

Abstract
Complement component C6 deficiency (C6D) was diagnosed in a 16-year-old African-American male with meningococcal meningitis. The patient's father and two brothers also had C6D, but gave no history of meningitis or other neisserial infection. By using exon-specific polymerase chain reaction (PCR)/single-strand conformation polymorphism as a screening step and nucleotide sequencing of target exons, we determined that the proband was a compound heterozygote for two C6 gene mutations. The first, 1195delC located in exon 7, is a novel mutation, while the second, 1936delG in exon 12, has been described before to cause C6D in an unrelated African-American individual. Both mutations result in premature termination codons and C6 null alleles. Allele-specific PCR indicated that the proband's two brothers also inherited the 1195delC mutation from their heterozygous mother and the 1936delG mutation from their homozygous father.
Keywords: complement deficiency, gene mutation, Neisseria meningitidis, single-strand conformation polymorphism, allele-specific PCR
INTRODUCTION
Human C6 is a 120-kD single polypeptide chain protein [1] consisting of 913 amino acid residues [2, 3]. It is encoded by a single-copy gene located on chromosome 5p13 in proximity to the genes encoding C7 and C9 [4, 5]. The C6 gene spans about 80 kb of DNA and contains 18 exons [6]. The protein has a mosaic modular structure which is similar to that of C7, C8 and C9 [2, 3]. The only known function of these four proteins is their participation in the formation of a cytolytically active, macromolecular complex usually termed the membrane attack complex (MAC). MAC assembly begins with proteolytic cleavage of C5 by a C5-convertase and proceeds through the formation of a complex between C5b and C6 and subsequent sequential binding of C7, C8 and C9 [7]. The MAC is capable of being inserted into biological membranes, thus disrupting their lipid organization. This results in osmotic lysis of susceptible cells or triggering of various signalling events in nucleated cells [7–9]. In bacteria, the MAC is inserted into the outer membrane, causing increased permeability and lethal changes in the inner membrane [10]. Killing by the MAC appears to be the principal host defence mechanism against neisserial infections [11].
Like most other complement protein deficiencies, deficiency of C6 (C6D) is inherited as an autosomal recessive trait [12]. Its prevalence among healthy Japanese was found to be 0·0027% [13]. In the USA, C6D appears to be much more prevalent among individuals of African ancestry than among other ethnic groups, although data on actual prevalence are not available [14, 15]. Individuals with C6D have a well documented predisposition to neisserial infections, particularly meningococcal disease [15–17]. However, the frequency of the association between C6D and meningococcal disease cannot be estimated from available data.
Recently we reported the molecular bases for C6D in two unrelated individuals [18]. An African-American male with C6D and repeated episodes of meningitis was found to be homozygous for a 1936delG mutation in exon 12, resulting in premature termination of the C6 polypeptide. A Japanese male with C6D and no history of infections was found to be heterozygous for a 291delC/292delC/293delC/294delC mutation in exon 2, also resulting in premature termination of C6. The latter individual was considered to be a compound heterozygote with an unidentified molecular defect in the other C6 gene allele [18]. Here we report an African-American kindred with four C6D members. C6D in the father was apparently due to homozygosity for the previously described 1936delG mutation. In the three male siblings C6D was apparently due to compound heterozygosity for the 1936delG mutation and a novel 1195delC mutation, inherited from the C6D heterozygous mother. Only the propositus, one of the three sons, had a demonstrable susceptibility to meningococcal infection.
METHODS
Case report
The proband, a 16-year-old African-American male, developed fever, back pain, and headache 2 days before admission. Over the next 2 days fever continued and he developed worsening headache, back and neck pain, and dizziness. On the day of admission he became lethargic with slurred speech and was taken to the emergency room, where a complete blood count revealed 21 000 leucocytes/mm3 with 92% neutrophils. Cerebrospinal fluid (CSF) contained 5682 leucocytes/mm3 with 97% neutrophils. Gram stain of the fluid revealed no organisms; however, cultures grew Neisseria meningitidis. He was treated with penicillin G and made an uneventful recovery with no sequellae. The patient's past medical history was significant for a prior admission for meningitis 5 years earlier. At the time of that admission he was obtunded and had meningismus. CSF contained 3600 leucocytes/mm3 with 98% neutrophils. Gram stain did not reveal any organisms, but a Wright stain showed a few diplococci. Cultures were negative and the patient responded well to parenteral therapy with ampicillin and a third generation cephalosporin. His nuclear family includes two male siblings 13 and 6 years old and a female half-sibling, all of whom are in good health except for well controlled asthma in the sister. No family history of meningitis or other severe infections could be elicited.
Complement assays
Total serum haemolytic activity was measured as described [19]. C6 haemolytic activity was measured by using C6-depleted serum (Advanced Research Technologies, San Diego, CA), as described previously [18]. Using these assays the levels of detection are 10 CH50/ml and 2 U/ml for total and C6 haemolytic activity, respectively. Purified C6 for reconstitution experiments was purchased from Advanced Research Technologies and goat anti-human C6 serum for immunochemical analyses from Cappel (Durham, NC).
Polymerase chain reaction/single-strand conformation polymorphism analysis
Genomic DNA was isolated from the buffy-coat fraction of blood samples collected in EDTA [20]. Primers for amplifying all exons of the C6 gene and their boundaries have been described [18]. They were synthesized in a model 394 DNA/RNA synthesizer (Applied Biosystems, Foster City, CA). Polymerase chain reaction (PCR) was performed by using 0·2 μg genomic DNA, 25 pmol of each oligonucleotide primer, 5 μmol dNTP including 2 μCi α-32P-dCTP (Amersham, Arlington Heights, IL), and 0·5 U Taq polymerase in a 25-μl total reaction volume. Samples were overlaid with mineral oil to avoid evaporation and then subjected to 30 amplification cycles by using a Tempcycler (Coy Laboratory Products, Ann Arbor, MI). Each cycle consisted of 1 min denaturation at 95°C, 1 min annealing at different temperatures depending on the melting temperature (Tm) of the primers, and 1 min extension at 72°C, except for the last cycle which included a final 5-min extension at 72°C. For single-strand conformation polymorphism (SSCP) analysis, the PCR products were subjected to electrophoresis at room temperature in 6% non-denaturing acrylamide gels containing 5% glycerol or at 4°C without glycerol, using 44 mm Tris-borate, 1 mm EDTA buffer, pH 8·3. PCR products of exon 6, 7, 9, 10, 11, 14 and 16, which were longer than 300 bp, were restricted with Sac I, Bgl II, Sac I, Taq I, Rsa I, Rsa I and Dra I, respectively, before SSCP analysis. The single-stranded DNA fragments were visualized by autoradiography.
Nucleotide sequence analysis
PCR products amplified from exons 7 and 12 were purified by electrophoresis in 2·5% low melting point agarose gel (Sea plaque agarose; FMC Corp., Rockland, ME) and subcloned into the pCR II vector by using the TA cloning kit (Invitrogen, San Diego, CA). Nucleotide sequencing was performed by the dideoxy-chain termination method [21], using modified bacteriophage T7 DNA polymerase [22]. Two primers, 5′-AGATCTGCACCTTTCTGATGT and 5′-GTATGCAATGTGTACACATGT, were used to sequence the amplified exon 7 and exon 12, respectively. Sequence data were analysed using the MacVector Sequence Analysis Software (International Biotechnologies, Inc., New Haven, CT).
Allele-specific PCR
Allele-specific PCR [23] was used to analyse genomic DNA from family members for the presence of the 1195delC and 1936delG mutations of the C6 gene. For each mutation two oligonucleotide primers differing from each other only at the 3′ terminal nucleotide were synthesized (Table 1). In each pair one oligonucleotide corresponds to the mutant allele (M) and the other to the normal allele (N). For PCR, the M and N primers were paired separately with the same antisense or sense primer. PCR was performed in a total volume of 25 μl in 20 mm Tris–HCl pH 8·5, containing 2·0 mm MgCl2,6 mm (NH4)2SO4, and 100 μg/ml bovine serum albumin (BSA). Reaction mixtures contained 200 ng genomic DNA, 25 pmol of each oligonucleotide primer, 5 μmol of each dNTP, and 0·3 U of ΔTaq, Version 2·0, DNA polymerase (USB, Cleveland, OH). Samples were overlaid with mineral oil to avoid evaporation and then subjected to 30 amplification cycles. After an initial denaturation step at 95°C for 5 min, each cycle consisted of 1 min denaturation at 95°C, 1 min annealing at 57°C and 1·5 min extension at 72°C, except for the last cycle which included a final 5 min extension at 72°C. Reaction products were analysed by electrophoresis in 2·5% agarose gels and visualized by ethidium bromide staining.
Table 1.
Oligonucleotides used for allele-specific polymerase chain reaction (PCR)*

C6A and C6B typing
The C6 A/B polymorphism is due to an A→C transversion in exon 3 causing a Glu (C6A) for Ala (C6B) substitution at amino acid position 98 [24]. This polymorphism was detected by Hha I restriction digestion of PCR-amplified exon 3. Briefly, 223-bp fragments including exon 3 and its boundaries were amplified from genomic DNA [18], digested with HhaI, and analysed by agarose electrophoresis. C6A alleles lacking the HhaI site gave single 223-bp bands, while C6B alleles carrying the restriction site gave two, 101- and 122-bp, fragments.
RESULTS
Definition of C6D
Total and C6 haemolytic activities were undetectable in the proband's serum (II.1) (Fig. 1). In addition, double immunodiffusion analysis failed to detect antigenic C6 in the proband's serum. The proband's father (I.1) and two male siblings (II.2 and II.3) also had undetectable total and C6 serum haemolytic activities, while both activities were within the normal range in the sera of the proband's mother (I.2) and half sister (II.4). Addition of purified C6 (70 μg/ml) restored total haemolytic activity to 104, 135, 96 and 108 CH50/ml in the serum of the proband, his father, and two brothers, respectively. All these values are within the normal range. Thus, all four male members of this family are homozygous for C6D, while the mother is an obligate heterozygote.
Fig. 1.
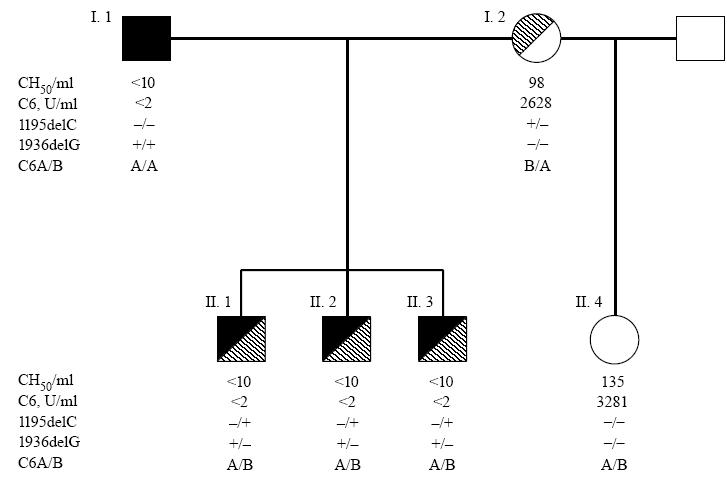
Pedigree of the reported family. Full and hatched symbols indicate C6 mutant alleles inherited from the father and mother, respectively.
Detection of gene defects by PCR/SSCP
PCR/SSCP analyses of all C6 gene exons of the proband in comparison with two C6-sufficient controls demonstrated aberrant bands in exons 7 and 12 of the proband. As shown in Fig. 2a, the exon 7-specific PCR product of the proband displayed three unique bands, indicated by arrows, which migrated differently from those of the controls. Distinct migration of these three bands was observed whether electrophoresis was performed at room temperature in glycerol-containing gels or at 4°C in the absence of glycerol (data not shown). All bands present in the PCR products of the two controls were also present in the proband's DNA, suggesting a heterozygous defect in exon 7 of the proband's C6 gene.
Fig. 2.
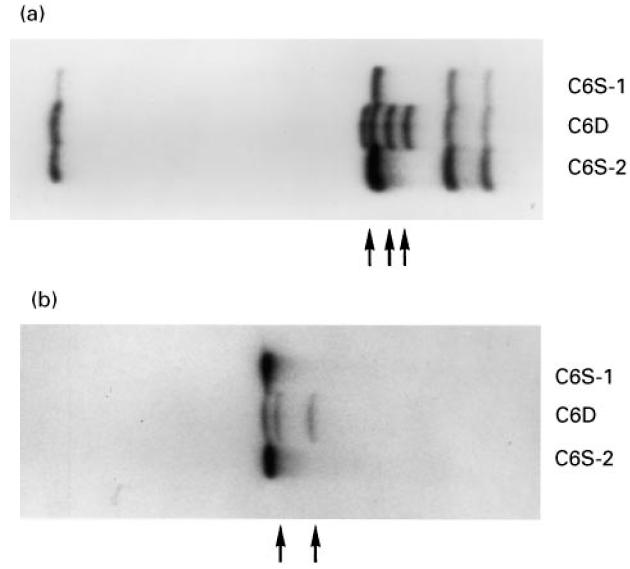
Polymerase chain reaction (PCR)/single-strand conformation polymorphism (SSCP) analysis of exon 7 (a) and exon 12 (b) of the proband's (C6D) C6 gene. C6S-1 and C6S-2 are genomic DNA samples from two unrelated C6-sufficient individuals. Arrows indicate bands unique to the proband.
Figure 2b shows that the exon 12-specific PCR product of the proband displayed three bands; one band migrated with the same mobility as the single one present in the controls, while two additional bands (arrows) were present only in the proband's PCR product. Again, this result suggests that the proband is heterozygous for a defect in exon 12 of the C6 gene.
Nucleotide sequencing of exons 7 and 12
PCR-amplified exons 7 and 12 of the C6 gene of the proband and of a C6-sufficient control individual were subcloned into the pCRII vector and individual clones subjected to nucleotide sequencing. A novel deletional mutation, 1195delC, was detected in three exon 7 clones of the proband (Fig. 3). As shown, the 1195delC mutation caused a shift in the reading frame, generating six codons for amino acids other than those present in normal C6, followed by a premature termination codon, TAA. The nucleotide sequence of four clones from the control individual was identical to that reported for the normal C6 cDNA [2, 3]. Nucleotide sequencing of two exon 12 clones from the proband's C6 gene demonstrated the presence of the 1936delG mutation described recently in an unrelated African-American kindred [18]. The 1936delG mutation causes a frameshift resulting in a premature termination codon corresponding to the codon for Met609 of the normal cDNA [18]. The nucleotide sequences of four additional exon 12 clones from the proband as well as all clones from the control DNA were identical to that reported for the normal C6 cDNA [2, 3]. The combined PCR/SSCP and nucleotide sequencing results indicate that the proband is compound heterozygote for two distinct molecular defects, both of which lead to C6D.
Fig. 3.
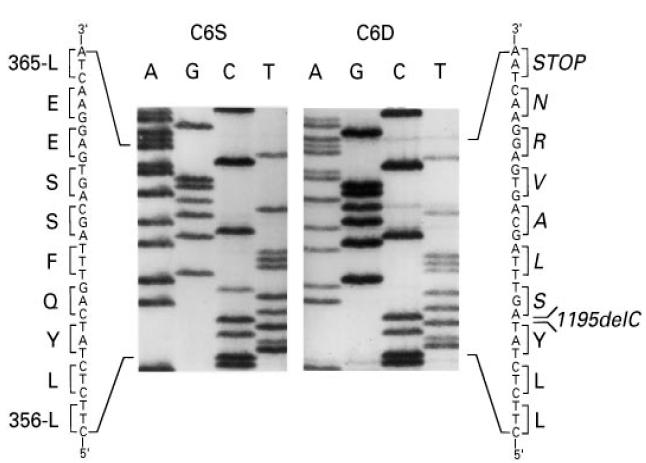
Partial nucleotide sequence of exon 7 amplified from genomic DNA of the proband (C6D) and a C6-sufficient individual (C6S). The position of the deletion (1195C) which leads to the premature TAA stop codon 18 nucleotides downstream in indicated. Encoded amino acids are listed in the single-letter code. Amino acids unique to the mutant allele are in italics.
Family studies
Allele-specific PCR analyses were used to investigate the distribution of the two C6 gene mutations among members of the proband's family. Using the primers listed in Table 1, 329- and 379-bp DNA fragments were amplified from exons 7 and 12, respectively. Under the conditions of the PCR, these fragments were amplified from normal or mutant C6 gene alleles when the N or M primers, respectively, were used. As shown in Fig. 4, allele-specific PCR for the 1195delC mutation (top panel) demonstrated that DNA from the C6-sufficient control and the proband's father (I.1) and half-sister (II.4) produced the 329-bp fragment only when the N primer was used. On the other hand, the proband (II.1), his two brothers (II.2 and II.3), and his mother (I.2) produced this fragment when either the N or the M primer were used. Therefore, the proband and his two brothers inherited the mutant C6 allele carrying the 1195delC mutation from their heterozygous mother.
Fig. 4.
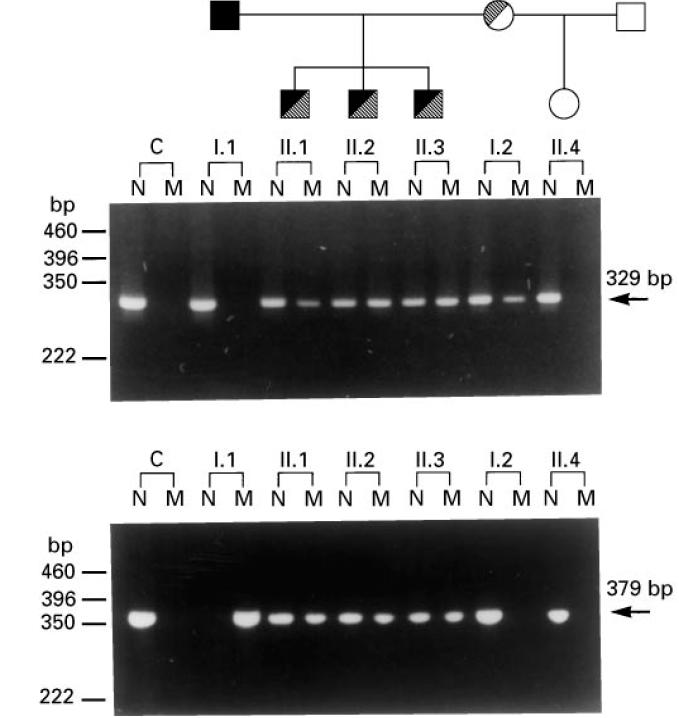
Allele-specific polymerase chain reaction (PCR) analysis of family members for the 1195delC (top panel) and 1936delG (bottom panel) C6 gene mutations. The family tree is shown at the top. C, C6-sufficient control; N and M, PCR products obtained by using primers corresponding to the normal and mutant C6 allele, respectively.
Allele-specific PCR for the 1936delG mutation (Fig. 4, bottom panel) demonstrated that the proband's father is homozygous for the mutation, while all three male siblings are heterozygous. The mother and half sister do not carry the mutant allele. Therefore, the proband and his two brothers inherited the mutant C6 allele carrying the 1936delG mutation from their father.
Analysis of C6 A/B polymorphism
PCR was used to amplify exon 3, which contains the E98A substitution causing the C6 A/B polymorphism [24, 25]. The 223-bp PCR product was restricted with Hha I, which allows discrimination between the two allelic forms, because only the C6B allele has a Hha I restriction site. As shown in Fig. 5, the proband's father is homozygous for C6A, while all other family members are A/B heterozygotes. Therefore, the novel 1195delC mutation segregates with the C6B allele, while, as we reported previously [18], the 1936delG mutation segregates with the C6A allele.
Fig. 5.
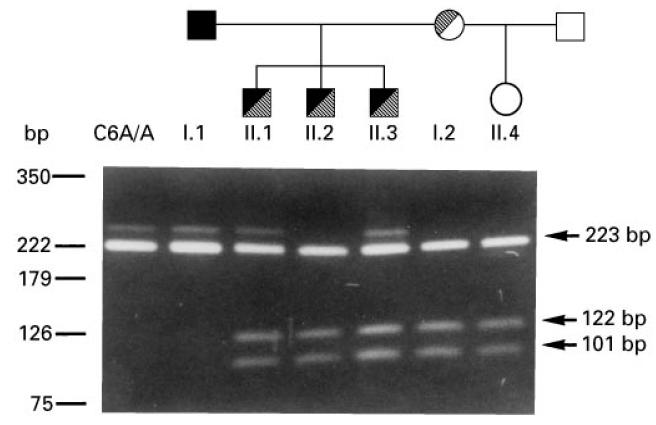
Analysis of C6 A/B allotypes. A 223-bp polymerase chain reaction (PCR)-amplified DNA fragment of exon 3 was restricted with HhaI. C6B alleles containing the restriction site are split into 122- and 101-bp fragments. C6A/A is a C6A homozygous control. The family tree is shown on the top.
DISCUSSION
We describe here two distinct molecular defects leading to C6D in an African-American family with four C6D members. The diagnosis of C6D in the father and three male offspring was established by the absence of total and C6 serum haemolytic activity, and by the restoration of the former to within normal range by addition of physiologic amounts of purified C6. To our knowledge this is only the second report of mutations leading to C6D.
To identify the molecular defects causing C6D in this family we employed an efficient two-step procedure which we described previously [18], that avoids sequencing the entire coding region of the gene. In a first step all exons of the C6 gene are amplified by PCR and analysed by SSCP. In a second step, exons exhibiting aberrantly migrating bands are sequenced. Using this procedure we identified two mutations, one on each allele of the proband's C6 gene. One mutation, 1195delC, located in exon 7, has not been described before. It causes a frameshift generating six codons for amino acids different from those present in normal C6, followed by a termination codon corresponding to residue Leu365 of normal C6. Thus, the predicted mRNA transcribed from the 1195delC mutant C6 gene would encode a polypeptide chain consisting of 364 residues instead of the 913 residues of normal C6 (Fig. 6). Even if secreted, the putative truncated polypeptide, missing the carboxyl 2/3 of normal C6, should not be able to participate in the assembly of the MAC. Analysis of all members of the nuclear family for the presence of the 1195delC mutation by using allele-specific PCR (Fig. 4, upper panel) demonstrated that the proband and his two brothers had inherited the mutant C6 allele from their mother, who also carried a normal C6 allele. The finding that all three male offspring of this family inherited the mutant allele from their heterozygous mother is of interest, in view of a previous report indicating preferential inheritance of C6 null alleles [17]. In that large study on the association between C6D and meningococcal disease in South Africa, it was found that C6D occurred much more frequently among siblings of index cases than would be expected for codominant inheritance. Although the statistical significance of the observation was borderline, the suggestion was made that C6D may confer certain biologic advantage to affected offspring.
Fig. 6.
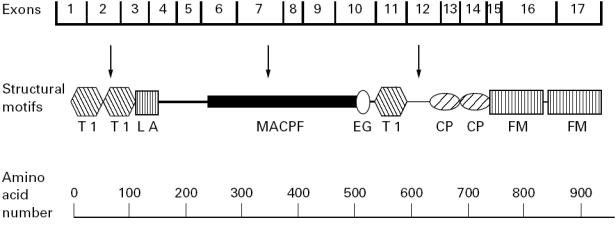
Schematic representation of the molecular structure of C6 (adapted from [18]). Protein modules are designated according to current convention [26], as follows: T1, thrombospondin type 1 (TSP1); LA, LDL-receptor type A (LDLRA); MACPF, MAC proteins/perforin; EG, epidermal growth factor-like (EGF); CP, complement control protein (CCP); FM, complement factor I/MAC proteins C6 and C7 (FIM). Arrows indicate the approximate locations of the three known molecular defects leading to C6D: 1, 291delC/292delC/293delC/294delC (exon 2); 2, 1195delC (exon 7); 3, 1936delG (exon 12).
The other mutant C6 allele of the proband carried the previously described 1936delG mutation [18]. This mutation is located in exon 12 and causes a shift in the reading frame, generating a stop codon corresponding to residue Met609 of normal C6. The putative encoded polypeptide is missing the carboxyl third of the C6 molecule, and apparently even if secreted expresses no C6 activity. All three male offspring inherited the C6 allele carrying the 1936delG mutation from their father who is homozygous for the mutant allele (Fig. 4, bottom panel).
The present study lends further support to published reports indicating a much higher prevalence of C6D among African-Americans than Americans of other ethnic origins [14, 15]. Among the complement-deficient cases assembled by Ross & Densen [14] there were 17 unrelated C6D individuals of known ethnic origin; of these 17 C6D subjects, 11 (65%) were of African descent. A higher than expected proportion of African-Americans was also found among C5-, C7- and C8-deficient individuals. The C6 null allele carrying the 1936delG mutation present in this family was originally described in another, unrelated African-American family. In addition, preliminary results of an ongoing study on the prevalence of this mutation among a random sample of patients admitted to this University Hospital with diagnoses other than neisserial infection have shown that it only occurs in African-Americans (K.T. Hovanky et al., unpublished). The apparent higher prevalence of late-acting complement component deficiencies among African-Americans suggests that these genetic defects may have endowed affected individuals with a biologic advantage. This hypothesis is consistent with the suggestion of Orren et al. [17], that individuals deficient in late-acting complement components may have the advantage of a more benign course of non-neisserial Gram-negative infections.
The association between usually recurrent meningococcal disease and C6D is well established [14–17, 26]. However, the actual prevalence of meningococcal disease among C6D individuals cannot be calculated from existing data, because ascertainment bias has probably contributed to the apparent high prevalence. An estimate of the overall prevalence of infectious complications may be obtained from historical data on siblings of C6D probands. Of the combined 21 non-proband C6D individuals reported by Ross & Densen [14] and by Orren et al. [17], only five (24%) gave a history of meningococcal disease. The finding that of the four C6D individuals of the family described here only the proband had had episodes of meningitis is consistent with this figure. However, it should be noted that at the time of the proband's last admission to the hospital, the ages of his brothers, 13 and 6 years, were below the median age of first attack of meningitis for C6D individuals [14, 17], which is between 14 and 17 years, compared with 3 years in the normal population. Thus, future meningitis in additional members of the present family remains a real possibility.
Acknowledgments
The authors express their gratitude to Dr Richard Whitley for referring this patient for evaluation. The expert secretarial assistance of Mrs Paula Kiley is gratefully acknowledged. This work was supported in part by USPHS grants AI21067 and AR44505. J.E.V. is Anna Lois Waters Chair of Medicine in Rheumatology.
References
- 1.DiScipio RG, Gagnon J. Characterization of human complement components C6 and C7. Mol Immunol. 1982;19:1425–31. doi: 10.1016/0161-5890(82)90189-4. [DOI] [PubMed] [Google Scholar]
- 2.Haefliger JA, Tschopp J, Vial N, Jenne DE. Complete primary structure and functional characterization of the sixth component of the human complement system. Identification of the C5b-binding domain in complement C6. J Biol Chem. 1989;264:18041–51. [PubMed] [Google Scholar]
- 3.DiScipio RG, Hugli TE. The molecular architecture of human complement component C6. J Biol Chem. 1989;264:16197–206. [PubMed] [Google Scholar]
- 4.Hobart MJ, Fernie BA, DiScipio RG, Lachmann PJ. A physical map of the C6 and C7 complement component gene region on chromosome 5p13. Hum Mol Genet. 1993;2:1035–6. doi: 10.1093/hmg/2.7.1035. [DOI] [PubMed] [Google Scholar]
- 5.Setién F, Alvarez V, Coto E, DiScipio RG, López-Larrea C. A physical map of the human complement component, C6, C7, and C9 genes. Immunogenetics. 1993;38:341–4. doi: 10.1007/BF00210475. [DOI] [PubMed] [Google Scholar]
- 6.Hobart MJ, Fernie B, DiScipio RG. Structure of the human C6 gene. Biochemistry. 1993;32:6198–205. doi: 10.1021/bi00075a012. [DOI] [PubMed] [Google Scholar]
- 7.Müller-Eberhard HJ. The membrane attack complex of complement. Annu Rev Immunol. 1986;4:503–28. doi: 10.1146/annurev.iy.04.040186.002443. [DOI] [PubMed] [Google Scholar]
- 8.Esser AF. Big MAC attack: complement proteins cause leaky patches. Immunol Today. 1991;12:316–8. doi: 10.1016/0167-5699(91)90006-F. [DOI] [PubMed] [Google Scholar]
- 9.Bhakdi S, Tranum-Jensen J. Complement lysis: a hole is a hole. Immunol Today. 1991;12:318–20. doi: 10.1016/0167-5699(91)90007-G. [DOI] [PubMed] [Google Scholar]
- 10.Dankert JR, Esser AF. Complement-mediated killing of Escherichia coli: dissipation of membrane potential by a C9-derived peptide. Biochemistry. 1986;25:1094–100. doi: 10.1021/bi00353a023. [DOI] [PubMed] [Google Scholar]
- 11.Nicholson A, Lepow IH. Host defense against Neisseria meningitidis requires a complement-dependent bactericidal activity. Science. 1979;205:298–9. doi: 10.1126/science.451601. [DOI] [PubMed] [Google Scholar]
- 12.Colten HR, Rosen FS. Complement deficiencies. Annu Rev Immunol. 1992;10:809–34. doi: 10.1146/annurev.iy.10.040192.004113. [DOI] [PubMed] [Google Scholar]
- 13.Inai S, Akagaki Y, Moriyama T, Fukumori Y, Yoshimura K, Ohnoki S, Yamaguchi H. Inherited deficiencies of the late-acting complement components other than C9 found among healthy blood donors. Int Arch Allergy Appl Immunol. 1989;90:274–9. doi: 10.1159/000235037. [DOI] [PubMed] [Google Scholar]
- 14.Ross SC, Densen P. Complement deficiency states and infection: epidemiology, pathogenesis and consequences of neisserial and other infections in an immune deficiency. Medicine. 1984;63:243–73. [PubMed] [Google Scholar]
- 15.Ellison Iii RT, Kohler PF, Curd JG, Judson FN, Reller LB. Prevalence of congenital or acquired complement deficiency in patients with sporadic meningococcal disease. N Engl J Med. 1983;308:913–6. doi: 10.1056/NEJM198304213081601. [DOI] [PubMed] [Google Scholar]
- 16.Lee TJ, Snyderman R, Patterson J, Rauchbach AS, Folds JD, Yount WJ. Neisseria meningitidis bacteremia in association with deficiency of the sixth component of complement. Infect Immun. 1979;24:656–60. doi: 10.1128/iai.24.3.656-660.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Orren A, Potter PC, Cooper RC, Du Toit E. Deficiency of the sixth component of complement and susceptibility to Neisseria meningitidis infections: studies in 10 families and five isolated cases. Immunology. 1987;62:249–53. [PMC free article] [PubMed] [Google Scholar]
- 18.Nishizaka H, Horiuchi T, Zhu ZB, et al. Molecular bases for inherited human complement component C6 deficiency in two unrelated individuals. J Immunol. 1996;156:2309–15. [PubMed] [Google Scholar]
- 19.Oglesby TJ, Ueda A, Volanakis JE. Radioassays for quantitation of intact complement proteins C2 and B in human serum. J Immunol Methods. 1988;110:55–62. doi: 10.1016/0022-1759(88)90082-8. [DOI] [PubMed] [Google Scholar]
- 20.Sykes BC. DNA in heritable disease. Lancet. 1983;II:787–8. doi: 10.1016/s0140-6736(83)92314-0. [DOI] [PubMed] [Google Scholar]
- 21.Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977;74:5463–7. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tabor S, Richardson CC. DNA sequence analysis with a modified bacteriophage T7 DNA polymerase. Proc Natl Acad Sci USA. 1987;84:4767–71. doi: 10.1073/pnas.84.14.4767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sarkar G, Cassady J, Bottema CDK, Sommer SS. Characterization of polymerase chain reaction amplification of specific alleles. Anal Biochem. 1990;186:64–68. doi: 10.1016/0003-2697(90)90573-r. [DOI] [PubMed] [Google Scholar]
- 24.Dewald G, Nöthen MM, Cichon S. Polymorphism of human complement component C6: an amino acid substitution (Glu/Ala) within the second thrombospondin repeat differentiates between the two common allotypes C6A and C6B. Biochem Biophys Res Commun. 1993;194:458–64. doi: 10.1006/bbrc.1993.1841. [DOI] [PubMed] [Google Scholar]
- 25.Fernie BA, Delbridge G, Hobart MJ. Correlation of a Glu/Ala substitution at position 98 with the complement C6 A/B phenotypes. Hum Mol Genet. 1993;2:591–2. doi: 10.1093/hmg/2.5.591. [DOI] [PubMed] [Google Scholar]
- 26.Lim D, Gewurz A, Lint TF, Ghaze M, Sepheri B, Gewurz H. Absence of the sixth component of complement in a patient with repeated episodes of meningococcal meningitis. J Pediatr. 1976;89:42–47. doi: 10.1016/s0022-3476(76)80924-9. [DOI] [PubMed] [Google Scholar]