

Abstract
There is evidence that TNF-α contributes to the pathogenesis of chronic viral hepatitis. The cellular effects of this cytokine are regulated by two specific receptors, and membranous shedding of these receptors reflects activation of the TNF system. We performed a study of TNF-α and functionally active soluble TNF-receptors (TNFR-p55 and -p75) in 105 patients with chronic HCV infection. In HCV RNA-positive patients a significant enhancement of TNF-α and both receptor types was observed compared with controls (TNF-α 83.8 ± 91.7 pg/ml versus18.8 ± 8.4 pg/ml, P < 0.001; TNFR-p55 1.4 ± 0.4 ng/ml versus 0.9 ± 0.2 ng/ml, P < 0.0001; TNFR-p75 6.4 ± 2.4 ng/ml versus 2.9 ± 0.6 ng/ml, P < 0.0001, respectively). The enhanced serum levels of TNF-α and TNFRs were reflected by a significant expression of TNFR-specific mRNA in peripheral mononuclear cells of HCV-infected patients (P < 0.001). Serum aminotransferases correlated with soluble TNFR-p75 (P < 0.001) but not with TNFR-p55 and TNF-α. We demonstrated an association of the degree of histological inflammation with both TNFRs (P < 0.01). Furthermore, enhanced hepatocellular expression of TNF-α and TNFRs could be demonstrated by immunohistochemical staining in HCV-infected patients. Sixty-eight out of 105 patients were treated with interferon-alpha (IFN-α) (3 × 106 U × 3/week). Pretreatment levels of TNF-α and TNFRs did not differ between responders and non-responders. Our results demonstrate that TNF-α and TNFRs are enhanced in chronic HCV infection and reflect histological activity of the disease. This up-regulation of TNFRs might modify host response and potentially contribute to liver damage in chronic HCV infection.
Keywords: chronic hepatitis C, tumour necrosis factor-alpha, TNF receptors
INTRODUCTION
HCV is the most important aetiologic agent of post-transfusion and community-acquired non-A, non-B hepatitis. HCV infection often results in chronic hepatitis (50–70% of patients), with 20% progressing to cirrhosis [1]. The pathogenic mechanisms responsible for the hepatocellular injury in viral hepatitis C are still poorly defined [2]. Recent experimental evidence supports both a direct cytopathic effect of HCV [3] as well as an immune-mediated cell damage triggered by HCV [4, 5].
TNF-α is a principal mediator of inflammation and cellular immune responses, with multiple biological functions including cytotoxicity towards virus-infected cells, several immunomodulatory actions and direct anti-viral activity [6]. TNF-α is produced primarily by activated macrophages/monocytes and Kupffer cells [7]. TNF-α is involved in the immunopathogenesis of various chronic liver diseases, including alcoholic and viral hepatitis [8, 9]. Furthermore, the induction of TNF-α in response to interferon-alpha (IFN-α) therapy in chronic viral hepatitis B infection reflects the induction of immune-mediated viral clearance [10].
TNF-α mediates its various cellular responses by binding to specific cell surface receptors. Two immunologically distinct membrane-bound TNF receptors (TNFRs) with molecular masses of ≈ 55 kD (TNFR-p55) and 75 kD (TNFR-p75) have been identified [11, 12]. Each TNFR signals different cellular pathways and is responsible for distinct TNF-α activities; TNFR-p55 mediates cytotoxicity, fibroblast proliferation and anti-viral activity, whereas TNFR-p75 is involved in thymocyte proliferation, cytotoxic T lymphocyte (CTL) proliferation and induction of granulocyte-macrophage colony-stimulating factor (GM-CSF) synthesis [13]. Soluble forms of both TNFRs (sTNFRs) are present constitutively in serum, derived after receptor shedding upon cellular activation to the same inflammatory stimuli that induce TNF-α production [14–16].
In vitro, high concentrations of sTNFRs were shown to inhibit TNF-α, whereas low concentrations stabilize TNF-α activity by preventing dissociation of the homotrimeric TNF protein to inactive molecules [14]. However, in vivo administration of sTNFRs can prevent the pathologic sequelae caused by enhanced TNF-α production [17].
To ascertain whether there is induction of the TNF-α/TNFR system in chronic HCV infection we studied the serum levels and the hepatic expression of both TNF-α and the sTNFR-p55 and -p75 in patients with chronic HCV infection.
PATIENTS AND METHODS
Patients
One hundred and five consecutive patients with chronic HCV infection (68 male, 37 female, age 24–66 years) were recruited for this study. All patients were positive for anti-HCV (second generation ELISA; Abbott, Wiesbaden, Germany), RIBA-2 (Ortho, Neckargemuend, Germany), and HCV RNA by reverse transcriptase-polymerase chain reaction (RT-PCR). All patients were negative for markers of hepatitis B virus infection (HBsAg and anti-HBc), of autoimmune hepatitis (LKM, ANA) and of HIV-1 and HIV-2.
A percutaneous liver biopsy was performed prior to treatment in all 105 patients as part of the diagnostic evaluation, with informed consent. For the cross-sectional analysis, paired liver specimens and serum samples were examined in all 105 patients. Liver histology was assessed blindly on formalin-fixed paraffin sections, with respect to serum aminotransferases, sTNFR levels and hepatic expression of TNF-α and TNFRs. Liver biopsies were graded for necroinflammatory activity and fibrosis according to the histological scoring system (H.A.I.) of Desmet et al. [18].
Sixty-eight patients had chronic persistent hepatitis (H.A.I. < 5) and 37 patients had chronic active hepatitis (H.A.I. > 4). None of the patients included in this study showed histological signs of liver cirrhosis. As a control group, sera from 48 healthy controls (anti-HCV-negative, 29 male, 19 female, age 18–65 years) with normal aminotransferases were tested (Table 1).
Table 1.
Biochemical and histological profile of patients and controls

Sixty-eight of the 105 HCV-infected patients were treated with recombinant leucocyte IFN-α2b for at least 3 months (Essex, Munich, Germany; 3 × 106 U × 3/week). Patients considered as responders (serum HCV RNA-negative, normalized ALT and AST < 20 U/l) at the end of 3 months treatment continued their therapy for a further 6 months on the same treatment schedule.
Amplification of HCV RNA and genotyping
Serum HCV RNA was detected by nested RT-PCR as described previously [19]. HCV genotyping was determined by a line-probe assay (Inno Lipa, Innogenetics, Zwijndrecht, Belgium).
Immunoassay for TNF-α
TNF-α levels were determined by ELISA [20, 21]. Briefly, 96-well microtitre plates (Maxisorp Nunc, Roskilde, Denmark) were coated with 5 μg/ml of murine MoAbs to TNF-α for 24 h, washed twice with destilled water and blocked with buffer solution, containing 10 g/l bovine serum albumin (BSA) in 0.2 mol/l Tris–HCl at pH 7.5 and kept at 2–8°C before use. Diluted serum samples were added to the wells and compared with serial dilutions of recombinant TNF-α (specific activity 2 × 107 U/mg protein) after adding 100 ng/ml peroxidase-labelled rabbit anti-TNF-α antibodies and incubation for 24 h at 4°C. Optical density (OD) readings were made after the addition of peroxidase-specific substrate at 450 nm. The lower limit of detection for this ELISA was 8.5 pg/ml and there was no cross-reaction with lipopolysaccharide (LPS), IFN-α or IFN-γ, TNF-β, IL-1α or IL-1β and IL-2.
TNF-receptor assays
Soluble TNFRs with biological activity were measured in diluted sera (1:5) by an enzyme-linked binding assay (ELIBA) with a TNF-α horseradish peroxidase (HRP) conjugate as detecting agent as described previously [22]. The first detecting agent was a MoAb directed against the TNFR-p55 and -75. In contrast to conventional ELISA techniques (using labelled MoAbs to recognize the bound target molecule as a protein), in this ELIBA the second step of the reaction was based on the natural binding of the soluble TNF receptor and its specific ligand TNF-α. Therefore our test system combined the immunological and biological detection of soluble TNF receptors. Briefly, 96-well microtitre plates (Maxisorp Nunc) were coated for 24 h at 20°C with TNF-binding non-inhibitory MoAbs against TNFR-p55 (clone htr-20) or -p75 (clone utr-4). The plates were saturated with 1% BSA, 0.1% phenol and 0.1% Tween-20. In a single-step reaction, sera were incubated with TNF-α enzyme conjugate in the microtitre plates. Bound TNF-α was measured enzymatically with tetramethyl-benzidene as substrate and reading was done at 450 nm. Standard curves were generated with recombinant sTNFR-p55 or -p75 and the concentrations of sTNFRs were determined by interpolation from the standard curve. The lower limit of detection for both types of the receptors was 60 pg/ml. There was no alteration of the assay after adding free and unlabelled TNF-α in concentrations up to 30 ng/ml. The assays showed no cross-reactions to cytokines (e.g. TNF-β) or cytokine receptors (IFN-γR, IL-6R, IL-2R).
PCR-assisted mRNA amplification of TNFRs
To characterize the cellular activation of the TNFRs we determined mRNA synthesis in peripheral blood mononuclear cells (PBMC) by RT-PCR in a representative subgroup of patients (H.A.I. 1–4, n = 13; H.A.I. 5–9, n = 15) and compared with healthy controls (n = 11). PBMC were isolated from heparinized blood by Ficoll–Hypaque gradient centrifugation [23]. One million PBMC were lysed in 0.5 ml of 4 m guanidine-isothiocyanate, 25 mm sodium-citrate, 0.5% lauroylsarcosine, 100 mm 2-mercaptoethanol (2-ME). RNA preparation was performed using an acid phenol-chloroform extraction method [24]. The RNA concentration was quantified by absorbance at 260 nm, RNA degradation and purity were assessed by electrophoresis on a 1.5% agarose gel containing formaldehyde. Total cellular RNA (1 μg) was used to generate cDNA as previously described [25]. Cytokine primer pairs were designed spanning exon–exon conjunctions and are therefore mRNA/cDNA-specific and non-reactive with genomic DNA. The following oligonucleotide 5′ and 3′ primer sequences were used: TNFR-p55 (amplification product 587 bp): 5′ ATTTGCTGTACCAAGTGCCACAAAGGAACC; 3′ GTCGATTTCCCACAAACAATGGAGTAGAGC; TNFR-p75 (amplification product 403 bp): 5′ GAATACTATGACCAGACAGCTCAGATG-TGC; 3′ TATCCGTGGATGAAGTCGTGTTGGAGAACG; glyceraldehyde phosphate dehydrogenase (GPDH; product 987 bp): 5′ TGAAGGTCGGAGTCAACGGATTTGGT; 3′ CATGTGGGCCATGAGGTCCACCAC.
Five microlitres of the cDNA (representing 5 × 104 PBMC) were used for PCR and amplification was performed for 30 cycles [25]. The amplified product was separated on an 2% agarose gel and visualized using ethidium bromide staining. A 100-bp DNA ladder was run in parallel as molecular size marker.
Immunohistochemistry
The expression of TNF-α and both cellular TNFR-p55 and -p75 was analysed in eight biopsies from control patients with either fatty liver disease negative for hepatitis B and C virus or patients with minimal elevated liver enzymes and normal liver histology and in 12 frozen liver biopsies of HCV-infected patients with various degrees of hepatic inflammation (seven biopsies of patients with H.A.I. < 5 and five biopsies of patients with H.A.I. > 4 as previously described [22]). Liver biopsies were embedded in OCT (optimal cutting temperature) compound, snap-frozen in liquid nitrogen-cooled isopentane, and stored at −70°C. Serially cut sections (5 μm) were mounted on 0.01% poly l-lysine-coated glass slides, air dried for 4–6 h at room temperature and stored at −20°C until use.
TNF-α and TNFR expression
TNF-α expression was detected in cryostat sections using a two-step immunoperoxidase detection system as described elsewhere [26]. The anti-TNF-α MoAb was donated by Dr A. Meager (National Institute for Biological Standards and Control, Mill Hill, UK). For TNFR expression, cryostat sections were thawed and fixed in acetone just before immunostaining with a primary MoAb using a three-step indirect alkaline phosphatase anti-alkaline phosphatase (APAAP) procedure [27]. The rehydrated sections were blocked for non-specific binding with 10% normal rabbit serum/1% BSA in 0.05 m Tris-buffered saline pH 7.6. The sections were incubated overnight at 4°C with either anti-TNFR-p55 moab or anti-TNFR-p75 moab (htr-9 and utr-1, respectively, provided by Dr M. Brockhaus, Hoffmann-La Roche, Basel, Switzerland), both at 20 μg/ml. The secondary and tertiary antibodies were alkaline phosphatase-conjugated rabbit anti-mouse antibody (1:50 dilution) and mouse APAAP complex (1:100 dilution), respectively (Dako Ltd, High Wycombe, UK). Incubation with these antibodies was carried out for 30 min at room temperature. The alkaline phosphatase reaction was developed using naphthol and fast red TR salt. The specificity of the immunoreactivity of the two moabs (htr-9 and utr-1) was verified using primary moabs with different specificities (anti-HBsAg, anti-HLA-DR, anti-p53) on cryostat sections from identical patients and by staining sections of normal human liver with both htr-9 and utr-1 moabs.
Statistical analysis
Data are expressed as mean ± s.d. The non-parametric Mann–Whitney U-test was used to compare data between two groups. Correlation analysis was calculated using the non-parametric Spearman rank-regression analysis. The χ2 was used to compare the prevalence of two variables in the same group. P < 0.05 was considered statistically significant.
RESULTS
TNF-α and sTNFR levels in sera of patients with chronic hepatitis C infection
Serum levels of TNF-α were significantly higher in HCV-infected patients compared with healthy controls (83.8 ± 91.7 pg/ml and 18.8 ± 8.4 pg/ml, respectively; P < 0.001, Mann–Whitney U-test). Serum levels of both sTNFRs -p55 and -p75 were also significantly elevated in patients with chronic HCV infection compared with controls (P < 0.0001; Mann–Whitney U-test). The mean serum levels (mean ± s.d.) of both TNFR-p55 (ng/ml) and -p75 (ng/ml) were 0.9 ± 0.2 and 2.9 ± 0.6 for the control group, 1.4 ± 0.4 and 6.4 ± 2.4 for HCV-infected patients.
To investigate the relationship between the level of serum sTNFRs and hepatic inflammation, hepatocellular damage and HCV infection, we studied the correlation between serum sTNFR, H.A.I., AST and ALT. Sixty-eight patients with minimal to moderate necroinflammatory activity (H.A.I. 1–4) showed significant lower serum levels for the two sTNFRs (TNFR-p55, 1.3 ± 0.3 ng/ml; TNFR-p75, 5.6 ± 1.8 ng/ml) compared with patients with moderate to severe necroinflammatory activity (H.A.I. 5–9) (TNFR-p55, 1.6 ± 0.5 ng/ml; TNFR-p75, 8.1 ± 2.8 ng/ml) (P < 0.01; Fig. 1). Both sTNFR-p55 and -p75 correlated with the histological activity index (Spearman r = 0.28/0.50, respectively; P < 0.01; Fig. 2), whereas only sTNFR-p75 was significantly correlated with both aminotransferases AST (r = 0.37, P < 0.001, Spearman rank correlation) and ALT (r = 0.34, P < 0.01, Spearman rank correlation) (Fig. 3). The necroinflammatory activity was reflected by the extent of elevated aminotransferases (Fig. 4) (H.A.I. versus AST, r = 0.52, P < 0.0001; H.A.I. versus ALT, r = 0.39, P < 0.0001; Spearman rank correlation). Different HCV genotypes were neither correlated with TNF-α, nor with serum levels of sTNFR-p55 or -p75.
Fig. 1.

Serum levels of TNF-α, sTNFR-p55 and TNFR-p75 in 105 patients with chronic HCV infection and different histological activity (68 patients with histological activity index (H.A.I.) 1–4 and 37 patients with H.A.I. 5–9, respectively) compared with 48 healthy controls. Data are presented using the box plot model. Significance is given as *P < 0.001 (Mann–Whitney U-test).
Fig. 2.

Correlation of sTNFR-p55 and -p75 levels with the histological activity index (H.A.I.) in HCV-infected patients. (Spearman rank regression analysis.)
Fig. 3.
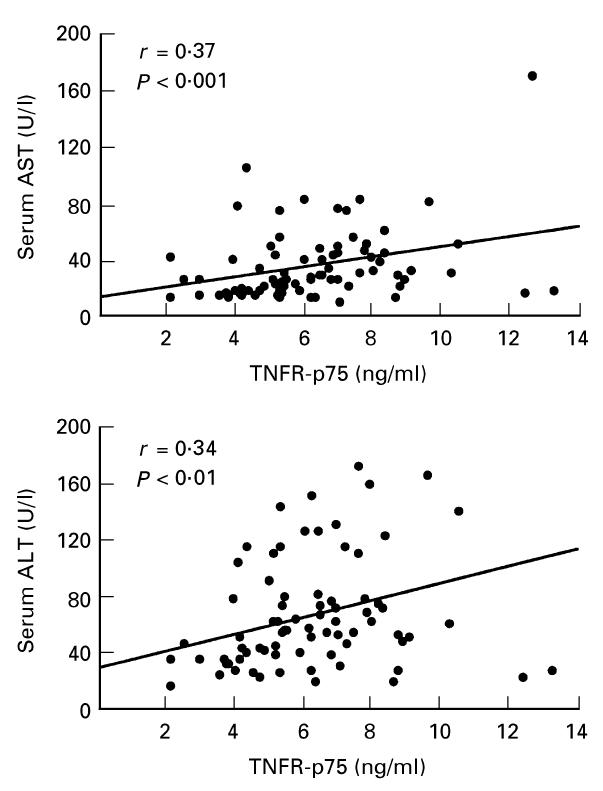
Correlation of sTNFR-p75 with aminotransferases AST and ALT in HCV-infected patients calculated by a Spearman rank regression analysis.
Fig. 4.
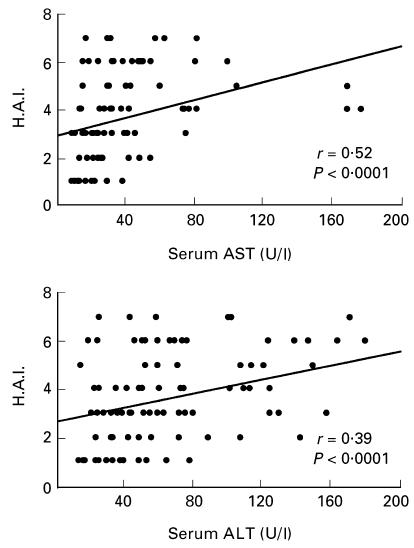
Correlation of aminotransferases AST and ALT with the histological activity index (H.A.I.) in HCV-infected patients calculated by Spearman rank regression analysis.
Out of 105 HCV-infected patients, 68 were treated with IFN-α. Of these patients, 26 (38%) were complete responders with normal aminotransferases and negative HCV RNA during therapy. There was no significant difference in respect to pretreatment serum levels of TNF-α, sTNFR-p55 and -p75 among responders and non-responders to IFN-α. Among the parameters tested (AST, ALT, HCV genotype) only HCV genotype 1 predicted a non-response to IFN-α therapy.
Amplification of TNFR transcripts in PBMC by PCR
To prove if enhanced serum TNFR levels of patients with chronic HCV infection were reflected by a significant induction of mRNA synthesis in PBMC, RT-PCR of TNFR mRNA was performed. Specific mRNA for both TNFRs -p55 and -p75 was detectable in 23/28 and 20/28 HCV-infected patients, respectively, compared with 2/11 and 1/11 healthy controls, respectively (χ2= 14.1 for TNFR-p55 (P < 0.001) and 12.1 for TNFR-p75 (P < 0.001)) (Fig. 5). Most patients with a high (H.A.I. 5–9) necroinflammatory activity demonstrated a significant induction of TNFR-p55- and TNFR-p75-specific mRNA (14/15 and 13/15 patients, respectively). There was no difference between HCV-infected patients with high (H.A.I. 5–9) and low (H.A.I 1–4) necroinflammatory activity.
Fig. 5.

Analysis of mRNA assisted polymerase chain reaction (PCR) amplification in peripheral blood mononuclear cells (PBMC) shows a significant expression of TNFR-p55 and -p75-specific RNA in HCV RNA-positive patients compared with healthy controls. GPDH mRNA analysis is given for comparison. Co, Control; H.A.I., histological activity index.
Immunohistochemical study
Control liver tissue showed no or very small amounts of TNF-α and both TNFRs on hepatocytes, bile duct epithelium, sinusoidal epithelial cells and lymphocytes (Fig. 6a–c). Liver specimens of HCV-infected patients demonstrated a marked expression of both TNFRs -p55 and -p75 (Fig. 7a–c). In liver biopsy specimens from patients with chronic HCV infection, the expression of cellular TNFR-p55 was much weaker than TNFR-p75. However, there was no correlation of receptor expression with the degree of the necroinflammatory activity. Furthermore, there was no strict topographical association between TNFR-p75+ hepatocytes and the presence of inflammatory infiltrates.
Fig. 6.

(See next page.) Hepatic expression of TNF-α (a), cellular TNFR-p55 (b) and cellular TNFR-p75 (c) in a patient with a normal liver histology.
Fig. 7.
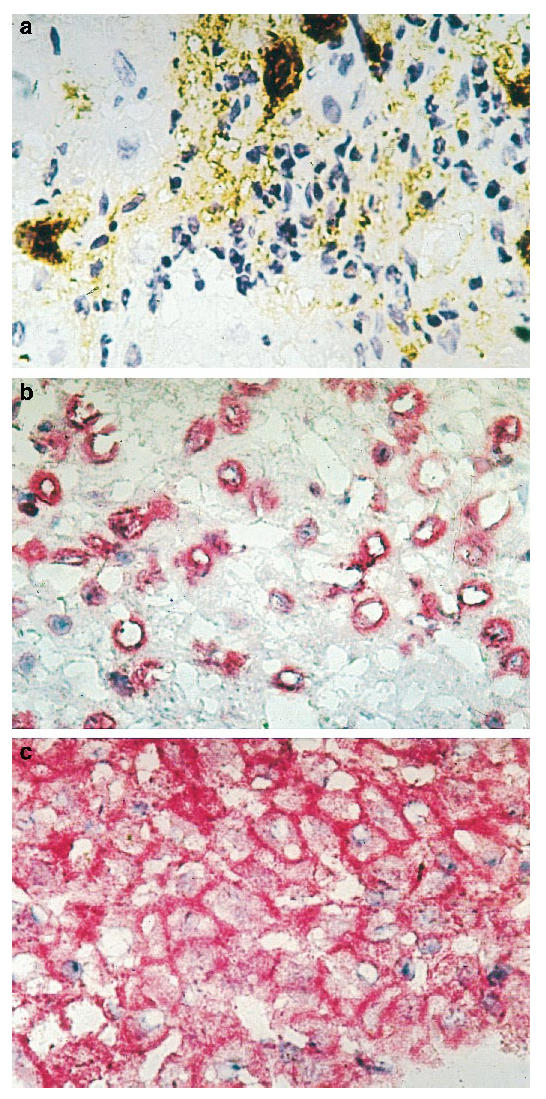
(See p. 275.) Hepatic expression of TNF-α (a), sTNFR-p55 (b) and TNFR-p75 (c) in a HCV-infected patient with a histological activity index (H.A.I.) > 6. TNFR-p55 could be demonstrated in the nucleus in a few hepatocytes, whereas TNFR-p75 showed a strong membranous expression.
DISCUSSION
Elevated plasma levels of TNF-α, a principal mediator in many inflammatory processes, have been documented in experimental liver injury, fulminant hepatic failure, alcoholic liver disease and chronic viral hepatitis B infection [8, 9, 28, 29]. In our study TNF-α was significantly elevated in patients with HCV infection. This is in accordance with previous results [30, 31] showing that HCV-related liver damage was reflected by systemically elevated TNF-α. However, in a more recent study Tilg et al. could not confirm enhanced TNF-α levels in HCV-infected patients, probably due to the small number of investigated individuals [32].
The biological effects of TNF-α are mediated by two membrane-bound specific receptor types TNFR-p55 and -p75. Our results demonstrate for the first time that the two functionally active soluble TNF receptors are increased in sera, PBMC and liver tissue of patients chronically infected with HCV. This is in contrast to chronic HBV infection, where the TNFR-p75 signalling pathway has been shown to be preferentially up-regulated and may contribute to liver damage and viral clearance [22].
We excluded HCV-infected patients with histological signs of liver cirrhosis, because previous studies demonstrated high levels of soluble TNFRs in cirrhotic patients irrespective of the aetiology [33, 34].
Elevated serum levels of soluble TNF receptors were neither correlated with response nor with non-response to IFN in chronic HCV infection, meaning that TNF receptors can not be used as predictive markers for response or non-response to IFN treatment. This is in contrast to elevated serum levels of TNFRs in chronic HBV infection, which were found to predict a successful response to treatment in conjunction with other predictive determinants, i.e. low levels of HBV DNA [22].
In patients with chronic HCV infection the significant up-regulation of the sTNFRs correlated with the degree of hepatic inflammation and the extent of elevated aminotransferases.
We therefore assume that serum levels of sTNFRs reflect ongoing disease activity and probably modulate some effects of endogenous TNF-α. This result is in accordance with previous findings in HBV infection demonstrating a significant enhancement of TNFR-p75 in association with hepatic inflammation and hepatocytolysis in chronic active hepatitis B infection [22].
However, it should be considered that the correlation between the expression of TNFR-p55 and the H.A.I. as well as between TNFR-p75 and aminotransferases (AST, ALT) was not very strong, but highly significant (P < 0.01).
In addition, we found a statistically significant relationship between the degree of serum ALT and AST elevation and the degree of liver injury based on the H.A.I. score in untreated HCV-infected patients. This observation is in agreement with a recent report [35], whereas most studies dealing with the same subject could not find a correlation between ALT levels and histological abnormalities in chronic HCV infection [36, 37].
Enhanced hepatocellular expression of sTNFRs was also found in various chronic liver diseases such as hepatitis B and HCV infection [38]. In our study, expression of mRNA specific for TNFR-p55 and TNFR-p75 was up-regulated in PBMC as well as in liver tissue of HCV-infected patients compared with controls.
In conclusion, we demonstrated an up-regulation of TNF-α, TNFR-p55 and TNFR-p75 in three different compartments: sera, PBMC and liver tissue. Whether the systemic enhancement of TNFRs is based on an increased receptor shedding from the liver and/or from circulating PBMC could not be completely clarified by this study. However, it should be discussed that probably part of the serum sTNFRs derived from an up-regulated mRNA synthesis of both TNFRs in activated PBMC.
The up-regulation of sTNFRs might modulate host response in chronic hepatitis C. High concentrations of sTNFRs potentially avoid harmful effects of TNF-α such as cell cytotoxicity and inflammation [14, 17], whereas low concentrations of sTNFRs might stabilize TNF-α activity [14].
Our results therefore support the hypothesis of immune-mediated cell damage by hepatitis C virus. As a consequence, future therapeutic studies in HCV infection should include both anti-viral aspects and anti-inflammatory strategies (e.g. IFN- and TNF-binding proteins).
Acknowledgments
This work was supported by grant 64/95 (to S.R.) from the Faculty of Clinical Medicine, Mannheim, Germany. These results are part of the thesis of K.H.
References
- 1.Alter HJ, Margolis HS, Krawczynski K, et al. The natural history of community acquired hepatitis C in the United States. N Engl J Med. 1992;327:1899–905. doi: 10.1056/NEJM199212313272702. [DOI] [PubMed] [Google Scholar]
- 2.Gonzalez-Peralta RP, Davis GL, Lau JY. Pathogenetic mechanisms of hepatocellular damage in chronic hepatitis C virus infection. J Hepatol. 1994;21:255–9. doi: 10.1016/s0168-8278(05)80405-2. [DOI] [PubMed] [Google Scholar]
- 3.Lau JY, Davis GL, Kniffen J, et al. Significance of serum hepatitis C virus RNA levels in chronic hepatitis C. Lancet. 1993;341:1501–4. doi: 10.1016/0140-6736(93)90635-t. [DOI] [PubMed] [Google Scholar]
- 4.Minutello MA, Pileri P, Unutmaz D, et al. Compartmentalization of T lymphocytes to the site of disease: intrahepatic CD4+ T cells specific for the protein NS4 of hepatitis C virus in patients with chronic hepatitis C. J Exp Med. 1993;178:17–25. doi: 10.1084/jem.178.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoffmann RM, Diepolder HM, Zachoval R, et al. Mapping of immunodominant CD4+ T lymphocyte epitopes of hepatitis C virus antigens and their relevance during the course of chronic infection. Hepatol. 1995;21:632–8. [PubMed] [Google Scholar]
- 6.Vilcek J, Lee TH. Tumour necrosis factor. New insights into the molecular mechanisms of its multiple actions. J Biol Chem. 1991;266:7313–20. [PubMed] [Google Scholar]
- 7.Tracey KJ, Vlassara H, Cerami A. Cachectin/tumour necrosis factor. Lancet. 1989;i:1122–6. doi: 10.1016/s0140-6736(89)92394-5. [DOI] [PubMed] [Google Scholar]
- 8.Bird GL, Sheron N, Goka AK, Alexander GJM, Williams R. Increased plasma tumor necrosis factor in severe alcoholic hepatitis. Ann Intern Med. 1990;112:917–20. doi: 10.7326/0003-4819-112-12-917. [DOI] [PubMed] [Google Scholar]
- 9.Sheron N, Lau JYN, Daniels H, et al. Increased production of tumour necrosis factor alpha in chronic hepatitis B virus infection. J Hepatol. 1991;12:241–5. doi: 10.1016/0168-8278(91)90945-8. [DOI] [PubMed] [Google Scholar]
- 10.Daniels HM, Meager A, Eddleston ALWF, Alexander GJM, Williams R. Spontaneous production of tumour necrosis factor α and interleukin-1β during interferon-α treatment of chronic HBV infection. Lancet. 1990;335:875–7. doi: 10.1016/0140-6736(90)90475-k. [DOI] [PubMed] [Google Scholar]
- 11.Smith CA, Davis T, Anderson D, et al. A receptor for tumour necrosis factor defines an unusual family of cellular and viral proteins. Science. 1990;248:1019–23. doi: 10.1126/science.2160731. [DOI] [PubMed] [Google Scholar]
- 12.Loetscher H, Pan Y-CE, Lahm H-W, et al. Molecular cloning and expression of the human 55 kd tumour necrosis factor receptor. Cell. 1990;61:351–9. doi: 10.1016/0092-8674(90)90815-v. [DOI] [PubMed] [Google Scholar]
- 13.Tartaglia LA, Goeddel DV. Two TNF receptors. Immunol Today. 1992;13:151–3. doi: 10.1016/0167-5699(92)90116-O. [DOI] [PubMed] [Google Scholar]
- 14.Aderka D, Engelmann H, Maor Y, Brakebusch C, Wallach D. Stabilization of the bioactivity of tumour necrosis factor by its soluble receptors. J Exp Med. 1992;175:323–9. doi: 10.1084/jem.175.2.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aderka D, Engelmann H, Shemer Avni Y, et al. Variation in serum levels of the soluble TNF receptors among healthy individuals. Lymphokine Cytokine Res. 1992;11:157–9. [PubMed] [Google Scholar]
- 16.Porteu F, Nathan C. Shedding of tumour necrosis factor receptors by activated human neutrophils. J Exp Med. 1990;172:599–607. doi: 10.1084/jem.172.2.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Van Zee KJ, Kohno T, Fischer E, Rock CS, Moldawer LL, Lowry SW. Tumor necrosis factor soluble receptors circulate during experimental and clinical inflammation and can protect against excessive tumor necrosis factor αin vitro and in vivo. Proc Natl Acad Sci USA. 1992;89:7845–9. doi: 10.1073/pnas.89.11.4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Desmet VJ, Gerber M, Hoofnagle JH, Manns M, Scheuer PJ. Classification of chronic hepatitis: diagnosis, grading and staging. Hepatol. 1994;19:1513–20. [PubMed] [Google Scholar]
- 19.Kallinowski B, Müller HM, Solbach C, Goeser T, Kabelitz D, Theilmann L. Hepatitis C virus RNA in different blood lymphocyte subsets. Europ J Gastroenterol. 1994;6:249–53. [Google Scholar]
- 20.Rossol S, Voth R, Brunner S, et al. Corynebacterium parvum (Propionibacterium acnes): an inducer of tumor necrosis factor-alpha in human peripheral blood mononuclear cells and monocytes in vitro. Eur J Immunol. 1990;20:1761–5. doi: 10.1002/eji.1830200821. [DOI] [PubMed] [Google Scholar]
- 21.Voth R, Rossol S, Klein K, et al. Differential gene expression of IFN-alpha and tumor necrosis factor-alpha in peripheral blood mononuclear cells from patients with AIDS related complex and AIDS. J Immunol. 1990;144:970–5. [PubMed] [Google Scholar]
- 22.Marinos G, Naoumov NV, Rossol S, et al. Tumor necrosis factor receptors in patients with chronic hepatitis B virus infection. Gastroenterol. 1995;108:1453–63. doi: 10.1016/0016-5085(95)90694-0. [DOI] [PubMed] [Google Scholar]
- 23.Böyum A. Separation of leukocytes from blood and bone marrow. Scand J Clin Lab Invest. 1968;21(Suppl. 97):77–89. [PubMed] [Google Scholar]
- 24.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium Thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–63. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 25.Ehlers S, Mielke ME, Blankenstein T, Hahn H. Kinetic analysis of cytokine gene expression in the livers of naive and immune mice infected with Listeria monocytogenes. The immediate early phase in innate resistance and acquired immunity. J Immunol. 1992;149:3016–22. [PubMed] [Google Scholar]
- 26.Hussain MJ, Mustafa A, Gallati H, Mowat AP, Mieli Vergani G, Vergani D. Cellular expression of tumour necrosis factor-α and interferon-γ in the liver biopsies of children with chronic liver disease. J Hepatol. 1994;21:816–21. doi: 10.1016/s0168-8278(94)80244-0. [DOI] [PubMed] [Google Scholar]
- 27.Cordell JL, Falini B, Erber WN, et al. Immunoenzymatic labelling of monoclonal antibodies using immune complexes of alkaline phosphatase and monoclonal anti-alkaline phosphatase (APAAP complexes) J Histochem Cytochem. 1984;32:219–28. doi: 10.1177/32.2.6198355. [DOI] [PubMed] [Google Scholar]
- 28.Tiegs G, Wolter M, Wendel A. Tumor necrosis factor is a terminal mediator in galactosamine/endotoxin-induced hepatitis in mice. Biochem Pharmacol. 1989;38:627–31. doi: 10.1016/0006-2952(89)90208-6. [DOI] [PubMed] [Google Scholar]
- 29.Nagakawa J, Hishinuma I, Hirota K, et al. Interleukin-1 alpha enhances hepatotoxicity of tumor necrosis factor-alpha in galactosamine-sensitized mice. Immunopharmacol Immunotoxicol. 1991;13:485–98. doi: 10.3109/08923979109019718. [DOI] [PubMed] [Google Scholar]
- 30.Tilg H, Wilmer A, Vogel W, et al. Serum levels of cytokines in chronic liver diseases. Gastroenterol. 1992;103:264–74. doi: 10.1016/0016-5085(92)91122-k. [DOI] [PubMed] [Google Scholar]
- 31.Larrea E, Garcia N, Qian C, Civeira MP, Prieto J. Tumor necrosis factor alpha gene expression and the response to interferon in chronic hepatitis C. Hepatol. 1995;23:210–7. doi: 10.1002/hep.510230203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tilg H, Vogel W, Dinarello CA. Interferon-alpha induces circulating tumor necrosis factor receptor p55 in humans. Blood. 1995;85:433–5. [PubMed] [Google Scholar]
- 33.Diez-Ruiz A, Tilz GP, Gutierrez-Gea F, et al. Neopterin and soluble tumor necrosis factor receptor type I in alcoholic cirrhosis. Hepatol. 1995;21:976–8. doi: 10.1002/hep.1840210414. [DOI] [PubMed] [Google Scholar]
- 34.Tilg H, Vogel W, Wiedermann CJ, et al. Circulating interleukin-1 and tumor necrosis factor antagonists in liver disease. Hepatol. 1993;18:1132–8. [PubMed] [Google Scholar]
- 35.Mc Cormick SE, Goodman ZD, Maydonovitch CL, Sjogren MH. Evaluation of liver histology, ALT elevation, and HCV RNA titer in patients with chronic hepatitis C. Am J Gastroenterol. 1996;91:1516–22. [PubMed] [Google Scholar]
- 36.Morales TG, Samplier RE, Bhattacharya A. Liver histology in individuals with positive markers for HCV and normal or minimally elevated aminotransferases. Am J Gastroenterol. 1994;89:1674–9. doi: 10.1097/00004836-199512000-00011. [DOI] [PubMed] [Google Scholar]
- 37.Haber MM, West AB, Haber AD. Relationship of aminotransferases to liver histology status in chronic hepatitis C. Am J Gastroenterol. 1995;90:1250–7. [PubMed] [Google Scholar]
- 38.Volpes R, van den Oord JJ, De Vos R, Desmet VJ. Hepatic expression of type A and type B receptors for tumor necrosis factor. J Hepatol. 1992;14:361–9. doi: 10.1016/0168-8278(92)90184-q. [DOI] [PubMed] [Google Scholar]