

Abstract
Neonates are at increased risk of infections compared to adults. To dissect the mechanisms that contribute to neonatal immune deficiency, we compared MHC-II antigen processing and presentation by monocytes from umbilical cord blood and unrelated adult controls. Antigen-specific, costimulation-independent murine T hybridoma cells were used to detect peptide:HLA-DR complexes. Relative to adult monocytes, neonatal monocytes were significantly defective in processing and presentation of protein antigens and presentation of exogenous peptide. Defects in responses to protein antigens and exogenous peptide were of similar magnitude (56–81% decrease), indicating that the defect lies in antigen presentation as opposed to intracellular antigen processing. Average surface MHC-II levels on neonatal monocytes were 38% less than on adult monocytes. However, there was no correlation between decreased MHC-II expression on individual neonatal monocyte samples and reduced T cell responses. We demonstrate for the first time that neonatal monocytes are defective in MHC-II antigen presentation by a mechanism not correlated with decreased MHC-II expression.
Keywords: neonates, MHC-II, antigen presentation, monocytes, human
Introduction
Worldwide, around 10.6 million children still die each year before reaching their fifth birthday. A majority of these deaths occur due to infections with around 40% of them occurring in newborns (1). Newborns become infected with bacteria and viruses that either colonize the birth canal or are prevalent in their immediate environment (2, 3). While these pathogens cause limited morbidity in adults, they can have devastating consequences in newborns. The high susceptibility of newborns to pathogens has been ascribed to the diminished CD4+ T helper cell type I (Th1) response observed during fetal and early neonatal life (4, 5). CD4+ T cells provide the help required for production of antibodies by B cells and for activation of CD8+ T cells that are involved in eliminating viral pathogens.
For activation of CD4+ T cells, antigens have to be processed and presented by class II major histocompatibility complex (MHC-II) molecules expressed by antigen presenting cells (APCs) (6, 7). Newly synthesized α and β chains of MHC-II molecules associate with the invariant chain (Ii) in the endoplasmic reticulum and are targeted to the endocytic pathway (8–11). There, Ii is proteolytically degraded until the MHC-II molecule is left associated with a fragment of Ii called CLIP (class II-associated Ii peptide) (12–15). CLIP is then replaced by an antigenic peptide, which may be derived from the degradation of an internalized “exogenous” antigen (either soluble or particulate) by vacuolar proteases. Removal of CLIP is often catalyzed by another MHC-encoded molecule, HLA-DM that resides in endosomes (16–18). Generation of peptide:MHC-II complexes is referred to as antigen processing. Peptide:MHC-II complexes then traffic to the cell surface for presentation to CD4+ T helper cells. While dendritic cells are the most potent APCs with their ability to activate naïve T cells, both monocytes and B cells are important for activating effector T cells.
Several studies have analyzed surface MHC-II expression in neonatal APCs. While a few studies concluded that MHC-II expression on neonatal APCs is increased (19) or equivalent (20, 21) to that of adult APCs, a majority of the studies found decreased MHC-II expression on neonatal APCs (22–32). It was speculated that decreased MHC-II expression might lead to reduced antigen processing and presentation function in neonatal APCs.
MHC-II antigen processing and presentation function in neonatal APCs has been analyzed mainly by the mixed lymphocyte reaction or by T cell proliferation and has generated mixed conclusions. MHC-II mediated antigen processing and presentation function in human neonatal APCs has been described as equivalent (25, 33, 34), decreased (24, 32, 35) or increased (36–39) when compared to that of adult APCs. The lack of consistent results may partly be from the use of adult or neonatal CD4+ T cells that are functionally different or from the analysis of dendritic cells of varying functional maturity. Whether decreased responses arise from specific defects in antigen processing or antigen presentation or both remain unaddressed. Correlation between surface MHC-II expression in neonatal APCs and their ability to process and present peptide:MHC-II complexes to CD4+ T cells remains unclear. It has been suggested that decreased surface MHC-II expression on neonatal monocyte-derived dendritic cells (32) and neonatal dendritic cells (24) contribute to decreased T cell responses.
To determine whether neonatal APCs are defective in antigen processing and/or presentation and whether reduced MHC-II is an explanation for this potential defect, we have compared MHC-II antigen processing and presentation function in negatively selected monocytes from umbilical cord blood and unrelated adult controls. Our analysis used antigen-specific, costimulation-independent, murine T hybridoma cells specifically generated to study MHC-II antigen processing and presentation function in human APCs (40, 41). This approach has allowed us to eliminate the use of allogeneic T cells derived from cord blood and adult blood that may be functionally different. It has also allowed us to analyze antigen processing and antigen presentation functions independently of each other. Finally, the use of a consistent T cell assay has allowed us to assess the correlation between MHC-II levels and T cell responses for the first time. Our data show that neonatal monocytes are selectively defective in antigen presentation as opposed to intracellular antigen processing. Neonatal monocytes have decreased surface MHC-II levels, but no statistical correlation was observed between decreased MHC-II levels on neonatal monocytes and magnitude of the defect in T cell responses. We demonstrate for the first time, that mechanisms other then decreased MHC-II expression contribute to the antigen presentation deficit of neonatal APCs.
Materials and Methods
Media
All cells were cultured at 37°C in 5% CO2 in standard medium composed of DMEM (Life Technologies, Grand Island, NY) supplemented with 10% decomplemented fetal calf serum (Hyclone, Logan, UT), 5 x 10−5 M 2-ME, L-arginine HCl (116 mg/l), L-asparagine (36 mg/l), NaHCO3 (2 g/l), sodium pyruvate (1 mM), 10 mM HEPES buffer and antibiotics (D-10-F).
Isolation and storage of PBMCs
Recruitment and participation of healthy blood donors for our research as well as collection of cord blood from consenting mothers was approved by the University Hospitals of Cleveland Institutional Review Board. HLA-matched healthy adult donors were identified among personnel at the university and hospital. Adult and cord blood were collected in tubes containing the anticoagulant citrate-phosphate-dextrose solution with adenine (Sigma, St. Louis). An aliquot of the cord blood was stored at −20°C, for HLA-DR typing (see below). PBMCs were isolated from both samples by layering blood diluted in DMEM over Ficoll Paque Plus (Amersham Biosciences, NJ). For cord blood samples, the PBMCs removed at the interface of the plasma and Ficoll were re-layered over Ficoll to help further eliminate RBCs. PBMCs were either used immediately for the isolation of monocytes or were stored in liquid Nitrogen in aliquots of 20 million cells/vial in freezing media containing 90% FCS/10% DMSO.
Isolation of monocytes
Untouched monocytes were isolated from freshly isolated PBMCs or from thawed aliquots of PBMCs using the Monocyte Isolation Kit II from Miltenyi Biotec (Germany).
Generation of T cell hybridomas
All animal experiments were done in accordance with the guidelines laid out by the Institutional Animal Care and Use Committee at Case Western Reserve University. Generation of the T cell hybridoma, ITIA has been described by Gehring et al. (41). The T cell hybridomas IACD5, ITC and A3 were similarly generated. Briefly, mice transgenic for HLA-DR1 (Dennis Zaller, Merck Laboratories), HLA-DR3 (Chella David, Mayo Clinic) or HLA-DR4 (Thomas Forstuber, University of Texas at San Antonio) were injected in the footpad with antigen prepared in complete Freund’s adjuvant (CFA) according to the manufacturer’s instructions. Popliteal lymph nodes were isolated 7 days later, re-stimulated in vitro for 5 days and the T cells subsequently fused with the TCR αβ negative BW1100 thymoma cell line. Fused cells were selected using medium containing hypoxanthine, aminopterin and thymidine (HAT) for 7–9 days and subsequently screened for antigen specificity by the T cell assay (described below).
PCR screening
The T cell hybridomas, IACD5 and ITIA, are restricted by HLA-DR1 (allele DRB1*0101), while the A3 T cell hybridoma is restricted by HLA-DR3 (allele DRB1*0301) and the ITC T cell hybridoma is restricted by HLA-DR4 (allele DRB1*0401). Therefore, the monocytes to be analyzed by the T cell assay have to be HLA-DR matched. Adult donors and cord blood that express HLA-DR1 (allele DRB1*0101) or HLA-DR3 (allele DRB1*0301) or HLA-DR4 (allele DRB1*0401) were identified by low and high-resolution PCR. DNA from donor’s blood was purified by DNeasy kit (Qiagen, CA). PCR was carried out initially using low-resolution HLA-DR PCR kits from BioSynthesis Inc. (Lewisville, TX) to identify individuals positive for HLA-DR1, HLA-DR3 and HLA-DR4 expression. Subsequently, high-resolution screening was performed on the positive heterozygous donors using high-resolution PCR kits (BioSynthesis Inc.) to identify individuals that were positive for the 01 allele.
T cell assay
Monocytes were plated in 96-well flat bottom plates in media containing 10% FCS at 5 x 104 cells/well. Antigen, reverse transcriptase (Kathryn Howard, CWRU) for IACD5, tetanus toxoid (Wyeth-Ayerst, Pearl River, NY) for ITIA and ITC or Antigen 85B (made according to Lakey, D.L. et al (42)) for A3 was serially diluted in cell culture medium and added to the monocytes. All antigen preparations were devoid of lipopolysaccaride (LPS) contamination. T hybridoma cells (1 x 105) were added to each well (200 μl total volume). After 24 h, supernatants were harvested and stored at −80°C and assessed for IL-2 by the CTLL assay or IL-2 ELISA.
In some of the T cell assays, corresponding peptide epitopes were used instead of the intact protein antigen (Table 1). In certain assays, monocytes were preincubated with control antibody or Anti-human CD80/86 (5 μg/ml, Ancell Corporation, Bayport, MN) for 10 mins prior to the addition of antigen and T hybridoma cells. In other assays, T hybridoma cells were incubated with control antibody or murine anti CD40L (10 or 25 μg/ml, eBioscience, San Diego, CA) for 10 mins prior to addition of antigen and monocytes.
All T cell assays involving fixation of monocytes were performed in 96-well round bottom plates to minimize cell loss. Monocytes were incubated with antigen alone for 24 h, washed once in media, fixed with 1% paraformaldehyde for 15 min, incubated with 0.2 M lysine (in PBS, pH 7.4) for 30 mins and then washed prior to the addition of T hybridoma cells for an additional 24 h.
Detection of IL-2
IL-2 levels in supernatants were assessed by CTLL assay or IL-2 ELISA. For the CTLL assay, 100 μls of supernatant were assessed for IL-2 using CTLL-2 cells. CTLL-2 proliferation was monitored by addition of Alamar blue (Trek Diagnostics, Cleveland, OH) as an indicator dye and measured as the difference between absorbance at 550 nm and 595 nm after 24 h (43). All analyses were performed in triplicate.
IL-2 ELISA was performed using the capture and detection anti-mouse IL-2 from eBioscience (San Diego, CA) in Nunc maxisorb flat-bottom ELISA plates (Nalge Nunc International, NY).
Flow analysis
Isolated monocytes were stained with phycoerythrin-conjugated anti-CD14 and Allophycocyannin-conjugated anti-HLA-DR antibody (L243, Pharmingen, CA) or isotype control antibody (Pharmingen) and analyzed on a clinically certified BDFacs Canto (BD Biosciences, CA). MHC-II levels on CD14+ monocytes was calculated by substracting geometric mean channel fluorescence for isotype control from geometric mean channel fluorescence for L243 staining.
Statistics
All data was analyzed with SAS® version 9.2 (Carey, NC). Mann-Whitney U-test was used to analyze differences in IL-2 production, generated by adult monocytes versus neonatal monocytes and differences in MHC-II levels between adult and neonatal monocytes. Pearson correlations was used to analyze correlation between MHC-II levels on neonatal and adult monocytes and IL-2 production by T hybridoma cells. p value less than 0.05 is considered statistically significant.
Results
T cell assay to assess antigen processing and presentation by human APCs
HLA-DR-restricted, antigen-specific murine T hybridoma cells were used to assess MHC-II antigen processing and presentation of protein antigens and presentation of exogenous peptides by neonatal monocytes and unrelated adult controls. Antigen-specific T hybridoma cells were generated in transgenic mice expressing human HLA-DR1 (allele DRB1*0101), HLA-DR3 (allele DRBI*0301) or HLA-DR4 (allele DRB1*0401) to specifically study antigen processing defects in human APCs (40, 41). Hybridomas were generated against a variety of antigens derived from bacterial and viral pathogens (tetanus toxoid, HIV-reverse transcriptase, M. tuberculosis Antigen 85, Table 1).
For the T cell assay, suitable donors were identified by low and high-resolution PCR based on polymorphisms in the β chain of human HLA-DR molecules. Negatively selected monocytes from HLA-matched donors were incubated for 24 h with protein antigen or exogenous peptide and corresponding HLA-restricted T hybridoma cells (see Table 1). Supernatants were assayed for IL-2 by the CTLL assay.
T hybridoma cells were characterized for co-stimulation (CD80/CD86 and CD40) dependence in a standard T cell assay. To test the requirements for CD80/CD86 co-stimulation, monocytes were pre-incubated with anti-CD80/CD86 antibodies prior to addition of antigen and T hybridoma cells. To test the requirement for CD40-CD40L interaction, T hybridoma cells were pre-incubated with anti-CD40L antibody prior to the addition of monocytes and antigen. Neither the anti-CD80/CD86 antibodies nor the anti-CD40L antibody blocked T cell hybridoma responses (Table 1). Therefore, all the hybridomas characterized for these studies are CD80/CD86 and CD40 independent.
Neonatal monocytes purified from fresh cord blood are deficient at MHC-II antigen processing and presentation
To analyze MHC-II antigen processing and presentation in neonatal monocytes, cord blood was immediately MHC-typed following receipt. Negatively selected monocytes were then purified from HLA-matched cord and adult blood and used in T cell assays with the corresponding MHC-restricted T hybridoma cells growing in passage. Purity of the monocyte population was 85–90% as assessed by CD14-positive staining with T cells comprising the main contaminant. For our initial analysis we compared antigen processing and presentation in pairs of monocytes: one derived from cord blood and the other from an HLA-matched adult donor (Fig. 1). Levels of T cell hybridoma generated IL-2 (Th1 cytokine) in supernatants was assessed by CTLL assay. As is clearly demonstrated in Fig. 1, HLA-DR1 and HLA-DR4 neonatal monocytes were deficient at processing and presentation of protein antigens.
Figure 1. Neonatal monocytes isolated from fresh cord blood are deficient at MHC-II antigen processing and presentation.
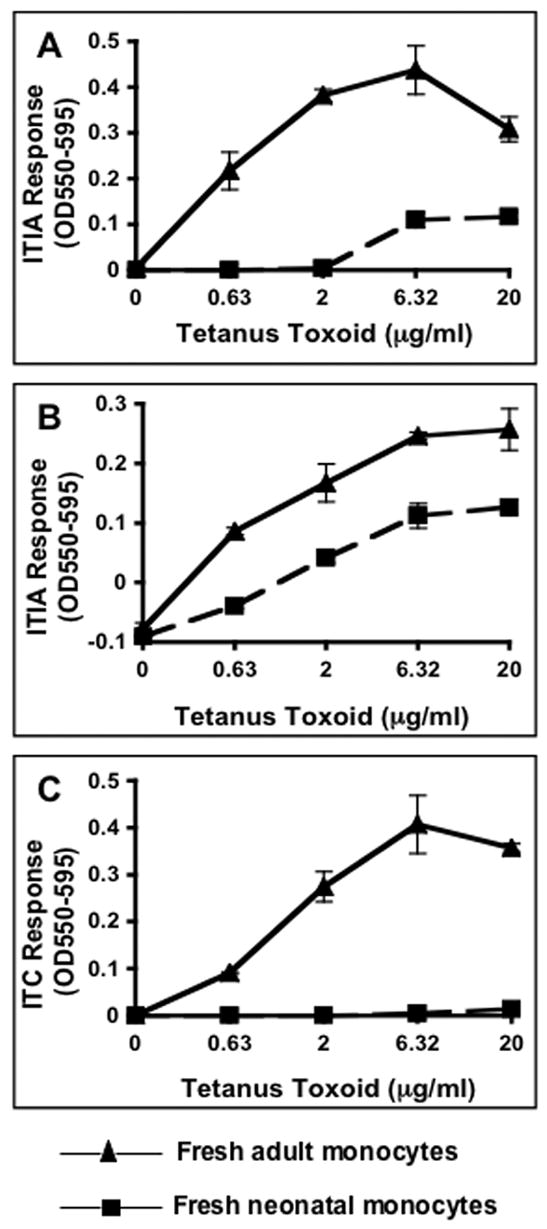
Negatively selected monocytes isolated from fresh HLA-matched cord and adult blood was incubated for 24 h with antigen and appropriate HLA-restricted T hybridoma cells. Supernatants were assessed for IL-2 content by a CTLL-2 proliferation assay that was monitored with Alamar blue, an indicator dye. A & B) HLA-DR1 (allele DRB1*0101) neonatal and adult monocytes incubated with tetanus toxoid and ITIA T hybridoma cells. C) HLA-DR4 (allele DRB1*0401) neonatal and adult monocytes incubated with tetanus toxoid and ITC T hybridoma cells. Data points are means of triplicate samples +/− S.D.
T cell assay for consistent analysis of multiple samples over time
To further quantify and assess the defect, a larger cohort of neonatal and adult monocytes had to be analyzed. Therefore, a quantitative readout that would ensure consistency over time and allow subsequent investigation of defects in the same monocytes that were analyzed in an initial T cell assay was needed. To ensure consistency in experiments done over time, all subsequent experiments have been performed using T hybridoma cells taken directly out of cryopreservation and derived from a single freeze. T hybridoma cells taken directly out of cryopreservation perform nearly as well as cells from a passage culture (data not shown). Analysis of IL-2 in supernatants was switched from the CTLL assay to the more quantitative IL-2 ELISA. Finally, monocytes isolated from fresh and frozen peripheral blood mononuclear cells (PBMCs) were evaluated in T cell assays. By freezing PBMCs one could evaluate monocytes isolated from frozen PBMCs in antigen processing and presentation assays and subsequently analyze these monocytes for mechanisms of defects at a later point in time. Using the T cell assay, monocytes purified from freshly isolated, HLA-DR1 (allele DRBI*0101) adult and cord blood samples were compared to monocytes purified from a frozen portion of the same PBMC sample. Adult and neonatal monocytes isolated from frozen PBMCs were functionally similar to monocytes isolated from freshly isolated PBMCs in T cell assays (data not shown).
Neonatal monocytes are selectively defective in MHC-II antigen presentation as opposed to intracellular antigen processing
Using our new strategy, antigen processing and presentation function was analyzed in negatively selected monocytes derived from frozen PBMC samples that expressed HLA-DR1 (allele DRB1*0101, Fig. 2) or HLA-DR3 (allele DRB1*0301, Fig. 3). HLA-DR1 monocytes isolated from 8 HLA-DR1 adult blood and 8 HLA-DR1 cord blood samples were incubated for 24 h with various concentrations of reverse transcriptase, reverse transcriptase peptide (350–364) or tetanus toxoid and the corresponding T hybridoma cells (see Table 1). Monocytes incubated with exogenous peptide by-pass the requirement for antigen processing, by directly presenting exogenous peptides in the context of surface MHC-II molecules to T hybridoma cells. Supernatants were analyzed for IL-2 by ELISA. The viability of neonatal and adult monocytes cultured overnight in media was found to be similar (around 90 % as determined by Trypan Blue exclusion). Majority of neonatal monocytes samples were defective for both processing and presentation of reverse transcriptase as well as presentation of exogenous reverse transcriptase peptide (Fig. 2). This defect was also observed in our analysis of processing and presentation of an unrelated antigen, tetanus toxoid, confirming that neonatal monocytes are generally defective in processing and presentation of protein antigens as well as presentation of exogenous peptide.
Figure 2. HLA-DR1 neonatal monocytes isolated from frozen PBMCs are deficient at MHC-II antigen processing and presentation of protein antigen and presentation of exogenous peptide.
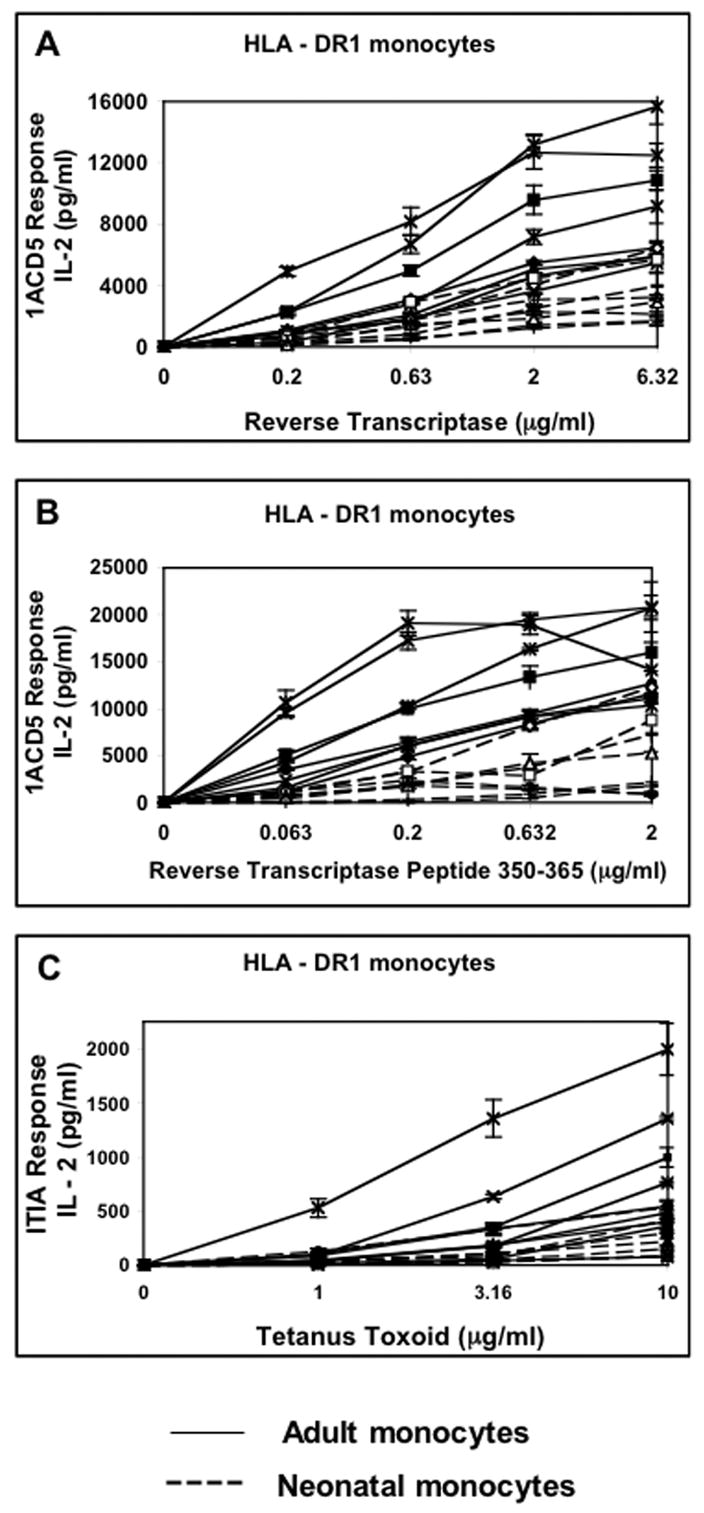
Negatively selected monocytes isolated from 16 frozen HLA-DR1 PBMCs (8 from cord blood, 8 from adult blood) were incubated for 24 h with A) reverse transcriptase and IACD5 T hybridoma cells, B) reverse transcriptase peptide(350–365) and IACD5 T hybridoma cells and C) tetanus toxoid and ITIA T hybridoma cells. Supernatants were assessed for IL-2 content by IL-2 ELISA. Neonatal monocytes are represented by dashed lines while adult monocytes are represented by intact lines. Data points are means of triplicate samples +/− S.D.
Figure 3. HLA-DR3 neonatal monocytes are also deficient at MHC-II antigen presentation.
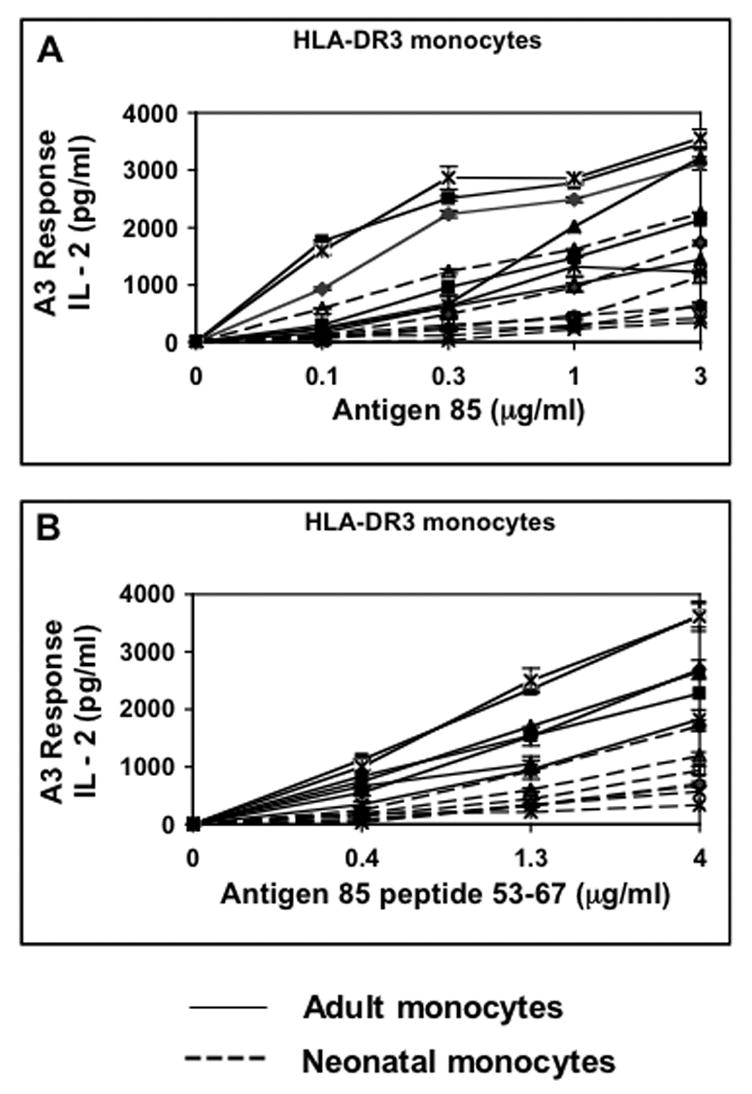
Negatively selected monocytes isolated from 14 frozen HLA-DR3 PBMCs (7 from cord blood, 7 from adult blood) were incubated for 24 h with A) Antigen 85 and A3 T hybridoma cells and B) Antigen 85 peptide(53–67) and A3 T hybridoma cells. Supernatants were assessed for IL-2 content by IL-2 ELISA. Neonatal monocytes are represented by dashed lines while adult monocytes are represented by intact lines. Data points are means of triplicate samples +/− S.D.
The magnitude of the processing and presentation defect was analyzed in HLA-DR1 neonatal monocytes by comparing IL-2 concentrations in T cell assay supernatants at specific antigen and peptide concentrations (Table 2; of all the different concentrations of antigen used in the T cell assay, only the concentration of antigen that gave close to a 1/2 maximal response is shown in Table 2). At reverse transcriptase concentrations of 0.2, 0.63 and 2 μg/ml, neonatal monocytes generated 76 % (p = 0.007), 67 % (p = 0.005) and 66 % (p = 0.001) less IL-2 respectively than adult monocytes (p values derived by non-parametric Mann-Whitney U test). At reverse transcriptase peptide concentrations of 0.06, 0.2 and 0.63 μg/ml, neonatal monocytes generated 86 % (p = 0.002), 82 % (p = 0.008) and 78 % (p = 0.0008) less IL-2 respectively than adult monocytes. At a tetanus toxoid concentration of 3.16 and 10 μg/ml, neonatal monocytes generated 72 % (p = 0.045) and 66 % (p = 0.027) less IL-2 respectively than adult monocytes. The magnitude of the defect in processing and presenting intact protein antigen was very similar to the magnitude of the defect in presenting exogenous peptide to T hybridoma cells. These observations suggest that neonatal monocytes are selectively defective in antigen presentation as opposed to intracellular antigen processing.
To determine whether the antigen presentation defect could be observed in neonatal monocytes expressing other HLA-DR molecules, our analysis was extended to additional monocytes that expressed HLA-DR3 (allele DRB1*0301, Fig. 3). HLA-DR3 monocytes isolated from 7 HLA-DR3 adult blood and 7 HLA-DR3 cord blood samples were incubated with various concentrations of M. tuberculosis Antigen 85 or Antigen 85 peptide(53–67) and the HLA-DR3 restricted A3 T hybridoma cells. Majority of neonatal monocyte samples were defective for both processing and presentation of Antigen 85 as well as presentation of Antigen 85 peptide confirming that the defect in neonatal monocytes is not restricted to only monocytes expressing HLA-DR1 molecules. Here as well, the magnitude of the processing and presenting defect was very similar to the defect seen in the presentation of exogenous Antigen 85 peptide to T hybridoma cells (Table 2; of all the different concentrations of antigen used in the T cell assay, only the concentration of antigen that gave close to a 1/2 maximal response is shown in Table 2). At Antigen 85 concentrations of 0.3, 1 and 3μg/ml, neonatal monocytes generated 79 % (p = 0.009), 70 % (p = 0.006) and 72 % (p = 0.002) less IL-2 respectively than adult monocytes. At Antigen 85 peptide concentration of 0.4, 1.3 and 4 μg/ml, neonatal monocytes generated 83 % (p = 0.002), 73 % (p = 0.002) and 70 % (p = 0.003) less IL-2 respectively than adult monocytes. These observations confirmed that the neonatal monocytes are selectively and significantly defective in antigen presentation to T cells.
While the majority of our cord blood samples were from newborns born by vaginal delivery, a few of the cord blood samples analyzed were from newborns delivered by C-section (Table 2). Although our sample size from newborns delivered by C-section is limited, the mode of delivery appears to have little impact on the magnitude of the defect.
Secretion of soluble factor(s) by neonatal monocytes could result in decreased T cell hybridoma responses in both the antigen processing and presentation assay as well as the antigen presentation assay. Therefore, the ability of fixed neonatal monocytes, that are incapable of secreting soluble factor(s), to present peptide:MHC-II complexes to T hybridoma cells was analyzed (Fig. 4). HLA-DR1 or HLA-DR3 neonatal and adult monocyte pairs were incubated with reverse transcriptase or Antigen 85 respectively for 24 h to allow for cell surface expression of peptide:MHC-II molecules. Cells were washed to remove all soluble factors in the supernatant and then fixed with paraformaldehyde to prevent secretion of any additional factors. Fixed monocytes were incubated with the corresponding T hybridoma cells for 24 h and supernatants were analyzed for IL-2 by ELISA. The fixed neonatal monocytes continued to be defective in antigen processing and presentation with no decrease in the magnitude of the defect. Thus, neonatal monocytes do not secrete soluble factors that inhibit T cell hybridoma responses.
Figure 4. Fixed neonatal monocytes are also defective in MHC-II antigen presentation.
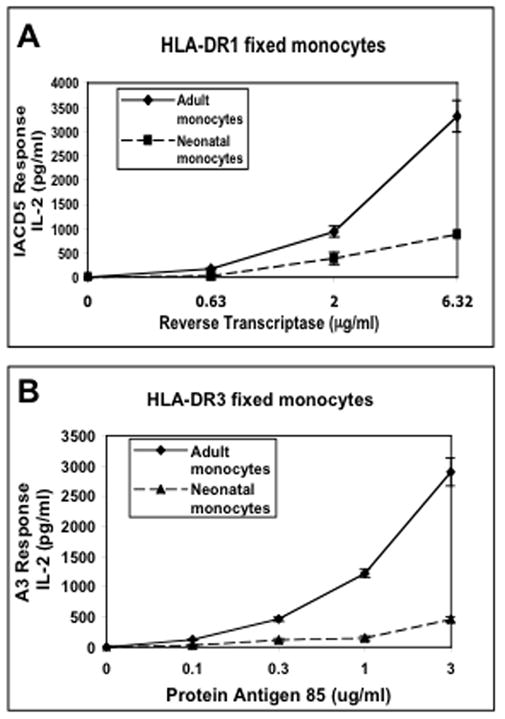
Negatively-selected monocytes isolated from HLA-DR1 or HLA-DR3 cord or adult blood-derived PBMCs were incubated for 24 h with reverse transcriptase and antigen 85 respectively. Cells were washed, fixed in 1% paraformaldehyde (to prevent secretion of any soluble factors) and then incubated for an additional 24 h with A) IACD5 T hybridoma cells or B) A3 T hybridoma cells. Supernatants were assessed for IL-2 content by IL-2 ELISA. Data points are means of triplicate samples +/− S.D.
The MHC-II antigen presentation defect in neonatal monocytes is not correlated with decreased surface MHC-II expression
Reduced surface MHC-II expression may result in decreased processing and presentation of intact protein antigen as well as reduced presentation of exogenous peptide. We initially tried to assess surface HLA-DR expression in neonatal and adult monocytes that had previously been analyzed in the T cell assay (Table 2) by using allele-specific antibodies. However, due to the low sensitivity of these antibodies, it was impossible to obtain consistent signal across a large number of donors of varying MHC-II expression. We therefore used the monoclonal antibody L243 to stain for surface HLA-DR. Although humans express many genetically distinct MHC-II molecules (HLA-DR, HLA-DP and HLA-DQ), L243 is specific for HLA-DR (44) and recognizes the two predominantly expressed HLA-DR heterodimers on the cell surface. Since the two HLA-DR heterodimers are expressed at equivalent levels, we conclude that L243 staining reflects the expression levels of either of the two HLA-DR heterodimers.
Mean channel fluorescence of surface MHC-II in adult monocytes ranged from 50 – 278 with an average of 140, whereas the mean channel fluorescence of surface MHC-II in neonatal monocytes ranged from 34–176 with an average of 87 (Table 3). The mean channel fluorescence of surface MHC-II was significantly lower (37.4 %) when comparing all neonatal monocytes to adult monocytes (Mann-Whitney U test; p = 0.008, Table 3).
We next analyzed whether decreased HLA-DR expression in neonatal monocytes correlated with reduced T cell responses (Fig. 5). Since the hybridomas differ in the magnitude of their responses (IL-2 generation), separate analysis of HLA-DR3 donors and HLA-DR1 donors was performed.
Figure 5. Surface MHC-II levels do not correlate with MHC-II antigen presentation defect.
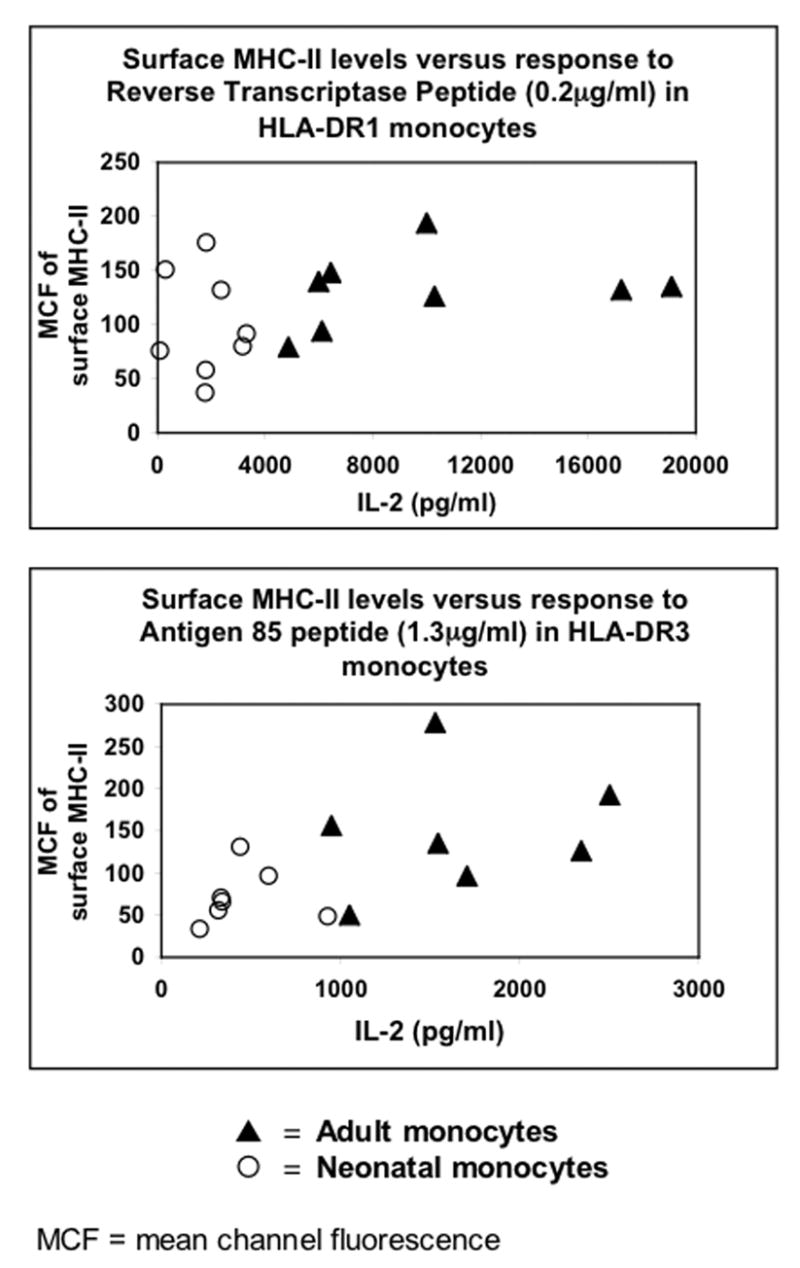
Mean channel fluorescence of surface MHC-II levels on adult and neonatal monocytes (from Table 3) plotted against concentration of IL-2 in supernatants from T cell assays (from Table 2).
Among HLA-DR3 monocytes (n = 14) there was a statistical difference in MHC-II levels between adult and neonatal monocytes (Mann Whitney U test; χ2 = 4.45, p = 0.035, Table 3). HLA-DR3 adult and neonatal monocytes also differed with regard to IL-2 generation (Figure 3, Table 2). Analysis of MHC-II levels versus log-transformed IL-2 levels (evaluated for all Antigen 85 peptide and Antigen 85 protein concentrations shown in Figure 3) demonstrated approximately normal distributions when plotted graphically. Pearson correlations were used to assess the relationship between these variables among adults and neonates. IL-2 generation was independent of MHC-II expression in both adult and neonatal monocytes (all p>0.05). A linear regression was performed to evaluate the difference in IL-2 response between adult and neonatal monocytes while controlling for the level of MHC-II expression. There was no significant correlation between MHC-II expression and IL-2 generation at any Antigen 85 peptide or Antigen 85 protein concentrations (e.g. at Antigen 85 peptide concentration of 1.3 μg/ml which gives close to the 1/2 maximal response in adults, p = 0.463), even when controlling for sample type (adult or neonate, Fig. 5). The level of IL-2 generation at all Antigen 85 peptide and Antigen 85 protein concentrations was significantly lower among neonatal monocytes than adult monocytes even when controlling for MHC-II expression (e.g. at Antigen 85 peptide concentration of 1.3 μg/ml, p = 0.0013).
Among HLA-DR1 positive monocytes (n = 16) there was no statistical difference in MHC-II levels between adult and neonatal monocytes (Mann-Whitney U test; χ2 = 1.726, p = 0.189, Table 3). Although, HLA-DR1 adult and neonatal monocytes did differ with regard to IL-2 generation (Figure 2, Table 2), no significant correlation was observed between MHC-II expression and IL-2 generation at any reverse transcriptase peptide or reverse transcriptase protein concentrations (e.g. at reverse transcriptase peptide concentration of 0.2 μg/ml which gives close to the 1/2 maximal response in adults, p = 0.559), even when controlling for sample type (adult or neonate, Fig. 5). Here as well the level of IL-2 generation at all reverse transcriptase peptide and reverse transcriptase protein concentrations was significantly lower among neonatal monocytes than adult monocytes even when controlling for MHC-II expression (e.g. at reverse transcriptase peptide concentration of 0.2 μg/ml, p = 0.008).
In conclusion, we have demonstrated that neonatal monocytes are selectively defective in antigen presentation as opposed to intracellular antigen processing. Although neonatal monocytes have decreased surface MHC-II levels, this decrease is not statistically correlated with the antigen presentation defect indicating significant contribution by other mechanisms that affect antigen presentation (e.g. cell adhesion, immunological synapse formation etc). Defects in antigen presentation may contribute to dampened CD4+ TH1 responses and susceptibility to infection observed during early neonatal life.
Discussion
The susceptibility of neonates to infection has often been ascribed to diminished CD4+ TH1 responses. The contribution of neonatal APCs to this defect remains unclear. MHC-II mediated antigen processing and presentation function in human neonatal APCs has been described as equivalent (25, 33, 34), decreased (24, 32, 35) or increased (36–39) when compared to that of adult APCs. This lack of consistent results may partly arise from the use of either adult or neonatal CD4+ T cells that are functionally different or from the analysis of dendritic cells of varying functional maturity. Most of the studies analyzing MHC-II antigen processing and presentation function in neonatal APCs have utilized mixed lymphocyte reactions. While mixed lymphocyte reactions may provide a general assessment of antigen processing and presentation function, they are not a suitable system for identifying specific processing or presentation defects.
For our analysis of MHC-II antigen processing and presentation function in neonatal APCs we have used antigen-specific, HLA-DR-restricted murine T hybridoma cells that have previously been used for detecting antigen processing defects in human APCs (41). This has allowed us to eliminate the use of allogeneic T cells derived from either cord or adult blood that may be functionally different. T cell hybridomas have provided us with several advantages. They can be grown indefinitely and are easy to maintain (unlike human T cell lines and clones). Freshly thawed cells can be used on the day of the experiment and this approach has helped eliminate variations in response that arise when using cells growing in culture. These T hybridoma cells are also CD80/CD86 and CD40-independent and are therefore ideal for independently analyzing antigen processing defects as well as specific antigen presentation defects in addition to correlating T cell responses to MHC-II levels.
Our studies of MHC-II antigen processing and presentation in neonatal monocytes have clearly demonstrated that neonatal monocytes generate reduced T cell activation. This defect was identified as an antigen presentation defect as opposed to an intracellular antigen processing defect. This defect was observed in several HLA-DR alleles and with different antigens, implying that it is a general antigen presentation defect that is not specific to a particular HLA-DR allele or antigen. The monocytes used in our assays were “untouched” and not activated prior to or during incubation with antigen (antigen preparations contained no detectable LPS). However, activated neonatal monocytes are also likely to be deficient at MHC-II antigen processing and presentation, since neonatal monocytes are less capable of LPS induced increases in HLA-DR expression than adult monocytes (30).
Studies in neonatal B cells have demonstrated that a surprising majority of MHC-II are “empty”, that is, devoid of detectable peptide in the binding groove (34). The authors suggested that neonatal B cells maybe defective in antigen processing, peptide loading or both. Surprisingly, the ability of these neonatal B cells to elicit a mixed lymphocyte reaction was similar to that of adult B cells. Whether any MHC-II molecules on neonatal monocytes are “empty” remains undermined. As speculated by Garban et al, the presence of “empty” MHC-II molecules suggests an antigen processing or peptide loading defect. This defect should potentially be overcome by incubating APCs with exogenous peptides, which bind surface MHC-II and by-pass the requirements for processing. Since neonatal monocytes incubated with exogenous peptides continue to be defective in presentation, we postulate that the contribution of any “empty” MHC-II molecules to the antigen presentation defect in neonatal monocytes is minimal. Alternatively, HLA-DR molecules in neonatal monocytes may be loaded with peptides (e.g. CLIP) that are difficult to displace with exogenous peptides. Reduced HLA-DM expression in neonatal monocytes may help generate HLA-DR molecules loaded with CLIP.
Surface MHC-II expression in neonatal APCs has been extensively analyzed by several investigators. A few studies found increased (19) or equivalent (20, 21) MHC-II expression on neonatal APCs (compared to adult APCs), while a majority of studies found decreased MHC-II expression on neonatal APCs (22–32). In agreement with many of the published findings, we also observed decreased surface MHC-II expression on our neonatal monocytes. Reduced MHC-II expression is thought to negatively impact both processing and presentation of intact protein antigen and presentation of exogenous peptide that directly bind surface MHC-II molecules. Therefore, the majority of studies have speculated that decreased MHC-II expression in neonatal APCs leads to reduced antigen processing and presentation function. However, none of these studies have tried to actually correlate reduced MHC-II levels in neonatal APCs to an antigen presentation defect. In the current work, we have analyzed correlation between decreased MHC-II levels in neonatal APCs and the antigen presentation defect for the first time. Contrary to the general assumption that decreased MHC-II levels contribute directly to decreased MHC-II antigen processing and presentation, we have found no significant correlation between MHC-II expression and IL-2 generation in either neonatal or adult monocytes. In addition, the level of IL-2 generation was significantly lower among neonatal monocytes than adult monocytes even when controlling for MHC-II expression. These results suggest that although MHC-II is required for the response, decreased or increased surface MHC-II expression cannot automatically be correlated to decreased or increased T cell hybridoma responses. We can conclude that other components that govern APC-T cell hybridoma interaction maybe defective in neonatal monocytes and contribute more significantly to the antigen presentation defect. These may include among others poor neonatal monocyte-T cell adhesion or altered/defective formation of the immunological synapse leading to reduced T cell hybridoma responses.
In conclusion, we have clearly demonstrated that neonatal monocytes are selectively defective in MHC-II antigen presentation. We also demonstrate for the first time that mechanisms other than decreased MHC-II expression contribute to the MHC-II antigen presentation defect in neonatal APCs. Defects in MHC-II antigen presentation may contribute to neonatal immune deficiency.
Table 1.
Characterization of T cell hybridomas
| Hybridoma | MHC Restriction | Antigen Specificity | Epitope | CDBO/CD86 dependence | CD40 dependence |
|---|---|---|---|---|---|
| ITIA | HLA-DR1 | Tetanus Toxoid | ND | Independent | Independent |
| 1ACD5 | HLA-DR1 | HIV-Reverse Transcriptase | aa350-364 | Independent | Independent |
| A3 | HLA-DR3 | Antigen 85 of Mycobacterium tuberculosis | aa53-67 | Independent | Independent |
| ITC | HLA-DR4 | Tetanus Toxoid | ND | ND | ND |
ND: Not determined
Table 2.
IL-2 concentration in supernatants from T cell assays described in Figures 2 and 3
| HLA-DR1 | IL-2 (pg/ml) | HLA-DR3 | IL-2(pg/ml) | |||||
|---|---|---|---|---|---|---|---|---|
| Adult monocytes | Mode of Delivery | RT (0.63μg/ml) | RT peptide (0.2μg/ml) | TT (3.16μg/ml) | Adult monocytes | Mode of Delivery | Ag85 (1 μg/ml) | Ag85 Peptide (1 .3 μg/ml) |
| AM1 | N/A | 6679 | 17227 | 633 | AM02 | N/A | 2485 | 1530 |
| AM14 | N/A | 3073 | 4861 | 334 | AM23 | N/A | 2773 | 2344 |
| AM19 | N/A | 4966 | 9994 | 352 | AM54 | N/A | 1005 | 1049 |
| AM21 | N/A | 2061 | 6098 | 179 | AM58 | N/A | 1313 | 949 |
| AM22 | N/A | 1885 | 5981 | 80 | AM62 | N/A | 2854 | 2502 |
| AM23 | N/A | 2766 | 10285 | 174 | AM67 | N/A | 1470 | 1545 |
| AM24 | N/A | 8178 | 19102 | 1356 | AM70 | N/A | 2017 | 1705 |
| AM34 | N/A | 1749 | 6432 | 59 | ||||
| Average: | 4130 | 9998 | 396 | Average: | 1083 | 1661 | ||
| Neonatal monocytes | Neonatal monocytes | |||||||
| NM24 | Vaginal | 550 | 291 | 30 | NM33 | Vaginal | 256 | 318 |
| NM25 | Vaginal | 466 | 85 | 25 | NM50 | Vaginal | 464 | 333 |
| NM32 | C-section | 815 | 1758 | 48 | NM52 | Vaginal | 1617 | 597 |
| NM39 | Vaginal | 1821 | 3156 | 76 | NM56 | ? | 426 | 439 |
| NM80 | Vaginal | 2906 | 3301 | 104 | NM60 | C-Section | 953 | 928 |
| NM81 | Vaginal | 1483 | 1803 | 109 | NM83 | Vaginal | 223 | 338 |
| NM93 | C-section | 1308 | 2351 | 327 | NM90 | C-section | 295 | 214 |
| NM97 | Vaginal | 1732 | 1784 | 187 | ||||
| Average: | 1385 | 1816 | 113 | Average: | 605 | 452 | ||
| % Difference P value | 67%↓ 0.005 | 82%↓ 0.0008 | 72%↓ 0.045 | % Difference Pvalue | 70%↓ 0.006 | 73%↓ 0.002 | ||
RT = Reverse transcriptase
TT = Tetanus toxoid
Table 3.
Surface MHC-II levels in neonatal and adult monocytes
| Adult Monocytes HLA-DR1 | MHC-II (MCF)** | Neonatal Monocytes HLA-DR1 | MHC-II (MCF)** |
|---|---|---|---|
| AM1 | 132 | NM24 | 151 |
| AM14 | 79 | NM25 | 76 |
| AM19 | 194 | NM32 | 37 |
| AM21 | 94 | NM39 | 80 |
| AM22 | 140 | NM80 | 92 |
| AM23* | 126 | NM81 | 176 |
| AM24 | 135 | NM93 | 132 |
| AM34 | 148 | NM97 | 58 |
| Average | 131 | Average | 100 |
| % Difference P value | 24% ↓ 0.189 | ||
| HLA-DR3 | HLA-DR3 | ||
| AM2 | 278 | NM33 | 56 |
| AM23* | 126 | NM50 | 71 |
| AM54 | 50 | NM52 | 97 |
| AM58 | 156 | NM56 | 131 |
| AM62 | 192 | NM60 | 49 |
| AMG7 | 135 | NM83 | 66 |
| AM70 | 96 | NM90 | 34 |
| Average | 148 | Average | 72 |
| % Difference P value | 51 % ↓ 0.035 | ||
| All adult monocytes (HLA-DR1 + HLA-DR3) | All neonatal monocytes (HLA-DR1 + HLA-DR3) | ||
| Average | 140 | Average | 87 |
| % Difference P value | 37.9% ↓ 0.003 |
AM23 expresses both HLA-DR1 and HLA-DR3
MCF = Mean Channel Fluorescence
Acknowledgments
This work was supported by a grant from Steris Corporation, start-up funds from Rainbow Babies and Children’s Hospital, Cleveland, OH, NIAID AI062368 to D.H.C., and AI35726 to C.V.H.
We thank Catherine Woods and the other nurses at the University Hospitals of Cleveland for collection and supply of cord blood and Dr. Mary Laughlin for providing us access to the cord blood.
We thank CFAR for flow cytometry services (supported by NIH Grant AI36219).
We thank Drs W. Henry Boom and Don Anthony for reading the manuscript.
Abbreviations
- MHC-II
class II MHC
- NM
neonatal monocytes
- AM
adult monocytes
- MCF
mean channel fluorescence
- APC
antigen presenting cell
- PBMC
peripheral blood mononuclear cell
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.WHO. 2005. [Google Scholar]
- 2.Cole FS. Avery's Diseases of the Newborn. W.B. Saunders Company; Philadelphia: 1998. [Google Scholar]
- 3.Cole FS. Avery's Diseases of the Newborn. W.B. Saunders Company; Philadelphia: 1998. [Google Scholar]
- 4.Lewis DB, Tu W. Immunologic Disorders in Infants and Children. Elsevier Saunders; Philadelphia: 2004. [Google Scholar]
- 5.Remington JS, Klein JO. Current concepts of infections of the fetus and newborn infant. W.B. Saunders Co.; Philadelphia: 2001. [Google Scholar]
- 6.Ramachandra L, Chu RS, Askew D, Noss EH, Canaday DH, Potter NS, Johnsen A, Krieg AM, Nedrud JG, Boom WH, Harding CV. Immunol Rev. 1999;168:217–239. doi: 10.1111/j.1600-065x.1999.tb01295.x. [DOI] [PubMed] [Google Scholar]
- 7.Harding CV, Ramachandra L, Wick MJ. Curr Opin Immunol. 2003;15:112–9. doi: 10.1016/s0952-7915(02)00008-0. [DOI] [PubMed] [Google Scholar]
- 8.Cresswell P. Annu Rev Immunol. 1994;12:259–293. doi: 10.1146/annurev.iy.12.040194.001355. [DOI] [PubMed] [Google Scholar]
- 9.Harding CV. Critical Reviews in Immunology. 1996;16:13–29. doi: 10.1615/critrevimmunol.v16.i1.20. [DOI] [PubMed] [Google Scholar]
- 10.Bakke O, Dobberstein B. Cell. 1990;63:707–716. doi: 10.1016/0092-8674(90)90137-4. [DOI] [PubMed] [Google Scholar]
- 11.Lotteau V, Teyton L, Peleraux A, Nilsson T, Karlsson L, Schmid SL, Quaranta V, Peterson PA. Nature. 1990;348:600–605. doi: 10.1038/348600a0. [DOI] [PubMed] [Google Scholar]
- 12.Cresswell P. Cell. 1996;84:505–507. doi: 10.1016/s0092-8674(00)81025-9. [DOI] [PubMed] [Google Scholar]
- 13.Nakagawa TY, Brissette WH, Lira PD, Griffiths RJ, Petrushova N, Stock J, McNeish JD, Eastman SE, Howard ED, Clarke SR, Rosloniec EF, Elliott EA, Rudensky AY. Immunity. 1999;10:207–217. doi: 10.1016/s1074-7613(00)80021-7. [DOI] [PubMed] [Google Scholar]
- 14.Riese RJ, Wolf PR, Brömme D, Natkin LR, Villadangos JA, Ploegh HL, Chapman HA. Immunity. 1996;4:357–366. doi: 10.1016/s1074-7613(00)80249-6. [DOI] [PubMed] [Google Scholar]
- 15.Shi GP, Villadangos JA, Dranoff G, Small C, Gu L, Haley KJ, Riese R, Ploegh HL, Chapman HA. Immunity. 1999;10:197–206. doi: 10.1016/s1074-7613(00)80020-5. [DOI] [PubMed] [Google Scholar]
- 16.Denzin LK, Cresswell P. Cell. 1995;82:155–165. doi: 10.1016/0092-8674(95)90061-6. [DOI] [PubMed] [Google Scholar]
- 17.Sherman MA, Weber DA, Jensen PE. Immunity. 1995;3:197–205. doi: 10.1016/1074-7613(95)90089-6. [DOI] [PubMed] [Google Scholar]
- 18.Sloan VS, Cameron P, Porter G, Gammon M, Amaya M, Mellins E, Zaller DM. Nature. 1995;375:802–806. doi: 10.1038/375802a0. [DOI] [PubMed] [Google Scholar]
- 19.Marwitz PA, Van Arkel-Vigna E, Rijkers GT, Zegers BJ. Clin Exp Immunol. 1988;72:260–266. [PMC free article] [PubMed] [Google Scholar]
- 20.Keever CA, Abu-Hajir M, Graf W, McFadden P, Prichard P, O'Brien J, Flomenberg N. Bone Marrow Transplant. 1995;15:409–419. [PubMed] [Google Scholar]
- 21.Langrish CL, Buddle JC, Thrasher AJ, Goldblatt D. Clin Exp Immunol. 2002;128:118–123. doi: 10.1046/j.1365-2249.2002.01817.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Durandy A, Brami C, Griscelli C. Am J Reprod Immunol Microbiol. 1985;8:94–100. doi: 10.1111/j.1600-0897.1985.tb00316.x. [DOI] [PubMed] [Google Scholar]
- 23.Stiehm ER, Sztein MB, Steeg PS, Mann D, Newland C, Blaese M, Oppenheim JJ. Clin Immunol Immunopathol. 1984;30:430–436. doi: 10.1016/0090-1229(84)90028-x. [DOI] [PubMed] [Google Scholar]
- 24.Hunt DW, Huppertz HI, Jiang HJ, Petty RE. Blood. 1994;84:4333–4343. [PubMed] [Google Scholar]
- 25.Roncarolo MG, Bigler M, Ciuti E, Martino S, Tovo PA. Blood Cells. 1994;20:573–585. [PubMed] [Google Scholar]
- 26.Petty RE, Hunt DWC. Vaccine. 1998;16:1378–1382. doi: 10.1016/s0264-410x(98)00095-4. [DOI] [PubMed] [Google Scholar]
- 27.Kotiranta-Ainamo A, Apajasalo M, Pohjavuori M, Rautonen N, Rautonen J. Clin Exp Immunol. 1999;115:309–314. doi: 10.1046/j.1365-2249.1999.00795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deneys VI, Mazzon AM, De Bruyere M, Cornu G, Brichard B. Eur J Haematol. 2001;66:107–114. doi: 10.1034/j.1600-0609.2001.00281.x. [DOI] [PubMed] [Google Scholar]
- 29.Birle A, Nebe CT, Gessler P. J Perinatol. 2003;23:294–299. doi: 10.1038/sj.jp.7210906. [DOI] [PubMed] [Google Scholar]
- 30.Drohan L, Harding JJ, Holm B, Cordoba-Tongson E, Dekker CL, Holmes T, Maecker H, Mellins ED. Hum Immunol. 2004;65:1356–1369. doi: 10.1016/j.humimm.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 31.Kanakoudi-Tsakalidou F, Debonera F, Drossou-Agakidou V, Sarafidis K, Tzimouli V, Taparkou A, Kremenopoulos G. Clin Exp Immunol. 2001;123:402–407. doi: 10.1046/j.1365-2249.2001.01471.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu E, Tu W, Law HK, Lau YL. Br J Haematol. 2001;113:240–246. doi: 10.1046/j.1365-2141.2001.02720.x. [DOI] [PubMed] [Google Scholar]
- 33.Chilmonczyk BA, Levin MJ, McDuffy R, Hayward AR. J Immunol. 1985;134:4184–4188. [PubMed] [Google Scholar]
- 34.Garban F, Ericson M, Roucard C, Rabian-Herzog C, Teisserenc H, Sauvanet E, Charron D, Mooney N. Blood. 1996;87:3970–3976. [PubMed] [Google Scholar]
- 35.Borras FE, Matthews NC, Lowdell MW, Navarrete CV. Br J Haematol. 2001;113:925–931. doi: 10.1046/j.1365-2141.2001.02840.x. [DOI] [PubMed] [Google Scholar]
- 36.Caux C, Massacrier C, Dezutter-Dambuyant C, Vanbervliet B, Jacquet C, Schmitt D, Banchereau J. J Immunol. 1995;155:5427–5435. [PubMed] [Google Scholar]
- 37.Caux C, Massacrier C, Vanbervliet B, Dubois B, Durant I, Cella M, Lanzavecchia A, Banchereau J. Blood. 1997;90:1458–1470. [PubMed] [Google Scholar]
- 38.Sorg RV, Kogler G, Wernet P. Bone Marrow Transplant. 1998;22:S52–54. [PubMed] [Google Scholar]
- 39.Sorg RV, Kogler G, Wernet P. Blood. 1999;93:2302–2307. [PubMed] [Google Scholar]
- 40.Canaday DH, Gehring A, Leonard EG, Eilertson B, Schreiber JR, Harding CV, Boom WH. J Immunol Methods. 2003;281:129–142. doi: 10.1016/j.jim.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 41.Gehring AJ, Dobos KM, Belisle JT, Harding CV, Boom WH. J Immunol. 2004;173:2660–2668. doi: 10.4049/jimmunol.173.4.2660. [DOI] [PubMed] [Google Scholar]
- 42.Lakey DL, Voladri RK, Edwards KM, Hager C, Samten B, Wallis RS, Barnes PF, Kernodle DS. Infect Immun. 2000;68:233–238. doi: 10.1128/iai.68.1.233-238.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ramachandra L, Noss EH, Boom WH, Harding CV. J Exp Med. 2001;194:1421–1432. doi: 10.1084/jem.194.10.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robbins PA, Evans EL, Ding AH, Warner NL, Brodsky FM. Hum Immunol. 1987;18:301–313. doi: 10.1016/0198-8859(87)90077-2. [DOI] [PubMed] [Google Scholar]