

Abstract
Nasal administration of μg doses of acetylcholine receptor (AChR) is effective in preventing the development of B cell-mediated EAMG in the Lewis rat, a model for human MG. In order to investigate whether nasal administration of AChR modulates ongoing EAMG, Lewis rats were treated nasally with AChR 2 weeks after immunization with AChR and Freund's complete adjuvant. Ten-fold higher amounts of AChR given nasally (600 μg/rat) were required to ameliorate the manifestations of EAMG compared with the amounts necessary for prevention of EAMG. In lymph node cells from rats receiving 600 μg/rat of AChR, AChR-induced proliferation and interferon-gamma (IFN-γ) secretion were reduced compared with control EAMG rats receiving PBS only. The anti-AChR antibodies in rats treated nasally with 600 μg/rat of AChR had lower affinity, reduced proportion of IgG2b and reduced capacity to induce AChR degradation. Numbers of AChR-reactive IFN-γ and tumour necrosis factor-alpha (TNF-α) mRNA-expressing lymph node cells from rats treated nasally with 600 μg/rat of AChR were suppressed, while IL-4, IL-10 and transforming growth factor-beta (TGF-β) mRNA-expressing cells were not affected. Collectively, these data indicate that nasal administration of AChR in ongoing EAMG induced selective suppression of Th1 functions, i.e. IFN-γ and IgG2b production, but no influence on Th2 cell functions. The impaired Th1 functions may result in the production of less myasthenic anti-AChR antibodies and contribute to the amelioration of EAMG severity in rats treated with AChR 600 μg/rat by the nasal route.
Keywords: mucosal tolerance, experimental autoimmune myasthenia gravis, Th1 cells, Th2 cells, cytokine
INTRODUCTION
Mucosal antigen exposure without adjuvant results in immunological unresponsiveness upon subsequent parenteral challenge, i.e. mucosal tolerance. Oral tolerance induction involves multiple mechanisms, including the induction of apoptosis and anergy of antigen-specific T cells following administration of high antigen doses, while induction of regulatory Th2 and Th3 cells follows low-dose antigen feeding (reviewed in [1]). Exploitation of the oral route for prevention and treatment of autoimmune disease has dealt with experimental models of cell-mediated conditions, such as experimental autoimmune encephalomyelitis (EAE), uveitis (EAU), or arthritis. Myasthenia gravis (MG) and its animal model EAMG are caused by autoantibodies against nicotinic acetylcholine receptors (AChR) at the neuromuscular junction [2]. The production of anti-AChR antibodies is mediated by cytokines produced by T helper (Th) [3,4]. Immunotherapies aimed at targeting disease-inducing AChR-specific Th cell populations should be effective in EAMG. We and others have used oral tolerance induction as a strategy to prevent EAMG, and found that oral administration of AChR inhibits clinical EAMG and suppresses both humoral and cellular immune responses [5–7]. AChR administration by the nasal route, although only 1/1000 of the dose of AChR needed, is still as effective as oral tolerance induction [8].
To determine whether nasal administration of AChR can be used to treat ongoing EAMG, i.e. a situation more likely to be encountered in human MG, we administered AChR nasally to Lewis rats 2 weeks after immunization with AChR + Freund's complete adjuvant (FCA). The effects of nasal administration of AChR in ongoing EAMG were evaluated following several important aspects: (i) clinical course of EAMG; (ii) AChR-induced T cell responses; (iii) anti-AChR antibody specificity; (iv) cytokine profiles.
MATERIALS AND METHODS
Antigen preparations
AChR was purified from the electroplax tissue of Torpedocalifornica (Pacific Biomarine, Venice, CA) by affinity chromatography on α-cobrotoxin-agarose resin (Sigma, St Louis, MO) [9]. The product was pure as judged by SDS–PAGE. The control antigen myelin basic protein (MBP) was purified from guinea pig spinal cord [10]. Purity was confirmed by SDS–PAGE.
Immunization
Female Lewis rats, 8 weeks of age, were purchased from Charles River Co. (Sulzfeld, Germany). Each rat was immunized subcutaneously in both hind footpads and base of tail with 50 μg of AChR emulsified in FCA in a total volume of 200 μl. The clinical severity of EAMG was blindly graded [11] as follows: 0, no weakness; 1 +, mildly decreased activity, weak grip or cry, with fatigue; 2 +, markedly decreased activity and body weight, hunched posture at rest, head down and forelimb digits flexed, tremulous ambulation; and 3 +, severe generalized weakness, no cry or grip. Rats were killed at day 49 post-immunization (p.i.).
Nasal tolerance induction
The schedule previously described for rats nasally tolerized with AChR before immunization [8] was modified. Two weeks p.i., rats received into each nostril 30 μl PBS pH 7.4 containing AChR at concentrations of 100 μg/ml, 500 μg/ml or 1000 μg/ml using a micropipette. Control rats received PBS only. At each administration, rats were gently anaesthetized with ether. The administrations were performed daily for 10 days. In all, each rat received AChR at amounts of 60, 300 or 600 μg.
Radioimmunoassay for muscle AChR content
Triplicate 2 pm aliquots of 125I-α-bungarotoxin (α-BT; Amersham Corp., Arlington Heights, IL)-labelled Triton X-100 solubilized rat muscle extract were mixed with standard pooled rat anti-AChR antiserum. After incubation, rabbit anti-rat immunoglobulin (Dakopatts, Copenhagen, Denmark) was added. The precipitates were counted in a Packard γ-counter. The percentage loss of muscle AChR in test rat carcass was calculated as described [12].
Enumeration of antigen-reactive interferon-gamma-secreting cells
The rats were killed on day 49 p.i. Popliteal and inguinal lymph node (PILN) cells were prepared and adjusted to a cell concentration of 2 × 106/ml. A solid-phase ELISPOT assay was adopted [13]. Nitrocellulose-bottomed microtitre plates (Microtiter-HAM plates; Millipore Co., Bedford, MA) were coated with 100 μl per well at 15 μg/ml of rat interferon-gamma (IFN-γ) capture antibody DB1 (Innogenetics, Genth, Belgium). Aliquots of 200 μl of cell suspension containing 4 × 105 mononuclear cells (MNC) were added to individual wells in triplicate, followed by antigen (AChR, MBP), or mitogen (concanavalin A (Con A); Sigma) in 10-μl aliquots to a final concentration of 10 μg/ml (AChR, MBP), or 5 μg/ml (Con A). The wells were emptied after 48 h of culture. Secreted and bound IFN-γ was visualized by sequential application of rabbit polyclonal anti-rat IFN-γ antibody (Innogenetics), biotinylated anti-rabbit IgG and avidin-biotin peroxidase complex (ABC; Dakopatts). After peroxidase staining, the red-brown spots which corresponded to the cells that had secreted IFN-γ were enumerated in a dissection microscope. To calculate the numbers of T cells responding to a particular antigen or mitogen, numbers of spots in culture without antigen (usually 1.5–2.4 per 105 MNC in this study) were subtracted from values obtained after antigen challenge. The data were expressed as numbers of spots per 105 cultured MNC.
Lymphocyte proliferation responses
Briefly, 200-μl aliquots of MNC suspensions with a cell density of 2 × 106/ml were applied in triplicate into round-bottomed wells of 96-well microtitre plates (Nunc, Copenhagen, Denmark). Ten-microlitre aliquots of AChR, MBP or Con A were added into appropriate wells at final concentrations of 10 μg/ml (AChR, MBP) or 5 μg/ml (Con A). After 60 h of incubation, the cells were labelled for an additional 12 h with 10-μl aliquots containing 1 μCi of 3H-methylthymidine (specific activity 42 Ci/mmol; Amersham, Aylesbury, UK). Cells were harvested onto glassfibre filters and thymidine incorporation was measured. The results were expressed as stimulation index (SI).
Determination of AChR-specific IgG isotype antibodies
Micotitre plates (Costar, Cambridge, MA) were coated with AChR at 5 μg/ml in a volume of 100 μl/well and incubated at 4°C overnight. After washing with PBS containing 0.05% Tween 20, non-specific binding was blocked with 1% bovine serum albumin (BSA). After washing, serum samples diluted 1:1000 were overlaid and incubated for 2 h at room temperature, followed by biotinylated mouse anti-rat IgG2a (1:2000; Ams, Frankfurt, Germany) or IgG1 (1:2000; Ams) and incubated for 2 h at room temperature. After washing and further incubation with avindin-biotin-alkaline phosphatase complex (Vector, Burlingame, CA), reaction was visualized after incubation with p-nitrophenyl phosphate (Sigma) solution and read at 405 nm.
Detection of relative affinity of serum anti-AChR IgG antibodies
The relative affinity of anti-AChR IgG antibodies was determined by ELISA using thiocyanate elution [14]. Briefly, microtitre plates (Costar) were coated with AChR and uncoated sites were blocked with 10% fetal calf serum (FCS; Gibco, Paisley, UK). Diluted serum with a predetermined amount of anti-AChR antibodies was added and incubated. Then, appropriate quantities of potassium thiocyanate (KSCN) were added in duplicate and incubated, followed by biotinylated goat anti-rat IgG and alkaline phosphatase-labelled avidin D. The colour was developed with p-nitrophenyl phosphate and expressed as absorbance at 405 nm. The relative affinity is expressed as affinity index, equal to the molarity of KSCN resulting in 50% of the absorbance obtained in the absence of KSCN.
Detection of functional ability of anti-AChR antibodies in muscle culture system
Cultures of limb muscles of 1–3-day-old Lewis rats were prepared as described [14]. Degradation of AChR was measured by an indirect method that depends on the specific labelling of the receptors with 125I-labelled α-BT (Amersham) and the release of radioactive material derived from degraded receptors into the culture medium. In order to test the effect of the antibody on receptor degradation in the cultures, the usual culture medium was replaced with 1 ml minimum essential medium (MEM; Gibco), to which 0.1 ml (2 mg/ml) of either control immunoglobulin or immunoglobulin from control EAMG rats was added, and EAMG rats were treated with AChR 600 μg/rat. The immunoglobulins were prepared [15] from pools of sera from control EAMG rats, and from EAMG rats treated with AChR 600 μg/rat. Immunoglobulin prepared from normal Lewis rats served as control. The radioactivity released into the medium, representing degraded receptors, was determined at intervals of up to 40 h, and degradation rates were calculated as described [16].
Detection of IFN-γ, tumour necrosis factor-alpha, IL-4, IL-10 and transforming growth factor-beta mRNA-expressing cells by in situ hybridization
In situ hybridization (ISH) was performed to detect cytokine mRNA expression in MNC as described [6]. Aliquots of 200 μl of cell suspensions from PILN containing 4 × 105 MNC were added into round-bottomed microtitre plates (Nunc) in triplicate. Separate cultures received no antigen, 10 μl aliquots of Con A (50 μg/ml), AChR (100 μg/ml), MBP (100 μg/ml). After culture for 24 h, cells were washed, counted and dried onto restricted areas of electrically charged glass slides (ProbeOn slides; Fisher Scientific, Pittsburgh, PA). Oligonucleotide probes (Scandinavian Gene Synthesiz AB, Köping, Sweden) were labelled using 35S-deoxyadenosine-5′-α-(thio)-triphosphate with terminal deoxynucleotidyl transferase (Amersham). The oligonucleotide sequences were obtained from GenBank using the MacVector System (Table 1). Control slides were hybridized with the same total amount of a sense probe with nucleotide sequence for exon 4 of rat IFN-γ. Cells were hybridized with 106 ct/min of labelled probe per 100 μl of hybridization mixture. After emulsion autoradiography, development and fixation, the coded slides were examined by dark field microscopy for positive cells containing > 15 grains per cell in a star-like distribution. In many positive cells, the grains were so heavily accumulated within and around the cells that it was not possible to count every single grain. In cells judged negative, the numbers of grains were mostly 0–2 per cell, and the grains were scattered randomly over the cells and not distributed in a star-like fashion. There were therefore no difficulties in differentiating between positive and negative cells. A control probe of the sense sequence for rat IFN-γ exon 4 was used in parallel with the cytokine probes, producing a uniformly weak background signal without revealing any positive cells. The number of cells used for ISH was not equal to the numbers that were ultimately detected on the slides. To compensate for cell losses, the total number of cells on the slides was counted. With the help of a microscope grid as a measuring unit, the radius (r) of the surface area (A) covered by cells was determined. The area A was calculated by the formula A = p × r2. Cells were usually counted in two grids at the periphery and one grid at the centre of the surface covered by cells. In case of uneven distribution, cells in additional grids were counted. The mean value of the number of the cells per grid was determined and multiplied by A. Variation between duplicates was < 10%. Results are expressed as numbers of labelled cells per 105 MNC.
Table 1.
Statistical analysis
Differences between pairs of groups were tested by Student's t-test. Differences between four groups were tested by one-factor analysis of variance (anova). Mann–Whitney's U-test was used to compare the maximal severity of clinical signs. The level of significance was set to α = 0.05 or 0.01. All tests were two-sided.
RESULTS
Treatment of EAMG with nasal AChR dose-dependently ameliorates muscular weakness and loss of muscle AChR
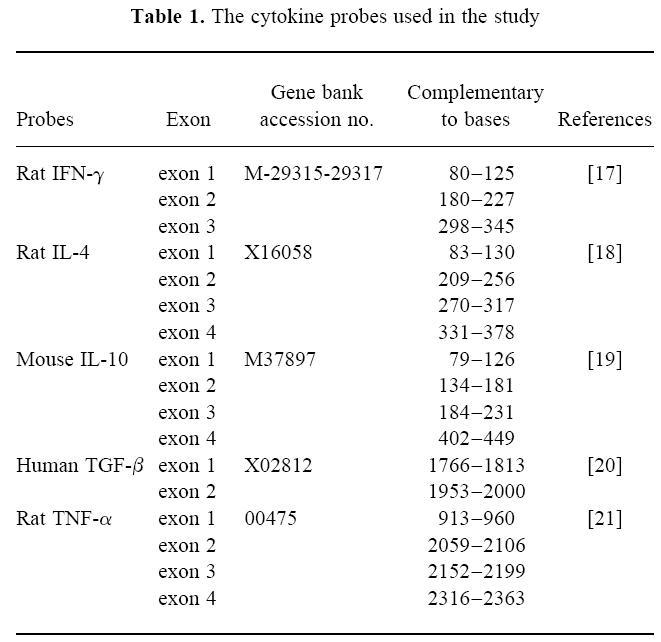
Control Lewis rats receiving PBS nasally after immunization with AChR + FCA developed typical two-phase EAMG, with an early phase of mild (+) or moderately severe (+ +) muscular weakness around 8 days p.i., and a late phase of progressive muscular weakness from about 27 days p.i., and afterwards most rats developed severe muscular weakness and four out of 10 rats died during the observation period of 7 weeks. Two weeks after immunization when AChR was administered nasally, rats had already recovered from the early phase of muscular weakness. The onset of late phase of muscular weakness was not affected by AChR nasal administration. In rats receiving 60 μg/rat and 300 μg/rat of AChR nasally, the muscular weakness was as severe as in control EAMG rats. In contrast, in rats receiving a high dose of AChR (600 μg/rat), five out of six rats developed relatively mild muscular weakness compared with control EAMG rats (mean clinical score at week 7 p.i.: 0.93 versus 3.12; P < 0.05) (Fig. 1). This group of rats also showed less pronounced loss of muscle AChR compared with control EAMG rats (Fig. 2).
Fig. 1.
Effects of nasal administration of acetylcholine receptor (AChR) on ongoing EAMG. Groups of 8–10 female Lewis rats were first immunized with AChR + Freund's complete adjuvant (FCA). Two weeks post-immunization (p.i.), rats were treated nasally with either different doses of AChR (total 60, 300 or 600 μg/rat) or PBS for 10 consecutive days. Arrows indicate days 14–24 p.i., when nasal treatment with AChR was conducted. Rats treated with 60 or 300 μg/rat of AChR exhibited a similar disease pattern as control EAMG rats treated nasally with PBS only. Rats treated nasally with 600 μg/rat of AChR developed relatively mild muscular weakness in comparison with control EAMG rats (mean clinical score at week 7 p.i.: 0.93 versus 3.12; P < 0.05). □, PBS; ▪, AChR 60 μg/rat; ○, AChR 300 μg/rat; ▵, AChR 600 μg/rat.
Fig. 2.

Loss of muscle acetylcholine receptor (AChR) content. The muscle carcasses from EAMG rats treated nasally with AChR and from control EAMG rats at week 7 post-immunization (p.i.) were examined for AChR content by radioimmunoassay (RIA). The percentage loss of muscle AChR was calculated as described in Materials and Methods. Data from eight rats in each group are shown. Symbols refer to mean values, bars to s.e.m. *P < 0.05.
Treatment of EAMG with nasal AChR suppresses AChR-specific Th1 responses
AChR-specific Th1 responses reflected by levels of AChR-reactive IFN-γ-secreting cells were also reduced in these rats (P = 0.02) compared with control EAMG rats (Fig. 3a). Proliferative responses to AChR were suppressed in EAMG rats treated nasally with AChR 600 μg/rat (P < 0.05) (Fig. 3b). There were no differences in proliferative responses to AChR or numbers of AChR-reactive IFN-γ-secreting cells between rats receiving 60 μg/rat or 300 μg/rat AChR compared with results in control EAMG rats. There were also no differences in T cell proliferative responses or numbers of IFN-γ-secreting cells between control EAMG rats and rats treated with the different doses of AChR nasally in control experiments when cells were cultured in the presence of MBP, Con A, or no antigen (data not shown).
Fig. 3.

(a) Numbers of acetylcholine receptor (AChR)-reactive IFN-γ-secreting cells per 105 lymph node mononuclear cells (MNC) from EAMG rats treated nasally with different doses of AChR and from control EAMG rats treated nasally with PBS only. Eight rats in each group were killed 7 weeks post-immunization (p.i.). Symbols refer to mean values and bars to s.d. *P < 0.05. (b) Proliferative responses to AChR on day 49 p.i. of lymph node cells from EAMG rats treated nasally with AChR and from control EAMG rats. Results are expressed as stimulation index (SI). Symbols refer to mean values and bars to s.e.m. *P < 0.05.
Anti-AChR IgG2b and IgG1 antibodies are reciprocally regulated by altered Th1 functions in EAMG rats nasally treated with AChR
Serum antibody isotype profiles can serve as relative indicators for Th subset activation in rats [22,23]. IgG1 and IgG2b were analysed at week 7 p.i. The rats treated with AChR 600 μg/rat had lower levels of AChR-specific IgG2b than control rats, suggesting that Th1 functions were suppressed in these AChR-treated rats. This group of rats also showed a borderline increase of serum IgG1 levels compared with control EAMG rats, suggesting the presence of Th2 functions (Fig. 4).
Fig. 4.
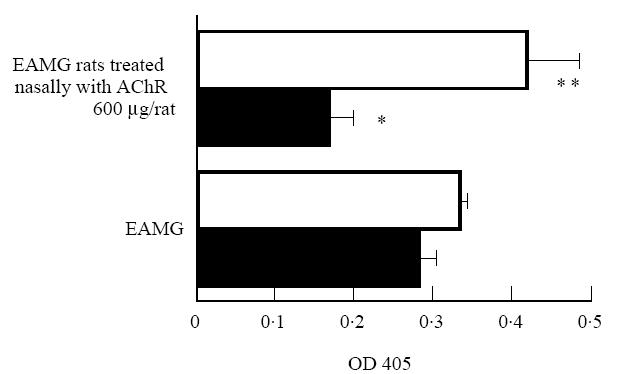
Serum anti-acetylcholine receptor (AChR) IgG isotype antibodies measured by ELISA. Sera from eight rats were analysed for IgG1 (□) and IgG2b (▪) at week 7 post-immunization (p.i.), when anti-AChR antibody IgG levels were higher in EAMG rats treated with AChR 600 μg/rat than in control EAMG rats treated with PBS only. Symbols refer to mean values and bars to s.e.m. *P < 0.05; **P = 0.0562.
Functional studies of anti-AChR antibodies
To characterize further the elevated anti-AChR antibodies resulting from nasal administration of AChR to treat ongoing EAMG, we measured the antibody affinity by KSCN-ELISA. The relative affinity of serum anti-AChR antibodies in control EAMG rats treated with PBS only increased gradually throughout the observation period. In rats treated with a high dose of AChR nasally, the relative affinity of serum anti-AChR antibodies was lower than in control EAMG rats. Significant differences between the two groups were noticed at 5 and 7 weeks p.i. (Fig. 5). These results indicate that serum anti-AChR antibodies at these time points had poor binding capacity and were thus less myasthenic. No such differences were found between rats treated with AChR 60, 300 μg/rat or control EAMG rats (data not shown).
Fig. 5.

The relative affinity of serum anti-acetylcholine receptor (AChR) antibodies measured at 1, 3, 5 and 7 weeks post-immunization (p.i.) by potassium thiocyanate (KSCN)-ELISA in EAMG rats treated nasally with AChR 600 μg/rat (▪) and control EAMG rats treated nasally with PBS only (□). Data from eight rats in each group are shown. Symbols refer to mean values and bars to s.e.m. *P < 0.05.
We also measured the functional ability of serum AChR antibodies to induce accelerated endocytosis and degradation of AChR in a muscle cell culture system. The degradation rates (% per hour) of control immunoglobulin, immunoglobulin from EAMG rats receiving high dose AChR and control EAMG rats were 3.21 ± 0.04, 5.32 ± 0.08, 8.78 ± 0.03, respectively. This effect was therefore less pronounced in the EAMG rats receiving high-dose AChR.
Treatment of EAMG by nasal AChR suppresses AChR-reactive Th1 but not Th2 and Th3 cytokine mRNA expression
In rats receiving 600 μg/rat AChR nasally, numbers of AChR-reactive IFN-γ and tumour necrosis factor-alpha (TNF-α) mRNA-expressing lymph node cells were lower than in control rats receiving PBS. No differences were found for AChR-reactive IL-4, IL-10 and transforming growth factor-beta (TGF-β) mRNA-expressing cells between the two groups. In control experiments when lymph node MNC were cultured without antigen or in the presence of the irrelevant antigen, MBP, or Con A, no differences were found for numbers of IFN-γ, TNF-α, IL-4, IL-10 or TGF-β mRNA-expressing cells between the two groups (data not shown). These data imply that nasal administration of AChR in ongoing EAMG actually suppressed antigen-specific Th1 cytokine mRNA expression, correlating with levels of AChR-reactive IFN-γ-secreting cells in the experiments (Fig. 6), while AChR-specific Th2 or Th3 mRNA-expressing cells were not affected.
Fig. 6.
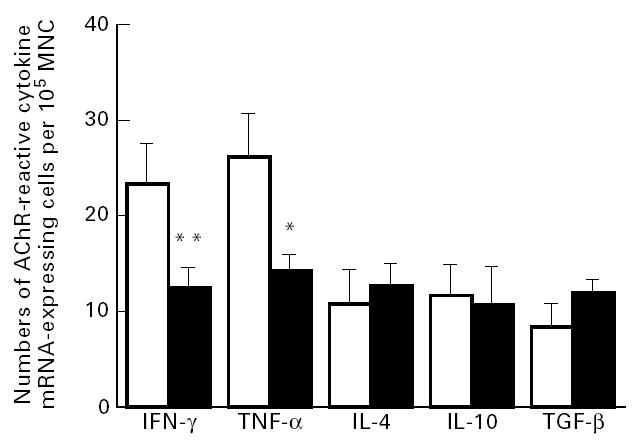
Numbers of acetylcholine receptor (AChR)-reactive cytokine mRNA-expressing cells per 105 lymph node mononuclear cells (MNC) at 7 weeks post-immunization (p.i.) from EAMG rats treated nasally with AChR 600 μg/rat (▪) and control EAMG rats treated nasally with PBS only (□). Data from eight rats in each group are shown. Symbols refer to mean values and bars to s.e.m. *P < 0.05; **P < 0.01.
DISCUSSION
Nasal administration of AChR in the Lewis rat before immunization with AChR in FCA resulted in markedly decreased severity of clinical EAMG, and suppressed AChR-specific T and B cell responses when a dose as low as 60 μg AChR/rat was used [8]. When administering AChR for the treatment of ongoing EAMG, we found that a 10-fold higher amount of AChR (600 μg/rat) is required to ameliorate muscular weakness of EAMG. These findings imply that when an abnormal immune response to AChR is established, much higher doses of AChR administered nasally are needed to arrest the progression of the autoimmune pathologic process.
AChR is highly immunogenic. The immune responses to AChR are heterogeneous, involving many myasthenic epitopes ([24], reviewed in [25]), associated with determinant spreading [26,27]. In the established EAMG, the broad range of T and B cell specificities may be more difficult to modulate. In fact, nasal AChR administration in ongoing EAMG is less effective compared with a prophylactic approach, and much higher doses are required.
High-dose AChR (600 μg/rat) resulting in incomplete abrogation of EAMG was associated with reduced lymphocyte proliferation and IFN-γ secretion in response to AChR, but with a concomitant augmentation of the production of anti-Torpedo and anti-rat AChR antibodies.
Upon activation by cognate ligand, naive Th cells differentiate into distinct functional subgroups which are characterized by their pattern of cytokine secretion [28]. It is likely that Th1- and Th2-type cells also exist in rats [29,30]. AChR-specific rat T cells have been shown to secrete Th1 and Th2 cytokines [31]. In the present study, nasal administration of AChR in ongoing EAMG with the current treatment protocol induces selective suppression of Th1 function (as reflected by reduced AChR-induced IFN-γ mRNA expression and secretion, and helper function for IgG2b production), but not of Th2 cells.
Tolerance induction, under certain circumstances, is associated with preferential inhibition and/or activation of either of these T cell subsets, resulting in a dichotomy between cellular and humoral immune responsiveness, reflecting influence of the reciprocal inhibition between Th1 and Th2 cells which maintains a specific balance among the two subpopulations [32–34]. Intrathymic injection of AChR in a rat model of EAMG resulted in the suppression of Th1 cell functions alone [35]. Tolerization of T cells, but not B cells, has previously been described after feeding [36,37] and injection of low antigen doses to experimental animals [38]. A gradient of sensitivity to tolerance induction with Th1 > Th2 > B cells has been proposed [37,39]. This implies that approaches that produce specific inhibition of cell-mediated conditions like EAE or EAU do not necessarily inhibit an antibody-mediated condition like MG. The manifestation of split tolerance is dependent on the nature of the antigen, route of antigen administration, and type of adjuvant used [34]. Data from the present study suggest that the degree of disease suppression is dose-dependent, higher antigen doses may be needed to induce tolerance of Th2 cells or B cells.
Besides Th2 cytokines, several lines of evidence implicate Th1 cytokines as contributing to the development of humoral immune responses in EAMG. Elevated levels of circulating cells secreting IFN-γ or expressing IFN-γ and TNF-α mRNA in response to AChR could reflect escalated AChR-specific Th1 responses in MG and EAMG [40–43]. Severe MG is associated with augmented spontaneous TNF-α production in cultures of MNC compared with mild disease [44]. Upon expression of IFN-γ within the neuromuscular junction, EAMG-resistant BALB/c mice exhibited clinical weakness and disruption of the neuromuscular junction, accompanied by autoantibody deposition at motor end plates, implying that IFN-γ in the milieu of the muscle tissue induces humoral autoimmunity without circulating anti-AChR antibodies [45]. We and others have recently shown that mice with either IFN-γ or IFN-γ receptor deficiency are less susceptible to EAMG, associated with lower levels and affinities of anti-AChR antibodies [46,47].
The decreased levels of IFN-γ and TNF-α in the present study, on the one hand, may have a serious impact on the balance of cytokine network (impacts on Th2 cytokines) and alter the amount of anti-AChR antibody. Since the serum antibody levels do not correlate with disease severity, either in EAMG or in human MG, differences in binding affinity, fine specificity for AChR epitopes, and isotype may determine the outcome of the disease [48,49]. IFN-γ and TNF-α direct B cell maturation, IFN-γ drives non-immunoglobulin-secreting B cells to active immunoglobulin secretion and isotype switches, and affinity maturation [50–53]. Therefore, the decreased levels of IFN-γ and TNF-α may contribute to the production of lower affinity antibodies and the reduced level of AChR-specific IgG2b, which are probably the only isotype triggering antibody-dependent cell-mediated cytotoxicity (ADCC) in rats [54]. These anti-AChR antibodies have less capacity in inducing AChR degradation, and contribute to the partial suppression of disease in rats receiving high-dose AChR.
In conclusion, our results imply that, in contrast to T cell-mediated conditions, the application of a mucosal tolerance strategy to an established antibody-mediated autoimmune disease such as EAMG has major implications for humoral immune responses. These results could provide the basis for designing antigen-specific therapy in MG.
Acknowledgments
We thank Anita Gustafsson and Birgitta Jonsson for technical help. The study was supported by grants from the Swedish Medical Research Council, Swedish MS Society (NHR), and Karolinska Institute Research Funds.
References
- 1.Weiner HL. Oral tolerance: immune mechanisms and treatment of autoimmune diseases. Immunol Today. 1997;18:335–243. doi: 10.1016/s0167-5699(97)01053-0. [DOI] [PubMed] [Google Scholar]
- 2.Drachman DB. Myasthenia gravis. N Engl J Med. 1994;330:1797–810. doi: 10.1056/NEJM199406233302507. [DOI] [PubMed] [Google Scholar]
- 3.Lennon VA, Lindstrom JM, Seybold ME. Experimental autoimmune myasthenia gravis: cellular and humoral immune responses. Ann NY Acad Sci. 1976;274:283–99. doi: 10.1111/j.1749-6632.1976.tb47693.x. [DOI] [PubMed] [Google Scholar]
- 4.Asthana D, Fujii Y, Huston Y, Lindstrom J. Regulation of antibody production by helper T cell clones in experimental autoimmune myasthenia gravis is mediated by IL-4 and antigen-specific T cell factors. Clin Immunol Immunopathol. 1993;67:240–8. doi: 10.1006/clin.1993.1071. [DOI] [PubMed] [Google Scholar]
- 5.Wang ZY, Qiao J, Link H. Suppression of experimental autoimmune myasthenia gravis by oral administration of acetylcholine receptor. J Neuroimmunol. 1993;44:209–14. doi: 10.1016/0165-5728(93)90045-z. [DOI] [PubMed] [Google Scholar]
- 6.Wang ZY, Link H, Ljungdahl Å, Höjeberg B, Link J, Qiao J, Melms A, Olsson T. Induction of interferon-γ, interleukin-4 and transforming growth factor-β in rats orally tolerized against experimental autoimmune myasthenia gravis. Cell Immunol. 1994;157:353–68. doi: 10.1006/cimm.1994.1233. [DOI] [PubMed] [Google Scholar]
- 7.Okumura S, McIntosh K, Drachman DB. Oral administration of acetylcholine receptor: effects on experimental myasthenia gravis. Ann Neurol. 1994;36:704–13. doi: 10.1002/ana.410360504. [DOI] [PubMed] [Google Scholar]
- 8.Ma CG, Zhang GX, Xiao BG, Link J, Olsson T, Link H. Suppression of experimental autoimmune myasthenia gravis by nasal administration of acetylcholine receptor. J Neuroimmunol. 1995;58:51–60. doi: 10.1016/0165-5728(94)00187-s. [DOI] [PubMed] [Google Scholar]
- 9.Lindstrom J, Seybold ME, Lennon VA, Whittingham S, Duane DD. Antibody to acetylcholine receptor in myasthenia gravis: prevalence, clinical correlates and diagnostic value. Neurol. 1976;26:1054–9. doi: 10.1212/wnl.26.11.1054. [DOI] [PubMed] [Google Scholar]
- 10.Deibler GE, Martensson RE, Kies MW. Large scale preparation of myelin basic protein from central nervous tissue of several mammalian species. Prep Biochem. 1972;2:139–64. doi: 10.1080/00327487208061467. [DOI] [PubMed] [Google Scholar]
- 11.Lennon VA, Lambert EH, Leiby KR, Okarma TB, Talib S. Recombinant human acetylcholine receptor α-subunit induces chronic experimental autoimmune myasthenia gravis. J Immunol. 1991;146:2245–8. [PubMed] [Google Scholar]
- 12.Christadoss P, Lindstrom J, Munro S, Talal N. Muscle acetylcholine receptor loss in murine experimental autoimmune myasthenia gravis: correlation with cellular, humoral and clinical responses. J Neuroimmunol. 1985;8:29–41. doi: 10.1016/s0165-5728(85)80045-x. [DOI] [PubMed] [Google Scholar]
- 13.Wang ZY, Link H, Huang WX. T-cell immunity to acetylcholine receptor and its subunits in Lewis rats over the course of experimental autoimmune myasthenia gravis. Scand J Immunol. 1993;37:615–22. doi: 10.1111/j.1365-3083.1993.tb02580.x. [DOI] [PubMed] [Google Scholar]
- 14.Macdonald RA, Hosking CS, Jones CL. The measurement of relative antibody affinity by ELISA using thicyanate elution. J Immunol Methods. 1988;106:191–4. doi: 10.1016/0022-1759(88)90196-2. [DOI] [PubMed] [Google Scholar]
- 15.Lindstrom J, Einarson B, Tzartos S. Production and assay of antibodies to AChR. Methods Enzymol. 1981;74:432–60. doi: 10.1016/0076-6879(81)74031-x. [DOI] [PubMed] [Google Scholar]
- 16.Kao I, Drachman DB. Myasthenic immunoglobulin accelerates acetylcholine receptor degradation. Sci. 1977;196:527–9. doi: 10.1126/science.850793. [DOI] [PubMed] [Google Scholar]
- 17.Dijkema R, van der Meide PH, Dubbeld M, Wubben J, Schellekens H. Cloning, expression and purification of rat IFN-γ. Methods Enzymol. 1986;119:453–8. doi: 10.1016/0076-6879(86)19065-3. [DOI] [PubMed] [Google Scholar]
- 18.McKnight AJ, Barclay AN, Mason DW. Molecular cloning of rat interleukin 4 cDNA and analysis of the cytokine repertoire of subsets of CD4+ T cells. Eur J Immunol. 1991;21:1187–94. doi: 10.1002/eji.1830210514. [DOI] [PubMed] [Google Scholar]
- 19.Moore KW, Vieia P, Fiorentino DF, Trounsteine ML, Khan TA, Mosmann TR. Homology of cytokine synthesis inhibitory factor (IL-10) to the Epstein–Barr virus gene BCRFI. Sci. 1990;248:1230–5. doi: 10.1126/science.2161559. [DOI] [PubMed] [Google Scholar]
- 20.Derynck R, Jarret JA, Chen EY, et al. Human transforming growth factor-β complementary DNA sequence and expression in normal and transformed cells. Nature. 1985;316:701–3. doi: 10.1038/316701a0. [DOI] [PubMed] [Google Scholar]
- 21.Shirai T, Shimizu N, Horiguchi S, Jto H. Rat TNF alpha. Agric Biol Chem. 1989;53:1733–9. [Google Scholar]
- 22.Alastair J, Bradley JA. Interleukin-12 induces interferon-γ-dependent switching of IgG alloantibody subclass. Eur J Immunol. 1996;26:1217–21. doi: 10.1002/eji.1830260605. [DOI] [PubMed] [Google Scholar]
- 23.Mussener Å, Lorentzen JC, Kleinau S, Klareskog L. Altered Th1/Th2 balance associated with non-major histocompatibility complex gene in collagen-induced arthritis in resistant and non-resistant rat strains. Eur J Immunol. 1997;27:695–9. doi: 10.1002/eji.1830270318. [DOI] [PubMed] [Google Scholar]
- 24.Hucho F, Tsetlin VI, Machold J. The emerging three-dimensional structure of a receptor: the nicotinic acetylcholine receptor. Eur J Biochem. 1996;239:539–57. doi: 10.1111/j.1432-1033.1996.0539u.x. [DOI] [PubMed] [Google Scholar]
- 25.Drachman DB. Immunotherapy in neuromuscular disorders: current and future strategies. Muscle Nerve. 1996;19:1239–51. doi: 10.1002/(SICI)1097-4598(199610)19:10<1239::AID-MUS1>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 26.Vincent A, Jacobson L, Shillito P. Response to human acetylcholine receptor α-139–199: determinant spreading initiates autoimmunity to self-antigen in rabbits. Immunol Letters. 1994;39:269–75. doi: 10.1016/0165-2478(94)90168-6. [DOI] [PubMed] [Google Scholar]
- 27.Aguis MA, Twaddle GM, Fairclough RH. Epitope spreading in experimental autoimmune myasthenia gravis (Abstr.). IX International Conference on Myasthenia Gravis and Related Disorders; 1997; Santa Monica. p. 4. [Google Scholar]
- 28.Mosmann TR, Sad S. The expanding universe of T-cell subsets: Th1, Th2 and more. Immunol Today. 1996;17:138–46. doi: 10.1016/0167-5699(96)80606-2. [DOI] [PubMed] [Google Scholar]
- 29.Papp I, Wieder KJ, Sablinski T, O'Connel PJ, Milford EL, Strom TB, Kupiec Weglinski JW. Evidence for functional heterogeneity of rat CD4+ T cells in vivo. Differential expression of IL-2 and IL-4 mRNA in recipient of cardiac allografts. J Immunol. 1992;148:1308–14. [PubMed] [Google Scholar]
- 30.McKnight AJ, Barclay AN, Mason DW. Molecular cloning of rat interleukin 4 cDNA and analysis of the cytokine repertoire of subsets of CD4+ T cells. Eur J Immunol. 1991;21:1187–94. doi: 10.1002/eji.1830210514. [DOI] [PubMed] [Google Scholar]
- 31.Fujii Y, Lindstrom JM. Regulation of antibody production by helper T cell clones in experimental autoimmune myasthenia gravis. J Immunol. 1988;141:3361–9. [PubMed] [Google Scholar]
- 32.Mosmann TR, Schumacher JH, Fiorentino DF, Leverah J, Moore KW, Bond MW. Isolation of monoclonal antibodies specific for IL-4, IL-5, IL-6 and a new Th2-specific cytokine (IL-10), cytokine synthesis inhibitory factor, by using a solid radioimmunoadsorbent assay. J Immunol. 1990;145:2938–45. [PubMed] [Google Scholar]
- 33.De Waal Malefyt R, Abrams J, Bennett B, Figdor CG, De Vries JE. Interleukin 10 (IL-10) inhibits cytokine synthesis by human monocytes: autoregulatory role of IL-10 produced by monocytes. J Exp Med. 1991;174:1209–20. doi: 10.1084/jem.174.5.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peterson JD, Karpus WJ, Clatch RJ, Miller SD. Split tolerance of Th1 and Th2 cells in tolerance to Theiler's murine encephalomyelitis virus. Eur J Immunol. 1993;23:46–55. doi: 10.1002/eji.1830230109. [DOI] [PubMed] [Google Scholar]
- 35.Ohtsuru I, Matsuo H, Fukudome T, Suenaga A, Tsujihata M, Nagataki S. ‘Split tolerance’ induction by intrathymic injection of acetylcholine receptor in a rat model of autoimmune myasthenia gravis; implication for the design of specific immunotherapies. Clin Exp Immunol. 1995;102:462–7. doi: 10.1111/j.1365-2249.1995.tb03838.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khoury SJ, Hancock WW, Weiner HL. Oral tolerance to myelin basic protein and nature recovery from experimental allergic encephalomyelitis are associated with downregulation of inflammatory cytokines and upregulation of TGF-β, IL-4 and prostaglandin E expression in the brain. J Exp Med. 1992;176:1355–64. doi: 10.1084/jem.176.5.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Husby S, Mesterdy J, Holland S, Elson CO. Oral tolerance in humans: T cell but not B tolerance after feeding. J Immunol. 1994;152:4664–70. [PubMed] [Google Scholar]
- 38.Burstein HJ, Shea CM, Abbas AK. Aqueous antigens induce in vivo tolerance selectively in IL-2 and IFN-gamma-producing cells. J Immunol. 1992;148:3687–91. [PubMed] [Google Scholar]
- 39.Melamed D, Fredman A. In vivo tolerization of Th1 lymphocytes following a single feeding with ovalbumin: anergy in the absence of suppression. Eur J Immunol. 1994;24:1974–81. doi: 10.1002/eji.1830240906. [DOI] [PubMed] [Google Scholar]
- 40.Link H, Olsson O, Sun JB, et al. Acetylcholine receptor-reactive T and B cells in myasthenia gravis and controls. J Clin Invest. 1991;87:2191–6. doi: 10.1172/JCI115253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Link J, Söderström M, Ljungdahl Å, et al. Organ-specific autoantigens induce IFN-γ and IL-4 mRNA expression in mononuclear cells in multiple sclerosis and myasthenia gravis. Neurol. 1994;44:728–35. doi: 10.1212/wnl.44.4.728. [DOI] [PubMed] [Google Scholar]
- 42.Matusevicius D, Navikas V, Palask W, Pirskanen R, Fredrikson S, Link H. Tumor necrosis factor-α, lymphotoxin, interleukin (IL)-6, IL-10, IL-12 and perforin mRNA expression in mononuclear cells in response to acetylcholine receptor is augmented in myasthenia gravis. J Neuroimmunol. 1996;71:191–8. doi: 10.1016/s0165-5728(96)00152-x. [DOI] [PubMed] [Google Scholar]
- 43.Shi FD, Zhang GX, Bai XF, van der Meide PH, Link H. Cellular mRNA expression of IFN-γ, IL-4 and IL-10 related to resistance to experimental autoimmune myasthenia gravis in young Lewis rats. Clin Exp Immunol. 1997;108:523–7. doi: 10.1046/j.1365-2249.1997.3881284.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ahlberg RE, Pirskanen R, Lefvert AK. Defective T lymphocyte function in nonthymectomized patients with myasthenia gravis. Clin Immunol Immunopathol. 1991;60:93–100. doi: 10.1016/0090-1229(91)90115-q. [DOI] [PubMed] [Google Scholar]
- 45.Gu D, Wogensen I, Calcutt NA, et al. Myasthenia gravis-like syndrome induced by expression of interferon-γ in the neuromuscular junction. J Exp Med. 1995;181:547–57. doi: 10.1084/jem.181.2.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang GX, Xiao BG, Bai XF, Örn A, Van der Meide PH, Link H. Mice with IFN-γ receptor deficiency are less susceptible to experimental autoimmune myasthenia gravis. J Immunol. in press. [PubMed] [Google Scholar]
- 47.Balasa B, Deng CS, Christadoss P, Bradley LM, Sarvetnick N. IFN-γ is necessary for the genesis of acetylcholine receptor induced clinical experimental autoimmune myasthenia gravis in mice. J Exp Med. 1997;168:385–91. doi: 10.1084/jem.186.3.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Graus YMF, van Breda Vriesman PJC, De Baets MH. Characterization of anti-AChR antibodies from mice differing in susceptibility for experimental autoimmune myasthenia gravis. Clin Exp Immunol. 1993;92:506–13. doi: 10.1111/j.1365-2249.1993.tb03429.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krolick KA, Zoda TE, Thompson PA. Examination of characteristics that may distinguish disease-causing from benign AChR-reactive antibodies in experimental autoimmune myasthenia gravis. Adv Neuroimmunol. 1994;4:475–93. doi: 10.1016/0960-5428(94)00033-k. [DOI] [PubMed] [Google Scholar]
- 50.Sidman CL, Marshall JD, Shultz LD, Gray PW, Johson HM. γ-interferon is one of the several direct B cell-maturing lymphokines. Nature. 1984;309:801–3. doi: 10.1038/309801a0. [DOI] [PubMed] [Google Scholar]
- 51.Snapper CM, Mond JJ. Toward a comprehensive view of immunoglobulin class switching. Immunol Today. 1993;14:15–17. doi: 10.1016/0167-5699(93)90318-F. [DOI] [PubMed] [Google Scholar]
- 52.Rizzo IV, DeKruyff RH, Umetsu DT. Generation of B cell memory and affinity maturation: induction with Th1 and Th2 cell clones. J Immunol. 1992;148:3733–8. [PubMed] [Google Scholar]
- 53.Jelinek DF, Lipsky PE. Enhancement of human B cell proliferation and differentiation by tumor necrosis factor alpha and interleukin 1. J Immunol. 1987;139:2970–9. [PubMed] [Google Scholar]
- 54.Keller R, Keist R, Bazin H, Joller P, Van der Meide PH. Binding of monomeric immunoglobulins by bone marrow-derived mononuclear phagocytes; its modulation by interferon-gamma. Eur J Immunol. 1990;20:2137–40. doi: 10.1002/eji.1830200937. [DOI] [PubMed] [Google Scholar]