

Summary
Arrestins regulate the activity and subcellular localization of G protein-coupled receptors and other signaling molecules. Here we demonstrate that arrestins bind microtubules (MTs) in vitro and in vivo. The MT-binding site on arrestins significantly overlaps with the receptor-binding site, but the conformations of MT-bound and receptor-bound arrestin are different. Arrestins recruit ERK1/2 and the E3 ubiquitin ligase Mdm2 to microtubules in cells, similar to the arrestin-dependent mobilization of these proteins to the receptor. Arrestin-mediated sequestration of ERK to MTs reduces the level of ERK activation. In contrast, recruitment of Mdm2 to microtubules by arrestin channels Mdm2 activity toward cytoskeleton-associated proteins, dramatically increasing their ubiquitination. The mobilization of signaling molecules to microtubules is a novel biological function of arrestin proteins.
Keywords: arrestin, microtubules, ERK, Mdm2, G-protein coupled receptor
Introduction
As their name implies, arrestins were originally described as proteins that terminate G protein-mediated signaling by binding the activated phosphorylated forms of their cognate G protein-coupled receptors (GPCRs) (reviewed in 1; 2; 3). Recent discoveries of their interactions with numerous other binding partners revealed the role of arrestins as multi-functional regulators of cell signaling (reviewed in 4; 5). Arrestins redirect GPCR signaling to G protein-independent pathways and determine the intracellular localization of key regulatory proteins. In particular, arrestin retains ERK2 and JNK3 in complex with the receptor in the cytoplasm and removes Mdm2 and JNK3 from the nucleus (reviewed in 4; 6; 7).
Structurally, arrestins are elongated two-domain molecules with an overall fold that is remarkably conserved between different subtypes. Receptor binding “unfastens” two critical “clasps” that hold the molecule in its basal state, inducing a global conformational change that involves the movement of the two arrestin domains 2. Most non-receptor partners bind the arrestin-receptor complex, engaging arrestin elements that are not involved in receptor binding 4; 7. Recently, we identified microtubules (MTs) as an interaction partner of visual (rod) arrestin 8; 9. The difference in microtubule affinity between the two splice variants of visual arrestin expressed in bovine rods 10 determines their differential subcellular localization 8.
Here we demonstrate that all arrestin subtypes bind microtubules and identify arrestin elements involved. Receptors and microtubules engage the same side of the arrestin molecule, leaving the interaction sites for non-receptor binding partners accessible. We found that arrestins recruit ERK1/2 and ubiquitin ligase Mdm2 to microtubules, differentially affecting their activity. Arrestin-dependent mobilization of signaling molecules to the cytoskeleton is an earlier unappreciated link in the network of cellular regulatory pathways.
Results
Arrestin binding to microtubules in living cells
Visual arrestin binds MTs in rod photoreceptors in vivo. This interaction determines its subcellular localization in membrane microdomains and in the compartments of the rod cell 8; 11. Remarkable structural homology between arrestin family members 12 suggests that other arrestin subtypes may also interact with MTs. To test this hypothesis, we fractionated HEK-293 cells expressing different arrestins. As expected, most arrestin was present in the cytosol, but a noticeable proportion of all arrestin subtypes co-fractionated with MTs. In fact, other arrestins bind microtubules better than rod (Fig. 1a, b). The proportion of arrestin in the cytoskeletal fraction depends on the subtype, with arrestin3 demonstrating the highest binding. The quantification of soluble and cytoskeleton-associated arrestins by Western blot demonstrates that about 2–3 % of wild type rod, cone, and arrestin2 are associated with MTs, and this proportion reaches ~8% for arrestin3 (Fig. 1b). The deletion of the arrestin2 C-tail (cf. Fig. 2c) (A2Tr), as well as a 7-residue deletion in its inter-domain hinge region (A2D7) that locks arrestin in its basal conformation 13 enhance its binding to microtubules (Fig. 1a).
Figure 1. Arrestins bind microtubules in cells.
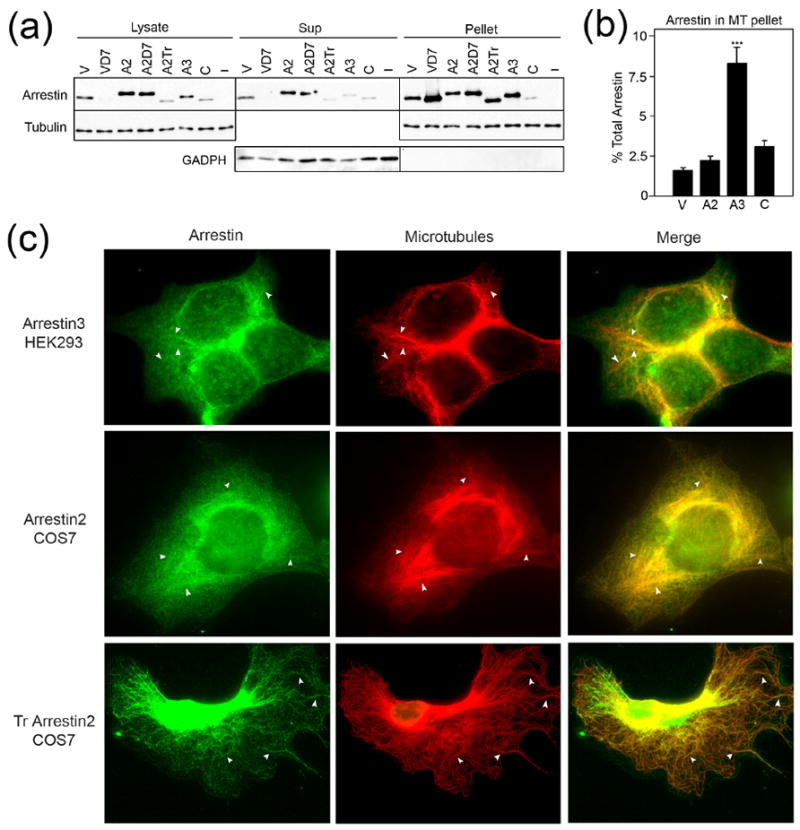
(a) HEK-293 cells transfected with the indicated arrestins or empty vector (−) were fractionated as described in the Methods and analyzed by Western blot. Representative blots from 6–8 experiments demonstrating the relative amount of each arrestin and tubulin present in the lysate, soluble (sup), and cytoskeletal (pellet) fractions are shown. Blots for the cytoplasmic marker, glyceraldehyde-3-phosphate dehydrogenase (GADPH), are also shown to demonstrate the quality of fractionation. When the same proportion of the soluble and pellet fraction is loaded, there is no appreciable GADPH signal in the pellet. (b) The percentage of arrestin in the cytoskeletal pellet fraction was quantified and analyzed by one-way ANOVA with arrestin type as the main factor. The percentage of arrestin3 in the pellet fraction is significantly greater than that of the other arrestins (*** p<0.001). (c) HEK293 cells expressing Flag-tagged arrestin3 or COS7 cells expressing untagged WT or Tr arrestin2 were fixed and stained as described in the Methods. White arrowheads indicate places where arrestin colocalization with microtubule bundles is most pronounced. Abbreviations: V, wild type visual rod arrestin; VD7, rod arrestin hinge deletion mutant; A2, wild type arrestin2; A2D7, arrestin2 hinge deletion mutant; A2Tr, truncated arrestin2(1–382); A3, wild type arrestin3; C, wild type cone arrestin. The mutants used are described in detail in supplemental Table S1.
Figure 2. Direct binding of arrestin to microtubules.
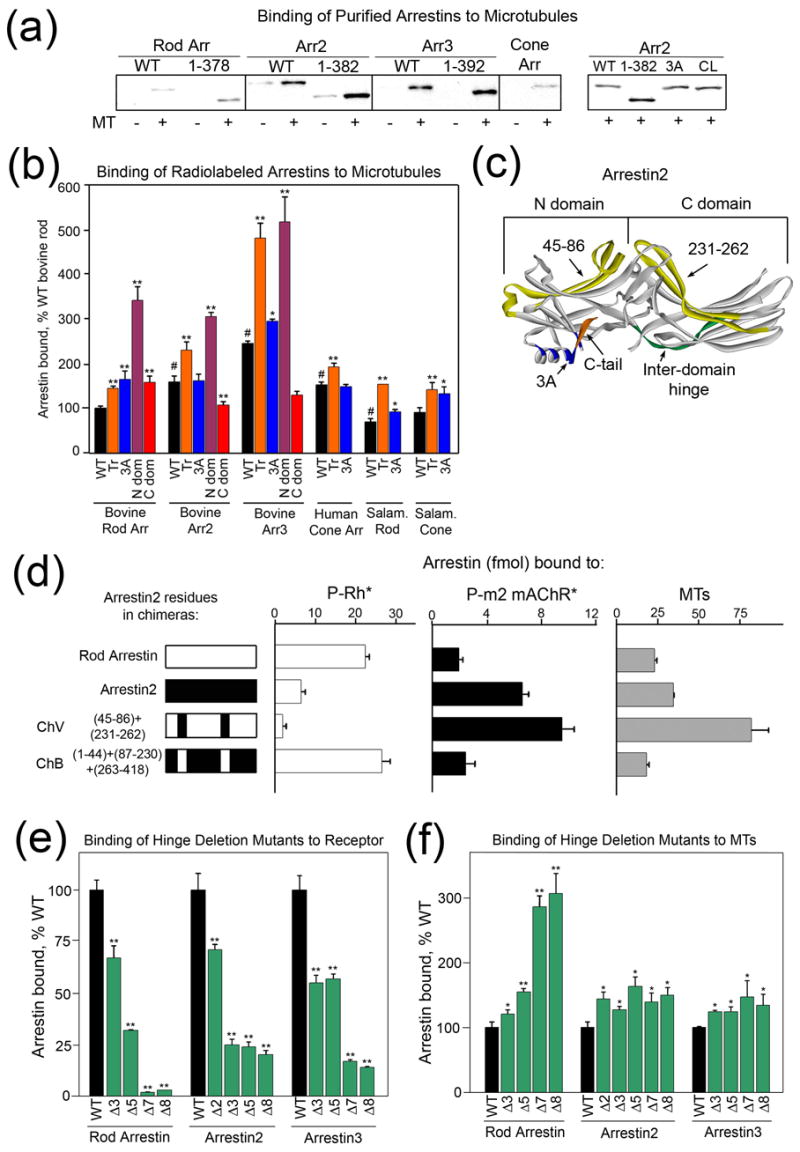
(a) Equal amounts of purified bovine rod, arrestin2, arrestin3, and salamander cone arrestin were incubated in the presence (+) or absence (−) of taxol-polymerized tubulin (MT) as described in the Methods. Arrestin bound to MTs was separated from free by centrifugation and quantified by Western blot. Representative results of 3 experiments are shown. WT, wild type; 3A, triple alanine substitution in the C-tail; CL, cysless arrestin2. (b, d–f) The binding of the indicated in vitro translated radiolabeled arrestins to MTs was determined as described in the Methods. (b) N-dom, N-domain; C-dom, C-domain; Tr, truncated; 3A, triple alanine substitution in the C-tail. (c) Arrestin2 crystal structure 24 highlighting important structural elements: orange, C-tail; green, inter-domain hinge; blue, hydrophobic residues that anchor the C-tail to the N-domain; yellow, residues that are important for MT and receptor binding (45–86 and 231–262). (d) Binding of arrestin to light-activated phosphorylated rhodopsin (P-Rh*), the phosphorylated carbachol-activated m2 muscarinic receptor (P-m2 mAChR*), or microtubules (MTs), respectively. ChV, rod arrestin-based chimera with residues 45–86 and 231–262 of arrestin2; ChB, arrestin2-based chimera with residues 45–86 and 231–262 of rod arrestin. (e, f) Binding of arrestin hinge deletion mutants to phosphorylated light-activated rhodopsin (e) or microtubules (f) was performed as described in the Methods. The number after Δ indicates how many hinge residues were removed. The structure of all mutants is described in detail in supplemental Table S1. Means +/− SD from at least 3 experiments are shown. In panels (b), (e), and (f), *p<0.05, **p<0.01, as compared to the respective WT arrestin, #p<0.01 as compared to WT bovine rod arrestin.
To visualize this phenomenon in intact cells, we performed colocalization experiments on HEK cells overexpressing arrestin3 using a fixing method that preserves MTs 14. The pattern of arrestin3 immunoreactivity partially overlaps with microtubule staining (Fig. 1c; arrows). The apparent extent of the colocalization is consistent with the proportion of this arrestin found in the cytoskeletal fraction (Fig. 1b). In contrast, when microtubules are depolymerized by incubation on ice, the disappearance of visible microtubule bundles is accompanied by the loss of the “structured” appearance of arrestin3 immunofluorescence (Fig. S1). To better visualize the cytoskeleton, for arrestin2 we also used COS7 cells which have a more extended cytoplasmic area. We found that truncated arrestin2, which binds MTs better than WT (Fig. 1a), colocalizes with microtubules to a much greater extent (Fig. 1c), demonstrating that co-sedimentation of this soluble protein with the cytoskeleton in our fractionation assay reflects its colocalization with microtubules in cells. Thus, the association with microtubules in cells is a common characteristic of all arrestin subtypes.
The conformation of microtubule-bound arrestin
Next, we used purified arrestins and MTs polymerized in vitro from pure tubulin to test whether this interaction is direct. This assay confirmed that both non-visual arrestins bind microtubules better than rod arrestin (Fig. 2A). The deletion of the arrestin C-tail (Fig. 2C) (Tr) or its detachment from the body of the molecule by a triple alanine substitution (3A) increases arrestin flexibility 15 and dramatically enhances its binding to receptors 16; 17; 18. Interestingly, the deletion of the C-tail also enhances MT binding (Fig. 2). Importantly, the relative binding of different purified arrestins and mutants reproduces our results in cells (compare Figs. 1 and 2). For further structure-function studies we used a relatively high-throughput direct binding assay with radiolabeled arrestins expressed in cell-free translation 9, similar to the assay we use to measure arrestin binding to purified GPCRs 19; 20. As shown in Fig. 2b, this method reproduces the relative binding of full-length and truncated forms of different arrestins that we detected in cells (Fig. 1) and with purified proteins (Fig. 2a).
The similar effects of C-tail deletion and its detachment by 3A mutation (Fig. 2c) on MT binding in all arrestins (Fig. 2b) suggest that the mechanism of microtubule binding is conserved in the arrestin family. Arrestin N- and C-domains are independent folding units which can be expressed separately and retain functional activity 19; 21 (Fig. 2c). We tested the relative role of the two arrestin domains in MT binding and found that the N-domains of rod, arrestin2, and arrestin3 bind substantially better than the full-length proteins, whereas the binding ability of the C-domains varies (Fig. 2b). Apparently, the arrestin N-domain binds MTs better when it is not impeded by the C-domain, although both domains clearly participate in this interaction.
Numerous lines of evidence demonstrate that the conformations of free and receptor-bound arrestin are substantially different 2; 22; 23. The N and C domains of arrestin are connected by a 12-residue loop termed the “hinge region” (Fig. 2c) 13. Progressive deletions in the inter-domain hinge severely impede receptor binding (Fig. 2e), indicating that the movement of the two domains relative to each other is required in this process. Interestingly, hinge deletions actually enhance MT binding of all arrestins (Fig. 2f), suggesting that the conformation of MT-bound arrestin differs from that of the receptor-bound form. The effect of hinge deletions on both receptor and MT binding is more dramatic in rod arrestin than in the non-visual subtypes, possibly due to the greater inherent flexibility of the non-visual arrestins 24. Importantly, the MT association of arrestin hinge deletion mutants was similarly enhanced in cells (Fig. 1a).
The microtubule-binding site in arrestin2
We took advantage of the significant difference between the MT binding of rod arrestin and arrestin2 and used a series of rod/arrestin2 chimeras 25 to further define the specific elements responsible for MT binding (Figs. 2d and S2). We found that the exchange of two elements on the concave sides of the N- and C-domains (Fig. 2c) simultaneously reverses receptor specificity and the relative ability of the two arrestins to bind microtubules (Fig. 2d), indicating that receptor- and MT-binding sites on arrestins significantly overlap. Replacement of several other rod arrestin elements with arrestin2 residues on the concave surfaces of the two domains supports this conclusion (Fig. S2). The positive charge in the loop between β-strands I and II unique for rod arrestin (R18) participates in binding to receptor-attached phosphates 12 and MTs 9. Introduction of this positive charge into arrestin2 in the context of wild type protein or any chimera invariably enhances MT binding (Fig. S2). Thus, even though MTs bind the same surface in rod and arrestin2, the contribution of different elements varies. Importantly, arrestin residues crucial for receptor binding play a key role in the MT interaction.
To study MT-binding elements in arrestin2 at dynamic equilibrium in solution, we used site-directed spin labeling (SDSL) electron paramagnetic resonance (EPR) spectroscopy. This method requires the elimination of reactive native cysteines from the protein, the introduction of a unique cysteine at the position of interest, and subsequent modification of the cysteine with a sulfhydryl-specific spin label to generate the side chain R1 (Fig. 3a) 26. The shape of the EPR spectrum of a spin label reflects its mobility and thus provides a means of mapping protein-protein interaction surfaces through changes in R1 motion 9; 22; 27; 28; 29. For example, a highly mobile spin label on the surface of a protein results in a narrow spectrum with three relatively sharp peaks, whereas the reduction of its mobility due to the presence of intramolecular interactions or involvement in a protein-protein interaction interface results in a broadening of the spectrum and/or the appearance of an additional peak indicating slower motion of the spin label.
Figure 3. Localization of the microtubule binding site in arrestin2.
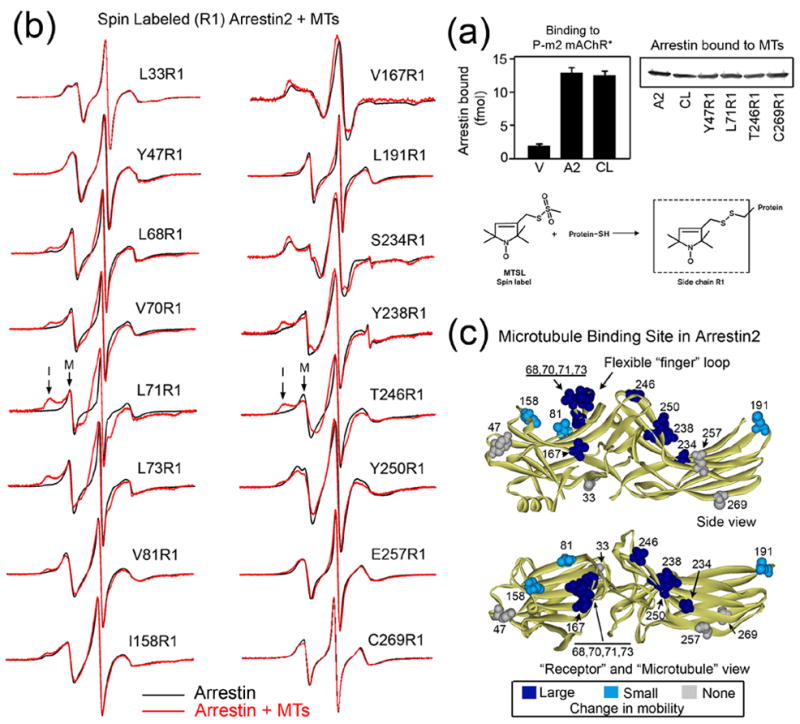
(a) Top left panel: binding of radiolabeled rod (V), arrestin2 (A2) or cysless arrestin2 (CL) to the phosphorylated carbachol-activated muscarinic receptor (P-m2 mAChR*). Top right panel: Purified arrestins with unique cysteines modified with spin label at the indicated position were tested for their ability to bind MTs as described in the Methods. Arrestin in the pellet fraction was quantified by Western blot. All spin-labeled mutants bound MTs similarly to wild type (A2) and cys-less arrestin2 (CL). Four representative R1 arrestins are shown. Lower panel: The R1 side chain generated by reacting the arrestin cysteine mutants with the methanethiosulfonate nitroxide reagent. (b) For each spin-labeled arrestin, the overlay of the EPR spectra in the absence (black) or presence (red) of MTs is shown. Arrows in the low-field region of the L71R1 and T246R1 spectra indicate examples of mobile (M) and immobile (I) peaks. EPR samples contained 10μg spin labeled arrestin2 and 150μg MTs in a final volume of 10μl. (c) The magnitude of microtubule-induced changes in spin label mobility is color-coded on the arrestin2 crystal structure 24 as follows: light blue/dark blue, small and large changes in mobility, respectively; gray, no change. Top panel: side view. Bottom panel: view of the concave sides of both arrestin domains.
We introduced unique cysteines in 16 different positions spanning the entire molecule on the background of fully functional cysteine-less arrestin2 (Fig. 3a). These spin-labeled proteins were examined by EPR to determine microtubule-induced changes in R1 mobility. The mobility of the spin label in all 16 positions in free arrestin2 (Fig. 3b, black traces) is consistent with the crystal structure 24 and R1 mobility/structure correlations previously established for several proteins 30; 31; 32; 33, including rod arrestin 9; 22. In particular, residues in flexible loops (e.g., 47, 68, 70, 71, 73, and 191) are highly mobile and are therefore very sensitive to potential decreases in R1 motion due to protein-protein contacts.
MT binding induced a substantial reduction in spin label mobility in several positions on the β-strands of the concave sides of both arrestin2 domains (167, 234, 238, 246, 250) (Fig. 3b, red traces). We observed some of the most dramatic changes at four positions (68, 70, 71, 73) in the flexible “finger loop” between β-strands V and VI (Fig. 3b, c), where the appearance of an additional peak (I) is indicative of a substantial reduction in mobility of the spin label due to the presence of MTs. Interestingly, this loop in rod arrestin was recently shown to play a key role in receptor binding 22. Smaller changes were detected on the concave surfaces at 81, 158, and 191, whereas spin label mobility at 33, 47, 257, and 269 did not appreciably change (Fig. 3b), even though these proteins bind MTs similarly to the other mutants (Fig. 3a). The positions with strong changes in the spin label mobility define an extensive microtubule “footprint” on the arrestin2 molecule (Fig. 3c) localized on the same surface that was previously implicated in receptor binding 22; 23; 25.
The changes in spin label mobility at several positions in arrestin2 are large enough to determine the affinity of the interaction. For this purpose we chose two arrestins spin labeled in different parts of the molecule and performed a titration with MTs to measure the extent of the spectral change as a function of MT concentration (Fig. 4). The data yielded a KD of 28 ± 5μM and 26 ± 7μM for arrestin2 labeled at positions 71 and 246, respectively (Fig. 4a). The numbers are in excellent agreement and indicate that arrestin affinity for MTs is sufficient to account for the observed levels of arrestin colocalization with the cytoskeleton (Fig. 1).
Figure 4. The affinity of arrestin2 for microtubules and tubulin dimer.
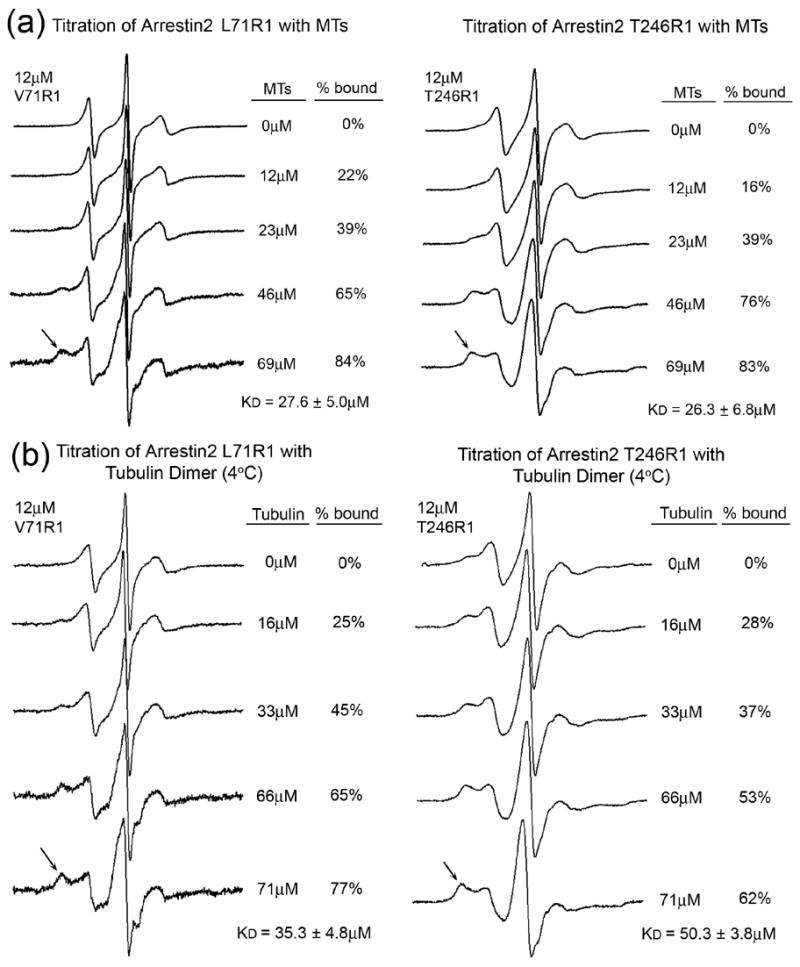
The spectra of 12 μM arrestin2 spin labeled at positions 71 (left panels) and 246 (right panels) were recorded in the presence of increasing concentrations of microtubules (a) or tubulin dimer (b). Final concentrations of tubulin are expressed as molar concentration of the tubulin dimer (Mr=110 kDa). Arrows indicate the location of the most immobilized population of spins that increases with tubulin concentration. The percent bound value for each spectrum was quantified by spectral subtraction of the corresponding free arrestin spectrum containing no tubulin from each of the composite spectra in the titration series. With the unbound arrestin spectrum subtracted out, each remaining spectrum included only the bound fraction. The percent arrestin bound values were calculated by comparison of the integrated intensities of each resulting bound spectrum with the corresponding composite spectrum. EPR spectroscopy of samples containing tubulin dimer was carried out at 4°C to prevent spontaneous polymerization. The mobility of the spin label side chain is decreased due to the decrease in temperature, thus the spectra of V71R1 and T246R1 free in solution in (b) differ from those recorded at room temperature in (a).
Cellular tubulin exists in dynamic equilibrium between αβ-dimer and microtubules 34; 35. If the arrestin binding site on microtubules is confined to a single αβ-dimer, arrestin should bind unpolymerized tubulin. To address this issue, we covalently attached unpolymerized tubulin to CNBr-activated Sepharose and tested arrestin binding. We found that all arrestins bind tubulin dimers (Fig. S3) suggesting that most of the arrestin-binding site is likely localized within a single αβ-dimer. To compare arrestin2 affinity for MTs and the αβ-dimers, we performed titration experiments using the same spin-labeled arrestins at 4°C to prevent spontaneous tubulin polymerization. We found that the KD of arrestin2 labeled in positions 71 and 246 for the αβ-dimer is 35 ± 5 μM and 50 ± 4 μM, respectively (Fig. 4b). Thus, arrestin2 affinity for the αβ-dimer and MTs is not dramatically different, supporting the conclusion that the bulk of the arrestin-binding site is localized within the αβ-dimer. Although arrestins bind both polymerized and unpolymerized tubulin (Figs. 3, 4, S3) they do not affect MT polymerization even at arrestin concentrations up to 100 μM (data not shown).
Functional consequences of the arrestin-microtubule interaction
Receptor-bound arrestins function as adaptors mobilizing the components of the trafficking machinery 36; 37 and recruiting a variety of signaling proteins to agonist-activated GPCRs 4; 7. Non-visual arrestins serve as scaffolds for the ASK1-MKK4-JNK3 and Raf-MEK-ERK1/2 MAP kinase cascades and target active ERK1/2 and JNK3 to specific subcellular locations 4; 38; 39; 40. Ubiquitin ligase Mdm2 interacts with receptor-bound 41 and free 42; 43 arrestins and plays a role in receptor ubiquitination 41; 44. The MT-binding site mapped by two different methods (Figs. 2, 3) covers a large portion of the concave surface of both arrestin domains and overlaps with the receptor-binding site 22; 23; 25. Thus, arrestin cannot interact with the receptor and microtubules simultaneously. Most importantly, in MT- and receptor-bound arrestin the docking sites for non-receptor partners may be equally accessible, enabling arrestin-dependent mobilization of signaling proteins to the cytoskeleton. To test this idea, we examined the localization of two known arrestin partners, ERK1/2 and Mdm2, in cells expressing different arrestins.
We found that the proportion of endogenous ERK1/2 present in the cytoskeletal fraction is significantly increased in cells expressing rod arrestin, arrestin2 or arrestin3 (Fig. 5a, c), indicating that these three subtypes bring ERK to the microtubules. Arrestin-dependent mobilization of ERK to the receptor results in ERK activation by upstream kinases, c-Raf-1 and MEK1 39. We tested whether this is also the case for ERK mobilized to MTs. The amount of active PP-ERK detected in the MT pellet fraction was negligible (Fig. 5a). However, the level of PP-ERK in the soluble fraction was significantly affected by arrestin expression (Fig. 5a, b). Rod and non-visual arrestins dramatically (by ~50%) reduced active ERK. Importantly, cone arrestin, which does not mobilize ERK to MTs, did not have this effect, suggesting that arrestin-dependent mobilization of ERK to MTs decreases the total level of active ERK in the cell by removing ERK from cellular compartments where it can be activated. Indeed we found that arrestins do not increase the proportion of upstream kinases MEK1 and c-Raf-1 in the cytoskeletal fraction (Fig. 5d), suggesting that the sequestration of an individual member of the MAP kinase cascade to MTs dampens signaling.
Figure 5. Arrestin-mediated sequestration of ERK to microtubules.
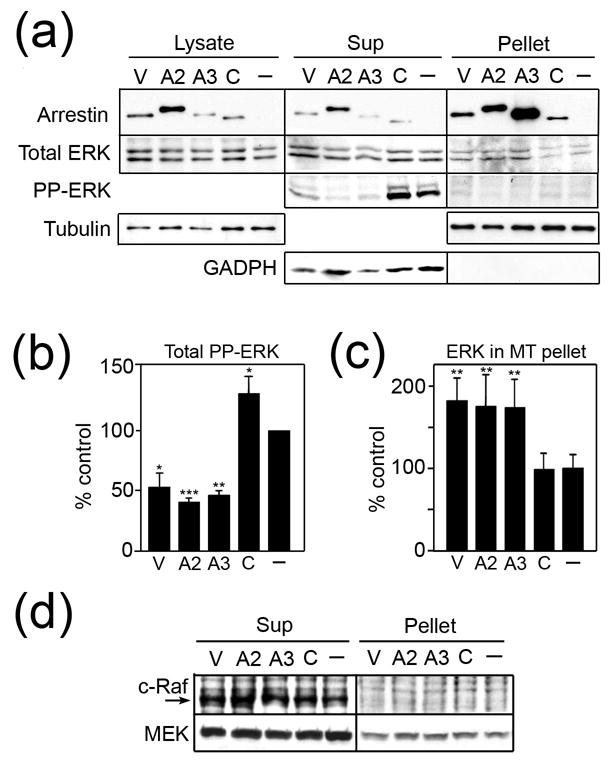
(a, d) HEK-293 cells transfected with the indicated arrestins or empty vector (−) were fractionated as described in the Methods and analyzed by Western blot. Representative blots from 5 experiments demonstrating the relative amount of each protein in the lysate, soluble (sup), and cytoskeletal (pellet) fractions are shown. To confirm the quality of fractionation, the distribution of the cytoplasmic marker GADPH was determined by subjecting the same proportion of sup and pellet to Western blot. A strong GADPH signal was detected in the sup, with no appreciable signal in the pellet. PP-ERK1/2 in the sup (b) and total ERK1/2 in the cytoskeletal fraction (c) was quantified. The data were analyzed by one-way ANOVA with arrestin type as the main factor. *p<0.05, **p<0.01, *** p<0.001, compared to control. The amount of active PP-ERK1/2 in the pellet was negligible in all cases. The percentage of ERK1/2 in the cytoskeletal fraction of control cells was 1.24 ± 0.41 % of the total ERK in the cell. Abbreviations: V, WT visual rod arrestin; A2, WT arrestin2; A3, WT arrestin3; C, WT cone arrestin; PP-ERK, phosphorylated active ERK.
The amount of endogenous Mdm2 present in the cytoskeletal fraction is also significantly increased in cells expressing rod and non-visual arrestins, but not cone arrestin (Fig. 6a, b). Overexpressed HA-Mdm2 follows the same pattern (Fig. 6b). Because Mdm2 is an E3 ubiquitin ligase, we tested whether its arrestin-dependent mobilization to microtubules affects the ubiquitination status of associated proteins. As shown in Figure 6, rod arrestin dramatically increases the ubiquitination of numerous proteins in the cytoskeletal fraction. Rod arrestin also increases the overall level of protein ubiquitination, whereas arrestin3 and cone arrestin reduce the total ubiquitination of most substrates, as compared to control cells (Fig. 6a, total=sup+pellet; Fig. 6c, left panel). Interestingly, even though arrestin3 significantly reduces total ubiquitination (more than any other arrestin), the percentage of the ubiquitinated proteins in the MT fraction of arrestin3-expressing cells is elevated to the same extent as in cells expressing rod arrestin (Fig. 6a, pellet; Fig. 6c, right panel). Thus, the four arrestin subtypes differentially affect the mobilization of Mdm2 to microtubules and the ubiquitination of soluble and cytoskeletal proteins.
Figure 6. Arrestin recruits Mdm2 to microtubules.
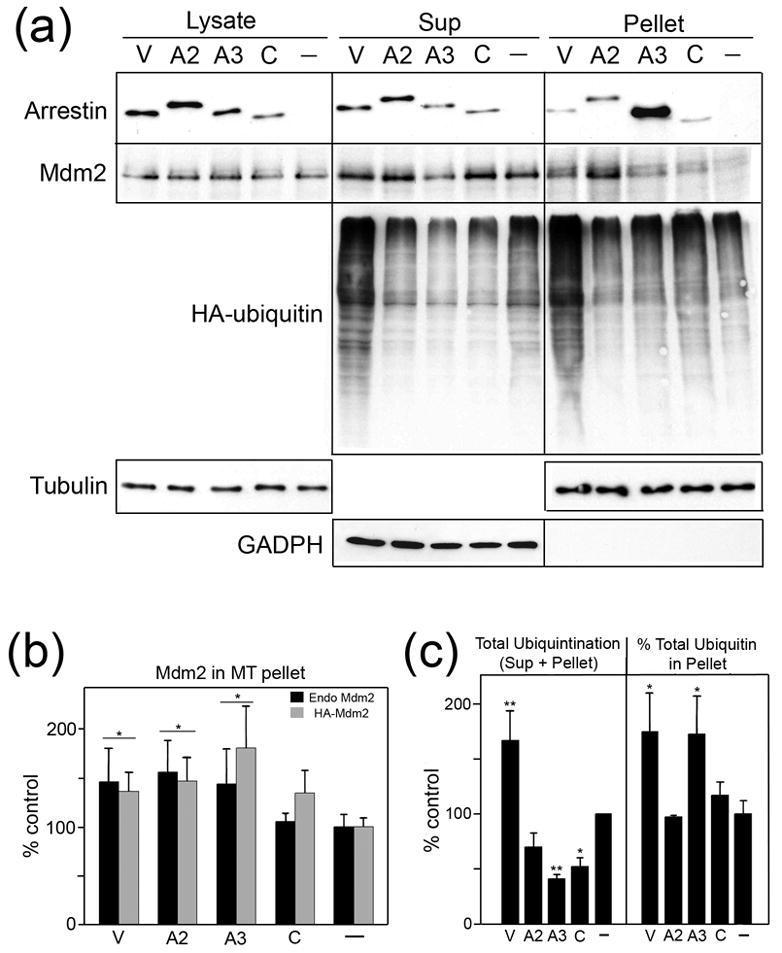
(a) HEK-293 cells transfected with the indicated arrestins or empty vector (−) were fractionated as described in the Methods and analyzed by Western blot. To confirm the quality of fractionation, the distribution of the cytoplasmic marker GADPH was determined by subjecting the same proportion of sup and pellet to Western blot. A strong GADPH signal was detected in the sup, with no appreciable signal in the pellet. To measure ubiquitination of soluble and cytoskeletal substrates, cells were co-transfected with HA-tagged ubiquitin and ubiquitinated proteins were visualized with anti-HA antibody. Representative blots from 3 experiments demonstrating the relative amount of each protein in the lysate, soluble (sup), and cytoskeletal (pellet) fractions are shown. Total ubiquitination was determined to be the amount of ubiquitinated substrate in the sup + pellet samples combined. (b) Endogenous Mdm2 in the cytoskeletal fraction in cells expressing different arrestins (black bars) (as shown in (a)) or endogenous plus overexpressed HA-Mdm2 (gray bars) was quantified and analyzed by one-way ANOVA with arrestin type as the main factor. *p<0.05, as compared to control. The amount of Mdm2 in the cytoskeletal fraction of control cells was 5.2 ± 0.7 % and 8.7 ± 0.8 % of the total Mdm2 for cells that did or did not express HA-Mdm2, respectively. (c) The total amount of ubiquitination (sup+pellet) (left graph) and percentage of protein-incorporated ubiquitin in the cytoskeletal fraction (pellet/(sup+pellet)) (right graph) was quantified by Western blot and analyzed by one-way ANOVA with arrestin type as the main factor. *p<0.05, **p<0.01, compared to control. Abbreviations: V, WT visual rod arrestin; A2, WT arrestin2; A3, WT arrestin3; C, WT cone arrestin.
Additional experiments showed that arrestins recruit signaling proteins to MTs more selectively than to the receptor. In particular, we observed no arrestin-dependent mobilization of a different MAP kinase JNK3, phospho-JNK3 38, or protein phosphatase 2A 45 (Fig. S4). In summary, among the six binding partners tested, arrestins only bring two (ERK1/2 and Mdm2) to the microtubules. Apparently, the distinct conformation of MT-bound arrestin can only interact with a subset of proteins that have been shown to bind the arrestin-receptor complex.
Discussion
Arrestins are multi-functional regulators of cellular signaling capable of interacting with more than 20 partners, often binding 2–3 different proteins simultaneously 3; 4; 5; 7. Two interconnected aspects of arrestin function are the regulation of the activity of its partners and their localization to a particular intracellular compartment. The first arrestin targets discovered, GPCRs, are restricted to the plasma membrane and endocytic vesicles. Therefore the interaction of numerous proteins (e.g., Src, JNK3, ERK1/2, Mdm2, etc) with receptor-bound arrestin localizes these molecules to receptor-rich membranes 4; 40. Here we describe a novel arrestin binding partner with a clearly defined cellular localization, microtubules, and demonstrate that the arrestin-MT interaction serves to localize certain signaling molecules to the cytoskeleton. Arrestin-dependent recruitment of protein kinase ERK and ubiquitin ligase Mdm2 to the microtubules differentially affects their activity, silencing ERK and directing Mdm2 to cytoskeletal substrates.
Our recent discovery of the light-regulated interaction of arrestin with MTs in rod photoreceptors 11 prompted us to test whether the other three members of the mammalian arrestin family interact with the cytoskeleton. We found that in intact cells and in vitro all four arrestin subtypes bind microtubules (Figs. 1, 3, 4). The affinity of arrestin2 for MTs (KD~26 μM; Fig. 4) is comparable to the normal tubulin concentration in many cell types 46; 47, which reaches 150 μM in mature neurons 46, suggesting that this interaction occurs in vivo. Indeed, our data show that an appreciable proportion of non-visual arrestins is bound to microtubules in cells (Fig. 1). Interestingly, several other regulators of GPCR signaling, such as G proteins 48 and GRK2 49; 50 also associate with microtubules. Considering the number of experimental studies of arrestins and microtubules, one might wonder why the connection between the two was not noticed before. In fact, both non-visual arrestins were previously found in the detergent-insoluble fraction 51, but the authors did not attach much significance to this observation.
Conceivably, arrestin association with MTs may serve three functions (which are not mutually exclusive): 1) to keep arrestins away from receptors, similar to its apparent role in rod photoreceptors 11; 2) to sequester arrestin binding partners to regulate their activation; 3) to mobilize signaling proteins to the cytoskeleton and direct their activity toward MT-associated substrates. The role of this interaction depends on the functional capabilities of MT-bound arrestin, and in particular, on the accessibility of the sites used by its other binding partners. The receptor binding site was mapped by numerous studies 22; 23; 25; 52 to the concave sides of both arrestin domains. The localization of the interaction sites for the non-receptor partners on the other side of the molecule allows arrestin to mobilize various signaling proteins to the receptor 4; 7. Our data show that microtubules bind to the same arrestin surface as the receptor (Figs. 2, 3)9, suggesting that the elements involved in the interactions with the non-receptor partners should be accessible in the MT-bound form. Indeed, our finding that arrestin mobilizes protein kinase ERK1/2 and ubiquitin ligase Mdm2 to the cytoskeleton (Figs. 5, 6) clearly shows that this is the case. However, the functional capabilities of MT- and receptor-bound arrestin are different: arrestin mobilizes ERK, Mdm2, but not c-Raf-1, MEK1, JNK3, or PP2A to microtubules, whereas all of these partners are recruited to the arrestin-receptor complex.
Receptor binding induces a global conformational change in arrestin 2 that is widely believed to facilitate the binding of clathrin, AP2, Src, MAP kinases, and other proteins to the complex 4; 7. The deletions in the inter-domain hinge impede arrestin transition into this active conformation, thereby dramatically reducing arrestin binding to the receptor (Fig. 2e) 13. In contrast, hinge deletions actually enhance arrestin binding to MTs, suggesting that the conformations of MT-bound and receptor-bound arrestins are different (Fig. 2f). Not surprisingly, the functional consequences of arrestin-dependent ERK mobilization to the MTs is opposite to its recruitment to GPCRs. Receptor-bound arrestin facilitates ERK activation 39, whereas cytoskeletal localization of ERK by arrestin keeps it inactive, likely because arrestins do not mobilize the upstream kinases c-Raf-1 and MEK1 to microtubules (Fig. 5). Conversely, the result of arrestin-dependent recruitment of Mdm2 to GPCRs and MTs is similar: Mdm2 ubiquitinates the receptor in one case 44; 53 and cytoskeletal proteins in the other (Fig. 6). Because arrestins bind another ubiquitin ligase, TRAF6 54, we cannot exclude the possibility that the mobilization of other ubiquitin ligases is partially responsible for the arrestin-dependent increase in the ubiquitination of cytoskeletal proteins. However, close correlation between arrestin-dependent Mdm2 mobilization and ubiquitination level in the MT pellet (Fig. 6) suggests that Mdm2 plays an important role. Thus, by recruiting Mdm2 to microtubules arrestin redirects its activity to a different set of substrates. Interestingly, rod arrestin successfully recruits Mdm2 to MTs and facilitates the ubiquitination of microtubule-associated proteins to the greatest extent (Fig. 6), suggesting that its association with the cytoskeleton may have earlier unappreciated functions in photoreceptors. Rod arrestin also enhances the ubiquitination of soluble proteins (Fig. 6), apparently by stabilizing Mdm2 in the cell.
The binding of arrestin proteins to microtubules with affinities that ensure their partial co-localization with the cytoskeleton in intact cells (Fig. 1) is a novel link in the network of cellular signaling pathways. Accumulating evidence shows that a number of signaling proteins that were believed to selectively interact with receptor-bound arrestin actually bind free arrestin in the cytoplasm 42; 43; 55 and the microtubule-bound form (Figs. 5, 6). Notably, two out of the six arrestin partners tested in our study are recruited to microtubules in an arrestin-dependent fashion. It is tempting to speculate that a number of other known binding partners may also be mobilized to the cytoskeleton via an arrestin-dependent mechanism with significant functional consequences. The recruitment of signaling molecules may affect their activation state (Figs 5, 6) and/or microtubule dynamics which is known to be regulated by post-translational modifications of tubulin and associated proteins 34. The full range of biological implications of the functional link between arrestins and the cytoskeleton remains to be elucidated.
Methods
In vitro transcription, translation, and evaluation of mutant stability
pGEM2-based plasmids containing the arrestin coding sequence with an “idealized” 5′-UTR 56 under the control of the SP6 promoter were linearized, transcribed and translated in vitro in the presence of [3H]leucine and [14C]leucine (Perkin Elmer Life Sciences) as previously described 57. The translation of every mutant used in this study produced a single labeled protein band with the expected mobility on SDS-PAGE. The relative stability of all mutants (assessed as described 18) exceeded 80% of WT. The construction of rod/arrestin2 chimeras is described in detail in 25.
Arrestin expression and purification
Arrestin was expressed in Escherichia coli, and purified as described 57. The cysteine-less arrestin2 (CL) (C59V, C125S, C140L, C150V, C242V, C251V, C269S) was used as a base mutant for the introduction of unique cysteines. The best substituting residue in each position was chosen based on our high-resolution crystal structure of arrestin2 24 to preserve existing intramolecular interactions as much as possible.
Direct binding of arrestin to microtubules
Microtubules with purified arrestins
Purified wild type (WT) or mutant arrestins (2μg) were incubated for 30 min at 30°C in 50mM Tris-HCl, pH 7.4, 100mM NaCl, 2mM EDTA, and 1mM EGTA with 20μg of purified tubulin (Cytoskeleton, Inc.) pre-polymerized with taxol according to the manufacturer’s instructions. Microtubules along with bound arrestin were pelleted by centrifugation for 10 min at 30°C at 100,000 rpm in a TLA 120.1 rotor in a Beckman TL100 ultracentrifuge. Parallel samples with the same amount of each arrestin without microtubules served as controls. The pellet was dissolved in Laemmli’s sample buffer (Sigma) and the amount of arrestin was quantified by Western blot. Microtubules with in vitro translated radiolabeled arrestins. 200 fmol of the indicated in vitro translated arrestins were incubated in 50mM Tris-HCl, pH 7.4, 0.5mM MgCl2, 1.5mM DTT, 1mM EGTA, and 50mM potassium acetate for 20 min at 25°C with 20 μg of pre-polymerized tubulin. Microtubules along with bound arrestin were pelleted. MT-arrestin pellets were not washed due to the low affinity (i.e. high off-rate) of the interaction. The pellet was dissolved in 0.1ml of 1% SDS, 50mM NaOH, and bound arrestin was quantified by liquid scintillation counting. Non-specific “binding” (arrestin pelleted without microtubules) was subtracted.
Arrestin binding to receptors
Arrestin binding to receptors was performed as described 21; 57; 58. Briefly, 50 fmol radiolabeled arrestin was incubated in 50mM Tris-HCl, pH 7.4, 100mM NaCl, 2mM EDTA, and 1mM EGTA with 0.3μg rhodopsin 21 in a final volume of 50μl for 5min at 37°C in room light. Alternatively, radiolabeled arrestin (50 fmol) was incubated with 50 fmol phosphorylated m2 mAChR 21; 58 in the presence of 100μM carbachol for 35min at 30°C. After incubation with either receptor, the samples were cooled and loaded onto 2ml Sepharose 2B columns. Bound arrestin eluted with receptor-containing membranes in the void volume.
Arrestin binding to microtubules in cells
HEK-293A cells were transfected with expression plasmids encoding untagged arrestins and/or HA-Mdm2 and/or HA-ubiquitin using Lipofectamine 2000. 48 hours post-transfection cells were incubated with 5 μM taxol for 1.5 hours at 37°C and washed with 80mM PIPES pH 6.8, 70mM NaCl, 1mM MgCl2 (PB). Cells were cross-linked by incubation in PB, 5 μM taxol, and 2 mM DSP (Pierce) for 30 min and quenched by 50mM Tris pH 7.4 for 15 min at RT before lysis in PB supplemented with 0.2% NP-40 and 1mM PMSF. Lysates were centrifuged at 400xg for 5min to remove cell debris. The supernatant was loaded onto a 60% glycerol cushion made in PB and microtubules were pelleted by centrifugation for 20 min at 25 °C at 90,000 rpm in a TLA 120.1 rotor in a Beckman TL100 ultracentrifuge. The pellet was dissolved in Laemmli’s sample buffer and aliquots of the lysate, supernatant, and pellet were analyzed by Western blot using arrestin (F4C1), β-tubulin, Mdm2, HA (Sigma), ERK1/2, PP-ERK, MEK1, c-Raf-1, JNK3, PP-JNK3 (Cell Signaling) or PP2A (BD Biosciences) antibodies. The cross-linking step was omitted in experiments designed to measure protein ubiquitination.
Immunocytochemistry
HEK 293A cells expressing arrestin3 Flag-tagged at the C-terminus 43 were incubated with taxol and crosslinked as described above. Cells were fixed in 100% methanol for 5 min at RT, rehydrated with PBS, and permeabilized with 0.2% Triton in PB for 5 min at RT. Cells were blocked with 3% BSA in PBS for 1 h at RT before application of primary and secondary antibodies. Alternatively, COS7 cells expressing untagged arrestin2 and truncated (1–382) arrestin2 were treated with taxol and fixed by incubation for 1 min at RT with 0.25% glutaraldehyde and 0.5% Triton, rinsed with PB, and further incubated with 1% glutaraldehyde for 15 min followed by 1 min incubation in 0.5% Triton at RT. To reduce autofluorescence, cells were incubated with 3 changes of 0.5mg/ml NaBH4 for 5–10min each 14. Untagged and flag-tagged arrestins and microtubules were visualized with F4C1 monoclonal anti-arrestin antibody 59, M2 anti-Flag antibody (Sigma), and ATN02 tubulin polyclonal antibody (Cytoskeleton), respectively, followed by the appropriate secondary antibodies (Molecular Probes, Eugene, OR). Cells were viewed and photographed on a Nikon EC2000 fluorescent microscope equipped with a digital camera.
Electron paramagnetic resonance spectroscopy
Arrestin cysteine mutants were dialyzed into 50 mM MOPS, 100 mM NaCl, pH 7.0 and labeled with a ten-fold molar excess of 2,2,5,5-tetramethylpyrroline-3-yl-methanethiosulfonate spin label (MTSL; Toronto Research Chemicals) overnight at 4°C as described 9. Polymerized MTs were blocked with methylmethanethiosulfonate and washed as described 9. Electron paramagnetic resonance (EPR) spectroscopy samples contained 10 μg spin labeled arrestin and 150 μg MTs in a final volume of 10μL. For the titration experiments final concentrations of MTs and unpolymerized tubulin are expressed as molar concentration of the tubulin dimer with Mr = 110 kDa.
X-band EPR spectra were recorded for samples in glass capillaries at room temperature over a 100 G range with an incident microwave power of 10mW on a Bruker ELEXSYS E500 fitted with a super high Q cavity. Spectra were typically the average of 25–36 scans, baseline corrected, and normalized to the same area for each graphic overlay in Figure 3. Titration experiments with tubulin dimer were carried out at 4°C using a variable Digital Temperature Controller (Bruker) to prevent spontaneous polymerization.
Supplementary Material
Acknowledgments
The authors are grateful to Drs. Brian E. Wadzinski and Hua Lu for expression constructs for HA-ubiquitin and HA-Mdm2, respectively. This work was supported by NIH grants EY11500 (VVG), AI58024 and GM70642 (CSK), and GM 060019 (VZS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Carman CV, Benovic JL. G-protein-coupled receptors: turn-ons and turn-offs. Curr Opin Neurobiol. 1998;8:335–344. doi: 10.1016/s0959-4388(98)80058-5. [DOI] [PubMed] [Google Scholar]
- 2.Gurevich VV, Gurevich EV. The molecular acrobatics of arrestin activation. Trends Pharmacol Sci. 2004;25:105–112. doi: 10.1016/j.tips.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 3.Gurevich VV, Gurevich EV. The structural basis of arrestin-mediated regulation of G protein-coupled receptors. Pharmacol Ther. 2006;110:465–502. doi: 10.1016/j.pharmthera.2005.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by beta-arrestins. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- 5.Xiao K, Shenoy SK, Nobles K, Lefkowitz RJ. Activation-dependent conformational changes in beta-arrestin 2. J Biol Chem. 2004;279:55744–55753. doi: 10.1074/jbc.M409785200. [DOI] [PubMed] [Google Scholar]
- 6.Marchese A, Chen C, Kim YM, Benovic JL. The ins and outs of G protein-coupled receptor trafficking. Trends Biochem Sci. 2003;28:369–76. doi: 10.1016/S0968-0004(03)00134-8. [DOI] [PubMed] [Google Scholar]
- 7.Gurevich VV, Gurevich EV. The new face of active receptor bound arrestin attracts new partners. Structure (Camb) 2003;11:1037–1042. doi: 10.1016/s0969-2126(03)00184-9. [DOI] [PubMed] [Google Scholar]
- 8.Nair KS, Hanson SM, Kennedy MJ, Hurley JB, Gurevich VV, Slepak VZ. Direct binding of visual arrestin to microtubules determines the differential subcellular localization of its splice variants in rod photoreceptors. J Biol Chem. 2004;279:41240–41248. doi: 10.1074/jbc.M406768200. [DOI] [PubMed] [Google Scholar]
- 9.Hanson SM, Francis DJ, Vishnivetskiy SA, Klug CS, Gurevich VV. Visual arrestin binding to microtubules involves a distinct conformational change. J Biol Chem. 2006;281:9765–9772. doi: 10.1074/jbc.M510738200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith WC, Milam AH, Dugger D, Arendt A, Hargrave PA, Palczewski K. A splice variant of arrestin. Molecular cloning and localization in bovine retina. J Biol Chem. 1994;269:15407–15410. [PubMed] [Google Scholar]
- 11.Nair KS, Hanson SM, Mendez A, Gurevich EV, Kennedy MJ, Shestopalov VI, Vishnivetskiy SA, Chen J, Hurley JB, Gurevich VV, Slepak VZ. Light-dependent redistribution of arrestin in vertebrate rods is an energy-independent process governed by protein-protein interactions. Neuron. 2005;46:555–567. doi: 10.1016/j.neuron.2005.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sutton RB, Vishnivetskiy SA, Robert J, Hanson SM, Raman D, Knox BE, Kono M, Navarro J, Gurevich VV. Crystal structure of cone arrestin at 2.3 Å: evolution of receptor specificity. J Mol Biol. 2005;354:1069–1080. doi: 10.1016/j.jmb.2005.10.023. [DOI] [PubMed] [Google Scholar]
- 13.Vishnivetskiy SA, Hirsch JA, Velez MG, Gurevich YV, Gurevich VV. Transition of arrestin in the active receptor-binding state requires an extended interdomain hinge. J Biol Chem. 2002;277:43961–43968. doi: 10.1074/jbc.M206951200. [DOI] [PubMed] [Google Scholar]
- 14.Wang K, Feramisco JR, Ash JF. Fluorescent localization of contractile proteins in tissue culture cells. Methods Enzymol. 1982;85:514–562. doi: 10.1016/0076-6879(82)85050-7. [DOI] [PubMed] [Google Scholar]
- 15.Carter JM, Gurevich VV, Prossnitz ER, Engen JR. Conformational differences between arrestin2 and pre-activated mutants as revealed by hydrogen exchange mass spectrometry. J Mol Biol. 2005;351:865–878. doi: 10.1016/j.jmb.2005.06.048. [DOI] [PubMed] [Google Scholar]
- 16.Vishnivetskiy SA, Schubert C, Climaco GC, Gurevich YV, Velez MG, Gurevich VV. An additional phosphate-binding element in arrestin molecule: implications for the mechanism of arrestin activation. J Biol Chem. 2000;275:41049–41057. doi: 10.1074/jbc.M007159200. [DOI] [PubMed] [Google Scholar]
- 17.Celver J, Vishnivetskiy SA, Chavkin C, Gurevich VV. Conservation of the phosphate-sensitive elements in the arrestin family of proteins. J Biol Chem. 2002;277:9043–9048. doi: 10.1074/jbc.M107400200. [DOI] [PubMed] [Google Scholar]
- 18.Gurevich VV. The selectivity of visual arrestin for light-activated phosphorhodopsin is controlled by multiple nonredundant mechanisms. J Biol Chem. 1998;273:15501–15506. doi: 10.1074/jbc.273.25.15501. [DOI] [PubMed] [Google Scholar]
- 19.Gurevich VV, Benovic JL. Visual arrestin interaction with rhodopsin: Sequential multisite binding ensures strict selectivity towards light-activated phosphorylated rhodopsin. J Biol Chem. 1993;268:11628–11638. [PubMed] [Google Scholar]
- 20.Gurevich VV, Benovic JL. Cell-free expression of visual arrestin. Truncation mutagenesis identifies multiple domains involved in rhodopsin interaction. J Biol Chem. 1992;267:21919–21923. [PubMed] [Google Scholar]
- 21.Gurevich VV, Dion SB, Onorato JJ, Ptasienski J, Kim CM, Sterne-Marr R, Hosey MM, Benovic JL. Arrestin interaction with G protein-coupled receptors. Direct binding studies of wild type and mutant arrestins with rhodopsin, β2-adrenergic, and m2 muscarinic cholinergic receptors. J Biol Chem. 1995;270:720–731. doi: 10.1074/jbc.270.2.720. [DOI] [PubMed] [Google Scholar]
- 22.Hanson SM, Francis DJ, Vishnivetskiy SA, Kolobova EA, Hubbell WL, Klug CS, Gurevich VV. Differential interaction of spin labeled arrestin with inactive and active phosphorhodopsin. Proc Natl Acad Sci USA. 2006;103:4900–4905. doi: 10.1073/pnas.0600733103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hanson SM, Gurevich VV. The differential engagement of arrestin surface charges by the various functional forms of the receptor. J Biol Chem. 2006;281:3458–3462. doi: 10.1074/jbc.M512148200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han M, Gurevich VV, Vishnivetskiy SA, Sigler PB, Schubert C. Crystal structure of beta-arrestin at 1.9 Å: possible mechanism of receptor binding and membrane translocation. Structure. 2001;9:869–880. doi: 10.1016/s0969-2126(01)00644-x. [DOI] [PubMed] [Google Scholar]
- 25.Vishnivetskiy SA, Hosey MM, Benovic JL, Gurevich VV. Mapping the arrestin-receptor interface: structural elements responsible for receptor specificity of arrestin proteins. J Biol Chem. 2004;279:1262–1268. doi: 10.1074/jbc.M308834200. [DOI] [PubMed] [Google Scholar]
- 26.Hubbell WL, Gross A, Langen R, Lietzow MA. Recent advances in site-directed spin labeling of proteins. Curr Opin Struct Biol. 1998;8:649–656. doi: 10.1016/s0959-440x(98)80158-9. [DOI] [PubMed] [Google Scholar]
- 27.Hubbell WL, Cafiso DS, Altenbach C. Identifying conformational changes with site-directed spin labeling. Nat Struct Biol. 2000;7:735–739. doi: 10.1038/78956. [DOI] [PubMed] [Google Scholar]
- 28.Crane JM, Mao C, Lilly AA, Smith VF, Suo Y, Hubbell WL, Randall LL. Mapping of the docking of SecA onto the chaperone SecB by site-directed spin labeling: insight into the mechanism of ligand transfer during protein export. J Mol Biol. 2005;353:295–307. doi: 10.1016/j.jmb.2005.08.022. [DOI] [PubMed] [Google Scholar]
- 29.Klug CS, Feix JF. Biomedical EPR - Part B: Methodology and Instrumentation. In: Eaton SS, Eaton GR, Berliner LJ, editors. SDSL: A Survey of Biological Applications in Biological Magnetic Resonance. Vol. 24. Kluwer Academic/Plenum Publishers; New York, NY: 2005. pp. 269–308. [Google Scholar]
- 30.Lietzow MA, Hubbell WL. Motion of spin label side chains in cellular retinol-binding protein: correlation with structure and nearest-neighbor interactions in an antiparallel beta-sheet. Biochemistry. 2004;43:3137–3151. doi: 10.1021/bi0360962. [DOI] [PubMed] [Google Scholar]
- 31.Columbus L, Hubbell WL. A new spin on protein dynamics. Trends Biochem Sci. 2002;27:288–295. doi: 10.1016/s0968-0004(02)02095-9. [DOI] [PubMed] [Google Scholar]
- 32.Mchaourab HS, Kalai T, Hideg K, Hubbell WL. Motion of spin-labeled side chains in T4 lysozyme: Effect of side chain structure. Biochemistry. 1999;38:2947–2955. doi: 10.1021/bi9826310. [DOI] [PubMed] [Google Scholar]
- 33.Mchaourab HS, Lietzow M, Hideg K, Hubbell WL. Motion of spin labeled side chains in T4 lysozyme. Correlation with protein structure and dynamics. Biochemistry. 1996;35:7692–7704. doi: 10.1021/bi960482k. [DOI] [PubMed] [Google Scholar]
- 34.Gundersen GG, Cook TA. Microtubules and signal transduction. Curr Opin Cell Biol. 1999;11:81–94. doi: 10.1016/s0955-0674(99)80010-6. [DOI] [PubMed] [Google Scholar]
- 35.Amos LA. Focusing-in on microtubules. Curr Opin Struct Biol. 2000;10:236–241. doi: 10.1016/s0959-440x(00)00070-1. [DOI] [PubMed] [Google Scholar]
- 36.Goodman OB, Jr, Krupnick JG, Santini F, Gurevich VV, Penn RB, Gagnon AW, Keen JH, Benovic JL. Beta-arrestin acts as a clathrin adaptor in endocytosis of the beta2-adrenergic receptor. Nature. 1996;383:447–50. doi: 10.1038/383447a0. [DOI] [PubMed] [Google Scholar]
- 37.Laporte SA, Oakley RH, Zhang J, Holt JA, Ferguson SG, Caron MG, Barak LS. The β2-adrenergic receptor/β-arrestin complex recruits the clathrin adaptor AP-2 during endocytosis. Proc Nat Acad Sci USA. 1999;96:3712–3717. doi: 10.1073/pnas.96.7.3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McDonald PH, Chow CW, Miller WE, Laporte SA, Field ME, Lin FT, Davis RJ, Lefkowitz RJ. Beta-arrestin 2: a receptor-regulated MAPK scaffold for the activation of JNK3. Science. 2000;290:1574–1577. doi: 10.1126/science.290.5496.1574. [DOI] [PubMed] [Google Scholar]
- 39.Luttrell LM, Roudabush FL, Choy EW, Miller WE, Field ME, Pierce KL, Lefkowitz RJ. Activation and targeting of extracellular signal-regulated kinases by beta-arrestin scaffolds. Proc Natl Acad Sci U S A. 2001;98:2449–2459. doi: 10.1073/pnas.041604898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luttrell LM. Location, location, location”: activation and targeting of MAP kinases by G protein-coupled receptors. J Mol Endocrinol. 2003;30:117–126. doi: 10.1677/jme.0.0300117. [DOI] [PubMed] [Google Scholar]
- 41.Shenoy SK, Lefkowitz RJ. Trafficking patterns of beta-arrestin and G protein-coupled receptors determined by the kinetics of beta-arrestin deubiquitination. J Biol Chem. 2003;278:14498–14506. doi: 10.1074/jbc.M209626200. [DOI] [PubMed] [Google Scholar]
- 42.Wang P, Gao H, Ni Y, Wang B, Wu Y, Ji L, Qin L, Ma L, Pei G. Beta-arrestin 2 functions as a G-protein-coupled receptor-activated regulator of oncoprotein Mdm2. J Biol Chem. 2003;278:6363–6370. doi: 10.1074/jbc.M210350200. [DOI] [PubMed] [Google Scholar]
- 43.Song X, Raman D, Gurevich EV, Vishnivetskiy SA, Gurevich VV. Visual and both non-visual arrestins in their “inactive” conformation bind JNK3 and Mdm2 and relocalize them from the nucleus to the cytoplasm. J Biol Chem. 2006;281:21491–21499. doi: 10.1074/jbc.M603659200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shenoy SK, McDonald PH, Kohout TA, Lefkowitz RJ. Regulation of receptor fate by ubiquitination of activated beta 2-adrenergic receptor and beta-arrestin. Science. 2001;294:1307–1313. doi: 10.1126/science.1063866. [DOI] [PubMed] [Google Scholar]
- 45.Hupfeld CJ, Resnik JL, Ugi S, Olefsky JM. Insulin-induced beta-arrestin1 Ser-412 phosphorylation is a mechanism for desensitization of ERK activation by Galphai-coupled receptors. J Biol Chem. 2005;280:1016–1023. doi: 10.1074/jbc.M403674200. [DOI] [PubMed] [Google Scholar]
- 46.Hiller G, Weber K. Radioimmunoassay for tubulin: a quantitative comparison of the tubulin content of different established tissue culture cells and tissues. Cell. 1978;14:795–804. doi: 10.1016/0092-8674(78)90335-5. [DOI] [PubMed] [Google Scholar]
- 47.Ackmann M, Wiech H, Mandelkow E. Nonsaturable binding indicates clustering of tau on the microtubule surface in a paired helical filament-like conformation. J Biol Chem. 2000;275:30335–30343. doi: 10.1074/jbc.M002590200. [DOI] [PubMed] [Google Scholar]
- 48.Wang N, Yan K, Rasenick MM. Tubulin binds specifically to the signal-transducing proteins, Gs alpha and Gi alpha 1. J Biol Chem. 1990;265:1239–1242. [PubMed] [Google Scholar]
- 49.Pitcher JA, Hall RA, Daaka Y, Zhang J, Ferguson SS, Hester S, Miller S, Caron MG, Lefkowitz RJ, Barak LS. The G protein-coupled receptor kinase 2 is a microtubule-associated protein kinase that phosphorylates tubulin. J Biol Chem. 1998;273:12316–12324. doi: 10.1074/jbc.273.20.12316. [DOI] [PubMed] [Google Scholar]
- 50.Carman CV, Som T, Kim CM, Benovic JL. Binding and phosphorylation of tubulin by G protein-coupled receptor kinases. J Biol Chem. 1998;273:20308–20316. doi: 10.1074/jbc.273.32.20308. [DOI] [PubMed] [Google Scholar]
- 51.Santini F, Penn RB, Gagnon AW, Benovic JL, Keen JH. Selective recruitment of arrestin-3 to clathrin coated pits upon stimulation of G protein-coupled receptors. J Cell Sci. 2000;113:2463–2470. doi: 10.1242/jcs.113.13.2463. [DOI] [PubMed] [Google Scholar]
- 52.Pulvermuller A, Schroder K, Fischer T, Hofmann KP. Interactions of metarhodopsin II. Arrestin peptides compete with arrestin and transducin. J Biol Chem. 2000;275:37679–37685. doi: 10.1074/jbc.M006776200. [DOI] [PubMed] [Google Scholar]
- 53.Girnita L, Shenoy SK, Sehat B, Vasilcanu R, Girnita A, Lefkowitz RJ, Larsson O. {beta}-Arrestin is crucial for ubiquitination and down-regulation of the insulin-like growth factor-1 receptor by acting as adaptor for the MDM2 E3 ligase. J Biol Chem. 2005;280:24412–24419. doi: 10.1074/jbc.M501129200. [DOI] [PubMed] [Google Scholar]
- 54.Wang YYT, Teng L, Wu Y, Zhao X, Pei G. Association of beta-arrestin and TRAF6 negatively regulates Toll-like receptor-interleukin 1 receptor signaling. Nat Immunol. 2006;7:139–147. doi: 10.1038/ni1294. [DOI] [PubMed] [Google Scholar]
- 55.Scott MG, Le Rouzic E, Perianin A, Pierotti V, Enslen H, Benichou S, Marullo S, Benmerah A. Differential nucleocytoplasmic shuttling of beta-arrestins. Characterization of a leucine-rich nuclear export signal in beta-arrestin2. J Biol Chem. 2002;277:37693–37701. doi: 10.1074/jbc.M207552200. [DOI] [PubMed] [Google Scholar]
- 56.Gurevich VV. Use of bacteriophage RNA polymerase in RNA synthesis. Methods Enzymol. 1996;275:382–397. doi: 10.1016/s0076-6879(96)75023-1. [DOI] [PubMed] [Google Scholar]
- 57.Gurevich VV, Benovic JL. Arrestin: mutagenesis, expression, purification, and functional characterization. Methods Enzymol. 2000;315:422–37. doi: 10.1016/s0076-6879(00)15859-8. [DOI] [PubMed] [Google Scholar]
- 58.Gurevich VV, Richardson RM, Kim CM, Hosey MM, Benovic JL. Binding of wild type and chimeric arrestins to the m2 muscarinic cholinergic receptor. J Biol Chem. 1993;268:16879–82. [PubMed] [Google Scholar]
- 59.Donoso LA, Gregerson DS, Smith L, Robertson S, Knospe V, Vrabec T, Kalsow CM. S-antigen: preparation and characterization of site-specific monoclonal antibodies. Curr Eye Res. 1990;9:343–355. doi: 10.3109/02713689008999622. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.