

Abstract
It has been previously reported that the expression of the complement receptor, CR1, on erythrocytes is reduced in patients with RA and that the reduced expression of CR1 is related to disease activity. In this study we investigate the role of other regulatory proteins, i.e. decay-accelerating factor (DAF) and CD59 (membrane inhibitor of reactive lysis), in the pathogenesis of RA by checking the expression of DAF and CD59 on erythrocytes of RA patients to establish whether reduced expression of DAF and CD59 on erythrocytes could be related to increased ability of erythrocytes to activate complement in RA. Flow cytometry was used to measure the expression of DAF and CD59 on erythrocytes from RA patients as well as the deposition of C3 fragments occurring in vivo or after in vitro complement activation. Significantly reduced expression of DAF and CD59 was observed on erythrocytes of RA patients. A significant inverse relationship was observed between DAF expression and in vitro complement activation, whereas no significant relationship between CD59 and complement activation was observed. Finally, we demonstrated an inverse relationship between CH50 activity and DAF expression. Thus, determination of DAF on erythrocytes can emerge as an additional tool in the assessment of extent of complement activation in RA.
Keywords: rheumatoid arthritis, decay-accelerating factor, CD59, erythrocytes, complement
INTRODUCTION
The complement system is considered to be important in the control of infections owing to its ability to act as an effector molecule to lyse many microorganisms and cells [1]. In autoimmune diseases like RA, complement produces tissue damage because it is activated under inappropriate circumstances. The uncontrolled activation of the complement system is regulated by several proteins, i.e. CR1/CD35, decay-accelerating factor (DAF)/CD55, membrane cofactor protein/CD46, CD59, C8 binding protein, Factor I, Factor H and C1 inhibitor [2].
Erythrocytes of many patients with RA display a relative deficiency of complement receptor 1 (CR1) [3], and one of the functional consequences of this may be impaired clearance of immune complexes from the circulation [4–7]. The deficiency of complement-regulatory proteins on erythrocytes of patients with RA is not restricted to the CR1, and the role of other complement-regulatory proteins in the pathogenesis of this disease requires investigation.
DAF is an ≈ 70-kD glycoprotein [8] that is expressed on peripheral blood, vascular endothelial and extravascular epithelial cell surfaces [9–12]. It inhibits the activity of C3 convertase of the classical and alternative pathways. Findings that affected cells of patients with paroxysmal nocturnal haemoglobinuria which lack DAF accumulate C3b in vivo have established a physiologic role for DAF in protection of cells from autologous complement [13–15]. CD59 is a 18-kD phosphatidylinositol-linked membrane protein that is expressed on erythrocytes, lymphocytes, monocytes, neutrophils, platelets, endothelia/epithelia, etc. It blocks C9 binding to C5b-8, preventing membrane attack complex formation and lysis [16–18].
The present study was conducted to understand the role of DAF and CD59 in the pathogenesis of RA. The study investigates the expression of DAF and CD59 on erythrocytes, in vivo deposition of C3 fragments and cellular uptake of C3 fragments in vitro in order to establish that reduced expression of complement-regulatory proteins is related to the decreased ability of erythrocytes to regulate complement pathway. Finally, to correlate the protein expression with the CH50 activity, CH50 activity in sera of normal controls and RA patients was calculated.
MATERIALS AND METHODS
Patients
Thirty-five patients (nine males and 26 females, mean age 35·9 years, range 17–56 years) were selected from the Rheumatology Clinic of AIIMS. All patients fulfilled the American Rheumatism Association (ARA) revised criteria for the classification of RA. There was no renal or skin involvement in any of the above mentioned patients. Each sample was separately tested for erythrocyte sedimentation rate (ESR) and rheumatoid factor (RF). RF was estimated by latex fixation test (LFT) using Rapi Tex-RF supplied by Behringwerke AG (Marburg, Germany). LFT was considered positive when agglutination was present in more than 1:12 dilution of serum corresponding to 40 U/ml. The normal population consisted of laboratory personnel and non-laboratory volunteers. Normal subjects were studied, four of whom were females (age 20–35 years, mean 27·5 years) and eight males (age 20–35 years, mean 27·5 years), all having no history of rheumatic diseases.
Cell preparation
Venous blood was collected in EDTA (10 mm, pH 7·5). Plasma and buffy coat were separated by centrifugation at 800 g for 10 min at 4°C. Erythrocytes were washed three times with PBS (0·1 m, pH 7·4) containing 1% bovine serum albumin (BSA) at 1500 g for 10 min and the cells were suspended in PBS–BSA.
Buffer and solution
PBS–BSA buffer consisted of PBS pH 7·4, containing 1% BSA (Sigma Chemical Co., St Louis, MO) and 0·05% NaN3. Veronal buffer (VB) was at pH 7·4, and VB2+ consisted of VB supplemented with 0·8 mm MgCl2and 0·15 mm CaCl2.
Antibodies
FITC-labelled rabbit anti-mouse IgG (Dako F313) and rabbit immunoglobulins to human C3d were obtained from Dakopatts Diagnostics (Copenhagen, Denmark). The anti-human C3d antibody preparation is known to react with epitopes expressed on native C3, C3b, iC3b and C3d,g. Murine MoAbs to DAF and CD59 were a generous gift from Professor M. R. Daha (Leiden, The Netherlands). FITC-labelled goat anti-rabbit IgG was obtained from the National Institute of Immunology (India).
Complement activation and measurement of C3 fragment deposition
In vitro complement activation was carried out by incubating the cell preparation at 37°C with 25% pool of normal human serum for 0·5 h in VB2+. The reaction was stopped by transfer of the samples to an ice bath and addition of EDTA at a final concentration of 20 mm. In vivo bound C3 fragments were measured on cells kept at 4°C after isolation. To detect the erythrocyte-bound C3 fragments, erythrocytes were incubated with rabbit anti-human C3d antibody for 2 h at 4°C. FITC-conjugated goat anti-rabbit IgG was used to detect the cell-bound C3 fragments. After incubation, the cells were washed at 4°C with PBS–BSA buffer and then fixed in 1% paraformaldehyde before they were analysed by flow cytometry.
Receptor quantification
Erythrocytes were washed, resuspended in PBS–BSA buffer at 2 × 106 cells/ml and incubated for 2 h at 4°C with mouse anti-DAF and anti-CD59 antibody. After washing with PBS–BSA buffer, cells were incubated with FITC-labelled anti-mouse antibody. The cells were analysed using a flow cytometer (Coulter, Hialeah, FL) after fixing in paraformaldehyde.
The amount of FITC-MoAb bound to CD59 and DAF on erythrocytes was determined from the means of fluorescence intensity.
CH50 assay
The total activity of complement components C1–C9 was measured using a CH50 assay [19] to assess disease activity in RA patients. Test sera in various dilutions were mixed with antibody-sensitized sheep erythrocytes. The samples were then incubated at 37°C for 1 h and centrifuged. The amount of lysis was determined by measuring the absorption of the supernatant at 541 nm and from that CH50 values were calculated.
Statistical analysis
Comparison between groups was performed by calculating confidence interval (CI) for the means using t-test. The correlations between the biological parameters (CD59 and DAF expression, C3 fragment deposition) were evaluated by linear regression analysis, using Student's t-test to determine whether the correlation was significant.
RESULTS
Quantification of CD59 and DAF on erythrocytes from RA patients
To study the role of CD59 and DAF in RA, expression of CD59 and DAF was studied on the erythrocytes of normal and RA patients. In the RA patients, the expression of CD59 and DAF was significantly reduced compared with expression on erythrocytes from healthy individuals. In RA patients, mean fluorescence of CD59 was 61% of CD59 on erythrocytes from normal individuals (P < 0·001) and the mean fluorescence of DAF on erythrocytes of RA patients was 44·3% of DAF (P < 0·001) on erythrocytes from normals (Fig. 1). To investigate whether reduced expression of DAF and CD59 was due to the masking by autoantibodies or immune complexes (IC), erythrocytes from normal control persons were incubated with patients' sera. No alteration in the expression of DAF and CD59 after incubation with the patients' sera was observed. Decreased expression of CD59 correlated significantly with that of DAF among the patients (r = 0·4776, P < 0·05, n = 35).
Fig. 1.
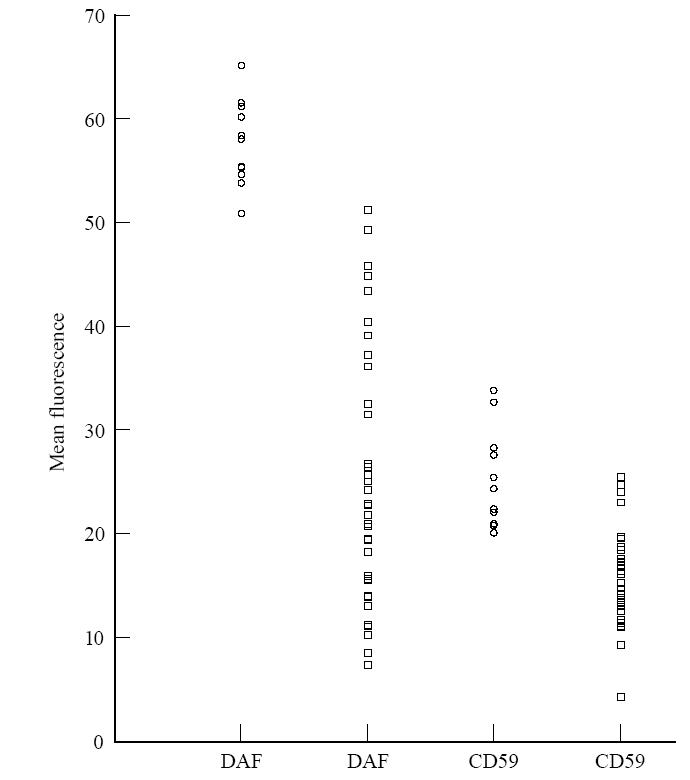
Decay-accelerating factor (DAF) and CD59 expression on erythrocytes from 12 healthy individuals (○) and 35 RA patients (□), using flow cytometry. Cells were gated on the forward and side scatter dot plot in regions containing erythrocytes, and the fluorescent histogram of that population was used for determining protein expression.
In vitro complement activation on erythrocytes
To show a correlation between the expression of complement-regulatory proteins on erythrocytes and the degree of complement activation, complement was activated on the erythrocytes of normal and RA patients. C3 deposition was checked on erythrocytes using anti-C3d antibody by flow cytometry analysis. Significantly increased amounts of C3 deposition were found on the erythrocytes of RA patients (157·9% of the mean of the normal group, P < 0·001) (Fig. 2).
Fig. 2.
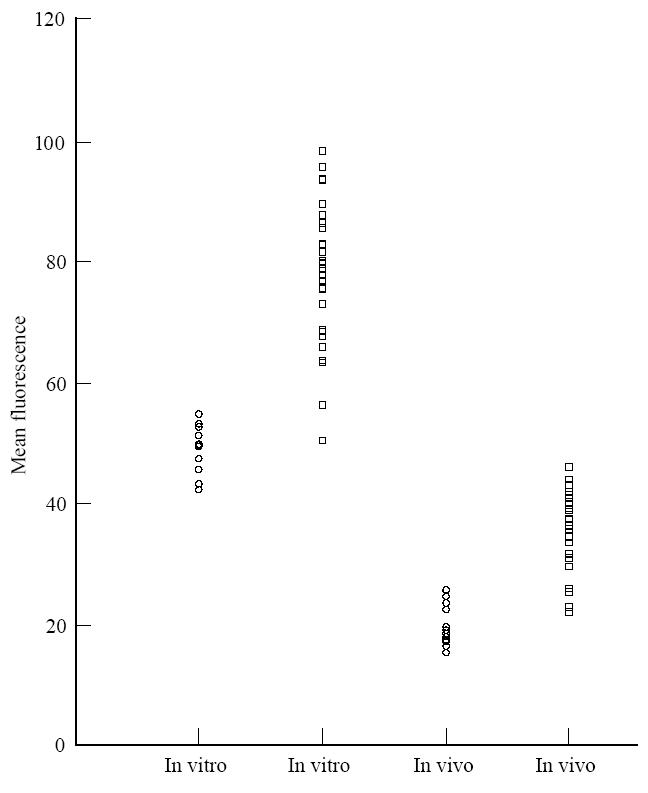
C3 fragment deposition on erythrocytes from RA patients (□) compared with normal controls (○) after in vitro complement activation in homologous normal human serum (NHS). In vivo bound C3 fragment deposition calculated on erythrocytes kept at 4°C after isolation. Erythrocytes incubated in buffer alone are taken as negative control.
In vivo deposition of C3 fragments on the erythrocytes was also calculated. Erythrocytes from RA carried raised levels of in vivo deposited C3 compared with normal individuals (161·4% of the mean of the normal group, P < 0·001) (Fig. 2), and this C3 was not readily dissociable from the surface during incubation with serum-free buffer for 30 min at 37°C.
Relationship between complement-regulatory protein expression, C3 deposition and CH50
The relationship between the expression of complement-regulatory proteins on erythrocytes, the extent of complement activation on erythrocytes and CH50 activity in patients' sera were investigated. A statistically significant inverse relationship was observed between DAF expression and complement activation (r = 0·694, P < 0·01). The relationship between CD59 expression and complement activation was not significant (Fig. 3). A significant inverse relationship was observed between DAF expression on erythrocytes and CH50 activity (r = 0·615, P < 0·01). On the other hand, there appeared to be a tendency for an inverse relationship between erythrocyte CD59 expression and CH50 activity, which, however, was not significant (Fig. 4). A highly significant (r = 0·755, P < 0·001) positive correlation was observed between in vitro C3 deposition and CH50 activity.
Fig. 3.
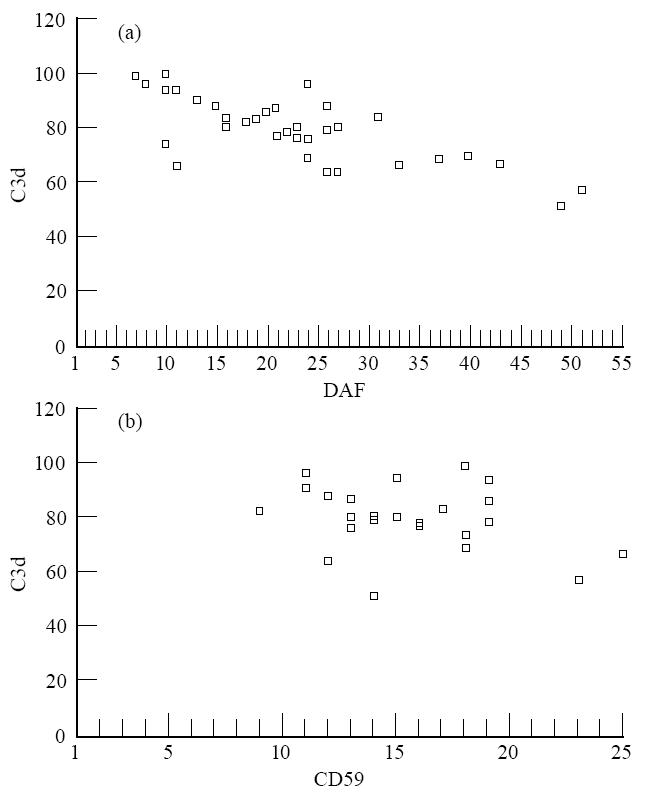
The correlation between decay-accelerating factor (DAF) levels (a) and CD59 levels (b) and C3 deposition after in vitro complement activation on erythrocytes from RA patients.
Fig. 4.
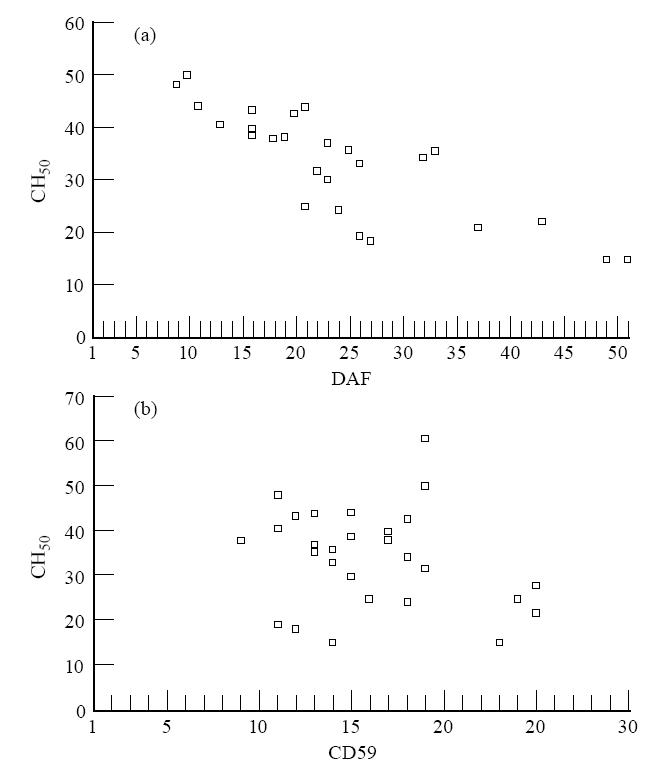
Scatter diagrams. Relation between expression of erythrocyte decay-accelerating factor (DAF) (a), erythrocyte CD59 (b) and CH50 assay.
DISCUSSION
Normal mammalian cells are protected from complement-mediated destruction by regulatory membrane proteins, including CR1, MCP, DAF and CD59. Our previous investigation, using ELISA, reported that patients of RA have reduced numbers of CR1 on their erythrocytes [3]. To elucidate the role of other regulatory proteins, i.e. DAF and CD59, in the pathogenesis of RA, we evaluated the levels of DAF and CD59 on erythrocytes of RA patients, and their relationship to CH50 and complement-activating ability of those erythrocytes was assessed. DAF and CD59 receptor expression on erythrocytes from RA patients was significantly lower than on those from normal individuals. This indicates abnormality in DAF and CD59 expression in RA patients, and the observed decreases could be the likely consequences of the disease process. Correlation between decreased CD59 and DAF in the RA patients was significant. Deficiency of DAF on the erythrocyte membrane, as in paroxysmal nocturnal haemoglobinuria [14], leads to unrestricted complement activation and haemolysis. Jones et al. showed significant reduction in the expression of DAF on blood polymorphonuclear neutrophils (PMN) from patients with RA compared with controls [20], but there is no known clinical example of altered DAF expression on erythrocytes from RA patients.
Small amounts of C3 fragments have previously been detected on erythrocytes from healthy individuals [21–23], while increased amounts of C3 have been demonstrated on erythrocytes from patients with inflammatory diseases such as systemic lupus erythematosus (SLE) or RA [21–23]. We observed that erythrocytes from RA patients carry raised levels of in vivo deposited C3 compared with normal individuals. No significant fall in the amount of detectable C3 fragments, in confirmation of the previous report [24], was observed after 30 min incubation in serum-free buffer at 37°C, which indicates that C3 fragments are primarily covalently attached to the cell surface.
The erythrocytes from RA patients displayed increased in vitro complement activation in the presence of normal serum compared with the in vitro complement-activating capacity of normal erythrocytes. Significant correlation was observed between in vivo deposited C3 and in vitro complement activation (P < 0·05). Furthermore, significant negative correlation was observed within the patient group between DAF expression on erythrocytes and in vitro complement activation. These findings support the results of Nussenzweig et al. [25], who demonstrated the inhibition of complement activation on the surface of cells after incorporation of DAF into their membranes. These findings indicate that the failure of the erythrocytes to act as a down-regulator of complement activation is associated with the reduced expression of DAF. However, no correlation was observed between CD59 expression on erythrocytes and in vitro complement activation.
Reduced expression of DAF on erythrocytes appeared to be predominantly an acquired phenomenon, because the reduced expression of DAF was significantly correlated to CH50 in RA (P < 0·01). However, only weak correlation was observed between CD59 expression and CH50 activity. Several explanations can be proposed to explain reduced expression of DAF and CD59 in RA: (i) a decreased synthesis; (ii) blockage by autoantibodies or IC; (iii) proteolytic cleavage in relation to activation of complement on erythrocyte surfaces [26–31]. Proposed masking by autoantibodies could not sustain the experimental evidence, since we observed no alteration in the expression of DAF and CD59 after incubating the normal erythrocytes with patients' sera. It has been suggested that spontaneous vesiculation occurred from erythrocytes incubated with C5b-9 [32], and these vesicles contain CR1, DAF and CD59 [33]. This might be the reason for acquired loss of erythrocyte DAF and CD59 in diseases associated with complement attack on erythrocytes, such as RA. The results of the present investigations suggest that erythrocyte DAF levels may emerge as an additional diagnostic tool.
Acknowledgments
M.A. is grateful to the University Grants Commission, New Delhi, for the award of a research fellowship. We are thankful to all the normal healthy subjects and patients for providing their precious blood for study. We would also like to thank Mr Rajbir Singh (Department of Biostastics, A.I.I.M.S.) for providing the statistical analysis of the data.
References
- 1.Johnston RB, Stoud RM. Complement and host defence against infection. J Paediatr. 1977;90:169–79. doi: 10.1016/s0022-3476(77)80625-2. [DOI] [PubMed] [Google Scholar]
- 2.Morgan BP, Meri S. Membrane proteins that protect against complement lysis. Springer Semin Immunopathol. 1994;15:369–96. doi: 10.1007/BF01837366. [DOI] [PubMed] [Google Scholar]
- 3.Kumar A, Malaviya AN, Srivastava LM. Lowered expression of C3b receptor on erythrocytes of rheumatoid arthritis patients. Immunobiol. 1994;191:9–20. doi: 10.1016/S0171-2985(11)80264-0. [DOI] [PubMed] [Google Scholar]
- 4.Cornacoff JB, Hebert LA, Smead WL, et al. Primate erythrocytes immune-complex clearing mechanism. J Clin Invest. 1983;71:236–47. doi: 10.1172/JCI110764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cosio FG, Hebert LA, Birmingham DJ, et al. Clearance of human antibody/DNA immune complexes and free DNA from the circulation of the non human primate. Clin Immunol Immunopathol. 1987;42:1–9. doi: 10.1016/0090-1229(87)90167-x. [DOI] [PubMed] [Google Scholar]
- 6.Lobatto S, Daha MR, Voetman AA, et al. Clearance of soluble aggregates of human immunoglobulin G in healthy volunteers and chimpanzees. Clin Exp Immunol. 1987;68:133–41. [PMC free article] [PubMed] [Google Scholar]
- 7.Schifferli JA, Ng YC, Peters DK, et al. The role of complement and its receptor in the elimination of immune complexes. N Engl J Med. 1986;315:488–95. doi: 10.1056/NEJM198608213150805. [DOI] [PubMed] [Google Scholar]
- 8.Nicholson WA, Burge J, Fearon DT. Isolation of a human erythrocyte membrane glycoprotein with decay accelerating activity for C3 convertases of the complement system. J Immunol. 1982;129:184–9. [PubMed] [Google Scholar]
- 9.Nicholson WA, March JP, Rosen CE, et al. Surface membrane expression by human blood leukocytes and platelets of DAF, a regulatory protein of the complement system. Blood. 1985;65:1237–44. [PubMed] [Google Scholar]
- 10.Kinoshita T, Medof ME, Silber R, et al. Distribution of decay accelerating factor in the peripheral blood of normal individuals and patients with paroxysmal nocturnal hemoglobinuria. J Exp Med. 1985;162:75–92. doi: 10.1084/jem.162.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Medof ME, Walter JL, Rutgers DM, et al. Identification of the complement decay accelerating factor on epithelium and glandular cells and in body fluids. J Exp Med. 1987;165:848–64. doi: 10.1084/jem.165.3.848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Asch AS, Kinoshita T, Jaffe EA, et al. Decay accelerating factor is present on cultured human umbilical vein endothelial cells. J Exp Med. 1986;163:221–6. doi: 10.1084/jem.163.1.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Medof ME, Kinoshita T, Silber R. Amelioration of lytic abnormalities of paroxysmal nocturnal hemoglobinuria with decay-accelerating factor. Proc Natl Acad Sci USA. 1985;82:2980–4. doi: 10.1073/pnas.82.9.2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pangburn MK, Schreiber RD, Muller-Eberhard HJ. Deficiency of an erythrocyte membrane protein with complement regulatory activity in paroxysmal nocturnal hemoglobinuria. Proc Natl Acad Sci USA. 1983;80:5430–4. doi: 10.1073/pnas.80.17.5430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nicholson WA, March JP, Rosenfeld SI, et al. Affected erythrocytes of patients with paroxysmal nocturnal hemoglobinuria are deficient in the complement regulatory protein, decay-accelerating factor. Proc Natl Acad Sci USA. 1983;80:5066–70. doi: 10.1073/pnas.80.16.5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holguin MH, Fredrick LR, Bernshaw NJ, et al. Isolation and characteristic of a membrane protein from normal human erythrocytes that inhibits the reactive lysis of the erythrocytes of paroxysmal nocturnal hemoglobinuria. J Clin Invest. 1989;84:7–17. doi: 10.1172/JCI114172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Okada N, Harada R, Fujita T, et al. A novel membrane glycoprotein capable of inhibiting membrane attack of homologous complement. Int Immunol. 1989;1:205–8. doi: 10.1093/intimm/1.2.205. [DOI] [PubMed] [Google Scholar]
- 18.Davies A, Simmons DL, Hale G, et al. CD59, an LY-6 like protein expressed in human lymphoid cells, regulates the action of the complement membrane attack complex on homologous cells. J Exp Med. 1989;170:637–54. doi: 10.1084/jem.170.3.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mayer MM. Complement and complement fixation. In: Kabat EA, Mayer MM, editors. Experimental immunology. Springfield: Thomas CC; 1967. pp. 133–240. [Google Scholar]
- 20.Jones J, Laffafian I, Cooper AM, et al. Expression of complement regulatory molecules and other surface markers on neutrophils from synovial fluid and blood of patients with rheumatoid arthritis. Br J Rheumatol. 1994;33:707–12. doi: 10.1093/rheumatology/33.8.707. [DOI] [PubMed] [Google Scholar]
- 21.Chaplin H, Nasonga M, Monroe MC. Quantitation of red blood cell-bound C3d in normal subjects and random hospital patients. Br J Haematol. 1981;47:69–78. doi: 10.1111/j.1365-2141.1981.00069.x. [DOI] [PubMed] [Google Scholar]
- 22.Freedman J, Barefoot C. Red blood cell-bound C3d in normal subjects and in random hospital patients. Transfusion. 1982;22:511–4. doi: 10.1046/j.1537-2995.1982.22683068615.x. [DOI] [PubMed] [Google Scholar]
- 23.Rasmussen JM, Jepsen HH, Teisner B, et al. Quantification by ELISA by erythrocyte-bound C3 fragments expressing C3d and/or C3c epitopes in patients with factor I deficiency and with autoimmune diseases. Vox Sang. 1989;56:262–9. doi: 10.1111/j.1423-0410.1989.tb02039.x. [DOI] [PubMed] [Google Scholar]
- 24.Marquart HV, Svehag SE, Leslie RGQ. CR2 is the primary acceptor site for C3 during alternative pathway activation of complement on human peripheral B lymphocytes. J Immunol. 1994;153:307–15. [PubMed] [Google Scholar]
- 25.Medof ME, Kinoshita T, Nussenzweig V. Inhibition of complement activation on the surface of cells after incorporation of decay-accelerating factor (DAF) into their membranes. J Exp Med. 1984;160:1558–78. doi: 10.1084/jem.160.5.1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ross GD, Yount WJ, Walport MJ, et al. Disease associated loss of erythrocyte complement receptors (CR1) in patients with systemic lupus erythematosus and other disease involving autoantibodies and/or complement activation. J Immunol. 1985;135:2005–14. [PubMed] [Google Scholar]
- 27.Walport MJ, Ng YC, Lachmann PJ. Erythrocytes transfused into patients with SLE and haemolytic anemia lose complement receptor type 1 from their cell surface. Clin Exp Immunol. 1987;69:501. [PMC free article] [PubMed] [Google Scholar]
- 28.Wilson JG, Jack RM, Wong WW, et al. Autoantibody to the C3b/C4b receptor and absence of this receptor from erythrocytes of patients with systemic lupus erythematosus. J Clin Invest. 1985;76:182–90. doi: 10.1172/JCI111944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cook JM, Kazatchkine MD, Burgeosis P, et al. Anti-C3b receptor antibodies in patients with SLE. Clin Immunol Immunopathol. 1986;38:135. doi: 10.1016/0090-1229(86)90131-5. [DOI] [PubMed] [Google Scholar]
- 30.Davies KA, Savill J, Walport MJ. In vitro transfer of immune complexes from erythrocytes to monocytes and macrophages. Complement Inflamm. 1989;6:328. [Google Scholar]
- 31.Ripoche J, Sim RB. Loss of CR1 on ageing of erythrocyte: studies of proteolytic release of the receptor. Biochem J. 1986;235:815. doi: 10.1042/bj2350815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iida K, Whitlow MB, Nussenzweig V. Membrane vesiculation protects erythrocytes from the destruction of complement. J Immunol. 1991;147:2638–42. [PubMed] [Google Scholar]
- 33.Pascual M, Lutz HU, Gteiger G, et al. Release of vesicles enriched in complement receptor 1 from human erythrocytes. J Immunol. 1993;151:397–404. [PubMed] [Google Scholar]