

Abstract
The activation of macrophages for antimicrobial responses is a multistage event involving numerous intracellular signalling cascades that makes possible target cell destruction by these effector cells. This study examined the effects of different potassium channel inhibitors and activators on the NO production of murine macrophage-like cell lines P388D.1 and B10-4(S). We found that the potassium channel inhibitors tetraethylammonium, 4-aminopyridine, and quinine caused dose-dependent reductions in the NO production of macrophages, and that the potassium channel activator, minoxidol, caused a dose-dependent enhancement of NO production. The inhibition of NO production was due to involvement of potassium channels in the priming stage of macrophage activation, since pretreatment with the priming agent interferon-gamma partially restored the NO response of the macrophages. The results of this study demonstrate a link between potassium channel activity and the activation of anitimicrobial functions of murine macrophages.
Keywords: nitric oxide, macrophage, potassium channels, activation, antimicrobial
INTRODUCTION
Production of NO molecules is an important cytotoxic function that macrophages use to resolve infection by several obligate intracellular protozoan and bacterial parasites [1]. The inducible form of the NO synthase enzyme (iNOS) produces large amounts of reactive nitrogen molecules by oxidizing the terminal guanidino nitrogen of l-arginine [2,3]. Since reactive nitrogen molecules can damage host tissues as well as the invading microbe, the activation of NO production by macrophages is a tightly regulated ligand-receptor mediated process that involves two stages [3]. The priming stage occurs when activating molecules, such as the cytokine interferon-gamma (IFN-γ), binds to receptors on the cell surface of the macrophages, inducing alterations in cytoplasmic calcium levels and initiating other intracellular signalling cascades [3–5]. These signalling cascades ultimately alter the expression of enzymes, including iNOS, and receptor proteins to prepare the metabolic machinery to initiate a cytotoxic response. The triggering stage is induced when another molecule, often a molecule of the intruding microbe such as bacterial lipopolysaccharide (LPS), binds to the receptors on macrophages, initiating another series of signalling cascades that activates the previously ‘primed’ cytotoxic machinery [3,5,6]. For the iNOS pathway it is believed that protein kinase C (PKC) is involved in the triggering step, but the exact cascades involved have yet to be fully determined [5].
Macrophages possess a number of types of voltage-gated potassium channels. Researchers using the patch clamp and whole cell techniques have found delayed outward rectifier type, calcium-dependent type, adenosine triphosphate (ATP)-dependent type, and inward rectifier type potassium channels within macrophages [7–10]. The major role of most ion channels within cells, particularly those specific for potassium ions, is the establishment and maintenance of the membrane potential [10]. There is growing evidence that alterations in the activity of ion channels and the resulting changes in membrane potential can have significant effects on other proteins and enzymes embedded in the plasma membrane [11,12]. This includes a number of intracellular signalling protein complexes and provides a mechanism for the inhibitors of ion channels to affect the cytotoxic function of immune cells. Indeed, experiments examining the role potassium channels play in the cytotoxic functions of a variety of leucocytes have suggested that potassium channels are involved in the signalling cascades responsible for inducing cytotoxic functions [10,12–14].
In this study we examined whether alterations in the activity of potassium channels caused by specific pharmacological blockers affected the NO production and cell viability of the P388D.1 and B10-4(S) macrophage-like cell lines.
MATERIALS AND METHODS
Reagents
Recombinant murine IFN-γ was purchased from Genzyme (Boston, MA) and LPS from Salmonella typhimurium was obtained from Difco Labs (Detroit, MI). Quinine hydrochloride, 4-aminopyridine, and tetraethylammonium chloride were obtained from Sigma Chemical Co. (St Louis, MO).
Cell lines and media
Two cell lines with different growth and cytotoxic activation characteristics were obtained for experimentation. The macrophage-like cell line P388D.1 consisted of slower growing cells at a later stage of differentiation, and was obtained from the American Type Culture Collection (ATCC, Rockville, MD) [15]. The B10-4(S) macrophage-like cell line consisted of rapidly dividing cells with morphology typical of less mature macrophages, and was a kind gift from Dr D. Radzioch (McGill University). The B10-4(S) cell line was immortalized from bone marrow-derived macrophages isolated from C57Bl/10.A (B10A.BcgS) mice that were susceptible to bacille Calmette–Guérin (BCG) [16]. Both cell lines were cultured at 37°C and 5% CO2 in Dulbecco's modified Eagles' medium containing 50 μg/ml gentamicin and 10% fetal bovine serum (complete DMEM).
Activation of macrophages and treatment with potassium channel inhibitors
Macrophages were seeded into the wells of 96-well tissue culture plates at a density of 1 × 105 cells/well and allowed to attach to the bottom of the wells at 37°C and 5% CO2 for 1 h. Macrophages in different culture plates were treated with quinine (5–250 μm), tetraethylammonium (TEA; 1–10 mm), 4-aminopyridine (4-AP; 50 μm to 2 mm), and minoxidol (10 nm to 5 μm) in conjunction with complete DMEM (control) or 1 μg/ml LPS and 25 U/ml IFN-γ. A NO assay and a viability assay were done on parallel cultures of macrophages for every treatment group. The macrophage cultures were incubated at 37°C and 5% CO2 for 24 h before performing the appropriate assays.
Time course of the NO production and respiratory viability
Macrophages were seeded at a density of 5 × 104 cells/well, and wells of triplicate macrophage cultures were treated with 10 mm TEA, 1 mm 4-AP, or 50 μm quinine in conjunction with complete DMEM (control) or 1 μg/ml LPS and 25 U/ml IFN-γ. A NO assay and a viability assay were done on parallel cultures of macrophages for each time point examined: 0, 6, 12, 24, 48 and 72 h. These experiments were repeated twice.
Pretreatment of macrophages with cytotoxic activators or potassium channel inhibitors
Macrophages were seeded at a density of 1 × 105 cells/well, and wells of triplicate macrophage cultures were pretreated for 6 h at 37°C and 5% CO2 with DMEM (control), 25 U/ml IFN-γ, 1 μg/ml LPS, LPS and IFN-γ or with the inhibitors 10 mm TEA, 1 mm 4-AP, and 50 μm quinine. The macrophage cultures were then fully activated by addition of 1 μg/ml LPS and/or 25 U/ml IFN-γ, as required, and potassium channel activity was inhibited by addition of 10 mm TEA, 1 mm 4-AP, or 50 μm quinine, again as required. The cultures were incubated at 37°C and 5% CO2 for a further 24 h. These experiments were repeated twice.
Nitric oxide assay
The NO production of the macrophages was indirectly determined using a colourimetric assay, the Griess reaction, to measure the accumulation of the stable end product of NO degradation, nitrite, within the superfusate of wells [17]. Briefly, 100-μl volumes of macrophage superfusates were transferred from the treatment plate wells to wells of a 96-well microtitre plate. An equal volume (100 μl) of 1% sulfanilamide (Sigma) in 2.5% phosphoric acid was added to each well. This was followed by 100 μl of 0.1% N-naphthyl-ethylenediamine (Sigma) in 2.5% phosphoric acid. The plate was gently tapped to mix the contents, allowed to sit for 2 min, and the optical density (OD) at 540 nm was determined using an automated spectrophotometer (Biotek, Winooski, VT). The approximate concentration of nitrite in samples was determined from a standard curve that was generated using known concentrations of sodium nitrite.
Cell respiration (XTT viability assay)
Changes in macrophage viability were assessed using the XTT viability assay. This assay measures the respiratory activity of the macrophages' mitochondria by determining the accumulation of a coloured formazan by-product within the superfusates of treated wells [18–20]. Briefly, a 50-μl volume of 1 mg/ml sodium 3,3′-[1[(phenylamino)carbonyl]-3,4-tetrazolium]-bis (4-methoxy-6-nitro)benzene sulphonic acid hydrate (XTT) (ICN Biomedicals Inc., Costa Mesa, CA) and 50 μg/ml 2,3-dimethoxy-5-methyl-1,4-benzoquinone (CoQ) (Sigma) in 1× Dulbecco's Phosphate Buffered Saline (Gibco BRL Life Technologies, Burlington, Ontario, Canada) were added to each well of the treatment plate. The plates were gently tapped to mix the contents, incubated for 1 h at 37°C and 5% CO2, and the OD at 450 nm was determined using an automated spectrophotometer (Biotek).
Statistical analysis
The data were analysed using Super-anova software for the Power Macintosh. Differences between experimental groups were determined using one-factor analysis of variance and a least square means test. P < 0.05 was considered statistically significant.
RESULTS
Nitric oxide production by activated macrophage-like B10-4(S) and P388D.1 cells
Treatment of the macrophages with 1 μg/ml LPS by itself for a 24-h period was sufficient to induce NO production in both macrophage-like cell lines (Table 1). The B10-4(S) cells consistently exhibited a higher NO response than the P388D.1 cells when seeded at identical cell densities (1 × 105/well) (Table 1). Treatment with 25 U/ml IFN-γ alone did not result in significant NO production within either macrophage cell line (Table 1). However, treatment of macrophages with both 1 μg/ml LPS and 25 U/ml IFN-γ resulted in a stronger NO response by both cell lines compared with treatment with LPS alone (Table 1).
Table 1.
Cytokine and bacterial lipopolysaccharide (LPS)-induced NO production of activated B10-4(S) and P388D.1 macrophage-like cell lines
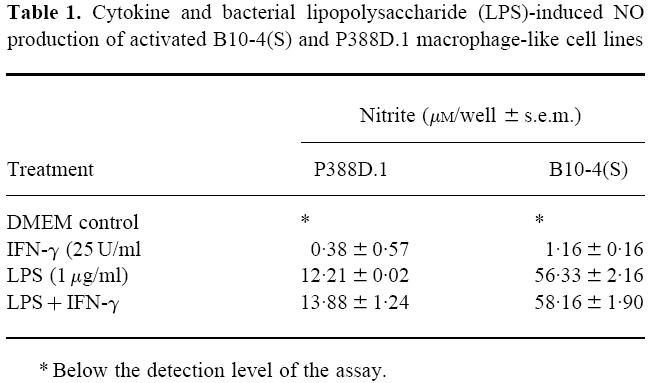
A time course of the NO production of LPS- and IFN-γ-activated P388D.1 macrophages showed that there was no significant nitrite accumulation in the supernatants of macrophage cultures until 12 h after activation (Fig. 1). Thereafter, there was a steady increase in nitrite accumulation until 72 h post-activation. (Fig. 1). The B10-4(S) macrophages had a similar time course of NO response, except that there was no significant nitrite accumulation until 24 h post-activation (Fig. 1). There was no significant nitrite accumulation in the supernatants of macrophage cultures that were not activated with LPS and IFN-γ. The time course of the respiratory viability of the activated B10-4(S) and P388D.1 macrophages showed a maintenance of respiratory viability for the first 6 h (P388D.1) or 12 h (B10-4(S)) followed by a steady increase in respiratory viability until 48 h after activation for P388D.1 or 72 h after activation for B10-4(S) (Fig. 1).
Fig. 1.
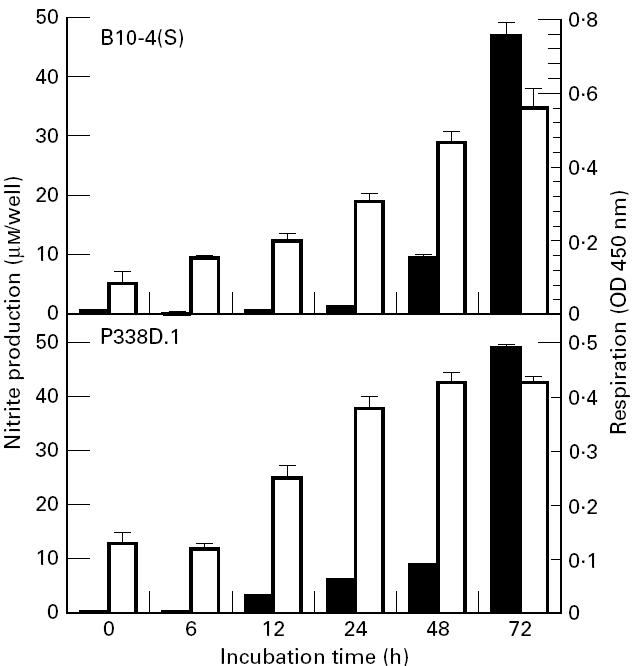
Time course of NO production and cell viability of B10-4(S) and P388D.1 macrophage-like cells activated with 1 μg/ml lipopolysaccharide (LPS) and 25 U/ml IFN-γ. ▪, NO production; □, respiration.
Potassium channel inhibitors decrease nitric oxide production of activated macrophages
The potassium channel inhibitors TEA, 4-AP and quinine caused a significant dose-dependent decrease in nitrite production of both the B10-4(S) and P388D.1 macrophages (Figs 2 and 3). This reduced NO output by the macrophages was not due to a reduced viability or cell number, because respiration of the cells was either at the same level as controls not treated with potassium channel inhibitors or slightly higher than control levels (Figs 2 and 3). There were differences in the sensitivity of both cell lines to the various potassium channel inhibitors. For the B10-4(S) macrophages, TEA was the least effective inhibitor, causing a 28.1% reduction in NO production at the highest concentration tested. In contrast, quinine caused a 55.4% reduction and 4-AP caused a 66.3% reduction in NO production of B10-4(S) macrophages. The P388D.1 macrophage NO response was similar for the three potassium channel inhibitors, causing a 55.4%, 48.2% and 55.3% reduction in NO production for TEA, 4-AP and quinine, respectively. None of the potassium channel inhibitors significantly reduced the viability of either the B10-4(S) cells or the P388D.1 cells at the concentrations examined (Figs 2 and 3).
Fig. 2.
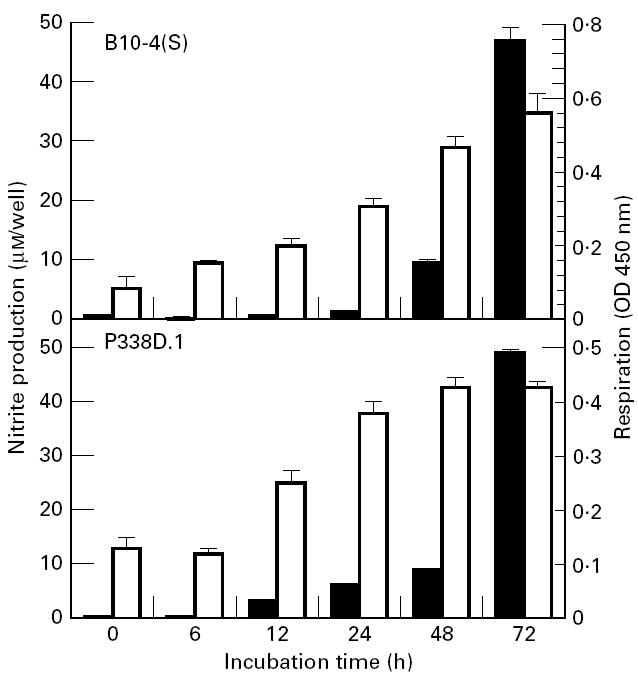
The effects of a 24-h treatment with the potassium channel inhibitors tetraethylammonium (a), 4-aminopyridine (b), and quinine (c) on NO production and cellular viability of B10-4(S) macrophage-like cells activated with 1 μg/ml lipopolysaccharide (LPS) and 25 U/ml IFN-γ. *P < 0.05. ▪, NO production; □, respiration.
Fig. 3.
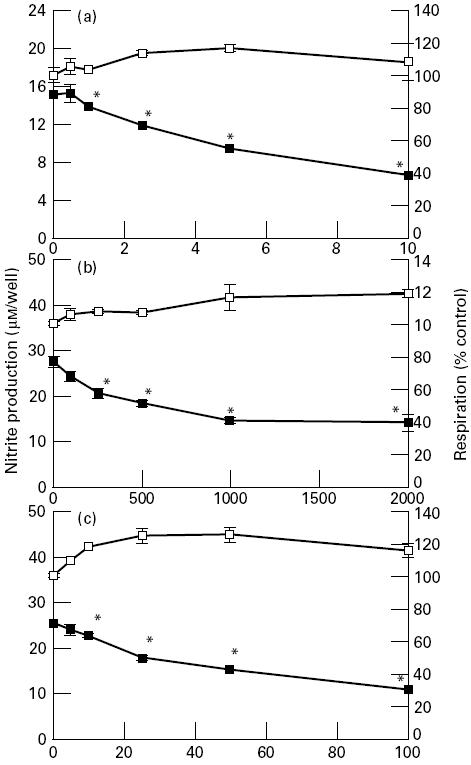
The effects of a 24-h treatment with the potassium channel inhibitors tetraethylammonium (a), 4-aminopyridine (b), and quinine (c) on NO production and cellular viability of P388D.1 macrophage-like cells activated with 1 μg/ml lipopolysaccharide (LPS) and 25 U/ml IFN-γ. *P < 0.05. ▪, NO production; □, respiration.
Time course of nitric oxide production of inhibitor-treated B10-4(S) and P388D.1 macrophages
Treatment of activated P388D.1 macrophages with 10 mm TEA caused a 50% decrease in NO production compared with control by 6 h after treatment (Fig. 4). By 12 h there was a slight recovery in NO production, but thereafter there was a steady decline in NO production to 72 h post-treatment (Fig. 4). The P388D.1 macrophages showed a greater reduction in nitrite production at 6 h after treatment with 1 mm 4-AP, but the cells exhibited a steady recovery to control levels between 12 h and 72 h post-treatment (Fig. 4). Quinine, at a concentration of 50 μm, caused a smaller reduction in NO production than either TEA or 4-AP by 6 h after treatment, but thereafter there was a steady decrease in nitrite production between 12 h and 72 h post-treatment. Examination of the respiration of the P388D.1 macrophages showed that the potassium channel inhibitors had minimal effects on the viability of the macrophages: a slight decrease in respiration at 72 h for TEA, a slight but steady increase in respiration for 4-AP, and no effect on respiration for quinine (data not shown).
Fig. 4.

Time course of the effects of ion channel inhibitor treatment on NO production of B10-4(S) and P388D.1 macrophage-like cells activated with 1 μg/ml lipopolysaccharide (LPS) and 25 U/ml IFN-γ. B10-4 control values (pm/cell ± s.e.m.): 0 h = 4.38 ± 1.92; 6 h = below detection level; 12 h = 57.8 ± 1.92; 24 h = 70.0 ± 4.00; 48 h = 722 ± 38.0; 72 h = 898 ± 8.46. P388D.1 control values (pm/cell ± s.e.m.): 0 h = 6.04 ± 1.92; 6 h = 20.5 ± 0.436; 12 h = 109 ± 0.438; 24 h = 296 ± 4.74; 48 h = 446 ± 16.6; 72 h = 1060 ± 26.2. Note that values of nitrite below the detection level of the assay were given values of zero. TEA, Tetraethylammonium; 4-AP, 4-aminopyridine.
Treatment of the B10-4(S) macrophages with 10 mm TEA, 1 mm 4-AP, and 50 μm quinine had a much more pronounced effect on their NO production than the same treatments had on the P388D.1 macrophages (Fig. 4). TEA caused a significant reduction in NO production, compared with control, by 12 h of incubation (there was no detectable nitrite at 6 h), followed by partial recovery at 24 h, and then a further decrease in NO response from 48 h to 72 h post-treatment (Fig. 4). Treatment of the B10-4(S) macrophages with 4-AP abolished detectable NO production for the first 12 h of incubation but, like the P388D.1 macrophages, with longer incubations there was some recovery of NO production by the B10-4(S) macrophages (Fig. 4). While not as effective as the other potassium channel inhibitors, quinine caused a large reduction in NO production by 12 h post-treatment, and longer incubation caused a further steady decrease (Fig. 4). The viability of the B10-4(S) macrophages was not affected by the potassium channel inhibitor incubations (data not shown).
Potassium channel agonist induces increased nitric oxide production by activated macrophages
To verify that the effect on NO production was due to a specific effect of the potassium channel inhibitors on potassium channels of the macrophages, the effects of the potassium channel agonist minoxidol on NO production and viability of the cells was assessed. Minoxidol caused a dose-dependent enhancement of nitrite production in both macrophage-like cell lines that was significant above a dose of 50 nm (Fig. 5). At the doses where there was the greatest enhancement of NO production there were no significant effects on respiration (Fig. 5).
Fig. 5.
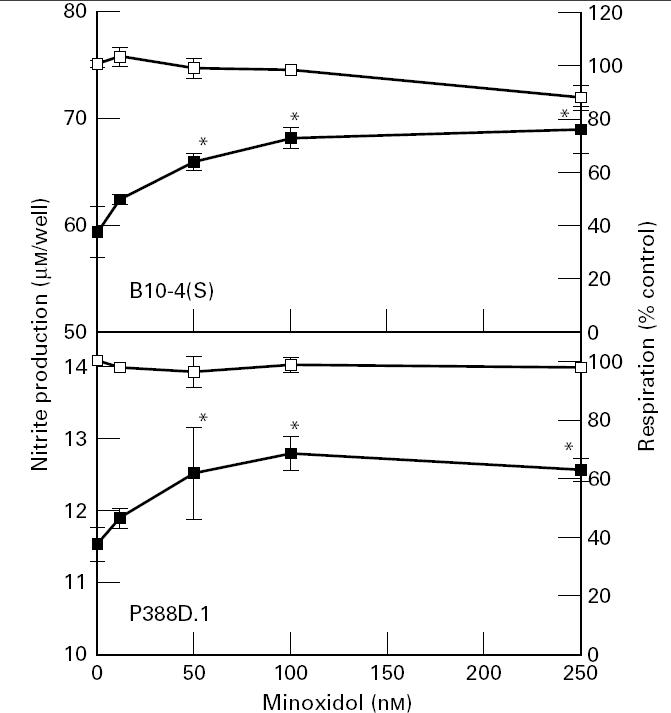
The effects of a 24-h treatment with minoxidol on the NO production and cellular viability of B10-4(S) and P388D.1 macrophage-like cells activated with 1 μg/ml lipopolysaccharide (LPS) and 25 U/ml IFN-γ. *P < 0.05. ▪, NO production; □, respiration.
Potassium channel inhibitors affect the ‘priming’ of macrophages for cytotoxic response
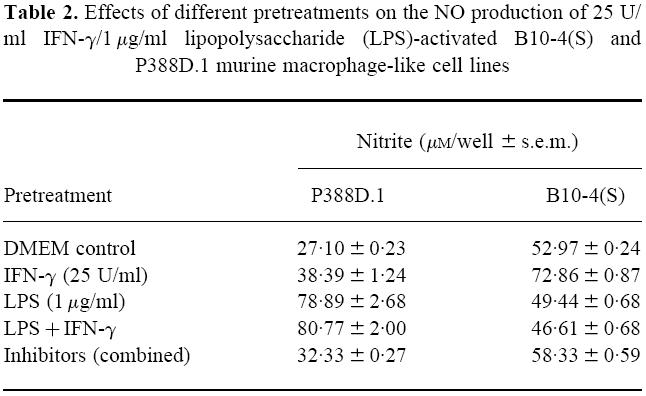
The effects of various activator and inhibitor pretreatments on NO production were assessed to determine which stage of activation was being affected by the potassium channel inhibitors. The control treatments consisted of cells that had only been treated with LPS and IFN-γ. For both the B10-4(S) and P388D.1 cell lines, the DMEM and inhibitor pretreatments had similar NO production, except that the inhibitor pretreatment plates had slightly higher nitrite values (Table 2). Pretreatment with IFN-γ caused significantly higher NO production than the DMEM control in both cell lines (Table 2). For the B10-4(S) cell line, pretreatment with either LPS or LPS and IFN-γ resulted in similar NO levels that were almost identical to the DMEM control (Table 2). The P388D.1 cell line also had similar NO production between macrophages treated with LPS or those treated with LPS and IFN-γ, but there was twice as much NO as in the DMEM control (Table 2).
Table 2.
Effects of different pretreatments on the NO production of 25 U/ml IFN-γ/1 μg/ml lipopolysaccharide (LPS)-activated B10-4(S) and P388D.1 murine macrophage-like cell lines
Pretreatment of the macrophages of the B10-4(S) and P388D.1 cell lines with potassium channel inhibitors for 6 h significantly decreased the NO production of both cell lines compared with the DMEM pretreatment (Fig. 6). This further reduction in NO production was not due to a reduction in viability, since the respiration was consistently either at the same level or slightly higher than the DMEM control for all pretreatments (data not shown). Pretreatment with IFN-γ or LPS and IFN-γ partially restored the NO production of potassium channel inhibitor-treated macrophages in both cell lines, with the LPS and IFN-γ pretreatment causing a larger restoration than IFN-γ alone (Fig. 6). Treatment with LPS alone consistently caused a much lower restoration of NO production within both cell lines than pretreatment with the other activators (Fig. 6). In fact, for the 10 mm TEA-treated B10-4(S) cells, pretreatment with LPS caused a reduction in NO production compared with the DMEM pretreatment (Fig. 6).
Fig. 6.
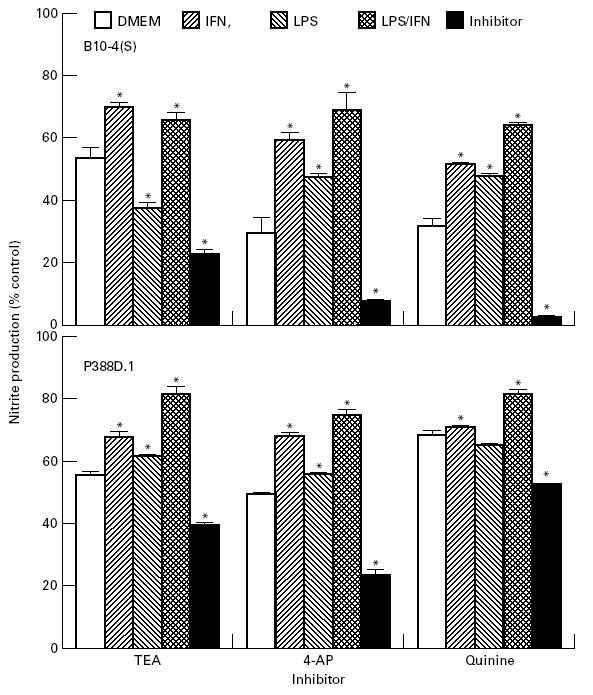
Effects of different 6-h pretreatments on the NO production of 1 μg/ml lipopolysaccharide (LPS)/25 U/ml IFN-γ-activated B10-4(S) and P388D.1 macrophage-like cells treated with 10 mm tetraethylammonium (TEA), 1 mm 4-aminopyridine (4-AP), or 50 μm quinine. B10-4 control values (pm/cell ± s.e.m.): Dulbeccp's modified Eagles' medium (DMEM) = 530 ± 2.37; IFN-γ = 729 ± 8.67; LPS = 494 ± 6.82; LPS and IFN-γ = 466 ± 6.82; inhibitor = 583 ± 0.589. P388 D.1 control values (pm/cell ± s.e.m.): DMEM = 271 ± 23.5; IFN-γ = 384 ± 12.4; LPS = 789 ± 26.8; LPS and IFN-γ = 808 ± 20.0; inhibitor = 323 ± 2.74.
DISCUSSION
The dose-dependent reductions in NO production of the B10-4(S) and P388D.1 cell lines induced by treating activated macrophages with the potassium channel inhibitors TEA, 4-AP and quinine show that potassium channel inhibitors affect NO production. In a different study using the ATP-sensitive potassium channel inhibitor glibenclamide, Wu et al. [8] concluded that while glibenclamide reduced the NO production of the macrophage-like J774.2 cells, it was not through any effects on the potassium channels of the macrophages [8]. Our results do not support this conclusion. By using a number of general inhibitors of potassium channels, we have established that the reduction in NO production by the inhibitors is due to an effect on the macrophages' potassium channels. We observed variations in the sensitivity of the B10-4(S) and P388D.1 cell lines to the different potassium channel inhibitors, especially to TEA. TEA caused a minimal reduction in NO production of B10-4(S) cells, but caused reductions equivalent to those for 4-AP and quinine in the P388D.1 cells. The inability of TEA to reduce the NO response in the Wu et al. [8] study may have been because the J774.2 cell line may be unresponsive to treatment with TEA.
The blockage of potassium channel inhibitors must in some way be involved in the induction or production of NO by iNOS. Our results show that treatment of activated macrophages with an agonist of potassium channels significantly increased the NO production of the cells, indicating that the alterations in the function of potassium channels can cause significant changes in NO production of activated macrophages. If the observed effects of potassium channel inhibitors on NO production were due only to non-specific toxicity that reduced either cell number or individual cell viability then a potassium channel agonist would not be expected to enhance NO production.
Potassium channels could be involved in the intracellular signals associated with either the priming step inducing formation of iNOS or the triggering step activating iNOS. Theoretically, the potassium channels could be responsible for producing conditions necessary for the iNOS enzyme to function at maximum efficiency. However, Wu et al. [8] convincingly showed glibenclamide, an ATP-sensitive potassium channel inhibitor, was not affecting the function of iNOS [8]. Glibenclamide was less effective at inhibiting the NO production of J774.2 cells when presented several hours after LPS activation, whereas the iNOS inhibitor L-NAME was equally effective at inhibiting NO production when presented from 2 h to 10 h after treating cells with LPS [8]. Therefore, the reductions in NO production were not due to a reduction in iNOS function, suggesting that potassium channels must be involved in inducing the formation and activation of iNOS.
The reduction of the NO response of macrophages caused by the potassium channel inhibitors is unlikely to be caused by the blockade of any single potassium channel type. The potassium channel inhibitors used in our study affect numerous types of potassium channels and affect those channel types through different mechanisms. In our experimental set-up, TEA was acting from outside the cell by binding to a specific receptor site on the extracellular side of the potassium channel and physically blocking the pore [7–9]. Delayed-rectifier type, inward rectifier type, calcium-sensitive type and ATP-sensitive type potassium channels are all affected by TEA, with only the exact concentration of TEA required to achieve half-maximal blockage of potassium channels varying from type to type [7]. The inhibitor 4-AP is lipid-soluble and membrane-permeant, allowing it to cross cell membranes to reach its site of action [7]. The binding site for 4-AP is probably within the pore of the potassium channel, and 4-AP probably binds the receptor by either directly diffusing out of the cell membrane or diffusing from the cytoplasmic side of the channel once the physical gate of the potassium channel opens [7,21]. Within macrophages, 4-AP physically blocks the pore of the delayed-rectifier type potassium channels. Quinine and quinidine are configurationally isomeric alkaloids isolated from plants of the Cinchona family that presumably block potassium channels through a similar mechanism [22]. Quinidine, like 4-AP, is a membrane-permeant molecule that blocks potassium channels by passing into the cytoplasm of cells or diffusing from the cell membrane to bind the receptor within the pore and physically block the passage of K+ ions [7,21–23]. Quinidine, and presumably quinine, blocks delayed-rectifier, calcium-sensitive type, and ATP-sensitive type potassium channels of macrophages, and, as with TEA, the exact concentration required for half-maximal inhibition varies from type to type [7,23]. Given the wide variety of potassium channel types being affected by the potassium channel inhibitors, it is possible that each potassium channel type contributes a small amount to the activation or signalling process responsible for inducing NO production.
The order of activator and inhibitor treatments was examined to establish whether potassium channels were involved in the priming stage or the triggering stage of macrophage activation. A 6-h pretreatment of the B10-4(S) and P388D.1 cells with the potassium channel inhibitors before activation with IFN-γ and LPS caused a greater reduction in the NO production of both cell lines compared with cells undergoing a 6-h preincubation in DMEM. Conversely, 6-h pretreatments with IFN-γ or IFN-γ and LPS resulted in significant restorations of NO production in macrophages treated with 10 mm TEA, 1 mm 4-AP, and 50 μm quinine. Taken together, our results suggest that potassium channels are involved in the priming stage of macrophage activation, because treatment with IFN-γ alone primes the macrophages for antimicrobial activity.
Alteration in the activity of potassium channels clearly results in an equivalent effect, whether inhibitory or enhancing, on the NO production of macrophage-like cell lines. This alteration in NO production is probably due to effects of altered potassium channel activity on one of the intracellular signalling steps in the cascade that primes the macrophage and induces iNOS. The exact intracellular signalling steps being affected and the mechanism of potassium channel involvement remain to be elucidated.
Acknowledgments
This research was supported by funding from the Alberta Heritage Foundation for Medical Research and the Natural Sciences and Engineering Research Council of Canada.
References
- 1.de Groote MA, Fang FC. NO inhibitions: antimicrobial properties of nitric oxide. Clin Infect Dis. 1995;21(Suppl. 2):S162–5. doi: 10.1093/clinids/21.supplement_2.s162. [DOI] [PubMed] [Google Scholar]
- 2.Dong Z, Yang X, Xie K, Juang S-H, Llansa N, Fidler IJ. Activation of inducible nitric oxide synthase gene in murine macrophages requires protein phosphatases 1 and 2A activities. J Leuko Biol. 1995;58:725–32. doi: 10.1002/jlb.58.6.725. [DOI] [PubMed] [Google Scholar]
- 3.Park YC, Jun CD, Kang HS, Kim HD, Kim HM, Chung HT. Role of intracellular calcium as a priming signal for the induction of nitric oxide synthesis in murine peritoneal macrophages. Immunol. 1996;87:296–302. doi: 10.1046/j.1365-2567.1996.456544.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Raddassi K, Berthon B, Petit J-F, Lemaire G. Role of calcium in the activation of mouse peritoneal macrophages: induction of NO synthase by calcium ionophores and thapsigargin. Cell Immunol. 1994;153:443–55. doi: 10.1006/cimm.1994.1041. [DOI] [PubMed] [Google Scholar]
- 5.Jun C-D, Yoon H-J, Park Y-C, Lee S-Y, Kang S-S, Kim H-M, Chung H-T. Synergistic cooperation between thapsigargin and phorbol ester for induction of nitric oxide synthesis in murine peritoneal macrophages. Free Rad Biol Med. 1996;20:769–76. doi: 10.1016/0891-5849(95)02157-4. [DOI] [PubMed] [Google Scholar]
- 6.Jun C-D, Choi B-M, Ryu H, et al. Synergistic cooperation between phorbol ester and IFN-γ for induction of nitric oxide synthesis in murine peritoneal macrophages. J Immunol. 1994;153:3684–90. [PubMed] [Google Scholar]
- 7.Hille B. Ionic channels of excitable membranes. 2. Sunderland: Sinauer Associates Inc; 1992. [Google Scholar]
- 8.Wu C-C, Thiemermann C, Vane JR. Glibenclamide-induced inhibition of the expression of inducible nitric oxide synthase in cultured macrophages and in the anaesthetized rat. Br J Pharmacol. 1995;114:1273–81. doi: 10.1111/j.1476-5381.1995.tb13343.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xie Q-W, Nathan C. The high-output nitric oxide pathway: role and regulation. J Leuk Biol. 1994;56:576–82. doi: 10.1002/jlb.56.5.576. [DOI] [PubMed] [Google Scholar]
- 10.Gallin EK. Ion channels in leukocytes. Physiol Rev. 1991;71:775–811. doi: 10.1152/physrev.1991.71.3.775. [DOI] [PubMed] [Google Scholar]
- 11.Artalejo CR, Rossie S, Perlman RL, Fox AP. Voltage-dependent phosphorylation may recruit Ca2+ current facilitation in chromaffin cells. Nature. 1992;358:63–66. doi: 10.1038/358063a0. [DOI] [PubMed] [Google Scholar]
- 12.Haslberger A, Romanin C, Koerber R. Membrane potential modulates release of tumor necrosis factor in lipopolysaccharide-stimulated mouse macrophages. Mol Biol Cell. 1992;3:451–60. doi: 10.1091/mbc.3.4.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schlicter L, Sidell N, Hagiwara S. Potassium channels mediate killing in human natural killer cells. Proc Natl Acad Sci USA. 1986;83:451–5. doi: 10.1073/pnas.83.2.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Damjanovich S, Pieri C, Tron L. Ion channel activity and transmembrane signaling in lymphocytes. Ann N Y Acad Sci. 1992;650:205–10. doi: 10.1111/j.1749-6632.1992.tb49123.x. [DOI] [PubMed] [Google Scholar]
- 15.Koren HS, Handwerger BS, Wunderlick JR. Identification of macrophage-like characteristics in a cultured murine tumour line. J Immunol. 1975;114:894–7. [PubMed] [Google Scholar]
- 16.Radzioch D, Hudson T, Boule M, Barrera L, Urbance JW, Varesio L, Skamene E. Genetic resistance/susceptibility to mycobacteria: phenotypic expression in bone marrow derived macropahges lines. J Leuk Biol. 1991;50:263–72. doi: 10.1002/jlb.50.3.263. [DOI] [PubMed] [Google Scholar]
- 17.Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR. Analysis of nitrate, nitrite and 15-nitrate in biological fluids. Anal Biochem. 1982;126:131–8. doi: 10.1016/0003-2697(82)90118-x. [DOI] [PubMed] [Google Scholar]
- 18.Scudiero DA, Shoemaker RH, Paul KD, et al. Evaluation of a soluble tetrazolium/formazan assay for cell growth and drug sensitivity in culture using human and other tumor cell lines. Cancer Res. 1988;48:4827–33. [PubMed] [Google Scholar]
- 19.Roehm NW, Rodgers GH, Hatfield SM, Glasebrook AL. An improved colorimetric assay for cell proliferation and viability using the tetrazolium salt XTT. J Immunol Methods. 1991;142:257–65. doi: 10.1016/0022-1759(91)90114-u. [DOI] [PubMed] [Google Scholar]
- 20.Stevens MG, Olsen SC. Comparative analysis of using MTT and XTT in colorimetric assays for quantiating bovine neutrophil bactericidal activity. J Immunol Methods. 1993;157:225–31. doi: 10.1016/0022-1759(93)90091-k. [DOI] [PubMed] [Google Scholar]
- 21.Hara N, Ichinose M, Sawach M, Maeno T. The activation of Ca2+-dependent K+ conductance by adrenaline in mouse macrophages. Pflugers Arch. 1991;419:371–9. doi: 10.1007/BF00371119. [DOI] [PubMed] [Google Scholar]
- 22.MacKinnon R. New insights into the structure and function of potassium channels. Curr Opin Neurobiol. 1991;1:14–19. doi: 10.1016/0959-4388(91)90005-r. [DOI] [PubMed] [Google Scholar]
- 23.Jan LY, Jan YN. Structural elements involved in specific K+ channel functions. Ann Rev Physiol. 1992;54:537–55. doi: 10.1146/annurev.ph.54.030192.002541. [DOI] [PubMed] [Google Scholar]