

Abstract
Cytokines such as TNF-α and interferon gamma (IFN-γ) are important for the elimination of infected hepatocytes during acute hepatitis B virus (HBV) infection. Two G versus A transitions in the TNF-α promoter region at positions −308 and −238 possibly influence TNF-α expression. We investigated these TNF-α polymorphisms in 71 patients with chronic HBV infection, in 32 subjects that had spontaneously recovered from acute HBV infection, and in 99 healthy controls. The −238 A promoter variant was present in 18 (25%) of 71 patients with chronic HBV infection compared with two (6%) of 32 subjects with acute infection (P < 0.04), and seven (7%) of 99 controls (P < 0.003). By contrast, the prevalence of the variant at position −308 was similar in all investigated groups. The observed differences could not be explained by linkage disequilibrium to HLA-B or -DRB1* alleles. These findings suggest an association between the TNF-α promoter polymorphism at position −238 and the development of chronic HBV infection. This promoter variant appears to be linked to defective viral clearance.
Keywords: hepatitis B virus, chronic infection, tumour necrosis factor-alpha, promoter polymorphism, MHC
INTRODUCTION
The hepatitis B virus (HBV) is non-cytopathic and causes acute necroinflammatory liver disease in infected subjects [1]. Most infected individuals clear the virus and only 5–10% develop chronic hepatitis. The reasons for viral persistence are still poorly understood, but host factors are likely to influence the disease outcome [1].
Data from an HBV transgenic mouse model have challenged the prevailing view that acute HBV infection can only be overcome by the elimination of infected hepatocytes by direct contact to cytotoxic T lymphocytes (CTL) [2]. In this model interferon-gamma (IFN-γ) and TNF-α secreted by CTL and antigen-non-specific macrophages can abolish HBV gene expression and replication without killing the hepatocytes. Viral clearance is also achieved by an unrelated viral infection of the liver that activates the production of IFN-γ and TNF-α by hepatic macrophages [3].
A number of studies could show that the TNF-α system is activated in the liver of patients with chronic HBV infection [4], including an up-regulation of both TNF-α receptors [5,6].
The gene for TNF-α is located within the class III region of the MHC between HLA-B and -DR. Its expression is tightly controlled at the transcriptional and post-transcriptional level. Two G versus A transitions in the promoter region at positions −308 [7] and −238 [8] have been shown to influence TNF-α expression. At position −308 TNF308.2 (A at −308) is associated with higher constitutive and inducible levels of TNF-α [9], whereas for the TNF238.2 allele (A at −238) functional consequences are not yet clear [10]. The TNF308.2 allele has been linked with susceptibility to cerebral malaria [11] and mucocutaneous leishmaniasis [12], whereas both TNF308.2 and TNF238.2 have been associated with tuberculosis and malarial anaemia [13].
The aim of this study was to determine whether TNF-α promoter polymorphisms show an association with the disease course in HBV infection.
PATIENTS AND METHODS
Study subjects
Seventy-one Caucasian patients with chronic HBV infection were prospectively enrolled in this study from our out-patient referral centre for liver diseases. The diagnosis of chronic hepatitis B was established by seropositivity for hepatitis B surface antigen (HBsAg), the lack of anti-HBs antibodies (anti-HBs) and the presence of anti-core antibodies (anti-HBcAb) of the IgG type. In addition, all patients had elevated serum alanine aminotransferase levels and were positive for HBV DNA on repeated testing. A second group of patients consisted of 32 healthy Caucasian blood donors who had spontaneously recovered from HBV infection. These subjects were positive for anti-HBs and anti-HBc IgG antibodies. They were also negative for anti-HCV and anti-HIV antibodies.
The normal control population consisted of 99 unrelated, healthy, white Caucasoid persons from routine consecutive paternity cases studied at the Institute of Legal Medicine. Patients came from the Mainz area. There were 47 female and 52 male individuals. All study subjects had been tested previously for HLA class I (serology) and HLA-DRB1* alleles [14]. DNA was isolated from all individuals using standard procedures and was subsequently stored at 4°C.
Serological testing
Plasma samples obtained from all patients were stored at −20°C. The serological status of every subject with regard to HBsAg, anti-HBs, anti-HBc (IgG and IgM), hepatitis B envelope antigen (HBeAg) and antibodies to hepatitis B envelope were determined by ELISA (IMX; Abbott Diagnostics, North Chicago, IL). All patients were also tested for HIV antibodies (Abbott Diagnostics) [14] and antibodies to HCV by a second generation ELISA (Abbott, Delkenheim, Germany). Serum HBV DNA was determined by a commercially available solution hybridization assay (Genostics; Abbott Diagnostics). Patients testing positive for HIV or HCV antibodies were excluded from the study.
TNF-α promoter polymorphisms
A 328-bp fragment spanning positions −396 to −69 of the 5′ untranslated region of the TNF-α gene was amplified using primers TNF-396 (5′ TTCCTGCATCCTGTCTGGAA 3′) and TNF-69 (5′ CAGCGGAAAACTTCCTTGGT 3′). Amplification was performed in a Perkin Elmer thermocycler (9600; Perkin Elmer Corp., Norwalk, CT) with 100 ng of genomic DNA, 25 pm of each primer, 200 μm total dNTP, 1.5 mm MgCl2, standard polymerase chain reaction (PCR) buffer and 2 U Taq polymerase (Boehringer, Mannheim, Germany). The following cycling conditions were used: 60 s 94°C, 60 s 55°C, 60 s 72°C for 30 cycles.
A panel of previously HLA-DR typed samples was initially typed for the polymorphisms by sequencing the entire 328-bp fragment. This led to the identification of a number of heterozygous and homozygous individuals for each of the four polymorphisms. TNF promoter alleles in the study subjects were detected by dot blot analysis of amplified DNA incorporating in every analysis a panel of sequenced reference samples. Four digoxigenin-labelled oligonucleotide probes were used for the identification of the promoter alleles [8]. Filters were hybridized at 40°C and washed with tetramethylammonium chloride (TMAC) at the temperatures described in [8]. Hybridization and colour detection with anti-digoxigenin antibody-coupled alkaline phosphatase were carried out according to the manufacturer's instructions (Boehringer).
Statistical analysis
The frequencies of TNF-α promoter alleles were compared between patients with chronic HBV infection, acute HBV infection and controls by χ2 tests with Yates' correction for small numbers. Relative risks associated with a particular allele were calculated using a χ2 distribution: RR = (n1 × n4)/(n2 × n3), where n1 is the number of patients with allele x, n2 is the number of controls with allele x, and n3 and n4 are the corresponding proportions of individuals in patient and control groups not carrying allele x [15]. In each group the distribution of TNF-α promoter genotypes was checked for deviations from Hardy–Weinberg equilibrium using an exact test [16]. Linkage disequilibrium between HLA-B, DRB1* and TNF-α was tested by χ2 test and by an exact test [17]. P values were corrected for the number of alleles tested (38 for HLA-B, 13 for DRB and four for TNF-α; 55 alleles tested). P< 0.05 was regarded as significant.
RESULTS
Observed TNF-α promoter genotype frequencies are given in Table 1. There were no differences in the frequencies of the TNF308 genotypes between the investigated groups.
Table 1.
TNF-α promoter genotype frequencies in the investigated groups
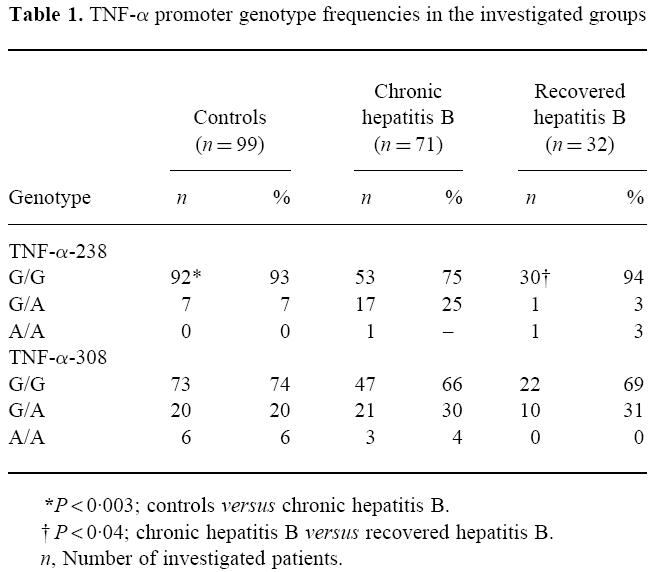
In subjects with chronic hepatitis B infection there was a significant increase of homozygous and heterozygous individuals for the TNF238.2 allele (P < 0.003). Eighteen (25%) subjects with chronic hepatitis B infection carried the TNF238.2 mutation, compared with seven (7%) controls and two (6%) individuals with acute hepatitis B.
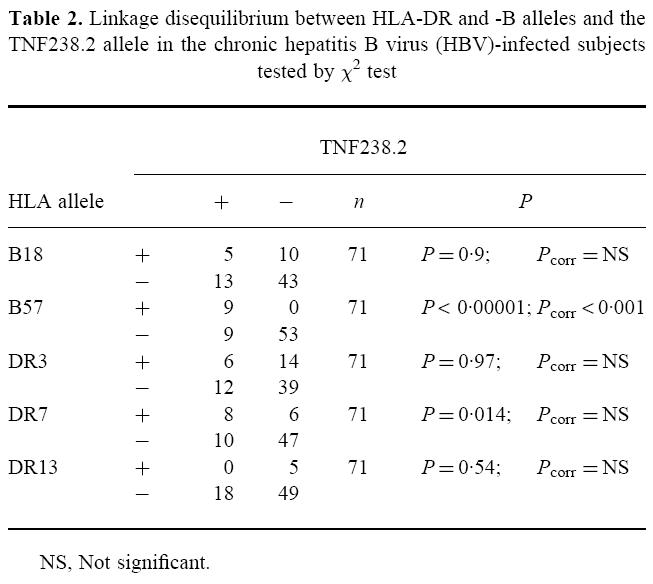
The TNF238.2 variant has been shown to be in linkage disequilibrium with B18, B57, DR7, DR17.2, and to be part of the extended haplotypes B18-BFF1-DR17.2 (DRB1*0301) and B57-C4 A6-DR7-DQw9 [7,9]. The previously published data on HLA class I and DRB1* antigens in our population [13] were used to test TNF-α polymorphisms at positions −238 and −308 for linkage disequilibrium to HLA-B and HLA-DRB1 (Table 2). Nine of 18 (50%) TNF238.2 alleles were observed in B57-positive patients (P < 0.00001; Pcorr < 0.001). Weak linkage disequilibrium was also observed between DRB1*0701 and TNF238.2, though this was not significant after correction. There were no significant differences in the frequencies of B57 (12.7% in chronic HBV infection and 6.9% in controls) and DRB1*0701 (18.6% in chronic HBV infection and 27.7% in controls) between subjects with chronic hepatitis B and patients with acute infections or controls [14]. DRB1*1301-02, which has been linked with protection against chronic HBV infection [14,18], was not associated with TNF238.2 either in the patients or in the controls (data not shown).
Table 2.
Linkage disequilibrium between HLA-DR and -B alleles and the TNF238.2 allele in the chronic hepatitis B virus (HBV)-infected subjects tested by χ2 test
DISCUSSION
Eighteen (25%) of 71 subjects with chronic hepatitis B were homozygous or heterozygous for the TNF238.2 allele, whereas only 6% of patients with acute infection and 7% of controls carried this allele. The observed association cannot be explained by linkage disequilibrium to the HLA-B or -DR genes, as the linked alleles B57 and DR7 were found with similar frequencies in the investigated groups. However, from the data of this study we cannot exclude linkage disequilibrium to a gene in closer proximity to TNF-α, e.g. the complement genes C4, factor B or C2 within the MHC class III region. However, in a previous study on patients with HBV infection we did not find any significant association of these genes with chronic hepatitis B (Schneider et al., manuscript in preparation).
The TNF238.2 polymorphism lies in a putative regulatory box (Y-box) of the TNF-α promoter region which binds to regulatory DNA-binding proteins like NF-Y [8]. This putative regulatory box is believed to contribute to optimal promoter activity [19]. Nucleotide position −238 is strongly conserved among different animal species, raising the possibility that it is functionally important [19]. A single base pair substitution in the Y-box of the HLA-DQA1 promoter causes significantly decreased transcription of the gene [20,21]. Similar observations have been reported for the human multidrug-resistance gene [22] and the human thymidine kinase promoter [23]. However, monocytes of TNFA-A/G heterozygous subjects show no significant difference in TNF-α production after lipopolysaccharide (LPS) treatment or allogenic stimulation [9].
The importance of the cytokines TNF-α and IFN-γ for the clearance of the hepatitis B virus has only recently been emphasized by a number of findings. TNF-α and IFN-γ inhibit the transcriptional activity of the HBV core promoter in vitro [24]. In an HBV transgenic mouse model [2] it could be shown that only a minority of infected hepatocytes are eliminated by direct contact with cytotoxic T cells. In the vast majority of infected cells HBV appears to be suppressed and eliminated by antigen-non-specific cytokines [2]. Coinfection with the lymphocytic choriomeningitis virus abolishes hepatocellular HBV replication non-cytopathically via TNF-α and IFN-α/β secretion by infected macrophages [3]. These data could explain the clinical observation that clearance of HBV occurs in patients coinfected with the hepatitis A, C or delta virus [25–27].
Viral infections can also influence the transcription of cytokine genes. Infection of hepatocytes with either the hepatitis B or C virus induces the expression of TNF-α in these cells [28]. During HBV infection HBcAg can suppress the transcription of IFN-β and the HBV polymerase protein has been shown to inhibit cellular responses to IFN-α and IFN-γ [29,30]. Transactivating/inhibiting factors of HBV might show differential interaction with the TNF238.2 allele and cause decreased transcription of the TNF gene. Failure to secrete adequate amounts of TNF-α could possibly prevent viral clearance and lead to chronic infection.
In a former study another group and ours have identified a protective effect of DRB1*1301 and 1302 against the development of chronic hepatitis B in Africans [18] and Caucasians [14]. Our data suggest that TNF238.2 and DRB1*1301-02 are independent factors encoded within the MHC that influence the outcome of acute hepatitis B infection. Disease susceptibility for infectious diseases is determined at such different functional levels as cytokine production and antigen presentation, as has been shown for malaria [11,13,31].
Acknowledgments
This project was funded by grants from the Deutsche Forschungsgemeinschaft DFG (SFB 311 A13 and A17).
References
- 1.Chisari FV, Ferrari C. Hepatitis B virus immunopathogenesis. Annu Rev Immunol. 1995;13:29–60. doi: 10.1146/annurev.iy.13.040195.000333. [DOI] [PubMed] [Google Scholar]
- 2.Guidotti LG, Ishikawa T, Hobbs MV, Matzke B, Schreiber R, Chisari FV. Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity. 1996;4:25–36. doi: 10.1016/s1074-7613(00)80295-2. [DOI] [PubMed] [Google Scholar]
- 3.Guidotti LG, Borrow P, Hobbs MV, Matzke B, Gresser I, Oldstone MBA, Chisari FV. Viral cross talk: intracellular inactivation of the hepatitis B virus during an unrelated viral infection of the liver. Proc Natl Acad Sci USA. 1996;93:4589–94. doi: 10.1073/pnas.93.10.4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sheron N, Lau L, Daniels H, Goka J, Eddleston A, Alexander GJ, Williams R. Increased production of tumor necrosis factor alpha in chronic hepatitis B virus infection. J Hepatol. 1991;12:241–5. doi: 10.1016/0168-8278(91)90945-8. [DOI] [PubMed] [Google Scholar]
- 5.Marinos G, Naoumov NV, Rossol S, Torre F, Wong PYN, Gallati H, Portmann B, Williams R. Tumor necrosis factor receptors in patients with chronic hepatitis B infection. Gastroenterol. 1995;108:1453–63. doi: 10.1016/0016-5085(95)90694-0. [DOI] [PubMed] [Google Scholar]
- 6.Fang JWS, Shen WW, Meager A, Lau JYN. Activation of the tumor necrosis factor-α system in the liver in chronic hepatitis B virus infection. Am J Gastroenterol. 1996;91:748–53. [PubMed] [Google Scholar]
- 7.Wilson AG, De Vries N, Pociot F, Di Giovine FS, Van der Putte LBA, Duff GW. An allelic polymorphism within the human tumor necrosis factor α promoter region is strongly associated with HLA A1, B8, and DR3 alleles. J Exp Med. 1993;177:557–60. doi: 10.1084/jem.177.2.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.D'Alfonso S, Richiardi PM. A polymorphic variation in a putative regulation box of the TNFA promoter region. Immunogenetics. 1994;39:150–5. doi: 10.1007/BF00188619. [DOI] [PubMed] [Google Scholar]
- 9.Wilson AG, Symons JA, McDowell TL, Di Giovine FS, Duff GW. Effects of a tumor necrosis factor (TNF-alpha) promoter base transition on transcriptional activity. Br J Rheumatol. 1994;33:89. [Google Scholar]
- 10.Pociot F, D'Alfonso S, Compasso S, Scorza R, Richiardi PM. Functional analysis of a new polymorphism in the human TNF α gene promoter. Scand J Immunol. 1995;42:501–4. doi: 10.1111/j.1365-3083.1995.tb03686.x. [DOI] [PubMed] [Google Scholar]
- 11.McGuire W, Hill AVS, Allsopp CEM, Greenwood BM, Kwjatkowski D. Variation in the TNF-α promoter region associated with susceptibility to cerebral malaria. Nature. 1994;371:508–11. doi: 10.1038/371508a0. [DOI] [PubMed] [Google Scholar]
- 12.Cabrera M, Shaw MA, Sharples C, Williams H, Castes M, Convit J, Blackwell JM. Polymorphism in tumor necrosis factor genes associated with mucocutaneous leishmaniasis. J Exp Med. 1995;182:1259–64. doi: 10.1084/jem.182.5.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hill AVS, Ruwende C, Mcguire W, et al. Association of the TNF-238 promoter polymorphism with susceptibility to tuberculosis and malaria in Africa. Hum Immunol. 1996;47:118. [Google Scholar]
- 14.Höhler T, Gerken G, Notghi N, et al. HLA-DRB1*1301 and *1302 protect against chronic hepatitis B. J Hepatol. 1997;26:503–7. doi: 10.1016/s0168-8278(97)80414-x. [DOI] [PubMed] [Google Scholar]
- 15.Dyer P, Warrens A. Design and interpretation of studies of the major histocompatibility complex in disease. In: Lechler R, editor. HLA and disease. London: Academic Press; 1994. pp. 93–121. [Google Scholar]
- 16.Guo SW, Thompson EA. Performing the exact test of Hardy–Weinberg proportion for multiple alleles. Biometrics. 1992;48:361–72. [PubMed] [Google Scholar]
- 17.Zaykin D, Shivotovsky L, Weir BS. Exact tests for associations between alleles at arbitrary numbers of loci. Genetica. 1995;96:169–78. doi: 10.1007/BF01441162. [DOI] [PubMed] [Google Scholar]
- 18.Thursz MR, Kwiatkowski D, Allsopp CEM, Greenwood BM, Thomas HC, Hill AVS. Association between an MHC class II allele and clearance of the hepatitis B virus in Gambia. N Engl J Med. 1995;332:1065–9. doi: 10.1056/NEJM199504203321604. [DOI] [PubMed] [Google Scholar]
- 19.Shakov AN, Collart MA, Vassalli P, Nedospasov SA, Jongeneel CV. κB-type enhancers are involved in lipopolysaccharide-mediated transcriptional activation of the tumor necrosis factor α gene in primary macrophages. J Exp Med. 1990;171:35–47. doi: 10.1084/jem.171.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haas JP, Kimura A, Truckenbrodt H, Suschke J, Sasazuki T, Volgger A, Albert ED. Early onset juvenile chronic arthritis is associated with a mutation in the Y-box of the HLA-DQA1 promoter. Tissue Antigens. 1995;45:317–21. doi: 10.1111/j.1399-0039.1995.tb02460.x. [DOI] [PubMed] [Google Scholar]
- 21.Morzycka-Wroblewska E, Munshi A, Ostermayer M, Harwood JI, Kagnoff MF. Differential expression of HLA-DQA1 alleles associated with promoter polymorphism. Immunogenetics. 1997;45:163–70. doi: 10.1007/s002510050185. [DOI] [PubMed] [Google Scholar]
- 22.Goldsmith ME, Madden MJ, Morrow CS, Cowan KH. A Y-box consensus sequence is required for basal expression of the human multidrug resistance (mdr1) gene. J Biol Chem. 1993;268:5856–60. [PubMed] [Google Scholar]
- 23.Lipson KE, Chen ST, Koniecki J, Ku DH, Baserga R. S-phase-specific regulation by deletion mutants of the human thymidine kinase promoter. Proc Natl Acad Sci USA. 1989;86:6848–52. doi: 10.1073/pnas.86.18.6848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Romero R, Lavine JE. Cytokine inhibition of the hepatitis B virus core promoter. Hepatol. 1996;23:17–23. doi: 10.1002/hep.510230103. [DOI] [PubMed] [Google Scholar]
- 25.Davis GL, Hoofnagle JH, Waggoner JG. Acute type A hepatitis during chronic hepatitis B virus infection: association of depressed hepatitis B virus replication with appearance of endogenous alpha interferon. J Med Virol. 1984;14:141–7. doi: 10.1002/jmv.1890140208. [DOI] [PubMed] [Google Scholar]
- 26.Sheen IS, Liaw YF, Lin DY, Chu CM. Role of hepatitis C and delta viruses in the termination of chronic hepatitis B surface antigen carrier state: a multivariate analysis in a longitudinal follow-up study. J Infect Dis. 1994;170:1358–61.. doi: 10.1093/infdis/170.2.358. [DOI] [PubMed] [Google Scholar]
- 27.Pastore G, Monno L, Santantonio T, Angarano G, Milella M, Gianelli A, Fiore JR. Hepatitis B virus clearance from serum and liver after acute hepatitis delta virus superinfection in chronic HBsAg carriers. J Med Virol. 1990;31:284–90. doi: 10.1002/jmv.1890310408. [DOI] [PubMed] [Google Scholar]
- 28.González-Amaro R, García-Monzón C, García-Buey L, et al. Induction of tumor necrosis factor α production by human hepatocytes in chronic viral hepatitis. J Exp Med. 1994;179:841–8. doi: 10.1084/jem.179.3.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Whitten TM, Quets AT, Schloemer RH. Identification of the hepatitis B virus factor that inhibits expression of the beta interferon gene. J Virol. 1991;65:4699–704. doi: 10.1128/jvi.65.9.4699-4704.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Foster GR, Ackrill AM, Goldin RD, Kerr IM, Thomas HC, Stark GR. Expression of the terminal protein region of hepatitis B virus inhibits cellular responses to interferons alpha and gamma and double stranded RNA. Proc Natl Acad Sci USA. 1991;88:2888–92. doi: 10.1073/pnas.88.7.2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hill AVS, Allsopp CEM, Kwiatkowski D, et al. Common West African HLA antigens are associated with protection from severe malaria. Nature. 1991;352:595–600. doi: 10.1038/352595a0. [DOI] [PubMed] [Google Scholar]