

Abstract
Annexin-1, a calcium-dependent phospholipid binding protein, has been shown to act as an endogenous central neuroprotectant, notably against cerebral ischaemic damage. In the present study we extend these findings to an animal model of multiple sclerosis, EAE, and report that endogenous annexin-1 is expressed in ED1+ macrophages and resident astrocytes localized within the lesions in the central nervous system (CNS). Intracerebroventricular (icv) administration of an NH2-terminal fragment spanning amino acids 1–188 of annexin-1 after the onset of the clinical symptoms significantly reduced both the neurological severity as well as weight loss of mild EAE. Immunoneutralization of endogenous brain annexin-1 failed to exacerbate the clinical features of EAE. Thus, although the role of endogenous annexin-1 in the pathogenesis of EAE remains to be determined, our findings suggest that annexin-1 may be of therapeutic benefit to the treatment of multiple sclerosis.
Keywords: annexin-1, lipocortin-1, experimental allergic encephalomyelitis, central nervous system, astrocytes, macrophages
INTRODUCTION
EAE represents an animal model for demyelinating diseases of the central nervous system (CNS) like multiple sclerosis (MS). EAE can be induced in Lewis rats after immunization with homogenized myelin, or myelin basic protein (MBP) in Freund's complete adjuvant (FCA). About 9–10 days after immunization, encephalitogenic T cells and impressive numbers of macrophages infiltrate the CNS, forming large perivascular inflammatory cuffs [1]. Oedema and demyelination are subsequently responsible for the neurological deficits, which include paresis and paralysis of the tail and the hind limbs. EAE is also accompanied by an impressive loss in body weight [1].
Corticosteroid treatment has proved beneficial in both MS and EAE [2, 3]. Elevated plasma concentrations of glucocorticoids are also necessary for the spontaneous recovery from clinical EAE [4]. How glucocorticoids suppress EAE is not clear, but it may be mediated in part by the local reduction of cellular infiltration and inflammatory reactions in the CNS. Glucocorticoids inhibit the production of proinflammatory molecules such as IL-1β, IL-6, tumour necrosis factor-alpha (TNF-α) and eicosanoids, but induce the expression of annexin-1 [5–8]. Annexin-1, also known as lipocortin-1, is a 37-kD member of a family of structurally related Ca2+ and phospholipid binding proteins [5–9]. Annexin-1 has evoked interest as a glucocorticoid-inducible protein that can suppress the production of eicosanoids by inhibition of phospholipase A2 (PLA2) and arachidonic acid release [10–17]. The protein is expressed constitutively in the immune system in macrophages and polymorphonuclear cells (PMN), and also in the CNS, where it is located in neurones, astrocytes and ependymal cells lining the cerebral ventricles [18, 19]. Furthermore, annexin-1 is present at nanomolar concentrations in human serum [20]. Glucocorticoid analogues can up-regulate the constitutive expression of annexin-1 in some cell types, and glucocorticoid receptor antagonists or adrenalectomy reduce constitutive expression of annexin-1 also in site- and cell type-dependent ways [7, 19, 21]. Annexin-1 is up-regulated in the brains of MS patients as well as of rats with EAE, possibly due to the rise in plasma cortisol and corticosterone levels during disease [4, 22, 23].
Functional studies using recombinant full length annexin-1, an NH2-terminal fragment spanning amino acids 1–188 of annexin-1 (annexin-1 (1–188)) or amino acids 2–26 (annexin-1 (2–26)), and neutralizing antibodies raised against annexin-1 (1–188) have revealed involvement of annexin-1 in inflammation and neurodegeneration. Annexin-1 (1–188) reduces febrile responses to cytokines [24, 25]. Also, neurodegeneration caused by cerebral ischaemia- or N-methyl-D-aspartate (NMDA) receptor-mediated tissue damage in the rat is reduced by annexin-1 (1–188) [26, 27]. In addition, anti-inflammatory actions of exogenous full length annexin-1 have been demonstrated in an animal model of lung perfusion and a model of paw oedema [28, 29], and migration of PMN to an inflamed peritoneal cavity is also greatly reduced by annexin-1 (2–26) as well as by annexin-1 (1–188) [30]. Anti-inflammatory effects of dexamethasone, like suppression of oedema and inhibition of the production of eicosanoids, TNF-α and nitric oxide (NO), can be reversed by annexin-1 antiserum [31–33]. Together with the induction of annexin-1 by dexamethasone, these findings support the hypothesis that annexin-1 can act as key mediator of the anti-inflammatory actions of glucocorticoids [31–33].
To gain insight into annexin-1 production and function in the CNS during EAE we investigated the spatial and temporal pattern of immunoreactive annexin-1 expression in the CNS of rats during EAE. Furthermore, we studied the effects of annexin-1 (1–188) on the clinical manifestation of EAE. Finally, we analysed the role of endogenous annexin-1 in the clinical expression of EAE using neutralizing antibodies.
Here we report the novel findings that: (i) annexin-1 is induced in the CNS during clinical manifestation of EAE in ED1+ macrophages as well as in reactive astrocytes in the lesions; and (ii) administration of annexin-1 (1–188) protected against clinical manifestations of mild EAE.
MATERIALS AND METHODS
Animals
Male Lewis rats were obtained from the Zentral Institut für Verzuchstierzucht (Hannover, Germany) or the Broekman Instituut (Someren, The Netherlands), and maintained under standard laboratory conditions. Water and food were available ad libitum. The animals weighed ≈ 200 g at the start of experiment.
Reagents
Annexin-1 (1–188) (amino acid sequence 1–188 of the NH2-terminal of human annexin-1) was kindly donated by Dr F. Carey (Zeneca Pharmaceuticals, Macclesfield, UK) [24]. Polyclonal anti-annexin-1 antibodies were raised in a rabbit against the human recombinant annexin-1 (1–188) [24]. A purified mouse MoAb to annexin-1 which had been raised against full-length bovine annexin-1 was obtained commercially from Zymed (San Francisco, CA) (clone ZO13). Mouse MoAb ED1, recognizing all rat macrophages, was produced in our laboratory and used after protein A purification [34].
Experimental allergic encephalomyelitis
Acute EAE was induced under ether anaesthesia by a single subcutaneous injection in the hind foot pad of 60 μl of the following emulsions: guinea pig spinal cord (GSC) homogenate (1 g GSC/ml saline; experiments 1 and 2), or 25 μg of MBP in saline (experiment 3), emulsified in an equal volume of FCA, to which 10 mg Mycobacterium tuberculosis H37 RA (Difco Labs, Detroit, MI) per ml of FCA were added. The rats were weighed and investigated daily for the development of neurological symptoms. Clinical signs were scored on a scale ranging from 1 to 5 as described previously [1].
Immunocytochemistry
For annexin-1 (1–188) and ED1 staining, rats were perfused transcardially with Ringer solution pH 6·9 followed by 4% paraformaldehyde (PFA) in 0·1 m phosphate buffer. MoAb ED1 is specific for all rat macrophages [34]. The specificity of the polyclonal antibody raised against annexin-1 (1–188) has been described earlier [19]. Preabsorption of the antibody with 10−6 m annexin-1 (1–188) resulted in absence of annexin-1 staining in the rat brain [19]. Furthermore, stainings in which the first antibody was omitted resulted in absence of annexin-1 immunoreactivity in sections of the CNS of rats with full-blown clinical EAE (data not shown). Brains were dissected and post-fixed for 24 h in 4% PFA. Cryostat sections of 10 μm were double-stained with anti-annexin-1 (1–188) polyclonal rabbit antibody and ED1 diluted 1:100 and 1:300, respectively, in 0·05 m Tris-buffered saline pH 7·6 to which 2·5% BSA was added (TBS–BSA) overnight at 4°C. Primary antibodies were visualized using FITC or TRITC-labelled secondary antibodies (Jackson ImmunoResearch Labs, West Grove, PA). To prepare 1-μm sections, vibratome sections of 50 μm were incubated with anti-annexin-1 (1–188) polyclonal rabbit antibody diluted 1:100 in TBS–BSA for 48–72 h at 4°C. After rinsing, sections were incubated with biotin-conjugated anti-rabbit antibodies (Dakopatts, Tilburg, The Netherlands), which were subsequently visualized using peroxidase-labelled avidin–biotin complex (ABC; Vector Labs, Burlingame, CA) and 3,3′ diaminobenzidine-tetra-hydrochloride (DAB; Sigma, St Louis, MO), respectively. Annexin-1-stained sections were embedded in epon as described previously [35]. Selected areas from the 50-μm epon-embedded sections were cut out and mounted on prepared epoxy beams and semi-thin 1-μm sections were prepared and counterstained with toluidine blue.
Intracerebroventricular injections
For intracerebroventricular (icv) treatment administration of saline, annexin-1 (1–188) or antibodies, a guide cannula (internal diameter 0·58 mm, external diameter 0·96 mm) was placed stereotactically into the lateral ventricle as described earlier at least 7 days before the induction of EAE [36]. Treatment solutions were injected icv at a rate of 2 μl/min using a stainless steel injector (external diameter 0·5 mm) and a microinjection pump (Harvard Apparatus, South Natick, MA).
Experimental design
Cellular localization of annexin-1 and ED1
Brains were taken 5, 12 (fluorescence double-labelling) or 15 days (semi-thin sections) after EAE induction.
EAE icv treatment studies
In experiment 1, annexin-1 (1–188) (0·48 μg/μl saline, n = 5) or saline (n = 5) was injected (5 μl, icv) once daily before noon at 8–12 days after induction of EAE.
In experiment 2, polyclonal antiserum to annexin-1 (1–188) (n = 6) or saline (n = 6) was injected (5 μl, icv) once before noon at 9 days after EAE induction.
In experiment 3, annexin-1 (1–188) (0·48 μg/μl, n = 6), a MoAb to annexin-1 (1 μg/μl, n = 5) or saline (n = 7) was injected repeatedly (5 μl, icv) once daily before noon at 9–13 days after EAE induction. This experiment included a non-cannulated control group (n = 7) that received no treatment after EAE induction.
Statistical analysis
Effects of annexin-1 (1–188) treatment on EAE were evaluated statistically by analysing the incidence of clinical disease of EAE using χ2 test. The duration of clinical disease of EAE was analysed using Student's t-test for unmatched data, and weight loss during EAE was analysed using the Mann–Whitney U-test. Statistical significance is defined as P< 0·05.
RESULTS
Annexin-1 immunoreactivity in the CNS of rats with EAE
In non-immunized healthy rats, no ED1 reactivity was seen in the CNS (not shown) and annexin-1 immunoreactivity was confined to ependymal cells and tanycytes, as described earlier [19]. Five days after induction of EAE, annexin-1 immunoreactivity was confined mainly to ependymal cells (Fig. 1C). At this stage of EAE, there were no detectable ED1+ macrophages in the CNS (Fig. 1A). Twelve days after the induction of EAE, when the clinical manifestation of the disease is maximal, many ED1+ macrophages (Fig. 1B) and annexin-1-immunoreactive cells (Fig. 1D) were found in the lesions. Double-staining demonstrated that many, but not all, annexin-1-immunoreactive cells were in fact ED1+ macrophages (Fig. 1B,D). Brain sections of inflammatory lesions 15 days after the induction of EAE revealed that, in addition to macrophages, astrocytes also displayed intense annexin-1 immunoreactivity. These were found predominantly around small vessels in the lesions (Fig. 2).
Fig. 1.
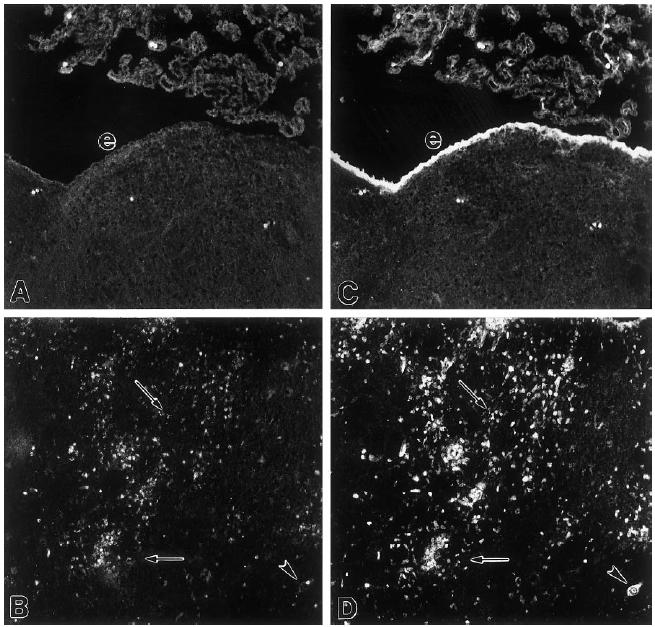
Microphotograph of a double-staining of ED1 (A,B) and annexin-1 (C,D) immunoreactivity in brains of rats with EAE. (A,C) Coronal section through the medulla oblongata 5 days after the induction of EAE. (B,D) Coronal section through the diencephalon 12 days after induction of EAE. e, Ependymal cells; arrowhead, neurone; arrows point at examples of annexin-1-positive cells that are not ED1+. Note that most ED1+ cells exhibit annexin-1 immunoreactivity. Mag. × 100.
Fig. 2.
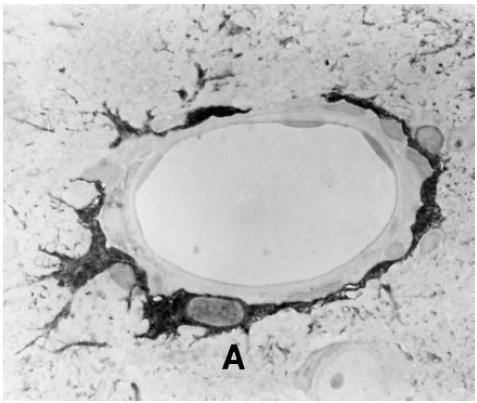
Microphotograph of annexin-1 immunoreactivity in a 1-μm section of a lesion in the brain 15 days after the induction of EAE. A, Astrocyte. Mag. × 400.
Experiment 1: effect of repeated icv administration of annexin-1 (1–188) on EAE
Saline-treated rats first developed neurological symptoms 9 days after the induction of EAE (Fig. 3). Incidence of clinical symptoms was 100% (5/5) in saline-treated rats and varied between paralysis of the tail (5/5) and paralysis of the hind limbs (2/5). Repeated daily icv administration of annexin-1 (1–188), starting 1 day before the anticipated onset of the clinical manifestation of the disease (i.e. 8–12 days after induction of EAE) markedly reduced the severity of EAE compared with saline-treated rats (P< 0·02, χ2 test, Fig. 3). Only three out of five animals treated with annexin-1 (1–188) developed clinical EAE, and severity of disease was confined to partial or complete paralysis of the tail. Duration of clinical disease was 5·6 ± 2·3 days in the control group and 2·3 ± 1·9 days in the annexin-1 (1–188)-treated group (P< 0·04, Student's t-test). Weight loss, which accompanies the clinical phase of EAE, was significantly reduced in the annexin-1 (1–188)-treated group compared with the control treated group 11–13 days after induction of EAE (P< 0·01 at day 11, P< 0·04 at day 12 and P< 0·05 at day 13; Mann–Whitney U-test, Fig. 4).
Fig. 3.
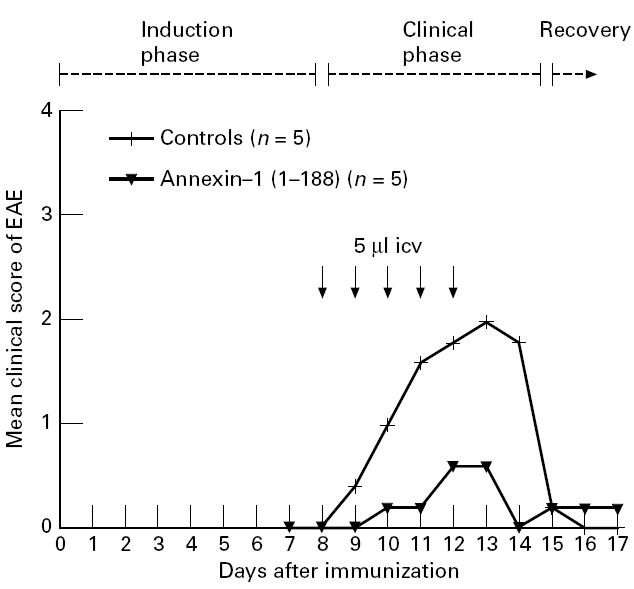
Effect of repeated intracerebroventricular (icv) administration of annexin-1 (1–188) on clinical signs of EAE (experiment 1). EAE was induced by injecting guinea pig spinal cord (GSC) homogenate. Rats were given once daily icv injections (arrows) of annexin-1 (1–188) (2.15 μg in 5 μl in 2.5 min, n = 5) or saline (controls, 5 μl in 2.5 min, n = 5) at 8–12 days after the induction of EAE. Treatment with annexin-1 (1–188) reduced the clinical severity of EAE (P< 0.02, χ2 test) as well as duration of EAE (P< 0.04, Student's t-test).
Fig. 4.
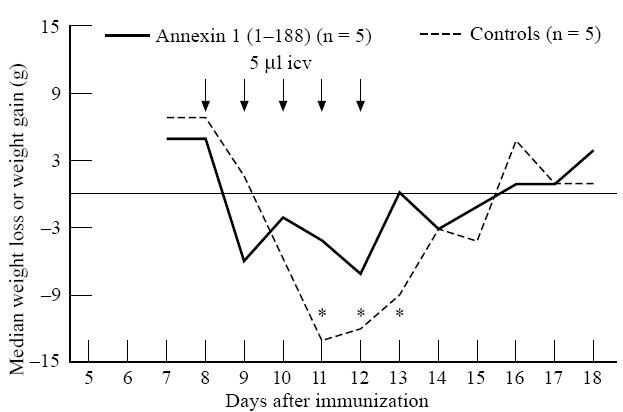
Effect of repeated intracerebroventricular (icv) administration of annexin-1 (1–188) on body weight during EAE (experiment 1). The median of daily weight loss or weight gain per group is given in grams. EAE was induced by injecting guinea pig spinal cord (GSC) homogenate. Rats were given daily icv injections (arrows) of annexin-1 (1–188) (2.15 μg in 5 μl in 2.5 min, n = 5) or saline (controls, 5 μl in 2.5 min, n = 5) at 8–12 days after induction of EAE. Treatment with annexin-1 (1–188) reduced weight loss during clinical disease of EAE (P< 0.01 at day 11, P< 0.04 at day 12, and P< 0.05 at day 13 after induction of EAE, Mann–Whitney U-test).
Experiment 2: effect of a single icv administration of antibodies to annexin-1 (1–188) on EAE
Clinical signs of EAE varied from paralysis of the tail to paralysis of the hind limbs in rats treated once either with saline or with anti-annexin-1 (1–188) antiserum at day 9 after induction of EAE (Fig. 5). No differences were observed in the severity of neurological disease or in weight loss during EAE between saline-treated and antibody-treated rats (data not shown).
Fig. 5.
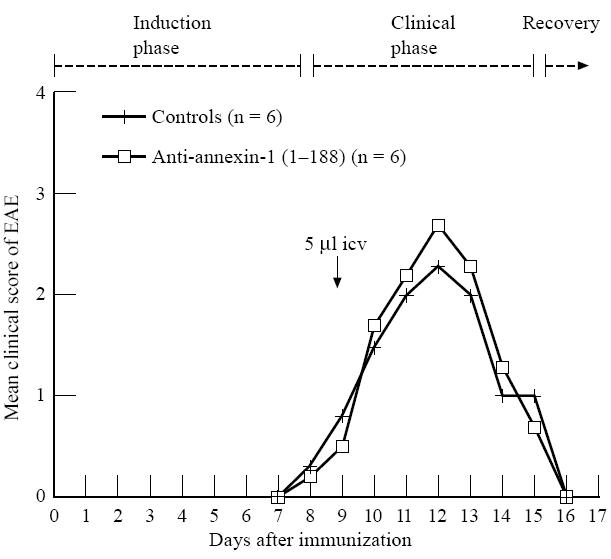
Effect of a single intracerebroventricular (icv) injection of polyclonal anti-annexin-1 (1–188) on EAE (experiment 2). EAE was induced by subcutaneous injection of guinea pig spinal cord (GSC) homogenate. Rats were treated once icv at day 9 (arrow) with polyclonal antibodies against annexin-1 (5 μl in 2.5 min, n = 6) or saline (controls, 5 μl in 2.5 min, n = 6).
Experiment 3: effect of repeated icv administration of annexin-1 (1–188) or a MoAb to annexin-1 on EAE
As illustrated in Fig. 6, clinical disease of EAE was more severe in control rats without an icv cannula compared with cannulated animals. Non-cannulated rats all developed paralysis of the complete lower part of the body (7/7, score 4). Based on the incidence of score 4 the clinical EAE was more severe in these non-cannulated rats compared with cannulated saline-treated rats (3/7 rats had score 4), anti-annexin-1 (1–188)-treated rats (2/5 had score 4, both P< 0·018, χ2 test) as wells as annexin-1 (1–188)-treated rats (2/6 had score 4, P< 0·007, χ2) (Fig. 6). Icv injections with annexin-1 (1–188) at 9–13 days after EAE induction did not reduce severity of clinical EAE (P< 0·18, Student's t-test). Anti-annexin-1 MoAbs injected at days 9–13 after EAE induction had no effect on clinical expression of EAE compared with control treated rats.
Fig. 6.
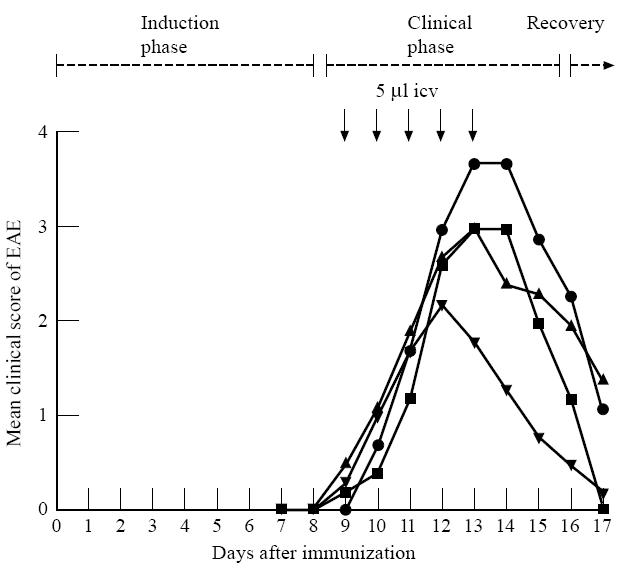
Effects of repeated intracerebroventricular (icv) injections of annexin-1 (1–188) or anti-annexin-1 MoAbs on EAE (experiment 3). EAE was induced by subcutaneous injection of myelin basic protein (MBP). Non-cannulated controls (•): rats that did not receive an icv cannula and that were not treated after induction of EAE (n = 7). Other groups of rats received icv cannulas and were treated once daily icv with annexin-1 (1–188) (2.15 μg in 5 μl in 2.5 min, n = 6; ▾), a MoAb against annexin-1 (5 μg in 5 μl in 2.5 min, n = 5; ▪) or saline (vehicle controls, 5 μl in 2.5 min, n = 7; ▴), at 9–13 days after induction of EAE. Note that the neurological symptoms of EAE were most severe in intact non-cannulated rats.
DISCUSSION
This is the first study to report suppressive effects in vivo of annexin-1 (1–188) in an experimental autoimmune model. Repeated icv administration of recombinant annexin-1 (1–188) during the first part of the clinical phase of EAE markedly suppressed the clinical manifestation of the disease. In addition, we demonstrate the appearance of annexin-1 immunoreactivity in ED-1+ macrophages and astrocytes in the CNS during the clinical and early recovery phase of EAE.
Annexin-1 has powerful immunosuppressant properties, as shown by in vitro and in vivo studies [26–30]. The immunosuppressive effects of annexin-1 may relate to its ability to inhibit PLA2 [15, 16], and the consequent reduction of proinflammatory eicosanoid production. Eicosanoids include prostaglandins, leukotrienes and thromboxanes, which can act on the vasculature to cause chemoattraction of immunocompetent cells, to increase blood flow and to enhance vascular permeability [37]. Indeed, blockade of eicosanoid synthesis suppresses EAE, emphasizing their proinflammatory role in EAE [38]. In addition, annexin-1 may interfere with other, non-eicosanoid-related, proinflammatory processes. This is indicated by the observation that inhibition of leucocyte migration to inflammatory sites by annexin-1 (2–26) could not be mimicked by selective eicosanoid blockers [30]. Recently, it has been demonstrated that annexin-1 (1–188) is also involved in the dexamethasone-induced suppression of TNF-α release from peripheral blood mononuclear cells [32]. Because a role of TNF-α in the clinical expression of EAE has been demonstrated [39], suppression of TNF-α release by annexin-1 in EAE can be expected to reduce clinical severity. In addition, it has been demonstrated that annexin-1 suppresses the clonal expansion of MBP-specific T cells [40]. Thus, icv-injected annexin-1 (1–188) may have suppressed EAE by interfering with the antigen-specific drive of the immune response in the CNS during EAE by inhibiting T cell expansion in the perivascular lesions on the one hand, and on the other hand by reduction of the release of proinflammatory mediators like eicosanoids and TNF-α by activated macrophages around the inflamed blood vessels.
Although more severe EAE tended to be reduced by annexin-1 (1–188) treatment, it was not statistically significant. Possibly, severe EAE induces higher levels of endogenous annexin-1 compared with mild EAE. In such a situation additional annexin-1 (1–188) may have relatively small effects. Alternatively, the proinflammatory drive during severe EAE may be too strong to be arrested by annexin-1 (1–188). Interestingly, chronic implantation of an icv cannula suppressed clinical severity of EAE significantly. The mechanisms underlying this suppression are not clear, but may involve neuroplasticity within the hypothalamic–pituitary–adrenal (HPA) axis induced by brain surgery, which can induce hyperresponsivitity of the axis and increased corticosterone responses to various stimuli [41, 42].
Administration of anti-annexin-1 antisera did not affect the clinical course of EAE. The polyclonal annexin-1 (1–188) antibody used in our study can bind and neutralize the annexin-1 fragment [24], and icv administration of this antibody reduces tissue damage after cerebral ischaemia and inhibits pyrogenic effects of cytokines at the same dose as used in the present study [24, 27]. In addition, the annexin-1 MoAbs have been shown to block the activity of rat annexin-1 in vitro, although this has not been tested in vivo [43]. It should be noted that the effects of annexin-1 antibodies on ischaemia and fever were studied over a much shorter time period and therefore might be more effective compared with the present study.
Glucocorticoids suppress EAE [3], and activation of the HPA axis during EAE is necessary for recovery [4]. Annexin-1 immunoreactivity has been demonstrated in neurones located in areas involved in the control of the HPA axis [19, 21] and both in vitro and in vivo studies suggest that annexin-1 is a mediator of the negative feedback actions of glucocorticoids after inflammatory mediator-induced activation of the HPA axis [43–45]. This implies that icv injection of annexin-1 (1–188) could mimic the negative feedback effects of glucocorticoids and therefore inhibit activation of the HPA axis, which would counteract anti-inflammatory effects of the annexin-1 treatment [4]. Conversely, anti-annexin-1 antibodies can suppress the negative feedback effects of glucocorticoids which would lead to sustained activation of the HPA axis. This phenomenon could have contributed to the absence of effects of anti-annexin-1 antibodies on the clinical course of EAE in this study.
In the healthy rat brain annexin-1 immunoreactivity is prominent in ependymal cells, choroid epithelial cells and a particular population of astrocytes called ‘tanycytes’, and is present in distinct neural cell populations [19]. Several studies describe up-regulation of annexin-1 in the CNS after brain damage or inflammation [22, 23, 46, 47]. This is mainly restricted to reactive astrocytes and infiltrating macrophages after haemorrhage, embolic or traumatic infarctions in human CNS and in Alzheimer's disease, as well as in kainate-induced lesions in the CNS of rats [46, 47]. The results of our study confirm earlier findings on the increase of annexin-1 immunoreactivity in the CNS during EAE [22], and identify ED-1+ macrophages and astrocytes as major production sites of annexin-1 in this experimental disease. The appearance of annexin-1 immunoreactivity in perivascular astrocytes may be of high strategic value in the inhibition of migration of proinflammatory leucocytes into the CNS. The mechanism responsible for the induction of annexin-1 in glial cells during the clinical phase of EAE is not clear. Dexamethasone up-regulates annexin-1 expression in many cell types [10–15], thus annexin-1 immunoreactivity in astrocytes could have been induced by circulating glucocorticoids which reach peak levels during the clinical phase of EAE [4, 14], although contradictory results have been published [48].
Promising new therapies for MS are currently being evaluated [49], but glucocorticoids are still used as therapy despite significant side-effects. Investigation of second messenger molecules of glucocorticoids such as annexin-1 may provide valuable information on the mechanism by which glucocorticoids modulate inflammation during MS.
Acknowledgments
The authors wish to thank Karin Sontrop, Wim van den Bovenkamp and Maarten van der Cammen for their technical assistance and Terence Smith (London, UK) for valuable discussion. This study was initiated by Frank Berkenbosch (who sadly died April 1993). Financial support for this study was provided by the Dutch foundation ‘Friends MS Research’ (grants 90-67 and 95-204 MS).
References
- 1.Huitinga I, Ruuls SR, Jung S, Van Rooijen N, Hartung H-P, Dijkstra CD. Macrophages in T cell line-mediated, demyelinating and chronic relapsing experimental autoimmune encephalomyelitis in Lewis rats. Clin Exp Immunol. 1995;100:344–51. doi: 10.1111/j.1365-2249.1995.tb03675.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Compston DAS, Milligan NM, Hughes PJ, Gibbs J, Morgan BP, Campbell AK. A double-blind controlled trial of high dose methylprednisolone in patients with multiple sclerosis: laboratory results. J Neurol Neurosurch Psych. 1987;50:517–22. doi: 10.1136/jnnp.50.5.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bolton C, Flower RJ. The effects of the anti-glucocorticoid RU 38486 on steroid-mediated suppression of experimental allergic encephalomyelitis. Life Sci. 1989;45:97–104. doi: 10.1016/0024-3205(89)90441-4. [DOI] [PubMed] [Google Scholar]
- 4.MacPhee IA, Antoni FA, Mason DW. Spontaneous recovery of rats from experimental allergic encephalomyelitis is dependent on the regulation of the immune system by endogenous adrenal corticosteroids. J Exp Med. 1989;169:431–45. doi: 10.1084/jem.169.2.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blackwell GJ, Carnuccio R, Di Rosa M, Flower RJ, Parente L, Perisco P. Macrocortin: a polypeptide causing the anti-phospholipase effects of glucocorticoids. Nature. 1980;287:147–9. doi: 10.1038/287147a0. [DOI] [PubMed] [Google Scholar]
- 6.Goulding NJ, Guyre PM. Glucocorticoids, lipocortins and the immune response. Curr Opinion Immunol. 1993;5:108–13. doi: 10.1016/0952-7915(93)90089-b. [DOI] [PubMed] [Google Scholar]
- 7.Flower RJ. Lipocortin and the mechanism of action of the glucocorticoids. Br J Pharmacol. 1988;65:987–1015. doi: 10.1111/j.1476-5381.1988.tb11614.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Flower RJ, Dale MM. The anti-inflammatory effects of corticosteroids. In: Dale MM, Foreman C, editors. Textbook of immunopharmacology. Oxford: Blackwell Publications; 1989. pp. 275–87. [Google Scholar]
- 9.Ahluwalia A, Mohamed RW, Flower RJ. Induction of lipocortin by topical steroid in rat skin. Biochem Pharmacol. 1994;48:1647–54. doi: 10.1016/0006-2952(94)90210-0. [DOI] [PubMed] [Google Scholar]
- 10.Goulding NJ, Godolphin JL, Sharland PR, Peers SH, Sampson M, Maddison PJ, Flower RJ. Anti-inflammatory lipocortin 1 production by peripheral blood leukocytes in response to hydrocortisone. Lancet. 1990;1:1416–8. doi: 10.1016/0140-6736(90)91445-g. [DOI] [PubMed] [Google Scholar]
- 11.Ambrose MP, Hunninghake GW. Corticosteroids increase lipocortin 1 in alveolar epithelial cells. Am J Resp Cell Mol. 1990;3:349–53. doi: 10.1165/ajrcmb/3.4.349. [DOI] [PubMed] [Google Scholar]
- 12.Errasfa M, Rothhut B, Fradin A, Billardon C, Junien JL, Bure J, Russo-Marie F. The presence of lipocortin in human embryonic skin fibroblasts and its regulation by anti-inflammatory steroids. Biochem Biophys Acta. 1985;847:247–54. doi: 10.1016/0167-4889(85)90027-8. [DOI] [PubMed] [Google Scholar]
- 13.McLeod JD, Bolton C. Dexamethasone induces an increase in intracellular and membrane-associated lipocortin 1 (annexin 1) in astrocyte primary cell cultures. Cell Mol Neurobiol. 1995;15:193–205. doi: 10.1007/BF02073328. [DOI] [PubMed] [Google Scholar]
- 14.Peers SH, Smillie F, Elderfield AJ, Flower RJ. Glucocorticoid induction of lipocortins (annexins) 1 and 2 in rat peritoneal leukocytes in vivo. Br J Pharmacol. 1993;108:66–72. doi: 10.1111/j.1476-5381.1993.tb13441.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flower RJ, Rothwell NJ. Lipocortin-1: cellular mechanisms and clinical relevance. Trends Pharmacol Sci. 1994;15:71–76. doi: 10.1016/0165-6147(94)90281-x. [DOI] [PubMed] [Google Scholar]
- 16.Kim KM, Kim DK, Park YM, Kim C-K, Na DS. Annexin-1 inhibits phospholipase A2 by specific interaction, not by substrate depletion. FEBS Letters. 1994;343:251–5. doi: 10.1016/0014-5793(94)80566-0. [DOI] [PubMed] [Google Scholar]
- 17.De Catarina R, Sicari R, Giannessi D, et al. Macrophage-specific eicoasonoid synthesis inhibition and lipocortin-1 induction by glucocorticoids. J Appl Physiol. 1993;75:2368–75. doi: 10.1152/jappl.1993.75.6.2368. [DOI] [PubMed] [Google Scholar]
- 18.Johnson MD, Kamso-Pratt JM, Whetsell WO, Pepinski RB. Lipocortin-1 immune reactivity in the normal human central nervous system and lesions with astrocytosis. J Clin Pathol. 1989;92:424–9. doi: 10.1093/ajcp/92.4.424. [DOI] [PubMed] [Google Scholar]
- 19.Strijbos Pjlm, Tilders FJH, Carey F, Forder R, Rothwell NJ. Localization of immunereactive lipocortin-1 in the brain and pituitary gland of the rat. Effects of adrenalectomy, dexamethasone and colchicine treatment. Brain Res. 1991;553:249–60. doi: 10.1016/0006-8993(91)90833-h. [DOI] [PubMed] [Google Scholar]
- 20.Uemura K, Inagaki H, Wada Y, Nakanishi K, Asai K, Kata T, Ando Y, Kannagi R. Identification of immuno-reactive lipocortin 1-like molecules in serum and plasma by an enzyme immunoassay for lipocortin 1. Biochim Biophys Act. 1992;1119:250–5. doi: 10.1016/0167-4838(92)90210-5. [DOI] [PubMed] [Google Scholar]
- 21.Smith T, Flower RJ, Buckingham JC. Lipocortins 1, 2 and 5 in the central nervous system and pituitary gland of the rat: selective induction by dexamethasone of lipocortin 1 in the anterior pituitary gland. Mol Neuropharmacol. 1993;3:45–55. [Google Scholar]
- 22.Bolton C, Elderfield A-J, Flower RJ. The detection of lipocortins 1, 2 and 5 in the central nervous system tissues from Lewis rats with acute experimental allergic encephalomyelitis. J Neuroimmunol. 1990;29:173–81. doi: 10.1016/0165-5728(90)90160-o. [DOI] [PubMed] [Google Scholar]
- 23.Elderfield A-J, Newcombe J, Bolton C, Flower RJ. Lipocortins 1, 2, 4 and 5 are increased in the central nervous system in multiple sclerosis. J Neuroimmunol. 1992;32:91–100. doi: 10.1016/0165-5728(92)90178-n. [DOI] [PubMed] [Google Scholar]
- 24.Carey F, Forder R, Edge MD, Greene AR, Horan MA, Strijbos Pjml, Rothwell NJ. Lipocortin 1 fragment modifies pyrogenic actions of cytokines in rats. Am J Physiol. 1990;259:R266–9. doi: 10.1152/ajpregu.1990.259.2.R266. [DOI] [PubMed] [Google Scholar]
- 25.Strijbos Pjlm, Horan MA, Carey F, Rothwell NJ. Impaired febrile responses in aging mice are mediated by endogenous lipocortin-1 (annexin-1) Am J Physiol. 1993;265:E289–97. doi: 10.1152/ajpendo.1993.265.2.E289. [DOI] [PubMed] [Google Scholar]
- 26.Black MD, Carey F, Crossman AR, Relton JK, Rothwell NJ. Lipocortin-1 inhibits NMDA receptor-mediated neuronal damage in the striatum of the rat. Brain Res. 1992;585:135–40. doi: 10.1016/0006-8993(92)91198-n. [DOI] [PubMed] [Google Scholar]
- 27.Relton JK, Strijbos Pjlm, O'Shaughnessy CT, Carey F, Forder RA, Tilders FJH, Rothwell NJ. Lipocortin-1 is an endogenous inhibitor of ischemic damage in the rat brain. J Exp Med. 1991;174:305–10. doi: 10.1084/jem.174.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cirino G, Flower RJ, Browning JL, Sinclair LK, Pepinsky RB. Recombinant human lipocortin 1 inhibits thromboxane release from ginea pig perfused lung. Nature. 1987;328:270–2. doi: 10.1038/328270a0. [DOI] [PubMed] [Google Scholar]
- 29.Cirino G, Peers SH, Flower RJ, Browning JL, Pepinsky RB. Human recombinant lipocortin 1 has acute local anti-inflammatory properties in the rat paw edema test. Proc Natl Acad Sci USA. 1992;86:3428–31. doi: 10.1073/pnas.86.9.3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perretti M, Ahluwia A, Harris JG, Goulding NJ, Flower RJ. Lipocortin-1 fragments inhibit neutrophil accumulation and neutrophil-dependent edema in the mouse. J Immunol. 1993;151:4306–14. [PubMed] [Google Scholar]
- 31.Wu C-C, Croxtall JD, Perretti M, Bryant CE, Thiemermann C, Flower RJ, Vane JR. Lipocortin 1 mediates the inhibition by dexamethasone of the induction by endotoxin of nitric oxide synthase in the rat. Proc Natl Acad Sci USA. 1995;92:3473–7. doi: 10.1073/pnas.92.8.3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sudlow AW, Carey F, Forder R, Rothwell NJ. The role of lipocortin-1 in dexamethasone-induced suppression of PGE2 and TNFα release from human peripheral blood mononuclear cells. Br J Pharmacol. 1996;117:1449–56. doi: 10.1111/j.1476-5381.1996.tb15305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duncan GS, Peers SH, Carey F, Forder R, Flower RJ. The local anti-inflammatory action of dexamethasone in the rat carageenan oedema model is reversed by an antiserum to lipocortin-1. Br J Pharmacol. 1993;108:62–65. doi: 10.1111/j.1476-5381.1993.tb13440.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dijkstra CD, Döpp EA, Joling P, Kraal G. The heterogeneity of mononuclear phagocytes in lymphoid organs: distinct macrophage subpopulations in the rat recognized by monoclonal antibodies ED1, ED2 and ED3. Immunol. 1985;54:589–99. [PMC free article] [PubMed] [Google Scholar]
- 35.Bauer J, Berkenbosch F, Van Dam A-M, Dijkstra CD. Demonstration of interleukin 1b in Lewis rat brain during experimental autoimmune encephalomyelitis at the light and ultrastructural level. J Neuroimmunol. 1993;48:13–21. doi: 10.1016/0165-5728(93)90053-2. [DOI] [PubMed] [Google Scholar]
- 36.Ruuls SR, Van der Linden S, Sontrop K, Huitinga I, Dijkstra CD. Aggravation of experimental allergic encephalomyelitis by administration of nitric oxide synthase inhibitors. Clin Exp Immunol. 1996;103:467–74. doi: 10.1111/j.1365-2249.1996.tb08304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davies P, Bailey PJ, Goldenberg MM, Ford-Hutchinson AW. The role of arachidonic oxygenation products in pain and inflammation. Ann Rev Immunol. 1984;2:335–57. doi: 10.1146/annurev.iy.02.040184.002003. [DOI] [PubMed] [Google Scholar]
- 38.Prosiegel M, Neu I, Mallinger J, Wildfeuer L, Mehlber S, Vogl G, Hoffmann G, Ruhenstroth-Bauer G. Suppression of experimental allergic encephalomyelitis by dual cyclo-oxygenase and 5-lipoxygenase inhibition. Acta Neurol Scand. 1989;79:223–6. doi: 10.1111/j.1600-0404.1989.tb03742.x. [DOI] [PubMed] [Google Scholar]
- 39.Ruddle NH, Bergman CM, McGrath KM, Lingenheld EG, Grunnet ML, Padula SJ, Clark RB. An antibody to lymphotoxin and tumor necrosis factor prevents transfer of experimental allergic encephalomyelitis. J Exp Med. 1990;172:1193–2000. doi: 10.1084/jem.172.4.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gold R, Pepinsky RB, Zettl UK, Toyka KV, Hartung H-P. Lipocortin-1 (annexin-1) suppresses activation of autoimmune T cell lines in the Lewis rat. J Neuroimmunol. 1996;69:157–64. doi: 10.1016/0165-5728(96)00086-0. [DOI] [PubMed] [Google Scholar]
- 41.Schmidt ED, Binnekade R, Janszen Awjw, Tilders FJH. Short stressor induced long-lasting increases of vasopressin stores in the hypothalamic corticotropin-releasing hormone (CRH) neurons in adult rats. J Neuroendocrinol. 1996;8:703–12. [PubMed] [Google Scholar]
- 42.Schmidt ED, Janszen Awjw, Wouterlood FG, Tilders FJH. Interleukin-1-induced long-lasting changes in hypothalamic corticotropin-releasing hormone (CRH)-neurons and hyperresponsiveness of the hypothalamus–pituitary–adrenal axis. J Neurosci. 1995;15:7417–26. doi: 10.1523/JNEUROSCI.15-11-07417.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taylor AD, Loxley HD, Flower RJ, Buckingham JC. Immunoneutralization of lipocortin 1 reverses the acute inhibitory effects of dexamethasone on the hypothalamo–pituitary–adrenocortical responses to cytokines in the rat in vitro and in vivo. Neuroendocrinol. 1995;62:19–31. doi: 10.1159/000126984. [DOI] [PubMed] [Google Scholar]
- 44.Buckingham JC. Stress and the neuroendocrine-immune axis: the pivotal role of glucocorticoids and lipocortin 1. Br J Pharmacol. 1996;118:1–19. doi: 10.1111/j.1476-5381.1996.tb15360.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Loxley HD, Cowell A-M, Flower RJ, Buckingham JC. Effects of liportin 1 and dexamethasone on the secretion of corticothropin-releasing factors in the rat: in vitro and in vivo studies. J Neuroendocrinol. 1993;5:51–61. doi: 10.1111/j.1365-2826.1993.tb00363.x. [DOI] [PubMed] [Google Scholar]
- 46.Mullens L, Marriott DR, Young KA, Tannahill L, Lightman SL, Wilkin GP. Up-regulation of lipocortin-1 and its mRNA in reactive astrocytes in kainate-lesioned rat cerebellum. J Neuroimmunol. 1994;50:25–33. doi: 10.1016/0165-5728(94)90211-9. [DOI] [PubMed] [Google Scholar]
- 47.Johnson MD, Kamso-Pratt J, Pepinsky RB, Whetsell WO. Lipocortin-1 immune reactivity in central and peripheral nervous system glial tumors. Hum Pathol. 1989;800:772–6. doi: 10.1016/0046-8177(89)90071-3. [DOI] [PubMed] [Google Scholar]
- 48.Gebicke-Haerter PJ, Schobert A, Dieter P, Honegger P, Hertting G. Regulation and glucocorticoid-independent induction of lipocortin 1 in cultured astrocytes. J Neurochem. 1991;57:175–83. doi: 10.1111/j.1471-4159.1991.tb02113.x. [DOI] [PubMed] [Google Scholar]
- 49.Polman CH, Hartung H-P. The treatment of multiple sclerosis: current and future. Curr Opin Immunol. 1995;8:200–9. doi: 10.1097/00019052-199506000-00008. [DOI] [PubMed] [Google Scholar]