

Abstract
A unified view of polymer, dumbbell, and oligonucleotide nearest-neighbor (NN) thermodynamics is presented. DNA NN ΔG°37 parameters from seven laboratories are presented in the same format so that careful comparisons can be made. The seven studies used data from natural polymers, synthetic polymers, oligonucleotide dumbbells, and oligonucleotide duplexes to derive NN parameters; used different methods of data analysis; used different salt concentrations; and presented the NN thermodynamics in different formats. As a result of these differences, there has been much confusion regarding the NN thermodynamics of DNA polymers and oligomers. Herein I show that six of the studies are actually in remarkable agreement with one another and explanations are provided in cases where discrepancies remain. Further, a single set of parameters, derived from 108 oligonucleotide duplexes, adequately describes polymer and oligomer thermodynamics. Empirical salt dependencies are also derived for oligonucleotides and polymers.
The application of the nearest-neighbor (NN) model to nucleic acids was pioneered by Zimm (1) and by Tinoco and coworkers (2–6). Subsequently, several experimental and theoretical papers on DNA and RNA NN thermodynamics have appeared (7–22). There has been disagreement concerning a number of issues, particularly differences between DNA polymer and oligonucleotide NN thermodynamic trends and the salt dependence of nucleic acid denaturation. These differences have led to the notion that there is a “length dependency” to DNA thermodynamics (18). In this article, I show that there is a length dependence to salt effects but not for the NN propagation energies. Instead, a single set of parameters derived from 108 oligonucleotide duplexes (22) adequately describes polymer and oligonucleotide behavior.
The major sources of confusion in the literature are that the different studies use different oligonucleotide and polymer design, different methods for determining thermodynamics, different methods for analyzing data, different salt conditions, and different formats for presenting the NN parameters. In this article, the results from seven studies (7, 10, 12, 17, 18, 20, 21) are presented in the same format so that direct comparisons can be made. These data are compared with the recently compiled “unified” oligonucleotide NN parameters based on a collection of 108 oligonucleotide duplexes from the literature (22). This work emphasizes the ΔG°37 parameters because ΔG°37 is more accurate than ΔH° or ΔS° due to compensating errors (22). Remarkably, there is consensus agreement among the parameters determined from six laboratories (7, 10, 17, 18, 20, 21).
Background
The NN Model.
The NN model for nucleic acids assumes that the stability of a given base pair depends on the identity and orientation of neighboring base pairs. Throughout this paper, the 10 NN dimer duplexes are represented with a slash separating strands in antiparallel orientation (e.g., AC/TG means 5′-AC-3′ Watson–Crick base-paired with 3′-TG-5′). For oligonucleotide duplexes, additional parameters for the initiation of duplex formation are introduced. Importantly, all other sequence-independent effects are also combined into the initiation parameter including differences between terminal and internal NNs (23) and counterion condensation (24, 25). To account for differences between duplexes with terminal A⋅T vs. terminal G⋅C pairs, two initiation parameters are introduced (22, 23): “initiation with terminal G⋅C” and “initiation with terminal A⋅T”. An additional entropic penalty (26) for the maintenance of the C2 symmetry of self-complementary duplexes is also included. The total ΔG°37 is given by:
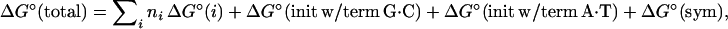 |
1 |
where ΔG°(i) are the standard free-energy changes for the 10 possible Watson–Crick NNs (e.g., ΔG°(1) = ΔG°37(AA/TT), ΔG°(2) = ΔG°37(TA/AT), … etc.), ni is the number of occurrences of each nearest neighbor, i, and ΔG°(sym) equals +0.43 kcal/mol (1 cal = 4.184 J) if the duplex is self-complementary and zero if it is non-self-complementary.
Application of the Unified NN Parameters.
Fig. 1 illustrates the calculation of ΔG°37 for the sequence CGTTGA⋅TCAACG using the unified NN parameters (22) in Table 1. The ΔH° and ΔS° parameters are analogously calculated from the parameters in Table 2 (22). The ΔG°37 can also be calculated from ΔH° and ΔS° parameters by using the equation:
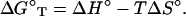 |
2 |
If large temperature extrapolation from 37°C is required, then the difference between the heat capacities of the folded and denatured states, ΔC°p, should be accounted for (27, 28). Previous data have indicated that ΔC°p is usually small for nucleic acids (29, 30). Due to enthalpy–entropy compensation, ΔG°37 is relatively insensitive to ΔC°p.
Figure 1.

Application of the unified nearest neighbor parameters (Table 1) and Eq. 1 to predict ΔG°37. Each arrow points to the middle of one of the NN dimers. The duplex CGTTGA⋅TCAACG is non-self-complementary and thus ΔG°(sym) is zero.
Table 1.
Comparison of published NN free energy parameters at 37°C
| Sequence | Parameter, kcal/mol
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Gotoh (ref. 7) | Vologodskii (ref. 10) | Breslauer (ref. 12) | Blake (ref. 17) | Benight (ref. 18) | SantaLucia (ref. 20) | Sugimoto (ref. 21) | Unified (ref. 22) | |
| AA/TT | −0.43 | −0.89 | (−1.66) | −0.67 | −0.93 | −1.02 | −1.20 | −1.00 |
| AT/TA | −0.27 | −0.81 | −1.19 | −0.62 | −0.83 | −0.73 | −0.90 | −0.88 |
| TA/AT | −0.22 | −0.76 | −0.76 | −0.70 | −0.70 | −0.60 | −0.90 | −0.58 |
| CA/GT | −0.97 | −1.37 | −1.80 | −1.19 | −1.26 | −1.38 | −1.70 | −1.45 |
| GT/CA | −0.98 | −1.35 | −1.13 | −1.28 | −1.52 | −1.43 | −1.50 | −1.44 |
| CT/GA | −0.83 | −1.16 | −1.35 | −1.17 | −1.03 | −1.16 | −1.50 | −1.28 |
| GA/CT | −0.93 | −1.25 | −1.41 | −1.12 | −1.56 | −1.46 | −1.50 | −1.30 |
| CG/GC | −1.70 | −1.99 | (−3.28) | −1.87 | (−1.65) | −2.09 | (−2.80) | −2.17 |
| GC/CG | −1.64 | −1.96 | (−2.82) | −1.85 | −2.44 | −2.28 | −2.30 | −2.24 |
| GG/CC | −1.22 | −1.64 | (−2.75) | −1.55 | −1.67 | −1.77 | −2.10 | −1.84 |
| Average | −0.92 | −1.32 | −1.82 | −1.20 | −1.36 | −1.39 | −1.64 | −1.42 |
| Init. w/term. G·C* | NA | NA | (+2.60) | NA | NA | 0.91 | (+1.70) | 0.98 |
| Init. w/term. A·T* | NA | NA | (+2.60) | NA | NA | 1.11† | (+1.70) | 1.03 |
| Sodium concentration, M | 0.0195 | 0.195 | 1.0 | 0.075 | 0.115 | 1.0 | 1.0 | 1.0 |
| Rank of stacking matrix | 8 | 8 | 11 | 8 | 9 | 10 | 11 | 12 |
Values in parentheses differ from the unified parameters by more than 0.5 kcal/mol. To compare the polymer parameters with the unified parameters apply the salt correction given by Eg. 9 (the salt corrections are −0.51 kcal/mol for the Gotoh parameters, −0.11 kcal/mol for the Vologodskii parameters, and −0.27 kcal/mol for the Blake parameters). NA, not applicable because these studies did not determine duplex initiation parameters. The values shown for the polymer studies of Gotoh and Tagashira (7), Vologodskii, [et al. (10)], and Blake [Delcourt and Blake (17)] are recalculated to reflect the minimal sequnce dependence of the 10 NN dimers, consistent with the eight invariants (see text). For ref. 12, ΔG°37 was calculated from the published ΔH° and ΔS° parameters by assuming ΔC°p is zero. The NNs were calculated from table 3 of ref. 18 (see text).
The initiation parameter is listed according to the format given in ref. 22.
This work suggested that oligonucleotides with terminal 5′-T-A-3′ base pairs should have a penalty of +0.4 kcal/mol but that no penalty should be given for terminal 5′-A-T-3′ pairs. To present this in the unified format, the average penalty of +0.2 kcal is added to the initiation energy. This work also indicated uncertainty in initiation at A·T pairs (2.8 ± 1 kcal/mol). The work of Sugimoto et al. (21) and Allawi and SantaLucia (22) indicate that initiation at A·T and G·C pairs are within experimental error of one another.
Table 2.
Unified oligonucleotide ΔH° and ΔS° NN parameters in 1 M NaCl (22)
| Sequence | ΔH° kcal/mol | ΔS° cal/k·mol |
|---|---|---|
| AA/TT | −7.9 | −22.2 |
| AT/TA | −7.2 | −20.4 |
| TA/AT | −7.2 | −21.3 |
| CA/GT | −8.5 | −22.7 |
| GT/CA | −8.4 | −22.4 |
| CT/GA | −7.8 | −21.0 |
| GA/CT | −8.2 | −22.2 |
| CG/GC | −10.6 | −27.2 |
| GC/CG | −9.8 | −24.4 |
| GG/CC | −8.0 | −19.9 |
| Init. w/term. G·C | 0.1 | −2.8 |
| Init. w/term. A·T | 2.3 | 4.1 |
| Symmetry correction | 0 | −1.4 |
Prediction of the Melting Temperature TM.
TM is defined as the temperature at which half of the strands are in the double-helical state and half are in the “random-coil” state. For self-complementary oligonucleotide duplexes, the TM is calculated from the predicted ΔH° and ΔS° and the total oligonucleotide strand concentration CT, by using the equation:
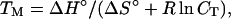 |
3 |
where R is the gas constant (1.987 cal/K⋅mol). For non-self-complementary molecules, CT in Eq. 3 is replaced by CT/4 if the strands are in equal concentration or by (CA − CB/2) if the strands are at different concentrations, where CA and CB are the concentrations of the more concentrated and less concentrated strands, respectively. Synthetic polymers with simple repeat sequences usually melt in a single cooperative transition (approximately two-state) that is concentration-independent so the TM = ΔH°/ΔS°. Natural polymers with heterogeneous sequences, on the other hand, usually melt with many stable intermediate states (non-two-state) and accurate prediction requires a statistical mechanical partition function approach (11, 31–33).
Methods
Formats of the NN Model.
Three formats for presenting NN thermodynamic data are as follows: (i) the 10 NN base pair dimer-stacking energies approach (4, 12, 13, 20, 21), (ii) the linearly independent sequences approach (also known as the “polymer approach”) (3, 10, 19), and (iii) the “independent short sequences” (ISS) approach, which presents the oligonucleotide NN data in an irreducible representation (23). All three of these methods are valid and provide equivalent predictions within round-off error (J.S. and D. M. Gray, unpublished result). In this work, the NN parameters from the literature are cast in the dimer-stacking format because it is easier for nonexperts to apply, because all 10 NN parameters are required to fully describe oligonucleotide melting, and because the ISS model does not apply to polymers.
The Rank of the Stacking Matrix.
An understanding of the effects of the rank of the stacking matrix is essential to reconcile the literature NN parameters derived from polymers with those derived from oligonucleotides. The stacking matrix S has dimensions M × N for a data set of M sequences and the columns N contain the number of occurrences in each DNA sequence of the 10 NN dimers plus initiation parameters for duplexes. The column rank of the stacking matrix determines the maximum number of parameters that can be uniquely determined (invariants) (19, 34). Because of constraints on the NN composition (23), there are only eight invariants for polymers that are usually expressed as linearly independent sequences. A convenient set of eight linearly independent sequences was provided by Vologodskii et al. (10):
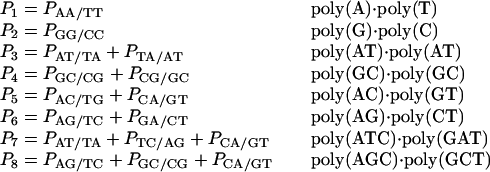 |
In this set, P is a measurable property such as ΔG°37, ΔH°, or ΔS°. Linear combinations of these eight invariants can be used to completely describe the behavior of any DNA polymer within the limits of the NN model. For oligonucleotide dumbbells with fixed termini but different lengths, nine invariants can be determined (18, 19). To fully characterize the thermodynamics of oligonucleotide duplexes, parameters for all 10 NN dimers (plus initiation parameters) are required. Importantly, a set of sequences with a rank of 8 (or 9 for dumbbells) can be used to derive a set of the 10 NN dimer energies that are a linear least-squares fit of the data set, but the solution is not unique. To verify that the solution is not unique, one can add a constant, C, to one of the dimers [other than AA/TT or GG/CC, which are uniquely determined for both polymers and oligomers (18)] and then add or subtract C or zero from the other dimers subject to the constraints of the eight linearly independent sequences. An alternative solution is obtained that makes exactly equal predictions as the first solution but with different trends in the NNs. The method of singular value decomposition (SVD) (20, 22, 34) provides the solution with C equal to zero and represents the minimum sequence dependence of the 10 dimers consistent with the eight invariants. Other methods for obtaining a linear least-squares fit of the data in terms of 10 parameters, particularly iterative methods (e.g., Gauss elimination or Gauss–Jordan iteration with back substitution) provide solutions with a nonzero and arbitrary value of C; the 10 dimers from these methods have an artificially larger sequence dependence than that determined by SVD. The solution obtained from these other methods, however, is a linear least-squares fit and makes predictions that are equal to those of the parameter set obtained with SVD. These points are at the heart of reconciling the NN data sets of Gotoh and Tagashira (7) and of Delcourt and Blake (17) with the oligonucleotide NN parameters (see below).
Converting Polymer Stability Temperatures to Energies.
Several studies (7, 10, 17, 18) present the NN stabilities in terms of Kelvin temperatures, T(i), where i are the NN dimers, instead of free energy changes, ΔG°(i). The dimer stacking ΔH°(i) can be calculated from T(i) with Eq. 4:
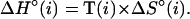 |
4 |
The polymer studies assume that the dimer propagation ΔS°(i) is −24.85 ± 1.74 cal/K⋅mol for all stacking dimers and is independent of the salt concentration (17, 18). The dimer ΔG°37 values are then calculated with Eq 2. For example, table 2 of Delcourt and Blake (17) lists T(TA/AT) = 56.31°C, which gives ΔH°(TA/AT) = (56.31 + 273.15) × −24.85 e.u. = −8,187 cal/mol (where e.u. is entropy unit). Using Eq. 2 gives ΔG°37 = −8,187 cal/mol − 310.15 × −24.85 e.u. = −0.48 kcal/mol. This conversion was performed for all of the NN parameters given in Delcourt and Blake (17), Vologodskii et al. (10), and Gotoh and Tagashira (7). To remove the C contribution, the literature NN parameters (7, 10, 17) were used to calculate the eight invariants given above (P1 through P8) and then SVD was used to produce a new set of NN parameters with C equal to zero (Table 1).
The studies of Vologodskii et al. (10) and Doktycz et al. (18) separated the dimer temperature stabilities into “hydrogen bonding” contributions THB(i) that are dependent on percent G+C content and “dimer-stacking” contributions TST(i) that are perturbations that contain the NN sequence dependence (these terms also contain the other fundamental interactions such as electrostatics and conformational entropy). The experimental work of Frank-Kamenetskii (35) indicated that hydrogen bonding contributions are salt-dependent and calculated as follows:
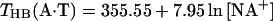 |
5a |
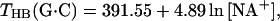 |
5b |
where THB(A⋅T) and THB(G⋅C) are the hypothetical melting temperatures of isolated A⋅T and G⋅C pairs without any stacking interactions and with the initiation parameter set to zero (polymer behavior). Thus, THB(AA/TT), THB(AT/TA), and THB(TA/AT) are given by Eq. 5a; THB(GG/CC), THB(GC/CG), and THB(CG/GC) are given by Eq. 5a; and THB(CA/GT), THB(AC/TG), THB(GA/CT), and THB(AG/TC) are given by the average of Eqs. 5a and 5b. Eqs. 5a and 5b were used to analyze the data of Vologodskii et al. (10). Doktycz et al. (18) performed their dumbbell study in 0.115 M Na+ and found experimentally that THB(A⋅T) and THB(G⋅C) were 339.67 K and 383.67 K, respectively. Table 3 of Vologodskii et al. (10) lists TST(i), and other works (17, 18) list the stacking perturbations as δΔG°(i). Eq. 6 allows calculation of stability temperatures T(i) from THB(i) and TST(i) or δΔG°(i).
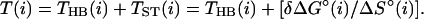 |
6 |
For example, the study of Doktycz et al. (18) reported δΔG°(AA/TT) as −196 cal/mol. Using Eqs. 6, 4, and 2 for the AA/TT stack gives:
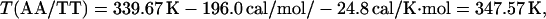 |
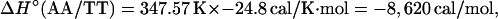 |
and
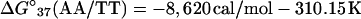 |
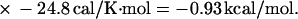 |
All other parameters in Table 1 in the columns for Gotoh and Tagashira (7), Vologodskii et al. (10), Delcourt and Blake (17), and Doktycz et al. (18) were calculated similarly.
Table 3.
Linear regression parameters for plots of literature ΔG°37 NN parameters vs. the unified NN parameters
| Parameter | Gotoh (ref. 7) | Vologodskii (ref. 10) | Breslauer (ref. 12) | Blake (ref. 17) | Benight (ref. 18) | SantaLucia (ref. 20) | Sugimoto (ref. 21) |
|---|---|---|---|---|---|---|---|
| Slope | 0.94 | 0.81 | 1.41 | 0.82 | 0.87 | 1.00 | 1.09 |
| Intercept, kcal/mol | 0.41 | −0.17 | 0.19 | −0.04 | −0.13 | 0.02 | −0.10 |
| R2 | 0.97 | 0.98 | 0.82 | 0.95 | 0.81 | 0.97 | 0.92 |
| SD, kcal/mol | 0.09 | 0.06 | 0.36 | 0.11 | 0.22 | 0.09 | 0.17 |
Fit to the equation ΔG°37 (unified NN) = ΔG°37 (literature NN) × slope + intercept (see text). See text for interpretation of the slope, intercept, and R2 (correlation coefficient). SD between the experimental NN and that predicted with the unified NN by using the linear least-squares slope and intercept shown. This reflects the agreement in the NN trend. See text for comparisons among the polymer studies.
Results and Discussion
Table 1 presents the NN ΔG°37 values for helix propagation and initiation from eight experimental studies. With all these data sets presented in a uniform format, a remarkable consensus is immediately evident. The qualitative trend observed in order of decreasing stability is GC/CG = CG/GC > GG/CC > CA/GT = GT/CA = GA/CT = CT/GA > AA/TT > AT/TA > TA/AT. To quantify the quality of the NN parameters, linear regression analysis was performed with the literature NN as the dependent variable (y axis) and the unified oligonucleotide NN parameters as the independent variable (x axis) (Table 3). The slope of this plot indicates how close the range (i.e., the difference between the largest and the smallest NN parameters) of the literature NN agrees with the range observed in the unified NN parameters. The intercept indicates the quality of the initiation parameter and salt dependence and also contains a contribution from the slope. The correlation coefficient R2 indicates the quality of the trend in NN parameters. The polymer studies (7, 10, 17) show a remarkable correlation with the unified oligonucleotide parameters (R2 = 0.97, 0.98, 0.95, respectively). The slopes are close to one for each of these studies, indicating that the ranges are in good agreement with the unified parameters. The intercepts show a systematic sodium concentration dependence (see below). The oligonucleotide-duplex-derived NN parameters of SantaLucia (20) and Sugimoto (21) are in excellent agreement with the unified parameters, which is not surprising because the data from these studies make up the majority of the unified data set. The poor agreement of the Breslauer NN parameters (12) (Tables 1 and 3) is discussed below. The oligonucleotide dumbbell parameters of Benight (18) also show good agreement with the unified parameters.
Why Do Rank-Deficient Polymer NN Parameters Agree with Rank-Determinant Oligonucleotide NN Parameters?
It is surprising that polymer parameters with a rank of 8 are observed to agree so well with the 10 oligonucleotide dimers. The rationale for this observation is that most of the sequence dependence of oligonucleotide DNA thermodynamics is captured in the first eight terms and the remaining two terms are small perturbations that are difficult to detect within the error limits of most measurement techniques. The slopes given in Table 3 reveal that the polymer NN have a slightly smaller range in NN ΔG°37 that is primarily due to the rank deficiency of the polymer parameters. For example, the range in the unified ΔG°37 parameters is 1.66 kcal/mol (TA/AT − GC/CG), whereas for ΔG°37 parameters from Vologodskii et al. (10), the range is only 1.32 kcal/mol (TA/AT − CG/GC). This extra sequence dependence for the oligomer NN has almost no effect on the eight invariants needed to predict polymer thermodynamics. A general result from this is that the oligomer NN can predict polymer behavior accurately, but the polymer NN data cannot be used to reliably predict oligomer behavior.
Salt Dependence of Oligonucleotides.
Recently, we have reanalyzed the literature thermodynamic data for 26 oligonucleotide duplexes dissolved in 0.01 M to 0.3 M NaCl (see ref. 20 and references therein). The salt correction for DNA is assumed to be independent of sequence but to be dependent on oligonucleotide length (36). The difference between the thermodynamics of 26 literature duplexes dissolved in different sodium concentrations and the NN predictions in 1 M NaCl was plotted vs. N × ln[Na+] with the intercept forced through zero. A linear least-squares fit of this plot gives:
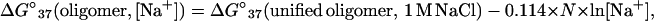 |
7 |
where ΔG°37(oligomer, [Na+]) is the ΔG°37 for an oligonucleotide duplex dissolved in a given sodium concentration, ΔG°37(unified oligomer, 1 M NaCl) is the ΔG°37 predicted from the unified NN parameters at 1 M NaCl, and N is the total number of phosphates in the duplex divided by 2 (e.g., for an 8-bp duplex without terminal phosphates, n = 7). The length dependence in Eq. 7 neglects differential cation binding in the middle vs. the ends of a duplex (36). The standard deviation in the slope (−0.114 kcal/mol) is 0.033 kcal/mol. Eq. 7 predicts the ΔG°37 of 26 oligonucleotide duplexes with fewer than 17 bp with a standard deviation of 0.60 kcal/mol. Eq. 7 gives ∂ΔG/∂ln[Na+] = −0.114 kcal/mol, which corresponds to a ∂TM/∂log[Na+] of 11.7°C (assuming a sequence independent ΔS° of −22.4 e.u. per base pair (see below)); this agrees with previously observed values for oligonucleotides (29, 30). The entropy correction is given by:
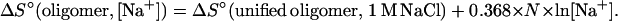 |
8 |
If the ΔH° is assumed to be salt-concentration-independent (30, 36), the TM values of the 26 oligonucleotides are predicted by using Eqs. 8 and 3 with an average deviation of 2.2°C. The salt corrections given in Eqs. 7 and 8 can be viewed as either length-dependent corrections to the initiation parameter or as corrections to the propagation parameters because there are N NNs in an oligonucleotide duplex.
Salt Dependence of Polymers.
Helix formation in polymers does not formally involve an initiation parameter so the salt dependence is by default incorporated into the NN propagation terms (35). The observation that polymer NN parameters and the unified oligonucleotide NN are highly correlated (Table 3) suggests a relationship could be determined that would allow prediction of polymer behavior from the unified NN parameters with an appropriate salt correction. The ΔG°37 differences of the three polymer NN data sets (Table 1) and the unified NN (1 M NaCl) data set were plotted vs. the ln[Na+] of the polymer data. From a least squares fit of this plot (30 data points) and the assumption that the salt correction is sequence independent, the following empirical equation was derived:
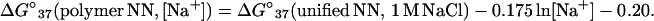 |
9 |
The standard deviations in the slope (−0.175 kcal/mol) and intercept (−0.20 kcal/mol) are 0.034 kcal/mol and 0.11 kcal/mol, respectively. Note that this correction is given in kcal/mol of base pairs. Alternatively, the ΔG°37 of each polymer NN at three salt concentrations can be individually plotted vs. ln[Na+] to test the sequence dependence of salt effects. Unfortunately, the ΔG°37(i) vs. ln[Na+] plots for the CT/GA, CG/GC, and TA/AT neighbors show correlation coefficients R2 that are less than 0.9. Nonetheless, for the seven NN that show a linear salt dependence of ΔG°37 (R2 > 0.95), it does appear that A+T-rich NNs show a larger salt dependence than the G+C-rich NNs, consistent with earlier observations (see Eq. 5a) (35). Blake (37) has provided a tentative salt dependence of the 10 dimers.
Eq. 9 gives ∂ΔG/∂ln[Na+] = −0.175 kcal/mol, which corresponds to a ∂TM/∂log[Na+] of 16.2°C (when a sequence-independent ΔS° of −24.85 e.u. is assumed), which agrees well with the widely used value for polymers of 16.6°C (38). Fig. 2 plots the NN stabilities observed in the three polymer studies (Table 1) (7, 10, 17) versus those predicted with Eq. 9. The slope and intercept are close to 1 and 0, respectively, and the correlation coefficient R2 is 0.96 (see Fig. 2). The standard deviation between the predictions with Eq. 9 and the experimental data is 0.12 kcal/mol (the eight polymer invariants are predicted within 0.09 kcal/mol). For comparison, when the polymer parameters of the three groups (7, 10, 17) are compared with each other, the least-squares fit produces standard deviations of 0.05, 0.9, and 0.9 kcal/mol for the comparisons of data from ref. 10 vs. ref. 17, ref. 10 vs. ref. 7, and ref. 17 vs. ref. 7, respectively. The experimental error reported for the unified parameters is ∼0.05 kcal/mol (22). Thus, the unified NN used with Eq. 9 provides predictions of polymers within experimental error of those obtained with the polymer parameters. A test of the validity of Eq. 9 is to use it to actually predict the stability of polymers. A plot of the experimental ΔG°37 for 27 different synthetic polymers dissolved in solutions ranging from 0.01 M to 0.20 M Na+ (16, 39) vs. those predicted with Eq. 9 gives a linear least-squares regression line of y = 1.043 × −0.040 with R2 = 0.894 (data not shown). The standard error between experiment and prediction is 0.14 kcal/mol. This level of agreement suggests the unified NN parameters accurately reflect the polymer NN trends.
Figure 2.

Experimental vs. predicted NN ΔG°37 parameters (Table 1) of three polymer studies (7, 10, 17). •, NN data of Gotoh and Tagashira (7); ▪, NN data of Vologodskii et al. (10); ▴, NN data of Delcourt and Blake (17). Predictions are based on the unified oligonucleotide parameters (22) with the salt concentration extrapolated according to Eq. 9. The linear least-squares regression line is shown (y = 1.101 × +0.115; R2 = 0.961). The standard deviation from the regression line is 0.12 kcal/mol.
It is interesting to compare the salt dependence for oligonucleotide duplexes and for polymers given by Eqs. 7 and 9. The slope of the ln[Na+] term is 54% larger for polymers than for oligonucleotides (0.175 vs. 0.114, respectively). When extrapolated to 1 M Na+, the polymer NN are more stable than oligomer NN by −0.20 kcal/mol. Qualitatively, these differences in salt dependence can be viewed simply as arising either from “end effects” present in oligonucleotides but not in polymers (36, 40) or from “polymer counterion condensation effects” (24, 25) that are reduced in oligonucleotides. On the basis of these data, there does appear to be a “length dependency” to the salt behavior of nucleic acids that is not yet completely understood (18, 36).
Sequence Dependence of the Propagation Entropy.
The polymer and dumbbell studies assume that the propagation entropy change, ΔS°(i) is independent of sequence and salt concentration (7, 10, 17, 18). The most reliable estimate for polymers is −24.85 ± 1.74 e.u. in 0.075 M NaCl (17). The sequence-independent ΔS°(i) assumption introduces error into ΔH° and ΔG°37 via Eqs. 4 and 2 of approximately 0.6 kcal/mol and 0.06 kcal/mol, respectively. The unified oligonucleotide ΔS°(i) in 1 M NaCl range from −19.9 to −27.2 e.u. (22) with an average of −22.4 e.u. and standard deviation of 2.1 e.u. The use of the polymer ΔS°(i) of −24.85 e.u. is not appropriate for predictions of oligonucleotides (>20% error in ΔS° predictions). However, the idea that the ΔS°(i) is sequence independent is nearly correct for DNA. A sequence independent ΔS°(i) of −22.4 e.u. predicts the ΔS° values of the unified oligonucleotide data set with an average deviation of 9.4%. This is close to the predictive capacity of the unified NN parameters themselves, which predict the unified data set with an average deviation of 8.4%.
Analysis of Gotoh and Tagashira (7).
Gotoh and Tagashira (7) measured the UV thermal denaturation curves of 11 DNA restriction fragments dissolved in 0.0195 M Na+. The curves were fit with the Poland partition function algorithm (32) using the Fixman–Freire approximation for the loop functions (33) and modified to incorporate heterogeneous stacking (7). Vologodskii et al. (10) critically evaluated the work of Gotoh and Tagashira (7) and concluded that the low salt concentration was responsible for the observed hysteresis and suggested that this indicated nonequilibrium conditions. This work, however, shows that with the proper salt extrapolation, the parameters of Gotoh and Tagashira (7) are in remarkable agreement with other polymer parameters (10, 17) and with the unified oligonucleotide NN parameters (Fig. 2). This suggests any nonequilibrium effects in Gotoh and Tagashira’s study must have been relatively small.
Analysis of Vologodskii et al.
(10). Vologodskii et al. (10) derived NN parameters by using the linearly independent sequences approach from eight natural DNA polymer restriction fragments dissolved in 0.195 M Na+. The high salt concentration used ensured equilibrium conditions throughout the melting curve. Vologodskii’s study used a partition function approach similar to that in the study of Gotoh and Tagashira (7). Vologodskii et al. (10) presented their NN parameters as both eight linearly independent sequences and 10 nonunique dimer parameters by using the assumptions that AT/TA = TA/AT and GC/CG = CG/GC. Oligonucleotide experiments reveal that these assumptions are approximately correct for DNA but not for RNA (13). The Vologodskii ΔG°37 NN parameters show remarkable agreement with the unified NN parameters (Tables 1 and 3). The results presented herein verify that the experimental design and analysis methods used in ref. 10 are fundamentally sound.
Analysis of Breslauer et al. (12).
Breslauer et al. (12) derived NN thermodynamic parameters by using differential scanning calorimetry and UV melting analysis of 19 oligonucleotide duplexes (dissolved in 1 M NaCl) and nine synthetic DNA polymers (dissolved in low salt with results extrapolated to 1 M Na+). It is not possible to rederive the reported parameters, however, because much of the primary thermodynamic data have not been published. This work demonstrated good insight in that the authors reasoned that polymer and oligomer NN trends should be similar. However, the assumption that the initiation ΔG°37 is 5.2 kcal/mol is most likely what led to the incorrect NN determined (Tables 1 and 3). Breslauer’s NN predict the ΔG°37, ΔH°, ΔS°, and TM of the unified data set with average deviations of 16.7%, 10.1%, 10.6%, and 6.0°C, respectively. Predictions are particularly poor for oligonucleotides shorter than 8 bp. For example, the TM of the sequence CACAG⋅CTGTG (41) is incorrectly predicted by 31°C. Other groups have also been unable to reconcile the Breslauer parameters with experiments (18, 20, 21, 37, 42).
Analysis of Delcourt and Blake (17).
Delcourt and Blake (17) studied 41 restriction fragments of natural polymers dissolved in 0.075 M Na+ and expressed their NN parameters in terms of 10 nonunique dimers that make good predictions of polymers but do not represent the real trends in NN stability. The results presented herein verify that the experimental design and analysis methods used in Delcourt and Blake (17) are fundamentally sound.
Analysis of Doktycz et al. (18).
Benight and coworkers (18) recognized that there was consensus agreement among the polymer studies and their own dumbbell studies but could not reconcile the oligomer literature parameters (12). On the basis of this problem, the authors proposed that there must be a “length dependency” to DNA NN thermodynamics. Herein I show that the NN parameters themselves are not “length-dependent” but that the salt dependence is length-dependent in ways that are still not fully understood.
Benight and coworkers (18) used UV melting analysis of 17 oligonucleotide dumbbells with 14–18 bp to determine nine linearly independent sequences that follow a NN model. The experimental design for this study precluded measurement of an initiation parameter for duplex formation and a 10th NN parameter. The analysis was performed under four salt conditions, including 25, 55, 85, and 115 mM Na+. These data suggested that the NN model breaks down at salt concentrations of less than 85 mM but works well at 115 mM Na+. It is possible that the neglect of the length dependence of salt effects (Eq. 7) is what led to the apparent breakdown of the NN model at low salt concentrations. Doktycz et al. (18) assumed that the ΔS° for helix propagation was sequence- and salt-independent (−24.8 e.u. per bp) (17), which is incorrect for oligonucleotides. With the exception of the CG/GC neighbor, the dumbbell ΔG°37 NN parameters in 0.115 M Na+ show good agreement with the unified NN parameters (Table 1).
Analysis of SantaLucia et al. (20).
SantaLucia et al. (20) derived NN parameters from thermodynamics determined by a van’t Hoff analysis of UV melting data for 23 oligonucleotides combined with calorimetric or UV melting results from the literature for 21 other sequences. To minimize “fraying artifacts”, all sequences included in the linear regression analysis to determine NN parameters had terminal G⋅C pairs (12). The SantaLucia parameters are within experimental error of the unified parameters in Table 1.
Data were available in the literature for eight sequences with terminal T⋅A pairs. These data were included in the first fit of the NN parameters and the stacking matrix was not rank deficient but nonphysical results were obtained for six of the nearest neighbors (AT/TA, TA/AT, CA/GT, AC/TG, GA/CT, and AG/TC) (20). For example, the ΔH° parameters for AT/TA and TA/AT neighbors were found to be −10.80 and +1.16 kcal/mol, respectively, which is unlikely (J.S., unpublished results). We now know that four of the sequences with terminal T⋅A pairs exhibited non-two-state behavior (20, 21). Upon removal of the sequences with terminal T⋅A pairs, more reasonable results were obtained, but the rank of the stacking matrix was reduced to 10 (nine linearly independent sequences plus one initiation parameter) (23).
Analysis of Sugimoto et al. (21).
Sugimoto et al. (21) derived NN parameters from thermodynamics determined by a van’t Hoff analysis of UV melting data for 50 oligonucleotides combined with data for 15 sequences from other laboratories obtained by both calorimetry and UV melting. Except for the initiation ΔG°37, the CG/GC ΔG°37, and the GG/CC ΔH°, the Sugimoto parameters (21) are in good agreement with the unified NN parameters (Tables 1 and 3). With the proper linear regression analysis, Sugimoto’s data set provides NN and initiation parameters that are in excellent agreement with the unified parameters (22). Important results of this work are that separate parameters for terminal T⋅A base pairs and for initiation at A⋅T are not required.
Conclusion
A unified set of NN parameters is now available for making accurate predictions of DNA oligonucleotide, dumbbell, and polymer thermodynamics. The agreement among the various polymer and oligomer studies provides a great deal of confidence in their reliability.
Acknowledgments
I thank Douglas H. Turner and Hatim Allawi for stimulating conversations and for critical reading of the manuscript. I also thank Wayne State University and Hitachi Chemical Research for financial support.
ABBREVIATIONS
- SVD
singular value decomposition
- NN
nearest neighbor
- e.u.
entropy unit (cal/K⋅mol)
References
- 1.Crothers D M, Zimm B H. J Mol Biol. 1964;9:1–9. doi: 10.1016/s0022-2836(64)80086-3. [DOI] [PubMed] [Google Scholar]
- 2.DeVoe H, Tinoco I., Jr J Mol Biol. 1962;4:500–517. doi: 10.1016/s0022-2836(62)80105-3. [DOI] [PubMed] [Google Scholar]
- 3.Gray D M, Tinoco I., Jr Biopolymers. 1970;9:223–244. [Google Scholar]
- 4.Borer P N, Dengler B, Tinoco I, Jr, Uhlenbeck O C. J Mol Biol. 1974;86:843–853. doi: 10.1016/0022-2836(74)90357-x. [DOI] [PubMed] [Google Scholar]
- 5.Tinoco I, Jr, Borer P N, Dengler B, Levine M D, Uhlenbeck O C, Crothers D M, Gralla J. Nat New Biol. 1973;246:40–41. doi: 10.1038/newbio246040a0. [DOI] [PubMed] [Google Scholar]
- 6.Uhlenbeck O C, Borer P N, Dengler B, Tinoco I., Jr J Mol Biol. 1973;73:483–496. doi: 10.1016/0022-2836(73)90095-8. [DOI] [PubMed] [Google Scholar]
- 7.Gotoh O, Tagashira Y. Biopolymers. 1981;20:1033–1042. doi: 10.1002/bip.1981.360200514. [DOI] [PubMed] [Google Scholar]
- 8.Ornstein R, Fresco J R. Biopolymers. 1983;22:1979–2000. doi: 10.1002/bip.360220811. [DOI] [PubMed] [Google Scholar]
- 9.Otto P. J Mol Struct. 1989;188:277–288. [Google Scholar]
- 10.Vologodskii A V, Amirikyan B R, Lyubchenko Y L, Frank-Kamenetskii M D. J Biomol Struct Dyn. 1984;2:131–148. doi: 10.1080/07391102.1984.10507552. [DOI] [PubMed] [Google Scholar]
- 11.Wartell R M, Benight A S. Phys Rep. 1985;126:67–107. [Google Scholar]
- 12.Breslauer K J, Frank R, Blocker H, Marky L A. Proc Natl Acad Sci USA. 1986;83:3746–3750. doi: 10.1073/pnas.83.11.3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Freier S M, Kierzek R, Jaeger J A, Sugimoto N, Caruthers M H, Neilson T, Turner D H. Proc Natl Acad Sci USA. 1986;83:9373–9377. doi: 10.1073/pnas.83.24.9373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aida M. J Theor Biol. 1988;130:327–335. doi: 10.1016/s0022-5193(88)80032-8. [DOI] [PubMed] [Google Scholar]
- 15.Quartin R S, Wetmur J G. Biochemistry. 1989;28:1040–1047. doi: 10.1021/bi00429a018. [DOI] [PubMed] [Google Scholar]
- 16.Klump H H. In: Landolt-Bornstein, New series, VII Biophysics. Saenger W, editor. Vol. 1. Berlin: Springer; 1990. , Subvol. c, pp. 241–256. [Google Scholar]
- 17.Delcourt S G, Blake R D. J Biol Chem. 1991;266:15160–15169. [PubMed] [Google Scholar]
- 18.Doktycz M J, Goldstein R F, Paner T M, Gallo F J, Benight A S. Biopolymers. 1992;32:849–864. doi: 10.1002/bip.360320712. [DOI] [PubMed] [Google Scholar]
- 19.Goldstein R F, Benight A S. Biopolymers. 1992;32:1679–1693. doi: 10.1002/bip.360321210. [DOI] [PubMed] [Google Scholar]
- 20.SantaLucia J, Jr, Allawi H, Seneviratne P A. Biochemistry. 1996;35:3555–3562. doi: 10.1021/bi951907q. [DOI] [PubMed] [Google Scholar]
- 21.Sugimoto N, Nakano S, Yoneyama M, Honda K. Nucleic Acids Res. 1996;24:4501–4505. doi: 10.1093/nar/24.22.4501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Allawi H T, SantaLucia J., Jr Biochemistry. 1997;36:10581–10594. doi: 10.1021/bi962590c. [DOI] [PubMed] [Google Scholar]
- 23.Gray D M. Biopolymers. 1997;42:795–810. doi: 10.1002/(sici)1097-0282(199712)42:7<795::aid-bip5>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 24.Manning G. Q Rev Biophys. 1978;11:179–246. doi: 10.1017/s0033583500002031. [DOI] [PubMed] [Google Scholar]
- 25.Record M T, Anderson C F, Lohman T M. Quart Rev Biophys. 1978;2:103–178. doi: 10.1017/s003358350000202x. [DOI] [PubMed] [Google Scholar]
- 26.Cantor C R, Schimmel P R. Biophysical Chemistry Part III: The Behavior of Biological Macromolecules. San Francisco: Freeman; 1980. [Google Scholar]
- 27.Chaires J B. Biophys Chem. 1997;64:15–23. doi: 10.1016/s0301-4622(96)02205-3. [DOI] [PubMed] [Google Scholar]
- 28.Liu Y, Sturtevant J M. Biophys Chem. 1997;64:121–126. doi: 10.1016/s0301-4622(96)02229-6. [DOI] [PubMed] [Google Scholar]
- 29.Rentzeperis D, Ho J, Marky L A. Biochemistry. 1993;32:2564–2572. doi: 10.1021/bi00061a014. [DOI] [PubMed] [Google Scholar]
- 30.Erie D, Sinha N, Olson W, Jones R, Breslauer K. Biochemistry. 1987;26:7150–7159. doi: 10.1021/bi00396a042. [DOI] [PubMed] [Google Scholar]
- 31.Schmitz M, Steger G. Comput Appl Biosci. 1992;8:389–399. doi: 10.1093/bioinformatics/8.4.389. [DOI] [PubMed] [Google Scholar]
- 32.Poland D. Biopolymers. 1974;13:1859–1871. doi: 10.1002/bip.1974.360130916. [DOI] [PubMed] [Google Scholar]
- 33.Fixman M, Freire J J. Biopolymers. 1977;16:2693–2704. doi: 10.1002/bip.1977.360161209. [DOI] [PubMed] [Google Scholar]
- 34.Press W H, Flannery B P, Teukolsky S A, Vetterling W T. Numerical Recipes. New York: Cambridge Univ. Press; 1989. [Google Scholar]
- 35.Frank-Kamenetskii M D. Biopolymers. 1971;10:2623–2624. doi: 10.1002/bip.360101223. [DOI] [PubMed] [Google Scholar]
- 36.Record M T, Jr, Lohman T M. Biopolymers. 1978;17:159–166. [Google Scholar]
- 37.Blake R D. In: Encyclopedia of Molecular Biology and Molecular Medicine. Meyers R A, editor. Vol. 2. New York: VCH; 1996. pp. 1–19. [Google Scholar]
- 38.Schildkraut C, Lifson S. Biopolymers. 1965;3:195–208. doi: 10.1002/bip.360030207. [DOI] [PubMed] [Google Scholar]
- 39.Remeta D P, Mudd C P, Berger R L, Breslauer K J. Biochemistry. 1993;32:5064–5073. doi: 10.1021/bi00070a014. [DOI] [PubMed] [Google Scholar]
- 40.Olmsted M C, Anderson C F, Record M T., Jr Proc Natl Acad Sci USA. 1989;86:7766–7770. doi: 10.1073/pnas.86.20.7766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hall K B, McLaughlin L W. Biochemistry. 1991;30:10606–10613. doi: 10.1021/bi00108a002. [DOI] [PubMed] [Google Scholar]
- 42.Steger G. Nucleic Acids Res. 1994;22:2760–2768. doi: 10.1093/nar/22.14.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]