)RT-PCR analysis of RNA from lymphoblasts of unaffected (+/+), heterozygous (+/−), and homozygous (−/−) members of the RP01 pedigree. Nested RT-PCR was performed with mRNA that had been isolated from cultured lymphoblasts. The 314-bp product from the second PCR reaction was detected in patients heterozygous and homozygous for the Arg677Ter mutation, as well as in unaffected control members of the RP01 pedigree. There were no products in no-reverse-transcriptase (NRT) control experiments, indicating no contamination with genomic DNA. (B)Restriction enzyme digestion of the PCR products from (A). The final PCR products from three genotypes were gel pu-rified and digested with Taqα1. The undigested 314-bp fragment from the mutant RP1allele was present in both heterozygous and homozygous patients. Bands of 219 and 95 bp from the digested wild-type PCR product were detected in heterozygous and unaffected control subjects. (C)Sequence analysis of the nested RT-PCR products. The C→T (Arg667Ter) mutation was detected in the patients with Arg667Ter RP1allele.
Generation of Rp1-mycMutant Mice
To create a mouse model to use for studies of the function of the RP1 protein and the mechanism of disease, we produced a truncated Rp1allele in mice using gene-targeting techniques. The targeting construct was designed to truncate the mouse Rp1coding sequence after codon 662 in exon 4, which is analogous to codon 677 in human RP1(GenBank AF155141 mouse cDNA; NM_006269 human cDNA). A 10-amino-acid myctag was incorporated at the 3′ end of the truncated exon 4 to allow us to distinguish the mutant and wild-type Rp1 proteins (Fig. 2A). After transfection of the linearized Rp1-myctargeting vector into TL-1 mouse ES cells and selection with G418, a single ES cell clone with the correct homologous recombination at the Rp1locus was identified by Southern blot analysis (Fig. 2B). This ES cell clone was injected into C57BL/6 blastocysts, and eight highly chimeric mice obtained. Germline transmission of the mutant Rp1allele was obtained from seven of these eight chimeric founders. Homozygous (Rp1myc/myc) mice were produced from heterozygous crosses at the expected Mendelian ratio. Neither Rp1+/mycnor Rp1myc/mycmice showed any obviously abnormal behavioral phenotype. All the mice used for the experiments described herein were from generations F1 to F3.
Localization of the Truncated Rp1-myc Protein
To determine whether the mutant Rp1-mycallele produces a truncated Rp1-myc protein in mouse retina, equal amounts of total retinal protein from 4-week-old Rp1+/+, Rp1+/myc, and Rp1myc/mycmice were subjected to Western blot analysis. Probing with anti-myc and anti-C′-Rp1 antibodies revealed that a truncated Rp1-myc protein with a predicted molecular mass of ˜70 kDa is present in mice with a mutant Rp1allele, but not in the wild-type mice (Fig. 3). In contrast, the normal Rp1 protein was detected in wild-type and heterozygous mice, but no signal could be detected in the homozygous mice (Fig. 3). The level of the normal Rp1 protein in Rp1+/mycheterozygous mice was approximately half of that detected in the wild-type. Similarly, the level of truncated Rp1-myc protein in Rp1+/mycheterozygous mice was approximately half that in Rp1myc/mychomozygous mice.
FIGURE 3.
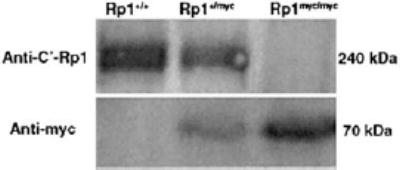
Western-blot analysis of wild-type Rp1 and mutant Rp1-myc protein expression. Total retinal proteins were extracted from 4-week-old Rp1+/+Rp1+/myc, and Rp1myc/mycmice. Equal amounts of total protein from each genotype were electrophoresed and transferred to polyvinylidene difluoride (PVDF) membrane and then probed with either antibodies to the C terminus of Rp1 (top: anti-C′-Rp1 antibody) or anti-myc (bottom). The normal mouse Rp1 protein (˜240 kDa) was detected in Rp1+/+and Rp1+/mycmice, but not in Rp1myc/mycmice. An ˜70-kDa protein corresponding to the predicted size of the mutant Rp1-myc protein was present in Rp1+/mycand Rp1myc/mycmice, but absent in Rp1+/+mice.
To identify the location of the truncated Rp1-myc protein in the retina, we performed confocal immunomicroscopic analysis of retinal sections from each group of mice. Antibodies against both Rp1 and myc were applied to all three sections. As was evident from the Western blot analysis, the wild-type protein was not detected in the Rp1myc/mycmice, whereas the truncated Rp1-myc protein was present only in mice with the mutant allele (Fig. 4A). Of note, the truncated Rp1-myc protein was found in the region of axoneme of the photoreceptor, in a location identical with that of the wild-type Rp1 protein. This was best demonstrated by coimmunostaining of the wild-type and Rp1-myc proteins in retinas of heterozygous mice (Fig. 4A). The distribution of the mutant protein matched that of the wild-type protein almost exactly, indicating that the signal(s) required for axoneme binding are present in the first 662 amino acids of Rp1. Close examination of the Rp1-myc immu-nofluorescence signal revealed that this mutant protein did not demonstrate the focus of intense signal at the junction between the inner segments and the outer segment that was observed with the wild-type Rp1 protein (Fig. 4B). This also could be seen in higher magnification confocal images (Fig. 4C).
FIGURE 4.
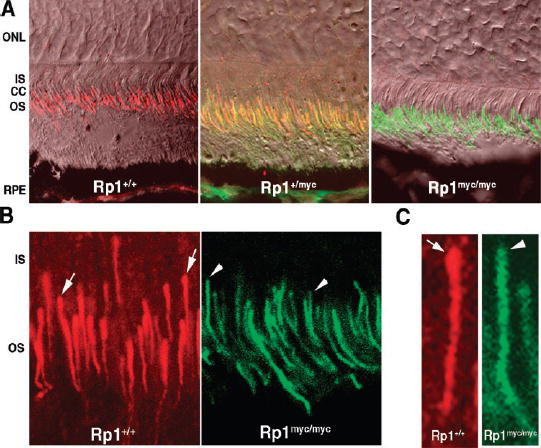
The Rp1-myc protein localizes to the axoneme of photoreceptor outer segments. (A)Immunofluorescence and Nomarski confocal images of retinal sections from 2-week-old mice after double labeling with antibodies against Rp1 (red)and myc (green). Left: wild-type retina. Strong Rp1 immuno-reactivity was observed along the photoreceptor axonemes; no myc immunoreactivity was detected. Middle: Rp1+/mycretina. Both Rp1 and myc signals were detected, and the distribution of the myc signal matched that of wild-type Rp1 signal (yellow). Right: Rp1myc/myc. The mutant Rp1-myc protein signal was located correctly in photoreceptor axonemes; no normal Rp1 was detected. (B)Close examination of the immunofluorescence signal revealed a subtle difference between the wild-type Rp1 and mutant Rp1-myc signals. The wild-type protein demonstrated a concentration at the proximal end of the signal, at the junction of the outer segment and connecting cilium (arrow, left). This distinct, intense signal is not observed for the mutant Rp1-myc protein (arrowheads, right). (C)Images of single photoreceptors from (B)at higher magnification show more clearly that the concentration of signal at the proximal end was present in wild-type Rp1 staining (arrow; left), but not in the mutant Rp1-myc staining (arrowhead; right).
Correct Outer Segment Protein Location in Rp1-mycMice
The ciliary axoneme of photoreceptors is thought to play an essential role in protein transport between the inner and outer segments.8To investigate the potential role of Rp1 in protein transport, retinal sections from 2-week-old light-adapted Rp1myc/mycmice and littermate control animals were immuno-stained with antibodies to known outer segment proteins. As shown in Figure 5, no mislocalization of rhodopsin or arrestin or α-CNGC was detected. Similar results were obtained for Rom1, ABCR, and cone opsins (data not shown).
FIGURE 5.
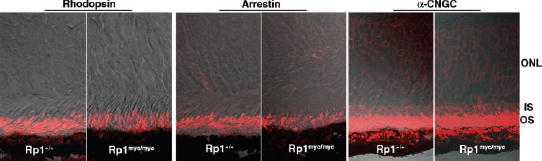
Outer segment proteins were correctly localized in Rp1-mycmice. Frozen retinal sections from 2-week-old wild-type (Rp1+/+) and homozygous mutant (Rp1myc/myc) mice were labeled with antibodies to rhodopsin, arrestin, and α-CNGC. No mislocation of rhodopsin was detected in the Rp1myc/mycmice. The distribution of arrestin and α-CNGC were also identical in the wild-type and mutant retinas.
Retinal Degeneration in Rp1-mycMice
To examine whether the truncated Rp1 protein affects photo-receptor function, we measured full-field ERGs of mice of the three genotypes at 4 to 5 weeks and 4 to 8 months of age, collecting and analyzing scotopic b-waves (Figs. 6A–C), a-waves (Figs. 6D–F), and cone-isolated b-waves (Table 1; see also Ref. 38). The principal finding is that the saturating amplitude of the rod a-wave (amax) of Rp1myc/mycmice was reduced by 58% at 4 to 5 weeks of age, and was practically absent in 4- to 8-month old animals (Figs. 6, 7). Perhaps surprisingly, the diminution of the a-wave in the 4- to 5-week-old animals was not accompanied by a major change in the amplification of the phototransduction process (Fig. 6; Table 1). The scotopic b-waves and cone-driven b-waves of Rp1myc/mycmice underwent a slower deterioration over the first 8-months than did the a-wave (Figs. 7A, 7B). Heterozygotes showed a smaller, and not statistically reliable, decrease in rod a-waves at both ages tested. The rod b-waves were significantly reduced at both 4 to 5 weeks and 4 to 8 months in Rp1+/mycmice, as were the cone b-waves at 4 to 8 months (Fig. 7). In a small population of 10- to 12-month-old animals, there was no reliable difference between Rp1+/+and Rp1+/mycin the saturating amplitudes of the three major ERG components (data not shown).
FIGURE 6.
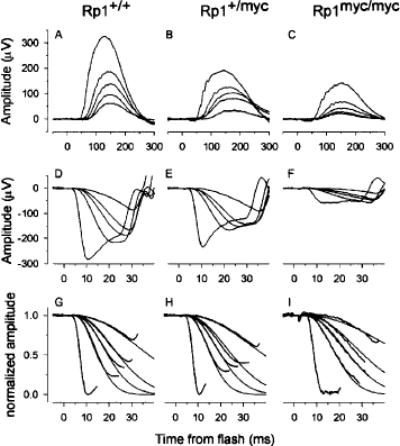
Electroretinograms of mice of the three genotypes at 4 to 5 weeks of age. (A–C)Scotopic b-waves obtained in response to a series of 510-nm flashes with luminances of 3.2 × 10−4, 9.2 × 10−4, 1.6 × 10−3, 3.4 × 10−3, and 1.8 × 10−2scot cd-s . m−2estimated to produce 0.16, 0.46, 0.79, 1.7, and 9.0 photoisomerizations/rod, respectively, in a normal mouse. Each trace is the average of nine individual records (the traces of the two eyes were also averaged during analysis). The saturating amplitude of the b-wave was extracted by fitting a hyperbolic saturation function to the intensity versus amplitude function.36(D–F)ERG a-waves, obtained in response to flashes having luminances of 0.74, 1.9, 4.0, 87.0, and 220 scot cd-s . m−2, estimated to produce 370, 960, 2,000, 4,000, and 110,000 photoisomerizations/rod, respectively in a wild-type mouse. The saturating amplitude of the a-wave was obtained directly from the response to the most intense flash. (G–I)The a-wave data of panels (D–F)are presented normalized by the saturating a-wave amplitude and fitted with a model of the activation phase of the phototransduction cascade to extract the amplification constant.37With flash intensities expressed in luminance units, the amplification parameters are KA2000, 1800, and 1000 seconds−2(scot cd-s. m−2)−1for the data of the Rp1+/+, Rp1+/myc, and Rp1myc/mycmice, respectively. For further details see Table 1.
TABLE 1.
Parameters of ERGs of Rp1-mycMice
| Rod-Driven b-Wave | Rod a-Wave | Cone-Driven b-Wave | |||
|---|---|---|---|---|---|
| Age/Genotype (n) | bmax,rod(μV) | I0.5(scot cd-s ∙ m–2) | amax(μV) | KAs–2(scot cd-s ∙ m–2)–1 | bmax,cone(μV) |
| 4–5 Weeks | |||||
| Rp1myc/myc(10) | 141 ± 64 | (2.1 ± 0.9) × 10–3 | 88 ± 45 | 1478 ± 690 | 81 ± 24 |
| Rp1+/myc(11) | 188 ± 60 | (1.4 ± 1.0) × 10–3 | 199 ± 54 | 1060 ± 390 | 130 ± 40 |
| Rp1+/+(10) | 251 ± 89 | (1.4 ± 0.8) × 10–3 | 233 ± 68 | 1700 ± 490 | 153 ± 66 |
| 4–8 Months | |||||
| Rp1myc/myc(10) | 109 ± 69 | (1.3 ± 0.6) × 10–3 | 36 ± 21 | 651 ± 250 | 49 ± 23 |
| Rp1+/myc(24) | 138 ± 55 | (1.5 ± 0.5) × 10–3 | 191 ± 64 | 1680 ± 600 | 89 ± 28 |
| Rp1+/+(24) | 203 ± 85 | (1.9 ± 0.6) × 10–3 | 233 ± 53 | 1700 ± 720 | 106 ± 24 |
The first column gives the age/genotype of the mice and the number of mice tested. The entries in each column are the mean ± SD of the parameter, measured for the group of animals identified in the first column. The parameters are as follows:bmax,rodis the saturating amplitude, and I0.5the intensity that produces a half-saturating amplitude, respectively, of the scotopic b-wave response, extracted as illustrated in Figs. 6A–C; amaxis the saturating amplitude of the a-wave, measured as illustrated in Figs. 6D–F; KAdescribes the efficacy of the activation steps of the rod transduction cascade (Methods, Figs. 6G–I); bmax,coneis the saturating amplitude of the cone-driven b-wave, obtained in the presence of a rod-saturating steady background.34
FIGURE 7.
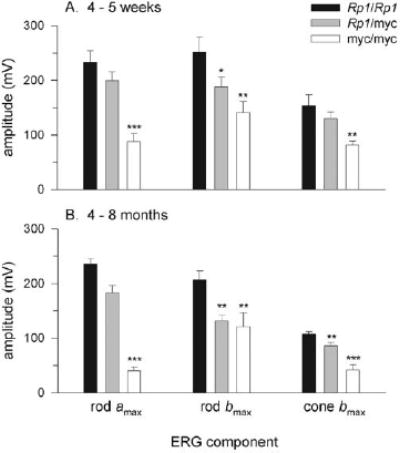
Saturating amplitudes of the principal components of the electroretinograms. Each panel presents the mean saturating amplitudes (±SEM) of the a-wave (rod amax), the scotopic b-wave (rod bmax),and the cone-driven b-wave (conebmax)obtained from populations of animals of the indicated genotypes of ages 4 to 5 weeks (A)and 4 to 8 months (B). Differences between the components of the wild-type (Rp1+/+) mice and mice of the other two genotypes that are statistically significant are indicated by *P< 0.05, **P< 0.01, and ***P< 0.001. See also Table 1.
Defective Outer Segment Formation in Rp1-mycMice
The retinal morphology of heterozygous and homozygous mice and wild-type littermate control animals was examined at P10, P14, 1 month, and 4 to 10 months of age. Eyes from three to seven Rp1+/mycand Rp1myc/mycmice and an equal number of littermate control animals were evaluated at each time point. Sections from the central retina, midperiphery, and periphery were examined from each eye. At the light microscopic level, the Rp1myc/mycmice demonstrated a progressive degeneration of photoreceptor cells. At 30 days of age, the outer nuclear layer (ONL) of retinas from the Rp1myc/mycmice was two to three rows thinner than in the control wild-type mice (Fig. 8A). Retinal degeneration was nearly complete in 10-month-old Rp1myc/mycmice, with only two rows of nuclei remaining at that date (Fig. 8A). Immunofluorescence showed that these two rows of photoreceptor nuclei contained both rods and cones and that the outer segments of both cell types were abnormal (Fig. 8C). In contrast, the morphology of the retinas in 10-month-old heterozygous Rp1+/mycmice was similar to that of age-matched control animals in all sections examined (Fig. 8B).
FIGURE 8.
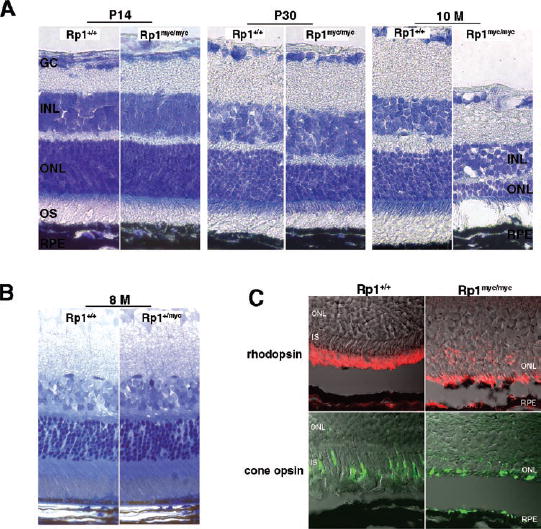
Progressive degeneration of photoreceptors in the Rp1myc/mycmice. (A)The extent of photoreceptor loss in Rp1myc/mycmice was estimated by observing the thickness of the outer nuclear layer (ONL) in sections of frozen retina from mutant and control mice at different ages. Lens-optic nerve sections were stained with Richard-son stain. At age P14, the thickness of the ONL was comparable between Rp1myc/mycand wild-type littermates (left). By 30 days of age, retinas in Rp1myc/mycmice showed significant degenerative changes. The thickness of the ONL was reduced by approximately two to three rows compared with wild-type littermate control animals (middle). By 10-months of age, only two rows of nuclei remained in the retinas of Rp1myc/mycmice (right). (B)Toluidine blue staining of Epon-embedded retinal sections from 8-month-old Rp1+/+and Rp1+/mycmice shows that the thickness of ONL and the length of outer segments did not differ significantly between Rp1+/+and Rp1+/mycmice. (C) Immunofluorescence and Nomar-ski confocal images of retinas in 10-month-old Rp1+/+and Rp1myc/mycmice. Antibodies against rhodopsin (red, top)and red/greencone opsin (green, bottom)show that both rods and cones remain in the degenerated Rp1myc/mycretinas. Mislocalization of both proteins to the inner segment and ONL was detected in the mutant mice.
Electron microscopy was used to evaluate the ultrastructure of photoreceptors in Rp1-mycmice. At 14 days of age, photo-receptor outer segments were grossly abnormal in homozygous Rp1myc/mycmice. As shown in Figure 9A, the outer segments of Rp1myc/mycmice lacked the organized arrangement of stacked discs seen in control animals. Small packets of enlarged discs were seen in place of intact outer segments. These smaller packets contained approximately 20 to 40 discs, and the stacks of discs tended to be oriented obliquely or parallel to the long axis of photoreceptor cells, instead of perpendicularly. The discs of Rp1myc/mycmice were two to three times larger than in control eyes, although they appeared to be complete, with identifiable rims.
FIGURE 9.
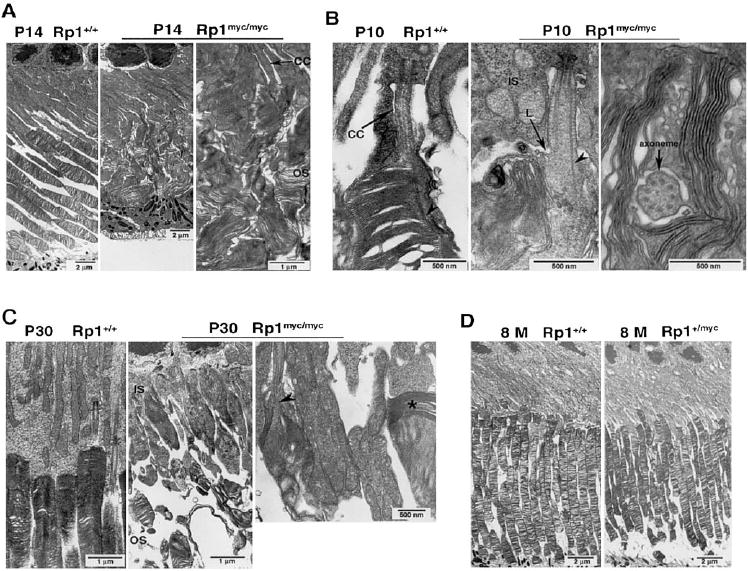
Ultrastructure of photoreceptors in Rp1-mycmice. Eyes of mice with indicated ages and genotypes were embedded in Epon, sectioned at 60- to 80-nm thickness and evaluated by electron microscopy. (A)Outer segments in the P14 wild-type retina appeared as straight, parallel cylinders from inner segments to the RPE. The stacks of discs were organized and perpendicular to the long axis of the cell (left). In contrast, outer segments in P14 Rp1myc/mycmice were disorganized. Small packets of discs were scattered in the space between the inner segments and RPE (middle). Close examination of the disorganized discs (right)showed that these small packets contained approximately 20 to 40 discs with diameters two to three times normal (2–4 μm), oriented obliquely or parallel to the long axis of the cell. Although abnormally oriented, the discs in Rp1myc/mycretinas were intact, with identifiable rims. (B)The morphology of the photoreceptor axoneme and newly forming discs at the base of the outer segments were examined at P10. The nascent discs were tightly aligned along the axoneme (arrowhead)at the base of the outer segment in the wild-type mice (left). In the Rp1myc/mycmice, even the early lip (L) of new membrane evagination at the base of the outer segment was misoriented, and nascent discs were parallel to the axoneme (arrowhead, middle). In addition, the disc rims were no longer associated with the axoneme. A cross section showed a normal axoneme structure, but also demonstrated discs oriented parallel to the axis of the outer segment (right). (C)At age P30 the outer segment dysplasia was more severe, with shortening and disorganization of outer segments structures in the photoreceptors of Rp1myc/mycmice (middle), compared with the wild-type control (left). Misoriented disc stacks (arrowhead)and short packets of discs (*)were still observed (right). (D)The ultrastructure of photoreceptors in 8-month-old Rp1+/mycmice (right)was grossly normal compared with that of wild-type mice (left). CC, connecting cilium; IS, inner segment; OS, outer segment.
Examination of micrographs from 10-day-old Rp1myc/mycmice shows that the abnormal outer segment structure observed at 14 days was due to a defect in disc morphogenesis (Fig. 9B). New membrane evaginations at the base of the outer segment started out incorrectly oriented and quickly lost the perpendicular orientation seen in wild-type or Rp1+/mycmice (9B, middle). This was also observed in cross sections. Although the axoneme structure appeared to be normal in cross sections in Rp1myc/mycmice, the presence of discs cut on edge in this view indicated that the discs were oriented parallel to the long axis of axoneme, instead of perpendicularly (Fig. 9B, right). The newly formed discs of Rp1myc/mycmice were not closely aligned along the axoneme, as normal discs uniformly are.
By 1 month of age, retinas from Rp1myc/mycmice exhibited severely shortened and disorganized outer segments compared with wild-type or Rp1+/mycmice (Fig. 9C). Again, the predominant feature of the abnormal discs in the Rp1myc/mycmice were small packets of intact but incorrectly oriented discs. For evaluation of the ultrastructure of photoreceptors in heterozygous Rp1+/mycmice, retinas from seven Rp1+/mycmice and seven littermate control animals at ages 6 to 8 months were examined extensively by electron microscopy. In contrast to the Rp1myc/mycmice, heterozygous Rp1+/mycmice had essentially normal photoreceptor ultrastructure up to 8 months of age, including normal cell bodies, as well as outer segments (Fig. 9D).
DISCUSSION
Rp1 Required for Correct OS Disc Orientation
The data presented above show that Rp1myc/mychomozygous mice have a profound defect in outer segment formation. Stacks of membrane discs in the Rp1myc/mycmice were oriented obliquely or parallel to the long axis of the outer segments, instead of perpendicularly. The discs were also large and were grouped in small packets of 20 to 40 discs, instead of the long arrays of mature discs found in healthy photorecep-tors. This defect in disc orientation appears to occur during disc formation. Even the early evaginations of membrane at the base of the outer segment were misdirected in the homozygous mutant mice (Fig. 9B). In control animals, the new discs always formed perpendicular to the axoneme, and discs retained their alignment with the axoneme after formation.39In homozygous mutant animals, the discs lost this association with the axoneme. Thus, it appears that Rp1 is required for the correct orientation and higher order stacking of outer segment discs.
Given the location of Rp1 in the axoneme and the abnormal disc orientation observed in the Rp1myc/mycmice, it is attractive to hypothesize that Rp1 functions as a connection between newly formed discs and the axoneme and that this interaction helps discs form in the correct orientation and stack up into outer segments. Proteins known to be located in the disc rims, such as rds/peripherin, Rom1, and ABCR are potential candidates for such an interaction.40-42Mutations in RP1, all of which cause loss of the C-terminal one half to two thirds of the protein, would then cause photoreceptor cell dysfunction and ultimately death by disrupting the interaction between the axoneme and the discs. We are currently searching for proteins that interact with RP1 and can mediate this interaction.
The importance of the connecting cilium and axoneme in disc formation has been recognized for many years. Indeed, the photoreceptor outer segment is considered to be a modified cilium adapted to optimize light absorption.43As a testament to the importance of the connecting cilium and axoneme in disc formation, targeted deletion of other protein components of the cilium can also lead to defects in disc formation. For example, mice deficient in RPGRIP exhibit defective outer segment orientation, in a fashion similar to that observed in the Rp1-mycmice.18
Localization of the Truncated Rp1-myc Protein
It was interesting to find that the truncated Rp1-myc protein localized correctly to the photoreceptor axoneme. This indicates that at least part of the information required for association of Rp1 with the axoneme is contained in the N-terminal third of the protein. It is likely that the two tandem doublecor-tin domains (DCX) at the N terminus of RP1 participate in the interaction of RP1 with the axoneme (Fig. 2). These domains (codons 35-236 of Rp1) are homologous to the microtubule binding domain of the doublecortin protein.44Doublecortin is a microtubule-associated protein (MAP) that is essential in the proper development of the layers of the cerebral cortex, and mutations in doublecortin cause X-linked lissencephaly and double cortex syndrome, which are inherited defects of the cerebral cortex associated with arrest of migrating cerebral cortical neurons.45-47The two tandem DCX domains in dou-blecortin are thought to stabilize microtubules by tethering tubulin dimers together or by cross-linking adjacent microtu-bules.48Additional data show that lack of the DCX domains from the Rp1 protein in vitro or in vivo leads to altered localization of the Rp1 protein (Liu Q, et al. IOVS2003;44: ARVO E-Abstract 4528). These data imply that the DCX domains in the N-terminal region of RP1 are active and raise the possibility that RP1 is a photoreceptor-specific MAP.
Aside from tubulin, axonemes are known to contain MAPs, and the components of the protein transport system, including dynein, kinesin, and IFT proteins.12It is likely that many other proteins are also required for ciliary axoneme structure and function, as a recent proteomic analysis detected more than 240 protein components in ciliary axonemes from respiratory epithelia.49It is reasonable to assume that photoreceptor axonemes contain unique proteins, given the specialized functions required to produce and maintain outer segments. Although RP1 was detected in the proteomic analysis of cultured respiratory epithelial cells mentioned earlier, we have not been able to detect the Rp1 protein in tissues, or cultured cells, with motile or primary cilia by using immunofluorescence assays (Pierce, et al. IOVS2003;44:ARVO E-Abstract 1529). RP1 thus appears to be one of the photoreceptor-specific, or at least photoreceptor-enriched, axoneme components.
One subtle difference between the location of the Rp1-myc protein and the wild-type Rp1 protein is detected at the junction of the outer segment and connecting cilium. The immu-nofluorescence signal for the wild-type protein demonstrates a concentration in this area, whereas such a concentration is not observed with antibodies against the myc-tagged mutant protein (Figs. 4B, 4C). This finding suggests that the C-terminal two thirds of Rp1 interacts with a protein or proteins found at this junction. Data from Rp1-exon 2/3 deletion mice support this concept. In these mice, abnormal splicing between exons 1 and 4 occurs, and a mutant Rp1 protein without the DCX domains is produced. The mutant exon 2/3 deletion protein is found at the junction between inner segments and outer segments, but only as a punctate signal.23Additional experiments show that the punctate signal is located at the base of the axoneme in outer segments (Liu Q, et al. IOVS2003;44:ARVO E-Abstract 4528). Actin and myosin II are known to be concentrated in this region at the base of the OS, and are thought to be important in disc formation, although their exact roles remain unclear.17,19,21A photoreceptor-specific form of cad-herin (prCAD) has been identified recently and found to be localized in the superficial aspects of newly evaginated disc membranes at the base of the outer segment. Targeted deletion of prCAD also results in fragmentation and disorganization of outer segments.22It is possible, therefore, that interactions between the C-terminal portion of RP1 and actin, myosin, cadherin, or another protein located at the base of the outer segment, are part of the mechanism by which Rp1 helps discs form in the correct orientation and stack up into outer segments.
The 10-amino-acid myctag was chosen for these studies because it is commonly used as an epitope tag and is recognized with high affinity by the well-characterized antibody 9E10.50Although we cannot rule out definitively that the myctag did not alter the function of the truncated Rp1-myc protein, the myctag itself does not generally affect protein function.51For example, introduction of the myctag into vascular endothelial growth factor (VEGF) did not alter its biological activity.52
Retinal Degeneration in Rp1myc/mycHomozygous Mice
The Rp1myc/mychomozygous mice undergo a rapid retinal degeneration. The amplitude of their rod a-waves is decreased by 4 weeks of age and is almost undetectable by 4 to 8 months of age. Rod and cone b-waves were also decreased in homozygous animals, although not to the same extent as the rod a-waves. Because the a-wave is the field potential produced by light suppression of the rod-circulating current,36,53the loss of the saturating a-wave amplitude in the Rp1myc/mycmice can be understood as a consequence of both loss of rods and disorder of the residual outer segments. Nonetheless, the proteins underlying the activation phase of rod phototransduction appear to be present in their normal ratios, because the amplification of the a-wave appears largely unchanged in the 4- to 5-week-old mice (Fig. 6; Table 1).54,55These observations are consistent with the conclusion that the primary defect in the rods is the misorientation of the outer segment discs and loss of large-scale outer segment structure, but not in the molecular mechanisms that deliver the lipid and protein components required for outer segment synthesis and for phototransduction. A similar phenotype was observed in the prCAD null mice.22
The finding that the components of the phototransduction cascade are present in normal ratios, even though outer segments are abnormal in the Rp1myc/mycmice, is consistent with the immunofluorescence data showing the correct location of outer segment proteins in the retinas of mutant mice (Fig. 5). No rhodopsin mislocalization was noted in the retinas of 2-week-old Rp1myc/mycmice, in contrast to findings with Rp1-exon 2/3 deletion mice.23The reason for this difference between the two animal models is not clear. It is possible that the differently truncated Rp1 proteins lead to different defects in protein function. It is also possible that protein transport is not a major function of Rp1, because the amount of rhodopsin mislocalization observed in the Rp1-exon 2/3 deletion mice was quite small compared with that observed in the retinas of mice with mutations in transport proteins, such as kinesin II or myosin VIIa.10,11
Mutant RP1mRNA and Nonsense-Mediated Decay
We chose to make the Rp1-mycallele after detecting the mutant RP1mRNA as an illegitimate transcript in lymphoblasts derived from patients with RP1 disease. We used lymphoblasts as a proxy for retina because a suitable sample of retina from a patient with RP1 was not available. Lymphoblasts of members of the RP01 pedigree were deposited in the Coriell Repository when this family was originally ascertained.35,56These lympho-blasts are a valuable resource, and can be used as an alternative to retinal tissue because there is a low level of illegitimate transcription of nonexpressed genes in many cell types, including lymphocytes.57RT-PCR data show that mutant RP1mRNA is present in lymphoblasts of patients heterozygous or homozygous for the Arg677Ter mutation in RP1. These data indicate that the mutant mRNA escapes nonsense-mediated mRNA decay, as predicted by the location of the truncation mutation in the final exon.25Although only indirect, the data also imply that truncated RP1 protein is made in the retina, because, when present, the mutant mRNA should be translated.
RP1 Disease Mechanism: Haploinsufficiency Versus Dominant Negative?
With the finding that mutant RP1mRNA can be detected in lymphoblasts, we hypothesized that mutations in RP1could lead to production of a truncated protein in the retina and may cause photoreceptor cell death through a dominant negative mechanism mediated by the truncated protein. We created the Rp1-mycallele in part to test this hypothesis. Our results indicate that the truncated Rp1-myc protein is nonfunctional, as animals homozygous for the mutant allele experience a rapid retinal degeneration. The lack of a major phenotype in the heterozygous animals, however, suggests that the truncated protein does not have a dominant negative effect. It is thus more likely that RP1 disease in humans is caused by decreased expression of functional RP1 protein, or haploinsuf-ficiency. In this case, the haploinsufficiency probably occurs because the Rp1-myc protein complement is nonfunctional. It still remains possible, however, that the mutant RP1mRNA is degraded in the retina. Additional evaluation of this question by using human tissue or point mutation knockin mice is warranted.
If mutations in RP1cause disease by haploinsufficiency, then the Rp1+/mycmice would still be expected to undergo retinal degeneration, because they have only one normal Rp1allele. However, the photoreceptors of the heterozygous mice remained relatively healthy. The lack of a major phenotype in the Rp1+/mycmice may be due to differences between mouse and human photoreceptors or to the genetic background of the Rp1-mycmice. It is known that the phenotype of patients with RP can vary dramatically, even among family members with the same mutation. This is true of patients with mutations in RP1.58The difference in disease severity has been suggested to be due to variations in disease-modifying genes. Effects of genetic background have also been observed in mice. A dramatic example involves the so called “rumpshaker” mutation of the proteolipid protein (Plp)gene, which causes dysmyelination in humans and mice. The phenotype of mice with this mutation depends critically on the genetic background in which the mutation is expressed. On the C3H background, there is normal longevity, whereas changing to a C57BL/6 strain results in seizures and death by postnatal day 30.59Similarly, the extent of neovascularization observed in response to an identical experimental stimulus varies up to 10-fold in different strains of mice.60We are currently outcrossing the Rp1-mycallele onto several different mouse strains to assess the effect of genetic background on the retinal phenotype.
Although photoreceptor structure appeared to be normal in the Rp1+/mycmice at 8 months of age, the Rp1-myc protein appeared to cause some alterations in photoreceptor function, because rod b-waves were significantly decreased at 4 to 8 months of age in the heterozygous animals. This finding suggests alterations in the signaling between rods and their bipolar cells.36However, given the apparently normal ultrastructure of photoreceptor cells in the retinas of these mice, it is unclear why the b-wave is diminished. At present, however, it is not certain whether these changes are the early signs of a slow retinal degeneration. The ERGs of a small number of 1-year-old F1 Rp1+/mycmice were not different from those of control subjects, implying that Rp1+/mycmice do not have a progressive retinal degeneration phenotype (data not shown). Additional investigations of the retinal function of older Rp1+/mycmice are in progress to address these questions.
To determine definitively whether haploinsufficiency of RP1can cause disease, we are preparing mice with a complete knockout of the Rp1coding region. We are hopeful that comparison of Rp1-mycmice and Rp1null mice will help clarify the mechanism of RP1 disease. This is an important issue, because the answer will dictate which potential therapeutic approaches are applicable to RP1 disease. Members of the RP1 Consortium are also preparing Rp1knockin mice, with a single nonsense mutation at Arg662 of the Rp1gene, but no other alterations, to further investigate the mechanism by which mutations in RP1cause photoreceptor cell death. These knockin mice will be especially useful for determining whether the mutant Rp1mRNA survives to produce a truncated protein. We are also hopeful that use of these different animal models will help elucidate the pathway(s) by which disorganized outer segments lead to photoreceptor cell death.
Acknowledgments
The authors thank Romaica Omarrudin, Suzanne Pavluk, David Pugh, Andrei Savchenko, and Xinyu Zhao for their technical assistance; Kathy Battaglia and Ann Milam for their comments on the manuscript; the other members of the RP1 Consortium for their advice; and Peter Sterling for use of his electron microscope.
Footnotes
Supported by National Eye Institute Grants EY12910 and EY02660, Bethesda, Maryland; Research to Prevent Blindness, New York, New York; the Foundation Fighting Blindness, Owings Mills, Maryland; the Rosanne Silbermann Foundation, Livingston, New Jersey; and the F. M. Kirby Foundation, Morristown, New Jersey.
Disclosure: Q. Liu, None; A. Lyubarsky, None; J.H. Skalet, None; E.N. Pugh, Jr, None; E.A. Pierce, None
References
- 1.Bowne SJ, Daiger SP, Hims MM. Mutations in the RP1 gene causing autosomal dominant retinitis pigmentosa. Hum Mol Genet. 1999;8:2121–2128. doi: 10.1093/hmg/8.11.2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Payne A, Vithana E, Khaliq S. RP1 protein truncating mutations predominate at the RP1 adRP locus. Invest Ophthalmol Vis Sci. 2000;41:4069–4073. [PubMed] [Google Scholar]
- 3.Berson EL, Grimsby JL, Adams SM, et al. Clinical features and mutations in patients with dominant retinitis pigmentosa-1 (RP1) Invest Ophthalmol Vis Sci. 2001;42:2217–2224. [PubMed] [Google Scholar]
- 4.Liu Q, Zhou J, Daiger SP, et al. Identification and subcellular localization of the RP1 protein in human and mouse photoreceptors. Invest Ophthalmol Vis Sci. 2002;43:22–32. [PMC free article] [PubMed] [Google Scholar]
- 5.Kaplan MW, Iwata RT, Sears RC. Lengths of immunolabeled ciliary microtubules in frog photoreceptor outer segments. Exp Eye Res. 1987;44:623–632. doi: 10.1016/s0014-4835(87)80134-3. [DOI] [PubMed] [Google Scholar]
- 6.Young RW. The renewal of photoreceptor cell outer segments. J Cell Biol. 1967;33:61–72. doi: 10.1083/jcb.33.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anderson DH, Fisher SK, Steinberg RH. Mammalian cones: disc shedding, phagocytosis, and renewal. Invest Ophthalmol Vis Sci. 1978;17:117–133. [PubMed] [Google Scholar]
- 8.Young RW. Passage of newly formed protein through the connecting cilium of retina rods in the frog. J Ultrastruct Res. 1968;23:462–473. doi: 10.1016/s0022-5320(68)80111-x. [DOI] [PubMed] [Google Scholar]
- 9.Whitehead JL, Wang SY, Bost-Usinger L, et al. Photoreceptor localization of the KIF3A and KIF3B subunits of the heterotrimeric microtubule motor kinesin II in vertebrate retina. Exp Eye Res. 1999;69:491–503. doi: 10.1006/exer.1999.0724. [DOI] [PubMed] [Google Scholar]
- 10.Liu X, Udovichenko IP, Brown SD, Steel KP, Williams DS. Myosin VIIa participates in opsin transport through the photoreceptor cilium. J Neurosci. 1999;19:6267–6274. doi: 10.1523/JNEUROSCI.19-15-06267.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marszalek JR, Liu X, Roberts EA, et al. Genetic evidence for selective transport of opsin and arrestin by kinesin-II in mammalian photoreceptors. Cell. 2000;102:175–187. doi: 10.1016/s0092-8674(00)00023-4. [DOI] [PubMed] [Google Scholar]
- 12.Pazour GJ, Baker SA, Deane JA, et al. The intraflagellar transport protein, IFT88, is essential for vertebrate photoreceptor assembly and maintenance. J Cell Biol. 2002;157:103–113. doi: 10.1083/jcb.200107108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kinney MS, Fisher SK. The photoreceptors and pigment epithelium of the adult Xenopus retina: morphology and outer segment renewal. Proc R Soc Lond B Biol Sci. 1978;201:131–147. doi: 10.1098/rspb.1978.0036. [DOI] [PubMed] [Google Scholar]
- 14.Steinberg RH, Fisher SK, Anderson DH. Disc morphogenesis in vertebrate photoreceptors. J Comp Neurol. 1980;190:501–508. doi: 10.1002/cne.901900307. [DOI] [PubMed] [Google Scholar]
- 15.Boesze-Battaglia K, Goldberg AF. Photoreceptor renewal: a role for peripherin/rds. Int Rev Cytol. 2002;217:183–225. doi: 10.1016/s0074-7696(02)17015-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chaitin MH, Schneider BG, Hall MO, Papermaster DS. Actin in the photoreceptor connecting cilium: immunocytochemical localization to the site of outer segment disk formation. J Cell Biol. 1984;99:239 –247. doi: 10.1083/jcb.99.1.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Williams DS, Linberg KA, Vaughan DK, Fariss RN, Fisher SK. Disruption of microfilament organization and deregulation of disk membrane morphogenesis by cytochalasin D in rod and cone photoreceptors. J Comp Neurol. 1988;272:161–176. doi: 10.1002/cne.902720202. [DOI] [PubMed] [Google Scholar]
- 18.Zhao Y, Hong DH, Pawlyk B, et al. The retinitis pigmentosa GTPase regulator (RPGR)-interacting protein: subserving RPGR function and participating in disk morphogenesis. Proc Natl Acad Sci USA. 2003;100:3965–3970. doi: 10.1073/pnas.0637349100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arikawa K, Williams DS. Organization of actin filaments and im-munocolocalization of alpha-actinin in the connecting cilium of rat photoreceptors. J Comp Neurol. 1989;288:640 –646. doi: 10.1002/cne.902880410. [DOI] [PubMed] [Google Scholar]
- 20.Wolfrum U. Centrin in the photoreceptor cells of mammalian retinae. Cell Motil Cytoskeleton. 1995;32:55–64. doi: 10.1002/cm.970320107. [DOI] [PubMed] [Google Scholar]
- 21.Chaitin MH, Coelho N. Immunogold localization of myosin in the photoreceptor cilium. Invest Ophthalmol Vis Sci. 1992;33:3103–3108. [PubMed] [Google Scholar]
- 22.Rattner A, Smallwood PM, Williams J, et al. A photoreceptor-specific cadherin is essential for the structural integrity of the outer segment and for photoreceptor survival. Neuron. 2001;32:775–786. doi: 10.1016/s0896-6273(01)00531-1. [DOI] [PubMed] [Google Scholar]
- 23.Gao J, Cheon K, Nusinowitz S, et al. Progressive photoreceptor degeneration, outer segment dysplasia, and rhodopsin mislocaliza-tion in mice with targeted disruption of the retinitis pigmentosa-1 (Rp1) gene. Proc Natl Acad Sci USA. 2002;99:5698 –5703. doi: 10.1073/pnas.042122399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dietrich K, Jacobi FK, Tippmann S, et al. A novel mutation of the RP1 gene (Lys778ter) associated with autosomal dominant retinitis pigmentosa. Br J Ophthalmol. 2002;86:328 –332. doi: 10.1136/bjo.86.3.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang J, Sun X, Qian Y, Maquat LE. Intron function in the nonsense-mediated decay of beta-globin mRNA: indications that pre-mRNA splicing in the nucleus can influence mRNA translation in the cytoplasm. RNA. 1998;4:801–815. doi: 10.1017/s1355838298971849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thorvaldsen JL, Duran KL, Bartolomei MS. Deletion of the H19 differentially methylated domain results in loss of imprinted expression of H19 and Igf2. Genes Dev. 1998;12:3693–3702. doi: 10.1101/gad.12.23.3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Labosky PA, Winnier GE, Jetton TL, et al. The winged helix gene, Mf3, is required for normal development of the diencephalon and midbrain, postnatal growth and the milk-ejection reflex. Development. 1997;124:1263–1274. doi: 10.1242/dev.124.7.1263. [DOI] [PubMed] [Google Scholar]
- 28.Subramanian A, Ranganathan P, Diamond SL. Nuclear targeting peptide scaffolds for lipofection of nondividing mammalian cells. Nat Biotechnol. 1999;17:873–877. doi: 10.1038/12860. [DOI] [PubMed] [Google Scholar]
- 29.Matise MP, Auerbach W, Joyner AL. In: Gene Targeting: A Practical Approach . Oxford, UK: Oxford University Press; 2000. Production of targeted embryonic stem cell lines; pp. 101–132. [Google Scholar]
- 30.Laemmli UK. Cleavage of structural proteins during assembly of the head of bacteriophage T4. Nature. 1970;227:680 –685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 31.Towbin H, Stahelen T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350 –4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Korf HW, Moller M, Gery I, Zigler JS, Klein DC. Immunocytochemical demonstration of retinal S-antigen in the pineal organ of four mammalian species. Cell Tissue Res. 1985;239:81–85. doi: 10.1007/BF00214906. [DOI] [PubMed] [Google Scholar]
- 33.Wang Y, Macke JP, Merbs SL, et al. A locus control region adjacent to the human red and green visual pigment genes. Neuron. 1992;9:429 –440. doi: 10.1016/0896-6273(92)90181-c. [DOI] [PubMed] [Google Scholar]
- 34.Lyubarsky AL, Falsini B, Pennesi ME, Valentini P, Pugh EN., Jr UVand midwave-sensitive cone-driven retinal responses of the mouse: a possible phenotype for coexpression of cone photopigments. J Neurosci. 1999;19:442–455. doi: 10.1523/JNEUROSCI.19-01-00442.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blanton SH, Heckenlively JR, Cottingham AW, et al. Linkage mapping of autosomal dominant retinitis pigmentosa (RP1) to the pericentric region of human chromosome 8. Genomics. 1991;11:857–869. doi: 10.1016/0888-7543(91)90008-3. [DOI] [PubMed] [Google Scholar]
- 36.Pugh EN, Falsini B, Lyubarsky AL. The origin of the major rod- and cone-driven components of the rodent electroretinogram and the effect of age and light-rearing history on the magnitude of these components In . In: Williams TP, Thistle AB, editors. Photostasis and Related Phenomena . New York: Plenum Press; 1998. pp. 93–128. [Google Scholar]
- 37.Lamb TD, Pugh EN., Jr A quantitative account of the activation steps involved in phototransduction in amphibian photoreceptors. J Physiol. 1992;449:719 –758. doi: 10.1113/jphysiol.1992.sp019111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lyubarsky AL, Chen C, Simon MI, Pugh EN., Jr Mice lacking G-protein receptor kinase 1 have profoundly slowed recovery of cone-driven retinal responses. J Neurosci. 2000;20:2209 –2217. doi: 10.1523/JNEUROSCI.20-06-02209.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nilsson SE. Receptor cell outer segment development and ultra-structure of the disk membranes in the retina of the tadpole (Rana pipens) J Ultrastruct Res. 1964;11:581–620. doi: 10.1016/s0022-5320(64)80084-8. [DOI] [PubMed] [Google Scholar]
- 40.Azarian SM, Travis GH. The photoreceptor rim protein is an ABC transporter encoded by the gene for recessive Stargardt’s disease (ABCR) FEBS Lett. 1997;409:247–252. doi: 10.1016/s0014-5793(97)00517-6. [DOI] [PubMed] [Google Scholar]
- 41.Bascom RA, Manara S, Collins L, et al. Cloning of the cDNA for a novel photoreceptor membrane protein (rom-1) identifies a disk rim protein family implicated in human retinopathies. Neuron. 1992;8:1171–1184. doi: 10.1016/0896-6273(92)90137-3. [DOI] [PubMed] [Google Scholar]
- 42.Molday RS, Hicks D, Molday L. Peripherin: a rim-specific membrane protein of rod outer segment discs. Invest Ophthalmol Vis Sci. 1987;28:50 –61. [PubMed] [Google Scholar]
- 43.Horst CJ, Johnson LV, Besharse JC. Transmembrane assemblage of the photoreceptor connecting cilium and motile cilium transition zone contain a common immunologic epitope. Cell Motil Cytoskel-eton. 1990;17:329 –344. doi: 10.1002/cm.970170408. [DOI] [PubMed] [Google Scholar]
- 44.Pierce EA, Quinn T, Meehan T, et al. Mutations in a gene encoding a new oxygen-regulated photoreceptor protein cause dominant retinitis pigmentosa. Nat Genet. 1999;22:248 –254. doi: 10.1038/10305. [DOI] [PubMed] [Google Scholar]
- 45.Gleeson JG, Allen KM, Fox JW, et al. Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell. 1998;92:63–72. doi: 10.1016/s0092-8674(00)80899-5. [DOI] [PubMed] [Google Scholar]
- 46.Gleeson JG, Lin PT, Flanagan LA, Walsh CA. Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron. 1999;23:257–271. doi: 10.1016/s0896-6273(00)80778-3. [DOI] [PubMed] [Google Scholar]
- 47.Taylor KR, Holzer AK, Bazan JF, Walsh CA, Gleeson JG. Patient mutations in doublecortin define a repeated tubulin-binding domain. J Biol Chem. 2000;275:34442–34450. doi: 10.1074/jbc.M007078200. [DOI] [PubMed] [Google Scholar]
- 48.Kim MH, Cierpicki T, Derewenda U, et al. The DCX-domain tandems of doublecortin and doublecortin-like kinase. Nat Struct Biol. 2003;10:324 –333. doi: 10.1038/nsb918. [DOI] [PubMed] [Google Scholar]
- 49.Ostrowski LE, Blackburn K, Radde KM, et al. A proteomic analysis of human cilia: identification of novel components. Mol Cell Pro-teomics. 2002;1:451–465. doi: 10.1074/mcp.m200037-mcp200. [DOI] [PubMed] [Google Scholar]
- 50.Evan GI, Lewis GK, Ramsay G, Bishop JM. Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol Cell Biol. 1985;5:3610 –3616. doi: 10.1128/mcb.5.12.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cravchik A, Matus A. A novel strategy for the immunological tagging of cDNA constructs. Gene. 1993;137:139 –143. doi: 10.1016/0378-1119(93)90262-2. [DOI] [PubMed] [Google Scholar]
- 52.Chavand O, Spilsbury K, Rakoczy PE. Addition of a c-myc epitope tag within the VEGF protein does not affect in vitro biological activity. Biochem Cell Biol. 2001;79:107–112. [PubMed] [Google Scholar]
- 53.Hagins WA, Penn RD, Yoshikami S. Dark current and photocurrent in retinal rods. Biophys J. 1970;10:380 –412. doi: 10.1016/S0006-3495(70)86308-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pugh EN, Lamb TD. Amplification and kinetics of the activation steps in phototransduction. Biochim Biophys Acta. 1993;1141:111–149. doi: 10.1016/0005-2728(93)90038-h. [DOI] [PubMed] [Google Scholar]
- 55.Lyubarsky AL, Pugh EN., Jr Recovery phase of the murine rod photoresponse reconstructed from electroretinographic recordings. J Neurosci. 1996;16:563–571. doi: 10.1523/JNEUROSCI.16-02-00563.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Spence MA, Sparkes RS, Heckenlively JR, et al. Probable genetic linkage between autosomal dominant retinitis pigmentosa (RP) and amylase (AMY2): evidence of an RPlocus on chromosome 1. Am J Hum Genet. 1977;29:397–404. [PMC free article] [PubMed] [Google Scholar]
- 57.Cooper DN, Berg LP, Kakkar VV, Reiss J. Ectopic (illegitimate) transcription: new possibilities for the analysis and diagnosis of human genetic disease. Ann Med. 1994;26:9–14. doi: 10.3109/07853899409147321. [DOI] [PubMed] [Google Scholar]
- 58.Jacobson SG, Cideciyan AV, Iannaccone A, et al. Disease expression of RP1 mutations causing autosomal dominant retinitis pig-mentosa. Invest Ophthalmol Vis Sci. 2000;41:1898 –1908. [PubMed] [Google Scholar]
- 59.Al Saktawi K, McLaughlin M, Klugmann M, et al. Genetic background determines phenotypic severity of the Plp rumpshaker mutation. J Neurosci Res. 2003;72:12–24. doi: 10.1002/jnr.10561. [DOI] [PubMed] [Google Scholar]
- 60.Rohan RM, Fernandez A, Udagawa T, Yuan J, D’Amato RJ. Genetic heterogeneity of angiogenesis in mice. FASEB J. 2000;14:871–876. doi: 10.1096/fasebj.14.7.871. [DOI] [PubMed] [Google Scholar]