

Abstract
EAE is an autoimmune disease of the central nervous system (CNS) that serves as an experimental model for the human inflammatory demyelinating disease multiple sclerosis (MS). Antigen-based immunotherapy including soluble antigen administration via feeding has been shown to be successful in treating EAE in rodents. In the present study, we explore nasal administration of small amounts of myelin basic protein (MBP) as a potential means of treatment of protracted, relapsing EAE (PR-EAE) in a novel DA rat system. We found that nasal administration of MBP prevented EAE induced with whole spinal cord homogenate + Freund's incomplete adjuvant (FIA), and strongly down-regulated levels of MBP-reactive interferon-gamma (IFN-γ)-secreting Th1-like cells. However, in rats with ongoing PR-EAE receiving the same regimen of MBP, a trend of aggravated disease was recorded, in conjunction with augmented levels of MBP-reactive IFN-γ-secreting Th1-like splenocytes during the acute phase of EAE. These data have implications for the clinical application of nasal tolerance to autoimmune diseases.
Keywords: experimental autoimmune encephalomyelitis, myelin basic protein, nasal tolerance, Th1 response, ELISPOT, interferon-gamma
INTRODUCTION
EAE in DA rats can be induced with crude central nervous system (CNS) preparations, myelin basic protein (MBP) or myelin proteolipid protein (PLP) [1, 2]. DA rats immunized with syngeneic spinal cord and Freund's incomplete adjuvant (FIA) show a protracted, relapsing EAE (PR-EAE) that is regarded as a model more closely related to multiple sclerosis (MS) than acute EAE in the Lewis rat [2]. PR-EAE therefore provides a good model for studying various strategies of immunomodulation in order to suppress the clinical disease [3–5].
Tolerance induction via mucosal membranes has been widely recognized as an effective means for the prevention of immune-mediated diseases in animal models like EAE [6–8] and experimental autoimmune myasthenia gravis (EAMG) [9], and has been found in preliminary studies to be a useful treatment for human disorders with probable autoimmune backgrounds (reviewed in [10]). Tolerance induction by nasal antigen administration is an active process analogous to oral tolerance induction [11–13]. Antigen inhalation as a strategy for tolerance induction to autoimmune diseases has evoked increasing interest [12, 14, 15]. Some studies have shown that the nasal pathway is more effective in terms of dosage used in induction of tolerance compared with the oral route [16, 17].
In the present study, we explored nasal administration of MBP as potential means of pre-inducing tolerance to PR-EAE versus treatment of PR-EAE. Nasal administration of MBP before EAE induction prevented clinical PR-EAE in DA rats, while PR-EAE was exacerbated when using the same dosage to treat ongoing EAE.
MATERIALS AND METHODS
Antigen preparation
MBP was purified from bovine spinal cord [18], and acetylcholine receptors (AChR) from the electroplax tissue of Torpedo californica (Pacific Biomarine, Venice, CA) [19]. Purity of both antigens was confirmed by SDS–PAGE.
Spinal cord homogenate
Freshly isolated syngeneic spinal cords from inbred DA rats were homogenized with a minimal amount of saline, frozen at −70°C and lyophilized.
Intranasal tolerance induction
The schedule for intranasal tolerance induction in naive rats was performed as previously described in other autoimmune disease models [11, 17]. Two groups of rats received into each nostril 30 μl of either PBS pH 7·4 containing bovine MBP at a concentration of 1 mg/ml, or PBS only, using a micropipette. At each administration, rats were gently anaesthetized with ether. The administrations were performed daily for 10 consecutive days and each rat received a total amount of 600 μg of MBP. For each administration, the dosage was 60 μg/rat. Rats rested for another 3 days before induction of EAE.
We used two different concentrations of MBP (1 mg or 2·7 mg/ml) to treat ongoing EAE in DA rats. A schedule similar to that used in naive rats was adopted, i.e. rats received different doses of MBP or PBS nasally from day 7 or 9 p.i. for 10 consecutive days. Thus, the total doses of MBP were 600 μg or 1·6 mg/rat. For each administration, the doses were 60 μg/rat or 160 μg/rat.
Induction of PR-EAE
For induction of PR-EAE, each rat was immunized subcutaneously in the dorsal tail base with 200 μl of inoculum containing 100 mg syngeneic spinal chord homogenate (SCH) and 100 μl FIA (Difco, Detroit, MI). The rats were weighed every other day and evaluated in a blinded fashion by at least two investigators every day for the presence of clinical signs. Clinical scores were graded according to the following criteria: 0, asymptomatic; 1, flaccid tail; 2, loss of righting reflex with or without partial hind limb paralysis; 3, complete hind limb paralysis; 4, moribund; and 5, dead.
Immunohistochemistry
Spinal cords from killed animals were dissected and segments of the lumbar spinal cord were snap-frozen in liquid nitrogen. Cryostat sections (10 μm) were fixed in acetone at −20°C. Non-specific binding was blocked with normal horse serum (Vector, Burlingame, CA) for 30 min. Macrophage and T lymphocyte infiltrates were characterized in serial sections by incubation with 5 μg/ml of the MoAbs ED1 and W3/13 (Serotec, Oxford, UK), which label macrophage and pan-T cells, respectively. Biotinylated horse anti-mouse antibody (Vector) was used to couple the primary MoAbs with an avidin–biotin–peroxidase complex (Vector). After washing in PBS, substrate amino-ethyl carbazole (AEC) was applied to the tissue and incubated for 5 min. The slides were rinsed in tap water, and mounted with Aquamount (Lerner Labs, New Haven, CT). Controls included the omission of the primary antibody and second antibody. Background staining was negligible.
Preparation of mononuclear cells from spleen
Spleen cell suspensions were prepared by grinding through a wire mesh. Erythrocytes in spleen cell suspension were osmotically lysed. Cells were then washed three times in complete modified Eagles' medium (CDME; Flow Labs, Irvine, UK) supplemented with 1% (v/v) non-essential amino acids (Flow), 50 U/ml penicillin, 60 μg/ml streptomycin (Gibco, Paisley, UK), 2 mm glutamine (Flow) and 3% (v/v) heat-inactivated fetal calf serum (FCS; Gibco). Splenocytes were then adjusted to 2 × 106/ml.
Enumeration of MBP-reactive IFN-γ-secreting cells
A solid-phase enzyme-linked immunospot assay (ELISPOT) was used to enumerate splenocytes secreting IFN-γ upon in vitro stimulation with MBP [20]. Briefly, 96-well microtitre plates with nitrocellulose bottoms (Microtiter-HAM; Millipore Co., Bedford, MA) were coated with 100 μl aliquots per well of mouse anti-rat IFN-γ MoAbs [21] at 15 μg/ml at 4°C overnight. After washing with PBS, aliquots containing 200 μl of cell suspension with 4 × 105 splenocytes were added to individual wells in triplicate, followed by adding antigen (AChR or MBP) or mitogen (concanavalin A (Con A); Sigma, St Louis, MO) in 10-μl aliquots to a final concentration of 10 μg/ml. After incubation at 37°C in 7% CO2 and humidified atmosphere for 48 h, the wells were extensively washed. The plates were incubated with 100 μl of rabbit polyclonal anti-rat IFN-γ antibody [21] diluted 1:2000 for 4 h at 20°C. After washing, the plates were incubated with biotinylated swine anti-rabbit IgG and with avidin–biotin–peroxidase complex followed by peroxidase staining. The red-brown immunospots which corresponded to the cells that had secreted interferon-gamma (IFN-γ) were counted in a dissection microscope. To calculate the numbers of T cells corresponding to a particular antigen or mitogen, numbers of spots in culture without added antigen or mitogen which are considered as cells spontaneously secreting IFN-γ, were subtracted from values obtained after antigen or mitogen challenge. Data are expressed as numbers of spots per 105 cultured splenocytes originally applied per well. The variation between triplicate values was < 10%.
Lymphocyte proliferation assays
For assessment of antigen-induced lymphocyte proliferation, a standard 3H-thymidine incorporation test was used. Briefly, 200-μl aliquots of splenocyte suspensions from EAE rats were set up in triplicate in round-bottomed 96-well polystyrene microtitre plates (Nunc, Copenhagen, Denmark) at a cell density of 2 × 106 cells/ml in culture medium. Antigen or Con A was added at the same final concentrations as in the IFN-γ ELISPOT assay. After 60 h of incubation, the cells were pulsed with 3H-methyl-thymidine (1 μCi/well; Amersham, Aylesbury, UK) for another 12 h. Cells were harvested onto glassfibre filters (Titertek, Skatron A/S, Lierbyen, Norway) and 3H-thymidine incorporation was measured in a liquid β-scintillation counter.
Statistical analysis
Differences between pairs of groups were tested by Student's t-test. Differences between three or more groups were tested by one-factor analysis of variance (anova). The level of significance was set to α = 0·05. All tests were two-sided.
RESULTS
Nasal administration of MBP prevents EAE in DA rats
As shown in Fig. 1, control rats immunized with SCH + FIA and receiving PBS nasally before immunization developed protracted, relapsing EAE, with mean onset on day 11·1 ± 0·9 p.i.; a mean maximal clinical score of 3·25 ± 1·7; and a mean maximal body weight loss of 27 ± 17·1 g. A typical protracted, relapsing course was observed in these rats, with a mean relapse number of 2 ± 0·7.
Fig. 1.

Nasal administration of myelin basic protein (MBP) prevented protracted, relapsing (PR)-EAE in DA rats. Two groups of 13 DA rats received nasally either PBS (□) or 600 μg/rat of MBP (▪) administered 1 week before immunization of 6–8-week-old male DA rats with syngeneic spinal cord homogenate (SCH) + Freund's incomplete adjuvant (FIA). Control rats receiving PBS by the nasal route developed typical PR-EAE, as reflected by high clinical scores (a) and body weight loss (b). In contrast, only one out of 13 rats receiving MBP nasally before induction of EAE developed mild and transient EAE.
In contrast to control rats, only one out of 13 rats pretreated with MBP nasally developed slight (score 1·0), and transient (duration 6 days) EAE.
The prevention of EAE was also confirmed at the tissue level. Rats pretreated with PBS nasally followed by induction of EAE with SCH + FIA showed extensive macrophage and T cell infiltration in spinal cord sections (data not shown). In contrast, rats pretreated with MBP nasally followed by immunization showed no or minimal infiltration of macrophages or T cells in spinal cord sections examined (data not shown).
Effects of nasal administration of MBP on ongoing EAE in DA rats
In studying the feasibility of nasal antigen administration for treatment of human autoimmune diseases, it becomes critical to determine whether nasal antigen administration can suppress an ongoing disease process. Since nasal administration of 600 μg/rat of MBP effectively prevented PR-EAE development in DA rats, the same as well as a higher dosage (total 1·6 mg/rat) were used to evaluate the therapeutic effects. Treatment was started on day 9 p.i., i.e. when the first clinical signs appeared. As shown in Fig. 2a and Table 1, untreated rats and rats receiving PBS nasally developed PR-EAE without revealing any differences. In contrast, rats receiving low-dose MBP (600 μg/rat) treatment developed more severe EAE in terms of earlier onset, increased accumulating score, mean maximal score, relapse numbers and higher body weight loss compared with PBS-treated rats. Thus, no ameliorative effects were observed in rats treated with low dose (total 600 μg/rat) MBP, but rather a trend of aggravated disease was recorded in the acute phase of EAE in rats thus treated. Compared with PBS-treated rats, no significant difference was observed in rats treated with higher dosage (1·6 mg/rat) of MBP regarding mean day of onset, mean maximal score and mean accumulating score, and one rat treated thus died of EAE in the acute phase (Table 1).
Fig. 2.
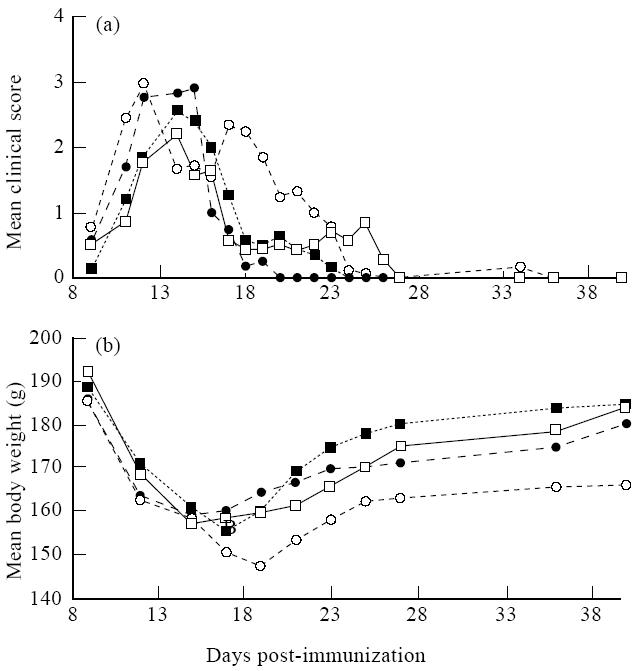
Effects of nasal administration of myelin basic protein (MBP) on ongoing EAE in DA rats. EAE was induced as described in Materials and Methods. Nasal administration of MBP was initiated at day 9 p.i. Control rats (untreated (□) or PBS-treated nasally (▪)) and MBP-treated rats (total 600 μg/rat (○) or 1.6 mg/rat (•)) were observed up to 40 days p.i. Earlier onset, more severe EAE (a) and more body weight loss (b) (P< 0.05 from days 20–40 p.i. compared with PBS-treated control rats) were observed in rats treated with low dose (600 μg/rat) of MBP.
Table 1.
Therapeutic effects of nasal administration of myelin basic protein (MBP) on incipient protracted, relapsing (PR)-EAE in DA rats

To test further whether nasal administration of MBP exacerbates ongoing EAE in DA rats during the acute phase, another group of 10 DA rats was immunized with reduced amounts of immunogen (50 mg SCH/rat), and the treatment was started on day 7 p.i. As shown in Fig. 3, rats receiving PBS nasally developed mild and transient EAE, with a mean onset on day 10·8 ± 1·1 p.i., and mean maximal score of 1·1 ± 0·2. In contrast, rats receiving MBP nasally developed severe EAE, with mean onset of EAE on day 10 ± 1·4 p.i., and mean maximal score of 3·2 ± 1·3 (P = 0·008 versus control rats). EAE was lethal in one of the five MBP-treated rats.
Fig. 3.

To define further whether nasal administration of myelin basic protein (MBP) exacerbates ongoing EAE, another group of 10 DA rats was immunized with reduced amount of immunogen (50 mg/rat of spinal cord homogenate (SCH) + Freund's incomplete adjuvant (FIA)). Five control rats receiving PBS nasally from day 7 to day 16 p.i. (□) developed slight EAE, while five rats receiving MBP nasally (▪, 600 μg/rat) from day 7 to day 16 p.i. developed enhanced EAE both in terms of enhanced mean clinical score (a) and mean body weight loss (b) (P<0.05 from day 11 to day 16 p.i.).
T cell responses in tolerized rats and rats with exacerbated EAE
To gain further insight into the suppression and exacerbation of EAE after nasal administration of MBP, we evaluated T cell responses in these rats. Since no proliferative responses to MBP were recorded in rats immunized according to the current protocol (data not shown), we used ELISPOT assays to evaluate frequencies of MBP-reactive splenocytes which, upon stimulation with MBP, secrete IFN-γ. As shown in Fig. 4a, rats pretreated with PBS nasally had high numbers of MBP-reactive IFN-γ-secreting cells both on day 13 and day 60 p.i. In contrast, rats pretreated with MBP nasally had strongly reduced numbers of MBP-reactive IFN-γ-secreting splenocytes when examined at both time points. MBP-reactive IFN-γ-secreting splenocytes were examined in rats receiving the same dosage of MBP nasally from day 9 p.i. and for 10 consecutive days, i.e. after onset of clinical EAE. Higher numbers of MBP-reactive IFN-γ-secreting splenocytes were detected in rats during the process of treatment (days 11 and 13 p.i., two time points chosen to represent acute EAE) compared with PBS-treated control rats. On day 26 p.i., i.e. 1 week after MBP treatment, no difference of numbers of such cells was recorded between MBP- and PBS-treated rats (Fig. 4b).
Fig. 4.

Influence of nasal administration of myelin basic protein (MBP) on levels of IFN-γ-secreting Th1-like splenocytes. Splenocytes were prepared from killed rats and ELISPOT assays were carried out as described in Materials and Methods. Using our protocol, control wells without added MBP or wells with the irrelevant control antigen acetylcholine receptor (AChR) showed very few spots (<5 per 105 splenocytes), and were subtracted from numbers of IFN-γ-secreting MBP-reactive cells. Results are expressed as mean numbers from five rats ± s.d. (a) Rats were pretreated nasally either with PBS (□) or 600 μg/rat of MBP (▪) before induction of EAE as scheduled in Materials and Methods. Groups of five rats were killed on day 13 or day 60 p.i. Strongly reduced numbers of MBP-reactive IFN-γ-secreting splenocytes were recorded at both time points in rats pretreated with MBP versus those treated with PBS nasally. (b) EAE was induced in DA rats by immunization with spinal cord homogenate (SCH) + Freund's incomplete adjuvant (FIA), followed by nasal administration of either PBS or 60 μg/rat of MBP from day 9 p.i. and for 10 consecutive days. Groups of five rats were killed at different time points during or after this treatment, and numbers of MBP-reactive IFN-γ-secreting splenocytes were evaluated by ELISPOT. No difference of numbers of MBP-reactive IFN-γ-secreting cells was observed between MBP-treated (▪) and PBS-treated control rats (□) on day 26 p.i., i.e. 1 week after treatment was stopped. However, higher numbers of MBP-reactive IFN-γ-secreting splenocytes were observed in MBP-treated rats (▪) when examined on days 11 and 13 p.i., i.e. during the early phase of MBP treatment, versus control rats (□). (c) EAE was induced by immunization of DA rats with SCH + FIA, followed by administration of either PBS or 160 μg/rat of MBP nasally from day 9 p.i. and for 10 consecutive days. Groups of five rats were killed at different time points during (day 11 p.i.) or after (day 26 p.i.) treatment, and numbers of MBP-reactive IFN-γ-secreting splenocytes were evaluated. Higher numbers of IFN-γ-secreting cells on day 11 p.i. (early phase of MBP treatment) but lower numbers of such cells on day 26 p.i. (1 week after MBP treatment was stopped) were observed in MBP-treated versus PBS-treated control rats. ***P< 0.001; **P< 0.01; *P< 0.05.
T cell responses were also evaluated in rats treated with a high dose (1·6 mg/rat) of MBP nasally from day 9 to 18 p.i. Higher numbers of MBP-reactive IFN-γ-secreting cells were detected during the early stage of treatment (day 11 p.i.), while numbers of such cells were lower on day 26 p.i. versus PBS-treated control rats examined at these two time points (Fig. 4c).
DISCUSSION
In this study we observed that nasal administration of MBP prevented PR-EAE in DA rats. In rats receiving a total dose of 600 μg/rat of MBP nasally before immunization, PR-EAE was completely blocked. The prevention of PR-EAE was accompanied by strongly reduced levels of MBP-reactive IFN-γ-secreting Th1-like splenocytes, which reflected down-regulated Th1 immunity in these tolerized rats. Our data confirm those from previous studies [16, 22], and suggest that (i) nasal administration of soluble antigens is highly effective in establishing naive T cell tolerance in vivo, and (ii) nasal administration requires much lower antigen amounts compared with the oral route [8, 9]. Furthermore, nasal administration of small amounts of MBP, i.e. a single antigen, prevented EAE induced with a spinal cord homogenate, a heterogeneous mixture of multiple potential autoantigens, each bearing a number of dominant, subdominant and cryptic determinants. This suggests that, in addition to an antigen-specific component, nasal tolerance evokes components of non-specific bystander regulation as previously suggested [23, 24]. Thus, these data have important implications for the design of antigen- or T cell-specific therapy of inflammatory diseases with putative autoimmune background such as MS, type I diabetes, and rheumatoid arthritis, where detrimental reactivities to multiple autoantigens in the target are suggested to occur.
However, when using the same schedule and dosage that had been shown to be highly effective in preventing EAE, in order to treat ongoing PR-EAE in DA rats, onset of treatment on day 9 p.i. revealed no beneficial effects, but rather a trend of aggravated disease compared with control rats. By reducing the amount of immunogen, mild EAE developed in control rats receiving PBS only by the nasal route, while rats treated with MBP (total dose 600 μg/rat) had severe and even lethal EAE in the acute EAE phase. ELISPOT assays revealed expansion of MBP-reactive Th1-like splenocytes in these nasally MBP-treated rats during the acute phase of EAE.
It is likely that inhalation tolerance would be more difficult to achieve after T cell priming, probably because: (i) the frequency of antigen-specific cells is higher; (ii) most antigen-specific T cells are activated; (iii) the immune response may have spread to include other T cell epitopes; and (iv) the effector arms of the immune response, such as inflammatory macrophages, have come into play. On the other hand, time is needed to establish tolerance, and before tolerance has been established, activated T cells may produce Th1 cytokines and proliferate in vivo after exposure to specific antigens, and consequently lead to exacerbation of ongoing disease. This is supported by our present data that nasal administration of small amounts of soluble antigen could lead to expansion of antigen-specific Th1 cells at the early phase of treatment (days 11 and 13 p.i.) and result in exacerbation of ongoing disease.
A recent study [25] indicates that oral administration of low-dose antigen induces activation of T cells followed by tolerization, while high-dose antigen induces tolerance without activation. Similar phenomena have also been observed in nasal tolerance induction, that transient activation of CD4+ T cells precedes the development of tolerance in vivo in mice administered house dust mite (HDM) antigen nasally [26]. Since inhalation tolerance is likely to be a low-dose tolerance, it is important and necessary to apply the induction of nasal tolerance to clinical medicine in such a manner that nasal tolerance is induced without activation of an adverse immune response.
Previous studies [27, 28] indicate that the frequency of reactive T cells versus the amount of antigen could be an important parameter in the induction of certain forms of tolerance. Thus, one possibility to induce tolerance in established disease is to increase the amount of tolerogen. Indeed, when we in the present study used a higher dosage (total 1·6 mg/rat) of MBP to treat ongoing EAE, reduced numbers of MBP-specific Th1 responses were observed on day 26 p.i. in treated rats, i.e. 1 week after treatment was stopped, which may indicate a long-term benefit of therapy using this dose. However, no ameliorative effect was observed in the acute phase of EAE in high-dose MBP-treated rats, and one rat even died of EAE. In line with the clinical observation, T cell expansion as reflected by ELISPOT assays was also observed in high-dose MBP-treated rats when examined on day 11 p.i., i.e. the acute phase of EAE. Thus, perhaps an even higher dosage of MBP is required to suppress acute EAE. Nonetheless, high concentrations of protein could induce an inflammatory reaction in the nasal mucosa and possibly in the lungs as well, and thereby preclude this modality for human therapeutic trials. Another possibility is to use altered peptide ligands (APL), that may have high tolerance induction efficacy. However, the effect of APL on the early phase of ongoing EAE is still unpredictable [22]. A recent study [29] has clearly indicated an alternative pathway to establish tolerance in ongoing disease, namely by employing strategies to reduce CD4+ T cell numbers and/or responsiveness before antigen is introduced, which could be a solution of the current problems.
In conclusion, we show that nasal administration of MBP before induction of EAE can prevent EAE in DA rats. However, when using this approach to treat ongoing EAE, a trend towards aggravated disease was recorded in the acute phase. This aggravation is likely to be related to the further activation of primed T cells during the early process of treatment. For the application of nasal tolerance in clinical medicine, it is important to identify measures that counteract, but do not activate, an adverse immune response.
Acknowledgments
This study was supported by grants from the Swedish Medical Research Council, the Swedish MS Society (NHR) and Karolinska Institute.
References
- 1.Lorentzen JC, Issazadeh S, Storch M, et al. Protracted, relapsing and demyelinating experimental autoimmune encephalomyelitis in DA rats immunized with syngeneic spinal cord and incomplete Freund's adjuvant. J Neuroimmunol. 1995;63:193–205. doi: 10.1016/0165-5728(95)00153-0. [DOI] [PubMed] [Google Scholar]
- 2.Stepaniak JA, Gould KE, Sun D, Swanborg RH. A comparative study of experimental autoimmune encephalomyelitis in Lewis and DA rats. J Immunol. 1995;155:2762–9. [PubMed] [Google Scholar]
- 3.Fairchild PJ, Thorpe CJ, Traver PJ, Wraith DC. Modulation of the immune response with T-cell epitopes: the ultimate goal for specific immunotherapy of autoimmune disease. Immunology. 1994;81:487–96. [PMC free article] [PubMed] [Google Scholar]
- 4.Brocke S, Gijbels K, Allegretta M, et al. Treatment of experimental encephalomyelitis with a peptide analogue of myelin basic protein. Nature. 1996;379:343–6. doi: 10.1038/379343a0. [DOI] [PubMed] [Google Scholar]
- 5.Waisman A, Ruiz PJ, Hirschberg DL, et al. Suppressive vaccination with DNA encoding a variable region gene of the T-cell receptor prevents autoimmune encephalomyelitis and activates Th2 immunity. Nature Med. 1996;2:899–905. doi: 10.1038/nm0896-899. [DOI] [PubMed] [Google Scholar]
- 6.Weiner HL, Friedman A, Miller A, et al. Oral tolerance: immunologic mechanisms and treatment of animal and human organ-specific autoimmune diseases by oral administration of autoantigens. Annu Rev Immunol. 1994;12:809–37. doi: 10.1146/annurev.iy.12.040194.004113. [DOI] [PubMed] [Google Scholar]
- 7.Whitacre CC, Gienapp IE, Meyer A, Cox KL, Javed N. Treatment of autoimmune disease by oral tolerance to autoantigens. Clin Immunol Immunopathol. 1996;80:S31–S39. doi: 10.1006/clin.1996.0139. [DOI] [PubMed] [Google Scholar]
- 8.Meyer AL, Benson JM, Gienapp IE, Cox KL, Whitacre CC. Suppression of murine chronic relapsing experimental autoimmune encephalomyelitis by the oral administration of myelin basic protein. J Immunol. 1996;157:4230–8. [PubMed] [Google Scholar]
- 9.Wang ZY, Qiao J, Link H. Suppression of experimental autoimmune myasthenia gravis by oral administration of acetylcholine receptor. J Neuroimmunol. 1993;44:209–14. doi: 10.1016/0165-5728(93)90045-z. [DOI] [PubMed] [Google Scholar]
- 10.Weiner HL. Oral tolerance: immune mechanisms and treatment of autoimmune diseases. Immunol Today. 1997;18:335–43. doi: 10.1016/s0167-5699(97)01053-0. [DOI] [PubMed] [Google Scholar]
- 11.Dick AD, Cheng YF, McKinnon A, Liversidge J, Forrester JV. Nasal administration of retinal antigens suppresses the inflammatory response in experimental allergic uveoretinitis. A preliminary report of intranasal induction of tolerance with retinal antigens. Br J Ophthalmol. 1993;77:171–5. doi: 10.1136/bjo.77.3.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dick AD, Cheng YF, Liversidge J, Forrester JV. Intranasal administration of retinal antigens suppresses retinal antigen-induced experimental autoimmune uveoretinitis. Immunology. 1994;82:625–31. [PMC free article] [PubMed] [Google Scholar]
- 13.McMenamin C, Holt PG. The natural immune response to inhaled soluble protein antigens involves major histocompatibility complex (MHC) class I-restricted CD8+ T cell-mediated but class II-restricted CD4+ T cell-dependent immune deviation resulting in selective suppression of immunoglobulin E production. J Exp Med. 1993;178:889–99. doi: 10.1084/jem.178.3.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Daniel D, Wegmann DR. Protection of nonobese diabetic mice diabetes by intranasal or subcutaneous administration of insulin peptide B-(9-23) Proc Natl Acad Sci USA. 1996;93:956–60. doi: 10.1073/pnas.93.2.956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wraith DC. Antigen-specific immunotherapy of autoimmune disease: a commentary. Clin Exp Immunol. 1996;103:349–52. doi: 10.1111/j.1365-2249.1996.tb08286.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Metzler B, Wraith DC. Inhibition of experimental autoimmune encephalomyelitis by inhalation but not oral administration of the encephalitogenic peptide: inhibition of MHC binding affinity. Int Immunol. 1993;5:1159–65. doi: 10.1093/intimm/5.9.1159. [DOI] [PubMed] [Google Scholar]
- 17.Ma CG, Zhang GX, Xiao BG, Link J, Olsson T, Link H. Suppression of experimental autoimmune myasthenia gravis by nasal administration of acetylcholine receptor. J Neuroimmunol. 1995;58:51–60. doi: 10.1016/0165-5728(94)00187-s. [DOI] [PubMed] [Google Scholar]
- 18.Deibler GE, Martensson RE, Kies MW. Large scale preparation of myelin basic protein from central nervous tissue of several mammalian species. Prep Biochem. 1972;2:139–64. doi: 10.1080/00327487208061467. [DOI] [PubMed] [Google Scholar]
- 19.Lindström J, Einarson B, Tzartos S. Production and assay of antibodies to acetylcholine receptors. Methods Enzymol. 1981;74:432–60. doi: 10.1016/0076-6879(81)74031-x. [DOI] [PubMed] [Google Scholar]
- 20.Link H, Olsson O, Sun J-B, et al. Acetylcholine receptor-reactive T and B cells in myasthenia gravis and controls. J Clin Invest. 1991;87:2191–6. doi: 10.1172/JCI115253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van der Meide PH, Dubbeld M, Vijverberg K, Kos T, Schellekens H. The purification of rat gamma interferon by use of two monoclonal antibodies. J Gen Virol. 1986;67:1059–71. doi: 10.1099/0022-1317-67-6-1059. [DOI] [PubMed] [Google Scholar]
- 22.Metzler B, Wraith DC. Mucosal tolerance in a murine model of experimental autoimmune encephalomyelitis. Ann NY Acad Sci. 1996;778:228–43. doi: 10.1111/j.1749-6632.1996.tb21131.x. [DOI] [PubMed] [Google Scholar]
- 23.Zhang ZJ, Lee CSY, Lider O, Weiner HL. Suppression of adjuvant arthritis in Lewis rats by oral administration of type II collagen. J Immunol. 1990;145:2489–93. [PubMed] [Google Scholar]
- 24.Al-Sabbagh A, Miller A, Santos LMB, Weiner HL. Antigen-driven tissue-specific suppression following oral tolerance: orally administered myelin basic protein suppresses proteolipid protein-induced experimental autoimmune encephalomyelitis in the SJL mouse. Eur J Immunol. 1994;24:2104–9. doi: 10.1002/eji.1830240926. [DOI] [PubMed] [Google Scholar]
- 25.Yoshida T, Hachimura S, Kaminogawa S. The oral administration of low-dose antigen induces activation followed by tolerization, while high-dose antigen induces tolerance without activation. Clin Immunol Immunopathol. 1997;82:207–15. doi: 10.1006/clin.1996.4319. [DOI] [PubMed] [Google Scholar]
- 26.Hoyne GF, Askonas BA, Hetzel C, Thomas WR, Lamb JR. Regulation of house dust mite responses by intranasally administered peptide: transient activation of CD4+ T cells precedes the development of tolerance in vivo. Int Immunol. 1996;8:335–42. doi: 10.1093/intimm/8.3.335. [DOI] [PubMed] [Google Scholar]
- 27.Kearney ER, Pape KA, Loh DY, Jenkins MK. Visualization of peptide-specific T cell immunity and peripheral tolerance induction in vivo. Immunity. 1994;1:327–39. doi: 10.1016/1074-7613(94)90084-1. [DOI] [PubMed] [Google Scholar]
- 28.Förster I, Hirose R, Arbeit JM, Clausen BE, Hanahan D. Limited capacity for tolerization of CD4+ T cells specific for a pancreatic beta cell neoantigen. Immunity. 1995;2:573–85. doi: 10.1016/1074-7613(95)90002-0. [DOI] [PubMed] [Google Scholar]
- 29.Lanoue A, Bona C, von Boehmer H, Sarukhan K. Conditions that induce tolerance in mature CD4+ T cells. J Exp Med. 1997;185:405–14. doi: 10.1084/jem.185.3.405. [DOI] [PMC free article] [PubMed] [Google Scholar]