

Abstract
Murine lupus in MRL mice has been strongly attributed to αβ T cell-dependent mechanisms. Non-αβ T cell-dependent mechanisms, such as γδ T cells, have been shown to drive antibody and autoantibody production, but they have not been considered capable of inducing end-organ disease. Here, we have expanded upon the findings of such previous work by examining the mechanism and extent of end-organ disease attainable via γδ T cells and/or non-αβ T cell-dependent mechanisms, assessing two prototypical lupus lesions, renal and skin disease, in TCR α−/− MRL mice that possessed either functional or defective Fas antigen (Fas + or lpr). Observed to 1 year of age, TCR α −/− MRL mice developed disease characterized by increased mortality, overt renal disease and skin lesions. While delayed in onset and/or reduced in severity compared with TCR α +/+ MRL/lpr animals, renal and skin lesions in αβ T cell-deficient animals were clearly increased in severity compared with age-matched control non-autoimmune mice. In contrast to TCR α +/+ MRL mice, whose disease reflected pan-isotype immune complex deposition with significant complement fixation, renal disease in TCR α −/− MRL animals reflected predominantly IgG1 immune complex deposition, with poor complement fixation. Thus, this study demonstrates conclusively that non-αβ T cell-dependent mechanisms can induce renal and skin injury in murine lupus, but at least in the kidney, only via humoral autoimmunity of a relatively non-pathological isotype which results in the delayed onset of end-organ damage.
Keywords: autoimmunity, T lymphocytes, mice, autoantibodies, nephritis, skin
INTRODUCTION
The MRL model of murine lupus is a particularly useful system to investigate systemic autoimmunity, since its disease closely resembles human systemic lupus erythematosus (SLE), including the development of autoantibodies and renal and skin disease [1–3]. The MRL/Mp-lpr/lpr (MRL/lpr) mouse, which develops a severely accelerated form of MRL lupus [1] due to a functional defect in the Fas apoptosis antigen [4], provides a convenient congenic strain to examine the role of Fas-mediated immune regulation in lupus. While B cells in MRL/lpr mice are intrinsically abnormal [5–8], many studies have used this model to establish the role of T cells in the pathogenesis of murine lupus, focusing upon the role of CD4+ αβ T cells as helpers for autoantibody production [9–14]. Some data suggest that γδ T cells may propagate systemic humoral autoimmunity [15–19]; however, none has found that γδ T cells serve as significant instigators of end-organ disease.
To evaluate the significance of γδ T cell- and/or other non-αβ T cell-dependent mechanisms in the induction of systemic disease, we assessed renal and skin end-organ disease in MRL mice made deficient in αβ T cells via genetic disruption of the T cell receptor (TCR) α locus [17, 20]. TCR α −/− MRL mice developed increased mortality, renal disease with compromised renal function, and skin disease in association with lupus autoantibodies, although their end-organ disease remained delayed and/or subdued in comparison with wild-type MRL/lpr animals. In addition, TCR α +/+ MRL mice developed pan-isotype immune complex deposition associated with complement fixation, while kidneys of TCR α −/− MRL animals had predominantly IgG1 isotype-restricted immune complex deposition associated with poor complement fixation. Thus, in comparison with previous studies which have shown that non-αβ T cells, particularly γδ T cells, can support autoantibody production [15–19], the current findings demonstrate in vivo that non-αβ T cell-dependent mechanisms are capable of inducing humoral autoimmunity, which, while less aggressive than αβ T cell-dependent mechanisms, nevertheless evolves into consequential autoimmune disease with end-organ dysfunction of the skin and kidneys.
MATERIALS AND METHODS
Mice
TCR α −/− (TCRα−) and TCR α +/+ (TCRα+) MRL mice bearing either functional (+/+) or defective (lpr/lpr) Fas antigen [17], as well as controls including age-matched B10.A mice (Jackson Laboratory, Bar Harbor, ME), were maintained under specific pathogen-free conditions at the Yale University School of Medicine. These animals, of the F4 backcross generation, reflect wild-type MRL lupus-prone animals, since MRL disease-inducing genes are dominant in genetic crosses [21] as confirmed by the penetrance of autoantibodies and end-organ disease in the TCR α +/+ mice of this cohort ([17] and this study). Indeed, in contrast to the present animals, which contain ≈ 87·5% MRL genes, previous studies have successfully utilized F2 animals, which contain only ≈ 50% MRL genes [19, 22]. Thus, it is notable that both F2 and F4 animals develop renal and skin lesions, as well as anti-snRNP antibodies, since MRL disease itself is stochastic, with incomplete penetrance of autoantibody production and end-organ disease [23–25]. Thus, while these mice may not technically reflect wild-type (‘100%’) MRL disease, their lupus syndrome closely resembles the severe MRL phenotype. Nevertheless, because of such issues, the various genetic groups in this study have been analysed as populations rather than individual mice: each genetic group as a whole being 87·5% MRL overall, and each population of animals being at similar risk for disease; in this study, we refer to these animals as TCRα+ MRL (+/+ or lpr) or TCRα− MRL (+/+ or lpr).
Antibody detection
Titres of IgM, IgA, and IgG were determined by ELISA on sera using an antibody isotyping kit (Pierce, Rockland, IL). IgE titres were determined by ELISA using separately purchased capture and detection antibodies (PharMingen, San Diego, CA). IgG anti-dsDNA and anti-snRNP, as well as κ anti-IgG, were determined as described elsewhere [17, 26–28].
Renal function tests
Sera were assayed for blood urea nitrogen (BUN) via clinical chemistry at Yale-New Haven Hospital. Proteinuria was assayed by collecting spot urine samples on 3 consecutive days. Total protein content was determined by quantitative Bradford assay [29]; creatinine content was determined by modified Jaffé reaction [30] (Sigma Chemical Co., St Louis, MO). Proteinuria indices were determined as (total urinary protein)/(total urinary creatinine), to normalize for glomerular filtration rate. Statistical significance was evaluated by unpaired, two-tailed Student's t-test.
Histopathology
Renal disease was assessed as described [31, 32]. Briefly, for light microscopy, tissue was fixed in 10% buffered formalin and stained with haematoxylin–eosin; glomeruli, tubular interstitium and vessels were examined. For immunofluorescence, specimens were frozen in OCT embedding medium, and sections were stained with fluorescein-conjugated goat anti-mouse IgG, IgG1, IgG2a, IgG2b or IgG3 (Southern Biotechnology Associates, Birmingham, AL), or fluorescein-conjugated goat anti-mouse C3 (Cappel, West Chester, PA). Immune deposits were sought in glomeruli, tubules, and renal cell nuclei. Individual specimens were read blinded by S.L.P, J.C. and/or M.P.M., and scored on a 0–4+ scale. Cutaneous lesions were processed similarly, and evaluated blinded by J.M.M.
Correlations of antibody titres with renal disease
Antibody indices for each mouse (for Fig. 4) were calculated on a 1–10 scale, using one point for each elevated immunoglobulin isotype in μg/ml (IgM, IgA, IgG and IgE; > 2 s.d. above average normal mice), and for each positive autoantibody titre (anti-dsDNA, anti-snRNP, or anti-immunoglobulin; > 3 s.d. above average optical density (OD) titres of normal mice). Renal disease indices were calculated as the sum of disease scores (0–4 + scale) as visualized by light microscopy [31, 32]. Pearson correlation coefficient was determined by Systat 5·2·1 software (Evanston, IL).
Fig. 4.
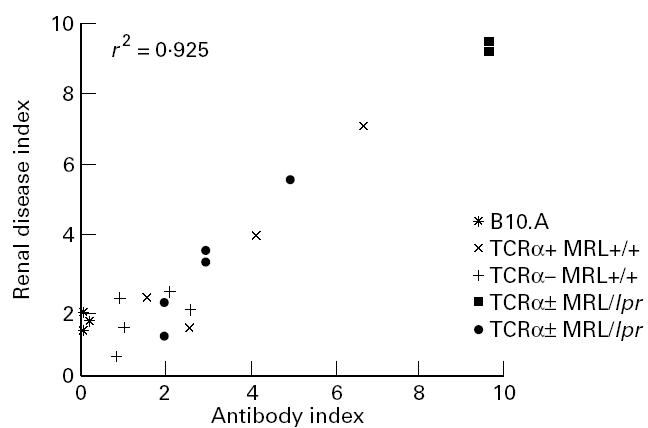
Antibody and autoantibody correlations with renal disease in TCRα− MRL mice, and in age-matched B10.A mice. Elevated serum immunoglobulins and/or autoantibodies show a correlation with renal disease severity (Pearson correlation coefficient, r, is shown). Renal disease indices were calcutated as the sum of light microscopic renal disease scores; antibody indices were calculated on a 1–10 scale, using one point for each elevated immunoglobulin level or positive autoantibody titre (see Materials anf Methods).
RESULTS
TCRα− MRL mice develop renal disease
End-organ disease was first assessed in 1-year-old mice by BUN and proteinuria (Fig. 1). In comparison with normal and TCRα+ MRL +/+ animals, both TCRα− MRL/lpr and TCRα− MRL +/+ mice contained elevated BUN, although levels in only the former group reached a statistically significant difference (P < 0·05). At the same time, neither TCRα− MRL group developed as high BUN as TCRα+ MRL/lpr mice (P < 0·05). In addition, all groups of lupus-prone mice, TCRα− MRL +/+ and TCRα− MRL/lpr, as well as TCRα+ MRL +/+ and TCRα+ MRL/lpr, developed elevated proteinuria indices in comparison with normal, age-matched B10.A mice (P < 0·05). In the TCRα+ MRL/lpr group, end-stage renal disease probably caused decreased protein excretion (data not shown), even though those animals remaining alive at this age probably represented a biased group with milder disease.
fig. 1.
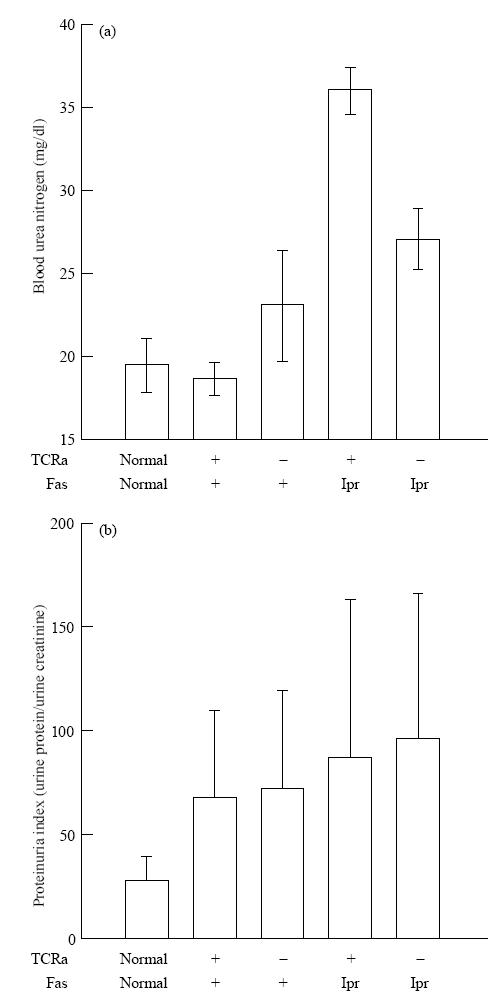
Renal function tests in 1-year-old TCRα+ MRL and TCRα− MRL mice. Mouse sera were measured for blood urea nitrogen levels. Urine samples were measured for total protein content and creatinine, and proteinuria index was calculated as protein/creatinine to normalize for glomerular filtration rate. Standard deviations are shown for five to seven mice in each group; normals are age-matched B10.A mice.
In accordance with the renal function studies, both MRL +/+ and MRL/lpr mice lacking αβ T cells developed glomerular, interstitial, and sometimes perivascular lesions (Table 1 and Fig. 2). While these were limited compared with their αβ T cell-intact MRL/lpr counterparts, they were still significant in comparison with age-matched normal mice. They also developed substantial renal immune deposits, sometimes comparable to the severe glomerular, tubular, and/or renal nuclear deposition characteristic of αβ T cell-intact disease (Table 2 and Fig. 3 and data not shown). Isotyping of the immune deposits in TCRα+ MRL/lpr animals consistently revealed pan-isotype accumulation by 12 weeks old, associated with significant complement (C3) deposition (Table 2 and Fig. 3). In contrast, deposits in TCRα− MRL/lpr animals consisted of predominantly IgG1 antibodies, which required ≈ 6 months or more to reach levels which were consistently comparable to TCRα+ MRL/lpr mice. TCRα− MRL/lpr animals furthermore had a relative paucity of IgG2a, IgG2b, IgG3 and C3 deposition, although these molecules were occasionally, but not predictably, detected. Nevertheless, histological abnormalities showed a correlation of light microscopic renal disease with serum antibody and autoantibody levels in both αβ T cell-intact and -deficient animals (Fig. 4), suggesting that immunoglobulin deposition was responsible for renal disease in all groups of mice.
Table 1.
Renal pathology in 1-year-old TCRα+ MRL and TCRα− MRL mice, and in normal B10.A controls
Fig. 2.

Renal disease in 1-year-old TCRα+ MRL and TCRα− MRL mice. Shown are light microscopic specimens from 48-week-old animals, including: (A) normal histology of a B10.A mouse; (B) focal glomerulonephritis and periglomerular infiltrate in a TCRα+ MRL/lpr mouse; (C) glomerular hypercellularity in a TCRα+ MRL +/+ mouse; (D) periglomerular infiltrate in a TCRα− MRL +/+ mouse; (E) focal glomerular hypercellularity in a TCRα− MRL/lpr mouse; and (F) perivasculitis in a TCRα− MRL/lpr mouse. Scale bar, 2 5 μm; all panels are of the same magnification.
Table 2.
Isotyping of immune deposits in TCRα− MRL/lpr mice

Fig. 3.
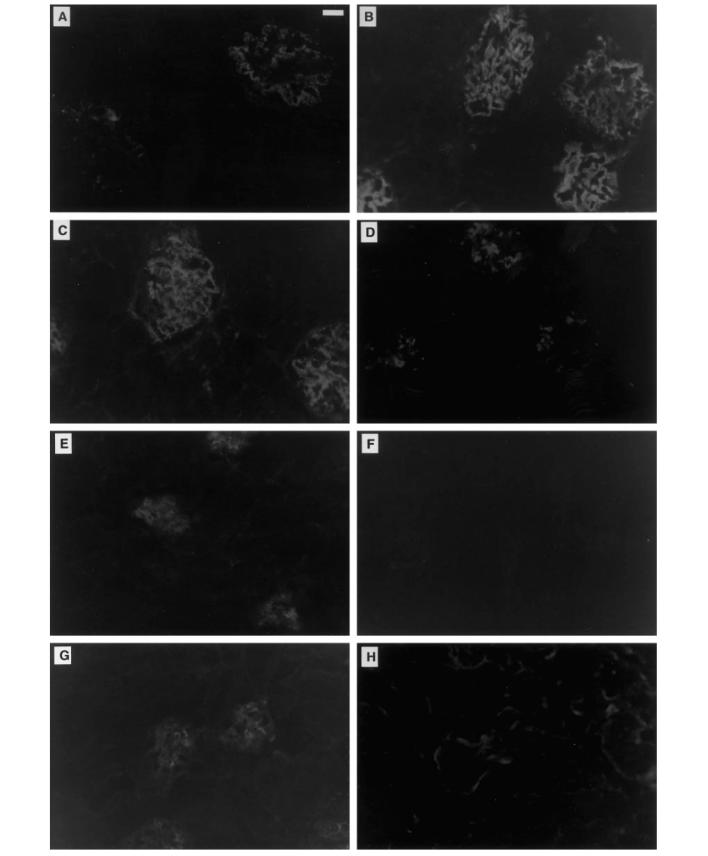
Isotyping of immune deposits in TCRα− MRL/lpr mice. Shown are representative kidney specimens from (A–D) TCRα+ MRL/lpr and (E–H) TCRα− MRL/lpr mice. All TCRα+ MRL/lpr animals consistently had (A) punctate mesangial IgG1 deposition, in contrast to the punctate and diffuse mesangial deposition of (B) IgG2a, (C) IgG2b and IgG3 (not shown) isotypes. Occasionally, tubular and/or antinuclear staining were evident as well in these animals (not shown). TCRα+ MRL/lpr mice also consistently developed substantial (D) punctate mesangial C3 deposits. TCRα− MRL/lpr mice, on the other hand, developed predominantly (E) mesangial IgG1 deposition, with a paucity of other IgG isotypes, resulting in negative (F) IgG2a as well as IgG2b or IgG3 (not shown) deposition. Occasionally, however, TCRα− MRL/lpr animals developed substantial immune deposits of IgG2a (not shown), (G) IgG2b or IgG3 (not shown) isotypes. (H) C3 deposition was generally absent in the mesangium of these animals, correlating with their relatively mild glomerular disease, and contrasting with the substantial C3 deposition and severe glomerular disease apparent in their TCRα+ MRL/lpr counterparts. Interestingly, the glomerular and tubular membranes of TCRα− MRL/lpr animals had C3 staining; the significance of this finding remains unclear. Shown here are samples from a 12-week-old TCRα+ MRL/lpr and a 42-week-old TCRα− MRL/lpr mouse, representative of four to six animals of each kind. Control 12-week-old TCRα+ MRL +/+ animals stained negative for all IgG isotypes and C3, similar to (F) (not Shown). Scale bar, 25 μm.
TCRα− MRL mice develop cutaneous disease
Skin disease developed in parallel with renal disease (Figs. 5 and 6). Whereas nearly all TCRα+ MRL/lpr mice characteristically developed severe hair loss, scab formation, and purpuric lesions of the dorsal neck and ears by 24 weeks old, TCRα− MRL mice of both Fas phenotypes appeared grossly normal. At this age, however, nearly all of TCRα− MRL/lpr mice developed subclinical lesions as determined by histology, which developed into grossly apparent cutaneous lesions and microscopic patterns resembling T cell-intact disease by 7–8 months old. Approximately 50% of TCRα− MRL +/+ mice also developed clinically apparent skin lesions by 8–9 months old, while none of the TCRα+ MRL +/+ animals developed skin lesions even beyond 1 year of age (Figs. 5 and 6 and data not shown). Thus, as determined by renal and skin lesions, TCRα− MRL mice develop end-organ disease, albeit with less intensity compared with age-matched TCRα+ MRL/lpr animals.
Fig. 5.
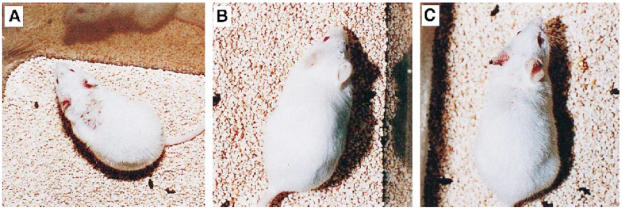
Cutaneous lesions in TCRα+ MRL and TCRα− MRL mice. (A) TCRα+ MRL/lpr mice generally developed severe hair loss, scab formation and purpuric lesions on the dorsal neck and ears by 24 weeks old. (B) In contrast, TCRα− MRL mice of either Fas genotype at the same age lacked gross skin disease, but soon developed visible lesions, as shown by (C) this 28-week-old TCRα− MRL/lpr mouse. TCRα− MRL/lpr mice generally developed gross lesions by 7–8 months of age, whereas TCRα− MRL +/+ animals generally developed lesions at 8–9 months of age (not shown). Within 2–3 months of onset, such TCRα− MRL +/+ lesions progressed to phenotypes indistinguishable from TCRα+ MRL/lpr disease, as in (A).
Fig. 6.
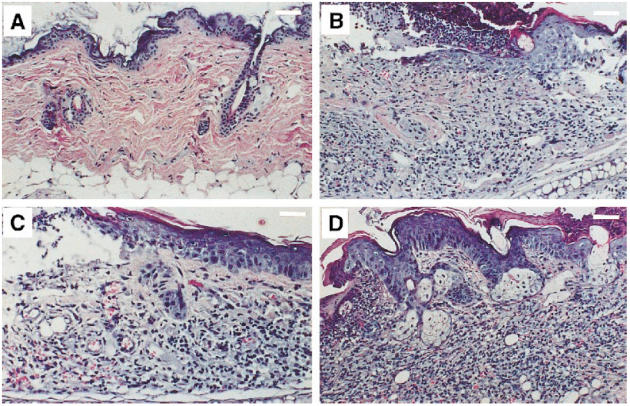
Dermatohistopathology in TCRα+ MRL and TCRα− MRL mice. (A) Some young TCRα− MRL/lpr animals had normal skin pathology, as seen in this neck specimen from a 24-week-old animal (scale bar, 200 μm). (B) In contrast, 24-week-old TCRα+ MRL/lpr animals consistently developed ulceration, hyperkeratosis, acanthosis, and mononuclear cell infiltration into the dermis and epidermis by 24 weeks old (here, a neck specimen; scale bar, 200 μm). (C) Many 24-week-old TCRα− MRL mice, however, developed subclinical ulceration, fibrosis, and mild dermal infiltration (here, a TCRα− MRL/lpr neck; 50–70% of TCRα− MRL/lpr versus 20–30% of TCRα− MRL +/+ animals developed such lesions; scale bar, 125 μm), which soon developed into (D) florid lesions, such as in this 28-week-old TCRα− MRL +/+ neck, which contained striking infiltrates, as well as hyperkeratosis, acanthosis, and dermal fibrosis (scale bar, 200 μm).
Mortality of TCRα− MRL mice
In conjunction with end-organ disease, TCRα− MRL mice of both Fas phenotypes demonstrated increased mortality in comparison with TCRα+ MRL +/+ mice (Fig. 7; P < 0·05 for MRL +/+, P < 0·01 for MRL/lpr), although they demonstrated decreased mortality in comparison with TCRα+ MRL/lpr animals (P < 0·05). The mortality rates correlated with renal and skin disease, since TCRα+ MRL/lpr mice, which had the highest mortality, developed the highest BUN and severity of skin disease, while TCRα− MRL/lpr mice, of second highest mortality, had the second most severe azotaemia and skin disease, and so forth (Figs. 1 and 7). These data establish a correlation between compromised renal function, skin disease and mortality in these animals, verifying the presence of significant, albeit limited, end-organ disease despite the absence of αβ T cells. Accordingly, more substantial renal and skin disease was incidentally noted in TCRα− MRL mice that died or were moribund (data not shown).
Fig. 7.
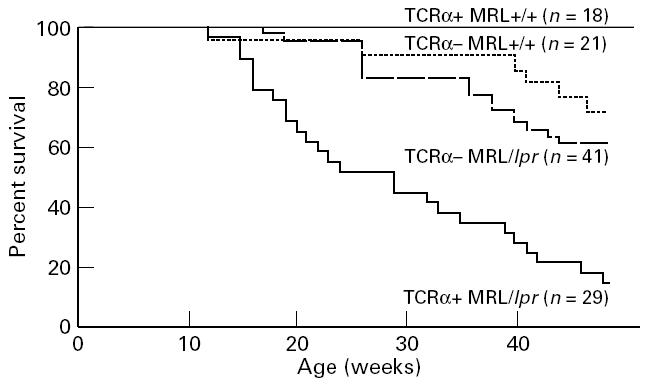
Life-table analysis of lupus-prone TCRα+ MRL and TCRα− MRL mice.
DISCUSSION
This study demonstrates that MRL mice without αβ T cells develop overt renal and skin disease, dispelling models that consider only these T cells capable of inducing end-organ disease in murine lupus, and suggesting that γδ T cell-dependent helper functions are capable of inducing lupus nephritis and cutaneous lesions. While a contribution to antibody production and disease by TCRα−β+ cells cannot be entirely ruled out [33, 34], previous studies have established that γδ T cells are the primary source of T-dependent help in αβ T cell-deficient mice [17–19]. In contrast to prior studies on non-αβ T cells in autoimmunity [15––19], this study provides a clear in vivo demonstration of the pathological consequences of such non-classical mechanisms.
The development of both renal and cutaneous lesions in TCRα− MRL mice contrasts with the conclusions of previous studies, which have strongly implicated a requirement for helperαβ T cells in the generation of pathogenic autoantibodies which may mediate end-organ disease [2, 9–14, 35, 36]. Indeed, TCRα− MRL mice contained delayed and/or subdued renal and cutaneous lesions, which may indicate an inability of non-αβ T cell-dependent mechanisms to promote pathogenic, affinity-matured autoantibodies [17]. Likewise, αβ T cells may be required for the development of complement-fixing pathogenic antibodies, particularly IgG of the IgG2a, IgG2b or IgG3 subclasses [37]. Thus, the T-dependent help in TCRα− MRL mice is notable for the presence of IgG antibodies, autoantibodies and end-organ immune deposits predominantly of the IgG1 subclass ([17] and Table 2 and Fig. 3), which fixes complement poorly ([37] and Table 2 and Fig. 3), suggesting that TCRα− MRL pathogenesis interestingly reflects a primarily complement-independent mechanism for glomerular disease. Accordingly, the apparently decreased renal interstitial infiltration in TCRα− MRL animals may reflect their relatively diminished glomerulonephritis, which may provide cytokines or other inflammatory mediators that recruit inflammatory cells to the interstitium. Regardless of the specific mechanism of renal injury, these results demonstrate that the TCRα− MRL phenotype reflects a delayed and subdued autoimmune disease syndrome in comparison with TCRα+ MRL/lpr animals, but nevertheless reflects a consequential pathological response. As an intriguing possibility, TCRα− MRL disease may in fact represent a protective form of T cell help, in which γδ T cells provide an IL-4-rich, Th2-like environment which protects against end-organ disease, skewing the immune response away from a disease-inducing, interferon-gamma (IFN-γ)-rich, Th1-like environment characteristic of murine lupus (which may induce complement-fixing IgG antibodies) [8]. Such a view correlates with the substantial development of autoantibodies and accumulation of renal immune deposits despite the delayed and limited progression of renal disease in TCRα– MRL mice, all in association with poor end-organ C3 accumulation, as well as with the results of a recent study describing the protective role of B cell-produced IL-4 in murine lupus [38].
This model also may account for the time course of cutaneous disease, which appears delayed but not necessarily subdued in TCRα− MRL animals. IgG1 autoantibodies have been implicated in the pathogenesis of cutaneous lesions, since they are found at disproportionately high frequencies in MRL/lpr skin [2]. At the same time, however, lesional skin does not contain IgG or complement deposits, probably because the histological architecture has been destroyed by the autoimmune processes ([2] and data not shown). It was therefore difficult to assess the degree of cutaneous immune complex deposition in TCRα− MRL mice, in order to draw conclusions regarding any relationship between autoantibody production, immune complex deposition, and/or cutaneous lesion development. The autoimmune skin disease in these mice therefore probably reflects the effects of autoantibodies of affinity and/or isotypes that are preserved despite the absence of αβ T cells; alternatively, these lesions may reflect primarily cellular autoimmune processes not previously described as important in these mice [36]. Regardless of the particular mechanisms of skin disease, these data emphasize the potential for non-αβ T cell-dependent mechanisms to enforce overt cutaneous lesions which may be virtually indistinguishable from wild-type disease when fully matured.
As an additional consideration, some components of the αβ T cell-independent autoimmunity seen here may result from the absence of regulatory αβ T cells [16, 17, 39]. Accordingly, TCRα− MRL +/+ mice demonstrated increased mortality in association with increased autoantibody production, renal dysfunction and skin disease in comparison with TCRα+ MRL mice, but TCRα− MRL/lpr mice demonstrated decreased mortality and autoantibody production in comparison with αβ T cell-intact counterparts. Such findings suggest a model for αβ T cells in MRL disease in which αβ T cells both help and regulate autoreactive B cells, the latter via a Fas-dependent mechanism [40–42]. As such, in MRL +/+ mice, αβ T cell deficiency may allow the proliferation of γδ T cell-driven B cell autoimmunity, which progresses unchecked in the absence of αβ T-dependent, Fas-dependent regulation. In contrast, MRL/lprαβ T cells, unable to regulate other cells via Fas, may only be capable of providing help to B cells; consequently, αβ T cell deficiency in the absence of Fas significantly reduces autoantibody production and end-organ disease, decreasing mortality. The revealed γδ T cell-dependent autoimmunity would then induce limited disease processes.
αβ T cell-independent autoimmunity therefore plays a significant role in murine lupus by maintaining the capability to drive systemic autoimmunity. These findings introduce a complexity in the αβ T cell-based models for lupus pathogenesis [11, 13, 14, 36], calling for a consideration of both αβ and γδ T cells in the T cell-directed therapy of human SLE [43].
Acknowledgments
This study was supported in part by grants from the NIH (AR40072 and AR44076 to J.C., and AI27855 to A.C.H.) and from the Arthritis and Lupus Foundations, their Connecticut chapters, and donations to Yale Rheumatology in the memories of Irene Feltman, Albert L. Harlow, and Chantal Marquis (to J.C.). S.L.P. was supported by the Medical Scientist Training Program, Yale University School of Medicine. This study is based partially upon a dissertation submitted to fulfil in part the requirements for the degree of Doctor of Philosophy in Yale University.
References
- 1.Andrews BS, Eisenberg RA, Theofilopoulos AN, et al. Spontaneous murine lupus-like syndromes. Clinical and immunopathological manifestations in several strains. J Exp Med. 1978;148:1198–215. doi: 10.1084/jem.148.5.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Furukawa F, Tanaka H, Sekita K, Nakamura T, Horiguchi Y, Hamashima Y. Dermatopathological studies on skin lesions of MRL mice. Arch Dermatol Res. 1984;276:186–94. doi: 10.1007/BF00414018. [DOI] [PubMed] [Google Scholar]
- 3.Hewicker M, Trautwein G. Glomerular lesions in MRL mice. A light and immunofluorescence microscopic study. Zentralblatt für Veterinarmedizin Reihe B. 1986;33:727–39. doi: 10.1111/j.1439-0450.1986.tb00093.x. [DOI] [PubMed] [Google Scholar]
- 4.Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature. 1992;356:314–7. doi: 10.1038/356314a0. [DOI] [PubMed] [Google Scholar]
- 5.Perkins DL, Glaser RM, Mahon CA, Michaelson J, Marshak-Rothstein A. Evidence for an intrinsic B cell defect in lpr/lpr mice apparent in neonatal chimeras. J Immunol. 1990;145:549–55. [PubMed] [Google Scholar]
- 6.Nemazee D, Guiet C, Buerki K, Marshak-Rothstein A. B lymphocytes from the autoimmune-prone mouse strain MLR/lpr manifest an intrinsic defect in tetraparental MRL/lpr in equilibrium DBA/2 chimeras. J Immunol. 1991;147:2536–9. [PubMed] [Google Scholar]
- 7.Sobel ES, Katagiri T, Katagiri K, Morris SC, Cohen PL, Eisenberg RA. An intrinsic B cell defect is required for the production of autoantibodies in the lpr model of murine systemic autoimmunity. J Exp Med. 1991;173:1441–9. doi: 10.1084/jem.173.6.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klinman DM, Steinberg AD. Inquiry into murine and human lupus. Immunol Rev. 1995;144:157–93. doi: 10.1111/j.1600-065x.1995.tb00069.x. [DOI] [PubMed] [Google Scholar]
- 9.Steinberg AD, Roths JB, Murphy ED, Steinberg RT, Raveche ES. Effects of thymectomy or androgen administration upon the autoimmune disease of MRL/Mp-lpr/lpr mice. J Immunol. 1980;125:871–3. [PubMed] [Google Scholar]
- 10.Seaman WE, Wofsy D, Greenspan JS, Ledbetter JA. Treatment of autoimmune MRL/lpr mice with monoclonal antibody to Thy-1.2: a single injection has sustained effects on lymphoproliferation and renal disease. J Immunol. 1983;130:1713–8. [PubMed] [Google Scholar]
- 11.Santoro TJ, Portanova JP, Kotzin BL. The contribution of L3T4+ T cells to lymphoproliferation and autoantibody production in MRL-lpr/lpr mice. J Exp Med. 1988;167:1713–8. doi: 10.1084/jem.167.5.1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Merino R, Iwamoto M, Fossati L, Izui S. Polyclonal B cell activation arises from different mechanisms in lupus-prone (NZB×NZW) F1 and MRL/MpJ-lpr/lpr mice. J Immunol. 1993;151:6509–16. [PubMed] [Google Scholar]
- 13.Jevnikar AM, Grusby MJ, Glimcher LH. Prevention of nephritis in major histocompatibility complex class II-deficient MRL-lpr mice. J Exp Med. 1994;179:1137–43. doi: 10.1084/jem.179.4.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koh D-R, Ho A, Rahemutulla A, Fung-Leung W-P, Griesser H, Mak T-W. Murine lupus in MRL/lpr mice lacking CD4 or CD8 T cells. Eur J Immunol. 1995;25:2558–62. doi: 10.1002/eji.1830250923. [DOI] [PubMed] [Google Scholar]
- 15.Rajagopalan S, Zordan T, Tsokos GC, Datta SK. Pathogenic anti-DNA autoantibody-inducing T helper cell lines from patients with active lupus nephritis: isolation of CD4−8− T helper cell lines that express the γδ T-cell antigen receptor. Proc Natl Acad Sci USA. 1990;87:7020–4. doi: 10.1073/pnas.87.18.7020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wen L, Roberts SJ, Viney JL, et al. Immunoglobulin synthesis and generalized autoimmunity in mice congenitally deficient in αβ(+) T cells. Nature. 1994;369:654–8. doi: 10.1038/369654a0. [DOI] [PubMed] [Google Scholar]
- 17.Peng SL, Madaio MP, Hughes DPM, Crispe IN, Owen MJ, Wen L, Hayday AC, Craft J. Murine lupus in the absence of αβ T cells. J Immunol. 1996;156:4041–9. [PubMed] [Google Scholar]
- 18.Wen L, Pao W, Wong FS, et al. Germinal center formation, immunoglobulin class switching, and autoantibody production driven by ‘non α/β’ T cells. J Exp Med. 1996;183:2271–82. doi: 10.1084/jem.183.5.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peng SL, Madaio MP, Hayday AC, Craft J. Propagation and regulation of systemic autoimmunity by γδ T cells. J Immunol. 1996;157:5689–98. [PubMed] [Google Scholar]
- 20.Philpott KL, Viney JL, Kay G, Rastan S, Gardiner EM, Chae S, Hayday AC, Owen MJ. Lymphoid development in mice congenitally lacking T cell receptor αβ-expressing cells. Science. 1992;256:1448–52. doi: 10.1126/science.1604321. [DOI] [PubMed] [Google Scholar]
- 21.Eisenberg RA, Craven SY, Fisher CL, Morris SC, Rapoport R, Pisetsky DS, Cohen PL. The genetics of autoantibody production in MRL/lpr lupus mice. Clin Exp Rheumatol. 1989;7:S35–40. [PubMed] [Google Scholar]
- 22.Shlomchik MJ, Madaio MP, Ni D, Trounstein M, Huszar D. The role of B cells in lpr/lpr-induced autoimmunity. J Exp Med. 1994;180:1295–306. doi: 10.1084/jem.180.4.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eisenberg RA, Craven SY, Warren RW, Cohen PL. Stochastic control of anti-Sm autoantibodies in MRL/Mp-lpr/lpr mice. J Clin Invest. 1987;80:691–7. doi: 10.1172/JCI113123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burlingame RW, Rubin RL, Balderas RS, Theofilopoulos AN. Genesis and evolution of antichromatin autoantibodies in murine lupus implicates T-dependent immunization with self antigen. J Clin Invest. 1993;91:1687–96. doi: 10.1172/JCI116378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takahashi S, Fossati L, Iwamoto M, Merino R, Motta R, Kobayakawa T, Izui S. Imbalance towards Th1 predominance is associated with acceleration of lupus-like autoimmune syndrome in MRL mice. J Clin Invest. 1996;97:1597–604. doi: 10.1172/JCI118584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rubin RL. Enzyme-linked immunosorbent assay for anti-DNA and antihistone antibodies including anti-(H2A-H2B) In: Rose NR, de Macario EC, Fahey JL, Friedman H, Penn GM, editors. Manual of clinical laboratory immunology. Washington, DC: American Society for Microbiology; 1992. pp. 735–40. [Google Scholar]
- 27.Shlomchik MJ, Zharhary D, Saunders T, Camper SA, Weigert MG. A rheumatoid factor transgenic mouse model of autoantibody regulation. Int Immunol. 1993;5:1329–41. doi: 10.1093/intimm/5.10.1329. [DOI] [PubMed] [Google Scholar]
- 28.Fatenejad S, Brooks W, Schwartz A, Craft J. Pattern of anti-small nuclear ribonucleoprotein antibodies in MRL/Mp-lpr/lpr mice suggests that the intact U1 snRNP particle is their autoimmunogenic target. J Immunol. 1994;152:5523–31. [PubMed] [Google Scholar]
- 29.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 30.Heinegard D, Tiderstrom G. Determination of serum creatinine by a direct colorimetric method. Clin Chim Acta. 1973;43:305–10. doi: 10.1016/0009-8981(73)90466-x. [DOI] [PubMed] [Google Scholar]
- 31.Vlahakos DV, Foster MH, Adams S, Katz M, Ucci AA, Barrett KJ, Datta SK, Madaio MP. Anti-DNA antibodies form immune deposits at distinct glomerular and vascular sites. Kidney Int. 1992;41:1690–700. doi: 10.1038/ki.1992.242. [DOI] [PubMed] [Google Scholar]
- 32.Vlahakos D, Foster MH, Ucci AA, Barrett KJ, Datta SK, Madaio MP. Murine monoclonal anti-DNA antibodies penetrate cells, bind to nuclei, and induce glomerular proliferation and proteinuria in vivo. J Am Soc Neph. 1992;2:1345–54. doi: 10.1681/ASN.V281345. [DOI] [PubMed] [Google Scholar]
- 33.Viney JL, Dianda L, Roberts SJ, Wen L, Mallick CA, Hayday AC, Owen MJ. Lymphocyte proliferation in mice congenitally deficient in T-cell receptor αβ+ cells. Proc Natl Acad Sci USA. 1994;91:11948–52. doi: 10.1073/pnas.91.25.11948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dianda L, Gulbranson-Judge A, Pao W, Hayday AC, MacLennan ICM, Owen MJ. Germinal center formation in mice lacking αβ T cells. Eur J Immunol. 1996;26:1603–7. doi: 10.1002/eji.1830260729. [DOI] [PubMed] [Google Scholar]
- 35.Kanauchi H, Furukawa F, Imamura S. Characterization of cutaneous infiltrates in MRL/lpr mice monitored from onset to the full development of lupus erythematosus-like skin lesions. J Invest Dermatol. 1991;96:478–83. doi: 10.1111/1523-1747.ep12470176. [DOI] [PubMed] [Google Scholar]
- 36.Mohan C, Datta SK. Lupus: key pathogenic mechanisms and contributing factors. Clin Immunol Immunopathol. 1995;77:209–20. doi: 10.1006/clin.1995.1146. [DOI] [PubMed] [Google Scholar]
- 37.Miletic VD, Frank MM. Complement–immunoglobulin interactions. Curr Opin Immunol. 1995;7:41–47. doi: 10.1016/0952-7915(95)80027-1. [DOI] [PubMed] [Google Scholar]
- 38.Santiago M-L, Fossati L, Jacquet C, Müller W, Izui S, Reininger L. Interleukin-4 protects against a genetically linked lupus-like autoimmune syndrome. J Exp Med. 1997;185:65–70. doi: 10.1084/jem.185.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mombaerts P, Mizoguchi E, Grusby MJ, Glimcher LH, Bhan AK, Tonegawa S. Spontaneous development of inflammatory bowel disease in T cell receptor mutant mice. Cell. 1993;75:274–82. doi: 10.1016/0092-8674(93)80069-q. [DOI] [PubMed] [Google Scholar]
- 40.Rathmell JC, Cooke MP, Ho WY, Grein J, Townsend SE, Davis MM, Goodnow CC. CD95 (Fas)-dependent elimination of self-reactive B cells upon interaction with CD4+ T cells. Nature. 1995;376:181–4. doi: 10.1038/376181a0. [DOI] [PubMed] [Google Scholar]
- 41.Rothstein TL, Wang JK, Panka DJ, et al. Protection against Fas-dependent Th1-mediated apoptosis by antigen receptor engagement in B cells. Nature. 1995;374:163–5. doi: 10.1038/374163a0. [DOI] [PubMed] [Google Scholar]
- 42.Scott DW, Grdina T, Shi Y. T cells commit suicide, but B cells are murdered! J Immunol. 1996;156:2352–6. [PubMed] [Google Scholar]
- 43.Lunardi C, Marguerie C, Bowness P, Walport MJ, So AK. Reduction in T gamma delta cell numbers and alteration in subset distribution in systemic lupus erythematosus. Clin Exp Immunol. 1991;86:203–6. doi: 10.1111/j.1365-2249.1991.tb05796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]