

Abstract
SLE is a disease characterized by the presence of multiple autoantibodies and high levels of circulating immune complexes. We studied the presence and functional relevance of autoantibodies directed against a receptor for the collagen-like stalks of the first subcomponent of complement, also known as calreticulin (cC1qR/CaR), in patients with SLE. In a cross-sectional study it was found that higher titres of antibodies against cC1qR/CaR are present in sera of SLE patients compared with normal donors. No association between anti-cC1qR/CaR titres and SLE disease activity was found. Following gel filtration of SLE serum it was found that anti-cC1qR/CaR reactivity is associated with the peak of monomeric IgG. Purified IgG from patients was able to specifically immunoprecipitate cC1qR/CaR. Since we have shown previously that cC1qR/CaR is able to inhibit the haemolytic activity of C1q, we determined a possible pathogenic role for anti-cC1qR/CaR on complement regulation. IgG derived from SLE serum reversed the inhibitory capacity of cC1qR/CaR in a dose-dependent fashion up to 63%, whereas IgG from normal donors had no significant effect. With respect to the capacity of anti-cC1qR/CaR antibodies to activate neutrophils, it was found that incubation of normal neutrophils with F(ab′)2 anti-cC1qR/CaR resulted in a very limited oxidative burst. However, cross-linking of F(ab′)2 anti-cC1qR/CaR on the neutrophils clearly induced neutrophil activation. Pre-incubation of the SLE-derived F(ab′)2 with cC1qR/CaR prevented activation of neutrophils up to 81 ± 5%. These results suggest that the presence of anti-cC1qR/CaR antibodies in patients with SLE may modulate complement and neutrophil activation.
Keywords: human, neutrophils, lupus, autoantibodies, complement, calreticulin, C1q receptor
INTRODUCTION
Circulating immune complexes (IC) are associated with the pathogenesis of different diseases such as SLE [1–4]. Deposition of IC generally results in complement activation [5–7], recruitment of other mediator systems [8] and finally tissue injury leading to development of diseases such as nephritis, vasculitis and arthritis [9]. It has been suggested that neutrophils play a significant role in inflammation by release of proteolytic enzymes and by induction of the oxidative burst. The interaction between neutrophils and IC is mediated by binding of immunoglobulins via specific Fc receptors (FcR) present on these cells [10,11]. However, since IC also may contain C1q [12,13], binding of IC to neutrophils may also be mediated by C1q receptors (C1qR) [14]. As described for FcR, it is also known that stimulation of neutrophils via C1qR on their surface can activate these cells, resulting in an enhanced oxidative metabolism [15,16]. Autoantibodies in SLE contribute to the formation of IC and are directed against different epitopes. For example, anti-C1q antibodies are associated with renal involvement, dermatitis, hypocomplementaemia and the presence of anti-dsDNA antibodies [17]. The mechanism underlying this process, however, is not fully understood. For other antibodies such as anti-CR1 the pathogenic mechanisms are more clearly defined [18]. Since IC may not only interact with phagocytic cells via FcR but also via C1qR, the possible presence of autoantibodies directed against C1qR might influence the binding of C1q containing IC to C1qR. Three types of C1qR have been described on neutrophils. The receptor for the globular domain of C1q (gC1qR [19,20]), the receptor for the collagen-like stalks of C1q which has high homology with calreticulin (cC1qR/CaR [21–24]), and the receptor for the collagen-like stalks that induces phagocytosis by neutrophils (C1qRp [14]). cC1qR/CaR is known to mediate IC binding [25] and oxidative bursts [24], which makes it a candidate to be an important mediator in autoimmune diseases. Autoantibodies against cC1qR/CaR were described to be present in many patients suffering from lupus disorders and Sjögren's syndrome (SS) [26–28] and were shown to interfere in binding of excreted cC1qR/CaR to IC [29]. Autoantibodies against cell surface-expressed cC1qR/CaR, on the other hand, may directly lead to activation of these cells. At present, however, it is not fully clear how cC1qR/CaR is involved in signal transduction. It is possible that cC1qR/CaR via interaction with a putative membrane protein, containing a transmembrane domain, may exert such effects.
To study the pathogenic effects of autoantibodies against cC1qR/CaR, we set up a specific ELISA for the detection of anti-cC1qR/CaR autoantibodies in sera from SLE patients and normal controls (ND). Furthermore, we studied the effect of anti-cC1qR/CaR autoantibodies isolated from SLE patients on the regulatory role of cC1qR/CaR in complement activation. In addition, the effect of these antibodies on neutrophil activation was assessed. Our results indicate that high anti-cC1qR/CaR titres are found in SLE patients and that these antibodies react specifically with purified cC1qR/CaR. Furthermore, these autoantibodies are able to reverse the inhibitory capacity of cC1qR/CaR on C1q haemolytic activity. F(ab′)2 anti-cC1qR/CaR are able to induce activation of polymorphonuclear neutrophils (PMN), and therefore we hypothesize that anti-cC1qR/CaR antibodies in SLE may potentially influence ongoing inflammatory reactions.
MATERIALS AND METHODS
Sera
Sera were collected from 56 patients with SLE and from 56 healthy individuals. SLE patients fulfilled the criteria for the classification of SLE [30]. The sera were stored at −70°C before use.
Anti-cC1qR/CaR ELISA
Either 10 μg/ml purified cC1qR/CaR, isolated from neutrophils as described [31], or bovine serum albumin (BSA), were coated on 96-well microtitre plates (Greiner BV, Alphen a/d Rijn, The Netherlands) for 2 h at 37°C in coating buffer (0.1 m sodium carbonate pH 9.6). After washing, wells were incubated for 1 h at 37°C with different sera obtained from either ND or SLE patients, diluted in PBS containing 0.05% (v/v) Tween, 1% fetal calf serum (FCS) in the presence or absence of 5 m NaCl. Although Bordin et al. [32] demonstrated complete inhibition of C1q binding to cC1qR/CaR with 2 m NaCl, we found even lower background intensity using 5 m NaCl. After washing, wells were incubated for 1 h at 37°C with a mouse IgG1 κ anti-human γ MoAb (HB43; ATCC, Rockville, MD) conjugated to digoxigenin (DIG; Boehringer, Mannheim, Germany). Then the wells were washed and incubated for 1 h at 37°C with a preformed mix of 10 parts of goat F(ab) anti-DIG avidin (Boehringer) and one part of biotin-peroxidase. After washing, wells were incubated for 1 h at 37°C with the substrate of peroxidase, 2,2′-azino-bis (3-ethylbenzthiazoline-6-sulphonic acid) (ABTS; Sigma, St Louis, MO). The optical density (OD) at 415 nm was measured using a Titertek Multiskan EL 312e (Bio-Tek Instruments Inc., Winooski, VT). Student's t-test for unpaired data was used to calculate P values. The mean anti-cC1qR/CaR titres + 2 s.d. measured in serum samples obtained from healthy individuals were considered to be the range of normal titres.
Isolation of IgG and detection of anti-cC1qR/CaR reactivity
One millilitre portions of either normal human sera or sera from patients with SLE were centrifuged at 10 000 g and the supernatant applied on a 90 × 1.5 cm Sephacryl S-300 SF column (Pharmacia, Roosendaal, The Netherlands). Fractions were collected and tested for IgG and anti-cC1qR/CaR antibodies by a standard ELISA, whereas C1q content in the fractions was determined using a haemolytic assay. Protein content was measured using the BCA protein assay (Pierce Chemical Co., Rockford, IL). In addition, IgG from sera of patients and controls was purified by DEAE anion exchange chromatography. F(ab′)2 were prepared by pepsin digestion [33] and assessed for reactivity with purified cC1qR/CaR in ELISA (data not shown).
Immunoprecipitation of cC1qR/CaR
cC1qR/CaR was isolated from neutrophils as described [31] and conjugated to biotin as indicated by the manufacturer's protocol (Zymed Labs Inc.). Biotinylated cC1qR/CaR was then precleared by incubation for 3 h at 4°C with Prot G Sepharose 4 FastFlow (Pharmacia). Precleared cC1qR/CaR–biotin was then incubated overnight at 4°C with either serum of ND or SLE patients or with purified IgG from the same donors. Alternatively, SLE IgG that was preincubated for 1 h at 4°C with two doses of purified cC1qR/CaR was incubated with cC1qR/CaR–Bio. After addition of 5 μl Prot G suspension and another incubation of 2 h at 4°C, samples were centrifuged and the pellet was washed 10 times with PBS. Then 10 μl of sample buffer were added and the mixture was boiled for 10 min. The samples were electrophoresed on a 10% polyacrylamide gel as described [34]. After the proteins were transferred to Imobilon P (Millipore, Bedford, MA), free sites were blocked by overnight incubation with PBS containing 5% Elk milk. The blot was incubated for 1 h at 4°C with streptavidin–horseradish peroxidase (HRP) in PBS containing 5% Elk milk and thereafter washed for 30 min with PBS. Finally, blots were incubated with diaminobenzidine tetrahydrochloride (DAB; Sigma) and after a few minutes the staining reaction was stopped by extensive washing with water.
Haemolytic assay for cC1qR/CaR activity
cC1qR/CaR activity was determined as described before [31]. To determine the effect of autoantibodies against cC1qR/CaR on complement inhibition, the following experiment was carried out. Antibody-sensitized erythrocytes (EA) were incubated with C1qD, a limited amount of C1q and such an amount of cC1qR/CaR that ≈ 60% inhibition of complement activation was obtained. Alternatively, cC1qR/CaR was preincubated for 30 min at 30°C followed by 10 min on ice with either buffer alone or with different concentrations of normal human IgG or IgG isolated from SLE serum. The percentage lysis of the triplicates was determined, relative to a reagent blank and 100% lysis, expressed as U/ml (Z) and converted to percentage inhibition.
Neutrophil isolation and activation
For the isolation of polymorphonuclear cells as described by Leid et al. [35], enriched leucocyte populations were resuspended in PBS containing 10 U/ml heparin (Sigma) and centrifuged for 15 min at 3500 g. Erythrocytes in the pellet were lysed twice by incubation for 20 min on ice with lysis buffer (167 mm NH4Cl, 10 mm KHCO3, 1 mm EDTA, pH 7.4). The leucocytes were washed with PBS containing 0.9 mm CaCl2, 0.5 mm MgCl2 and 5.5 mmd-glucose, and directly used in the experiments. Three experiments using triplicates of 1 × 107 neutrophils were incubated for 30 min at 37°C in the presence of 1 mm cytochrome C (Sigma) together with either buffer, normal donor F(ab′)2 or F(ab′)2 isolated from SLE serum. Samples without neutrophils served as negative controls, and the incubation of neutrophils with 1 μg/ml phorbol myristate acetate (PMA; Sigma) was used as a positive control. After centrifugation for 10 min at 500 g, the OD of the supernatant was measured at 550 nm.
Cross-linking of anti-cC1qR/CaR F(ab′)2 on the surface of PMN was performed as follows. Neutrophils were first incubated for 20 min at room temperature with normal donor F(ab′)2 or F(ab′)2 isolated from SLE patients, washed and then incubated with goat F(ab′)2 anti-human IgG together with cytochrome C for 30 min at 37°C. To determine whether the effect of SLE F(ab′)2 on PMN was dependent on specific antibodies against cC1qR/CaR, PMN were incubated with F(ab′)2 that were preincubated for 20 min at room temperature with either buffer or different concentrations of purified cC1qR/CaR and subsequently assessed for activation.
RESULTS
Detection of autoantibodies against cC1qR/CaR in patients with SLE
To determine the presence of antibodies directed against cC1qR/CaR, 56 sera from SLE patients and 56 ND were tested with a specific anti-cC1qR/CaR ELISA. Purified cC1qR/CaR, or equal amounts of BSA, were coated and incubated with diluted sera in the presence of 5 m NaCl. After washing, bound antibodies were detected with a DIG-conjugated MoAb followed by a mixture of anti-DIG avidin and biotin–peroxidase and incubation with ABTS, the substrate of peroxidase. This amplification step provided a three-fold enhancement in absorbance compared with the use of direct peroxidase-conjugated anti-human IgG antibodies (data not shown). It was found that sera of SLE patients contained significantly more reactivity against cC1qR/CaR (average absorbance 1.029) than controls (average absorbance 0.642) (P < 0.0001) (Fig. 1). When normal anti-cC1qR/CaR titres are considered as those that are below the mean absorbance of normal donors + 2 s.d., then 41% of the SLE patients had positive anti-cC1qR/CaR titres. Neither SLE sera nor ND sera showed an absorbance of more than 0.200 with BSA under these conditions. No relation was found between different treatments with immunosuppressive drugs and antibody titre.
Fig. 1.

Anti-cC1qR/CaR titres in normal human sera and SLE sera. Purified human cC1qR/CaR was coated and incubated with sera of normal donors (ND) or SLE patients. Subsequently, bound IgG was measured by incubation with an anti-human IgG MoAb conjugated to digoxigenin. The optical density was measured and plotted. —, Average; ----, ND average + 2 s.d.
Characterization of anti-cC1qR/CaR autoantibodies
To determine the specificity of the binding of autoantibodies to cC1qR/CaR, sera from three SLE patients or ND were selected and tested in different dilutions for binding to purified cC1qR/CaR or BSA. As depicted in Fig. 2, selected high titre sera from SLE patients bound to cC1qR/CaR in a dose-dependent fashion. Such a dose-dependent binding to cC1qR/CaR by antibodies from normal donors was not observed: in these cases the absorbance remained ≈ 0.100, which is the same absorbance as determined by anti-cC1qR/CaR-negative SLE patients. To determine the size of IgG reactive with cC1qR/CaR, positive SLE sera were subjected to gel filtration. The fractions were assessed for C1q haemolytic activity, IgG by ELISA and anti-cC1qR/CaR antibody activity. The results in Fig. 3 indicate that the anti-cC1qR/CaR activity is associated with the IgG-containing fractions without major overlap with C1q haemolytic activity. To find out whether anti-cC1qR/CaR IgG also reacted with cC1qR/CaR in the fluid phase, control IgG and patient IgG were used to immunoprecipitate biotinylated cC1qR/CaR (Fig. 4). It was found that IgG isolated from SLE serum immunoprecipitated a band of 66 kD, whereas IgG from ND did not. Pre-incubation of SLE IgG with 50 and 100 μg/ml of purified cC1qR/CaR prevented immunoprecipitation of biotinylated cC1qR/CaR, whereas preincubation with 100 μg/ml BSA was not able to do so (Fig. 4). No precipitation of cC1qR/CaR was seen in PBS.
Fig. 2.

Dose-dependent binding of anti-cC1qR/CaR antibodies to cC1qR/CaR. Purified cC1qR/CaR or bovine serum albumin (BSA) was coated on microtitre wells and incubated with increasing concentrations of three SLE sera in the presence of 5 m NaCl. Bound IgG was detected by incubation with a MoAb anti-human IgG conjugated to digoxigenin. The optical density was measured and plotted.
Fig. 3.
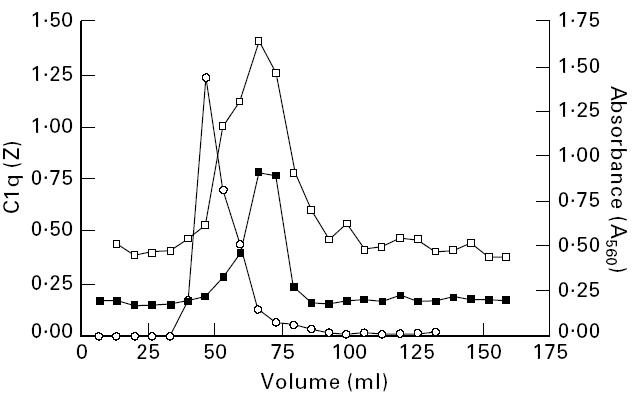
IgG isolated from SLE serum binds cC1qR/CaR. IgG from SLE serum was isolated by gel filtration using a Sepharose S-300 column. In the fractions IgG (□) and anti-cC1qR/CaR (▪) were determined by ELISA and C1q content (○) was determined by a haemolytic assay.
Fig. 4.
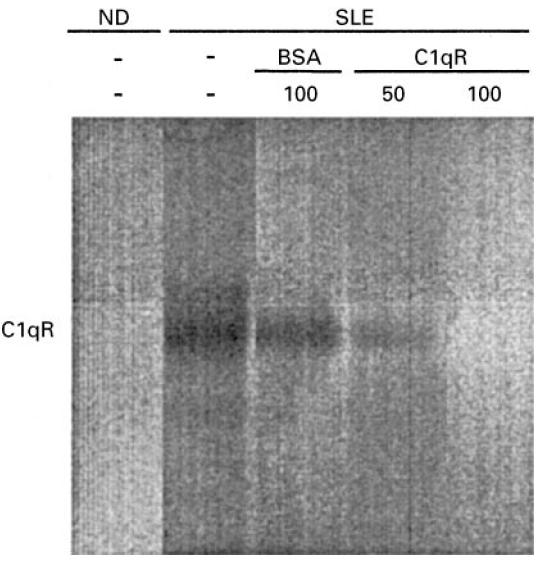
Immunoprecipitation of cC1qR/CaR by purified IgG from SLE. cC1qR/CaR was conjugated to biotin and incubated with purified IgG from SLE serum or normal donor (ND) serum. Alternatively, SLE IgG that was preincubated with increasing amounts of purified cC1qR/CaR was added to cC1qR/CaR-Bio and incubated. After precipitation using Prot G, the precipitate was electrophoresed on a polyacrylamide gel and blotted on PVDF membranes. CC1qR/CaR-Bio was demonstrated by subsequent incubation with streptavidin–horseradish peroxidase (HRP) and DAB.
Determination of the functional role of autoantibodies against cC1qR/CaR
The capacity of IgG from SLE patients to interfere in the complement regulatory role of cC1qR/CaR was determined next. For this experiment, cC1qR/CaR was incubated with different amounts of either SLE IgG or ND IgG and thereafter assessed for cC1qR/CaR activity in a haemolytic assay. The experiment was performed three times with similar results. The results in Fig. 5 demonstrate that IgG isolated from SLE serum reversed the inhibitory capacity of cC1qR/CaR in a dose-dependent fashion up to 63%, whereas ND IgG maximally reversed cC1qR/CaR activity by 14%. A control experiment without addition of cC1qR/CaR did not result in complement inhibition. Besides the capacity of SLE antibodies to react with cC1qR/CaR and thereby interfere with complement regulation, binding of the antibodies to cC1qR/CaR on cells might elicit cellular responses. To study the capacity of anti-cC1qR/CaR to activate PMN, F(ab′)2 fragments isolated from patients and ND were incubated with PMN, after which oxygen radical release was assessed. Again, three experiments revealed comparable data. ND F(ab′)2 induced a limited oxygen radical release of 8%, while F(ab′)2 from a SLE patient induced a maximal oxygen radical release of 23% (Fig. 6a). In order to induce significant cross-linking of cell-bound F(ab′)2, polyclonal F(ab′)2 anti-IgG antibody was employed. A significant dose-dependent release of up to 74% of oxygen radicals was found after incubation with SLE F(ab′)2 compared with PMA-induced release. Addition of a cross-linking antibody to neutrophils that were preincubated with ND F(ab′)2 did not result in increased oxygen radical release (Fig. 6a). Neutrophils incubated only with the polyclonal anti-F(ab′)2 antibody did not show any neutrophil activation (data not shown). To determine whether anti-cC1qR/CaR F(ab′)2 were responsible for the observed activation of the neutrophils, an experiment was performed in which SLE F(ab′)2 was preincubated with different concentrations of purified cC1qR/CaR before incubation of the antibodies with the neutrophils. The results shown in Fig. 6b demonstrate that preincubation of SLE F(ab′)2 with soluble cC1qR/CaR prevents neutrophil activation dependent on the dose of cC1qR/CaR used. No significant effect of cC1qR/CaR was seen on ND F(ab′)2-induced activation of PMN.
Fig. 5.
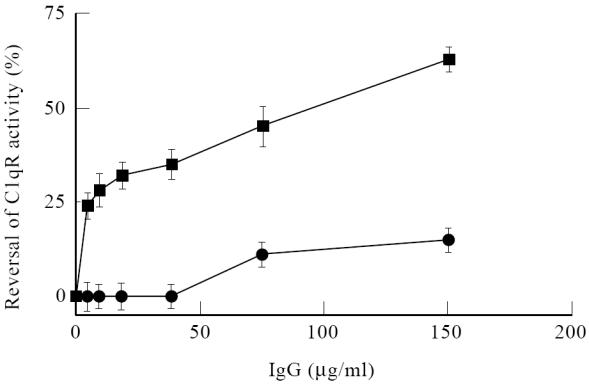
Reversal of cC1qR/CaR activity by SLE IgG. Purified cC1qR/CaR was incubated with either increasing concentrations of nornal donor (ND) IgG (•) or SLE IgG (▪). After incubation with antibody-sensitized erythrocytes (EA) and a limited amount of C1q in C1qD serum the percentage reversal on the inhibitory effect of cC1qR/CaR was plotted.
Fig. 6.

Neutrophil activation by SLE F(ab′)2. Freshly isolated neutrophils were incubated with different dilutions of F(ab′)2 isolated from normal donor (ND) serum or SLE serum in the presence of cytochrome C. As a positive control, neutrophils were stimulated with phorbol myristate acetate (PMA). Alternatively, neutrophils were incubated with either ND F(ab′)2 or SLE F(ab′)2. After washing, goat IgG anti-human IgG was added together with cytochrome C to X-link bound anti-cC1qR/CaR F(ab′)2. After incubation, the optical density (OD) of the supernatants was measured and the percentage oxygen radical release, compared with release induced by PMA stimulation, was plotted (a). To determine the specificity, F(ab′)2 isolated from SLE serum was preincubated with different concentrations of purified cC1qR/CaR before being added to neutrophils. After an incubation the OD of the supernatant was measured and the percentage release of oxygen radicals was calculated and plotted (b).
Analysis of the relation between SLE disease activity and anti-cC1qR/CaR autoantibody titres
To determine whether an association exists between anti-cC1qR/CaR titres and disease activity in SLE patients, titres of anti-cC1qR/CaR antibodies that were assessed at different time points were compared with the SLE disease activity index (SLEDAI) [36]. No association was found. Also, specific organ involvement in SLE was not associated with increased anti-cC1qR/CaR titres. However, individual patients may show a correlation between anti-cC1qR/CaR titre and SLEDAI. For example, one patient with fluctuating disease activity clearly showed a relation between anti-cC1qR/CaR titres and the SLEDAI. However, other patients with no clear relation between anti-cC1qR/CaR titres and disease activity were also found (data not shown).
DISCUSSION
This study describes the occurrence of autoantibodies against cell surface cC1qR/CaR in patients with SLE. It was found that the percentage of SLE patients having positive titres of autoantibody directed against cC1qR/CaR (41%, this study) is very similar to the percentage positives that was described earlier (40% [29]). By gel filtration we found that the anti-cC1qR/CaR antibodies were associated with the IgG fractions. To determine the capacity of the isolated antibodies to bind cC1qR/CaR in the fluid phase, immunoprecipitation of biotinylated cC1qR/CaR was carried out. It was clearly shown that IgG from SLE patients immunoprecipitated cC1qR/CaR. The capacity of SLE IgG to immunoprecipitate biotinylated cC1qR/CaR was inhibited by preincubation of the IgG with increasing amounts of unlabelled cC1qR/CaR. We described earlier that cC1qR/CaR is able to inhibit C1q haemolytic activity [31]. In the experiments to determine the capacity of SLE antibodies to interfere in this cC1qR/CaR-dependent regulation of complement activation, a dose-dependent effect was found with a maximal reversal of cC1qR/CaR activity of 63%. Recently, anti-cC1qR/CaR was shown also to act on a different level on complement activation. Kishore et al. [29] showed that anti-cC1qR/CaR is able to reverse the capacity of excreted cC1qR/CaR to inhibit C1q association with IC because both IgG and cC1qR/CaR compete for the same binding site on C1q.
Because cC1qR/CaR-bearing neutrophils are involved in tissue injury during inflammation, we investigated the possibility of anti-cC1qR/CaR antibodies to activate neutrophils. It was found that F(ab′)2 fragments of SLE patients induced limited neutrophil activation by itself. However, cross-linking of these membrane-bound F(ab′)2 resulted in a marked enhancement of the response. Preincubation of the anti-cC1qR/CaR F(ab′)2 with purified cC1qR/CaR reversed this effect, indicating that the neutrophils were predominantly activated by specific anti-cC1qR/CaR antibodies. The presence of high titres of circulating IC in patients with SLE together with specific anti-cC1qR/CaR antibodies may result in constitutive activation of neutrophils which may contribute to the severity of the disease. Although our results might provide insight, it is still not clear how cC1qR/C1R triggering may induce signal transduction. Only recently the homology between cC1qR and calreticulin (CaR), a calcium-binding protein found in many cell types with many different functions [23,37,38], was described [22]. Besides similarities, however, also clear differences exist between cC1qR and CaR. Whereas cC1qR, for example, is thought to be a transmembrane molecule on the cell surface, CaR has a ubiquitous cellular distribution and is present in compartments such as the endoplasmic reticulum (ER), secretory granules of T lymphocytes [39], supernatant of activated neutrophils [23] and calciosomes [40]. Until now, cell surface expression of CaR has only been described on lung fibroblasts [41] and mouse melanoma cells [42]. Furthermore, cC1qR binds to the collagen-like domain of C1q [21], whereas for CaR it is still debatable whether it binds the globular domains of C1q [43] or the collagen-like stalks [44].
In a cross-sectional study of patients with SLE no association between clinical parameters and anti-cC1qR/CaR levels was found. To determine whether, on an individual level, titres of anti-cC1qR/CaR antibodies were related to disease activity, a longitudinal analysis was performed in four patients. In one patient the anti-cC1qR/CaR titres varied over time and appeared to be related to SLE activity, but this relation was not found in the other three patients. In conclusion, further studies must be performed to determine whether the presence of harmful anti-cC1qR/CaR antibodies is an epiphenomenon in SLE, or is involved in the development of the disease.
Acknowledgments
This project was supported by the Netherlands Organization for Scientific Research.
References
- 1.Levinsky RJ, Cameron JS, Soothill JF. Serum immune complexes and disease activity in lupus nephritis. Lancet. 1977;1:564–7. doi: 10.1016/s0140-6736(77)91998-5. [DOI] [PubMed] [Google Scholar]
- 2.Lloyd W, Schur PH. Immune complexes, complement, and anti-DNA in exacerbations of systemic lupus erythematosus (SLE) Med. 1981;60:208–17. doi: 10.1097/00005792-198105000-00004. [DOI] [PubMed] [Google Scholar]
- 3.Abrass CK, Nies KM, Louie JS, Border WA, Glassock RJ. Correlation and predictive accuracy of circulating immune complexes with disease activity in patients with systemic lupus erythematosus. Arthritis Rheum. 1980;23:273–82. doi: 10.1002/art.1780230302. [DOI] [PubMed] [Google Scholar]
- 4.Andre-Schwartz J, Datta SK, Schoenfeld Y, Isenberg DA, Stollar BD, Schwartz RS. Binding of cytoskeletal proteins by monoclonal anti-DNA lupus autoantibodies. Clin Immunol Immunopathol. 1984;31:261–71. doi: 10.1016/0090-1229(84)90246-0. [DOI] [PubMed] [Google Scholar]
- 5.Schur PH. Complement in lupus. Clin Rheum Dis. 1975;1:519–43. [Google Scholar]
- 6.Schur PH. Complement and lupus erythematosus. Arthritis Rheum. 1982;25:793–8. doi: 10.1002/art.1780250715. [DOI] [PubMed] [Google Scholar]
- 7.Belmont HM, Hopkins P, Edelson HS, et al. Complement activity during systemic lupus erythematosus. Arthritis Rheum. 1986;29:1085–9. doi: 10.1002/art.1780290905. [DOI] [PubMed] [Google Scholar]
- 8.Erdei A, Fuest G, Gergly J. The role of C3 in the immune response. Immunol Today. 1991;12:332–7. doi: 10.1016/0167-5699(91)90011-H. [DOI] [PubMed] [Google Scholar]
- 9.Mannik M. Pathophysiology of circulating immune complexes. Arthritis Rheum. 1982;25:783–7. doi: 10.1002/art.1780250713. [DOI] [PubMed] [Google Scholar]
- 10.Schifferli JA, Ng YC, Peters DK. The role of complement and its receptors in the elimination of immune complexes. N Eng J Med. 1986;315:488–95. doi: 10.1056/NEJM198608213150805. [DOI] [PubMed] [Google Scholar]
- 11.Schifferli JA, Taylor RP. Physiological and pathological aspects of circulating immune complexes. Kidney Int. 1989;35:993–1003. doi: 10.1038/ki.1989.83. [DOI] [PubMed] [Google Scholar]
- 12.Loos M. The classical complement pathway: mechanism of activation of the first component by antigen–antibody complexes. Prog Allergy. 1982;30:135–92. [PubMed] [Google Scholar]
- 13.Cooper NZ. The classical complement pathway: activation and regulation of the first complement component. Adv Immunol. 1985;37:151–216. doi: 10.1016/s0065-2776(08)60340-5. [DOI] [PubMed] [Google Scholar]
- 14.Guan E, Robinson SL, Goodman EB, Tenner AJ. Cell-surface protein identified on phagocytic cells modulates the C1q-mediated enhancement of phagocytosis. J Immunol. 1994;152:4005–16. [PubMed] [Google Scholar]
- 15.Hamada A, Young J, Chmielewski RA, Greene BM. C1q enhancement of antibody-dependent granulocyte-mediated killing of nonphagocytosable targets in vitro. J Clin Invest. 1988;82:945–9. doi: 10.1172/JCI113702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tenner AJ, Cooper NR. Analysis of receptor-mediated C1q binding to human peripheral blood mononuclear cells. J Immunol. 1980;125:1658–64. [PubMed] [Google Scholar]
- 17.Siegert CEH, Daha MR, Westedt M-L, van der Voort EAM, Breedveld FC. IgG autoantibodies against C1q are correlated with nephritis, hypocomplementemia, and dsDNA antibodies in systemic lupus erythematosus. J Rheumatol. 1991;18:230–4. [PubMed] [Google Scholar]
- 18.Wilson JG, Jack RM, Wong WW, Schur PH, Fearon DT. Autoantibody to the C3b/C4b receptor and absence of this receptor from erythrocytes of a patient with systemic lupus erythematosus. J Clin Invest. 1985;76:182–90. doi: 10.1172/JCI111944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghebrehiwet B, Lim B-L, Peerschke EIB, Willis AC, Reid KBM. Isolation, cDNA cloning, and overexpression of a 33-kD cell surface glycoprotein that binds to the globular ‘heads’ of C1q. J Exp Med. 1994;179:1809–21. doi: 10.1084/jem.179.6.1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eggleton P, Ghebrehiwet B, Sastry KN, et al. Identification of a gC1q-binding protein (gC1q-R) on the surface of human neutrophils. J Clin Invest. 1995;95:1569–78. doi: 10.1172/JCI117830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peerschke EIB, Ghebrehiwet B. Human blood platelets possess specific binding sites for C1q. J Immunol. 1987;138:1537–41. [PubMed] [Google Scholar]
- 22.Malhotra R, Willis AC, Jensenius JC, Jackson J, Sim RB. Structure and homology of human C1q receptor (collectin receptor) Immunol. 1993;78:341–8. [PMC free article] [PubMed] [Google Scholar]
- 23.Eggleton P, Lieu TS, Zappi EG, et al. Calreticulin is released from activated neutrophils and binds to C1q and mannan-binding protein. Clin Immunol Immunopathol. 1994;72:405–9. doi: 10.1006/clin.1994.1160. [DOI] [PubMed] [Google Scholar]
- 24.Jonsson H, Sturfelt G. A novel assay for neutrophil clustering activity of human sera: relation to disease activity and neutropenia in systemic lupus erythematosus. Ann Rheum Dis. 1990;49:46–50. doi: 10.1136/ard.49.1.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van den Dobbelsteen MEA, van der Woude FJ, Schroeijers WEM, Klar-Mohamad N, van Es LA, Daha MR. C1q, a subunit of the first component of complement, enhances the binding of aggregated IgG to rat renal mesangial cells. J Immunol. 1993;151:4315–24. [PubMed] [Google Scholar]
- 26.Martinez-Lavin M, Vaughan JH, Tan EM. Autoantibodies and the spectrum of Sjögren's syndrome. Ann Intern Med. 1979;91:185–90. doi: 10.7326/0003-4819-91-2-185. [DOI] [PubMed] [Google Scholar]
- 27.Sontheimer RD, Maddison PJ, Reichlin M, Jordon RE, Stastny P, Gilliam JN. Serologic and HLA associations in subacute cutaneous lupus erythematosus, a clinical subset of lupus erythematosus. Ann Intern Med. 1982;97:664–71. doi: 10.7326/0003-4819-97-5-664. [DOI] [PubMed] [Google Scholar]
- 28.Rokeach LA, Haselby JA, Meilof JF, et al. Characterization of the autoantigen calreticulin. J Immunol. 1991;147:3031–9. [PubMed] [Google Scholar]
- 29.Kishore U, Sontheimer RD, Sastry KN, et al. The systemic lupus erythematosus (SLE) disease autoantigen-calreticulin can inhibit C1q association with immune complexes. Clin Exp Immunol. 1997;108:181–90. doi: 10.1046/j.1365-2249.1997.3761273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tan EM, Cohen AS, Fries JF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25:1271–7. doi: 10.1002/art.1780251101. [DOI] [PubMed] [Google Scholar]
- 31.van den Berg RH, Faber-Krol MC, van Es LA, Daha MR. Regulation of the function of the first component of complement by human C1q receptor. Eur J Immunol. 1995;25:2206–10. doi: 10.1002/eji.1830250814. [DOI] [PubMed] [Google Scholar]
- 32.Bordin S, Kolb WP, Page RC. C1q receptors on cultured human gingival fibroblasts: analysis of binding properties. J Immunol. 1983;130:1871–5. [PubMed] [Google Scholar]
- 33.Stad RK, Bruijn JA, van Gijlswijk-Jansen DJ, van Es LA, Daha MR. An acute model for IgA-mediated glomerular inflammation in rats induced by monoclonal polymeric rat IgA antibodies. Clin Exp Immunol. 1993;92:514–21. doi: 10.1111/j.1365-2249.1993.tb03430.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–5. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 35.Leid RW, Van der Heijden I, Ballieux Bepb. Cleavage and inactivation of human C1-inhibitor by the human leukocyte proteinase, proteinase-3. Eur J Immunol. 1993;23:2939–45. doi: 10.1002/eji.1830231132. [DOI] [PubMed] [Google Scholar]
- 36.Bombardier C, Gladman DD, Urowitz MB, Caron D, Hsing Chang C. Committee on prognosis studies in SLE. Derivation of the SLEDAI. Arthritis Rheum. 1992;35:630–40. doi: 10.1002/art.1780350606. [DOI] [PubMed] [Google Scholar]
- 37.Dedhar S. Novel functions for calreticulin: interaction with integrins and modulation of gene expression? Trends Biochem Sci. 1994;19:269–71. doi: 10.1016/0968-0004(94)90001-9. [DOI] [PubMed] [Google Scholar]
- 38.Wada I, Imai S, Kai M, Sakane F, Kanoh H. Chaperone function of calreticulin when expressed in the endoplasmic reticulum as the membrane-anchored forms. J Biol Chem. 1995;270:20298–304. doi: 10.1074/jbc.270.35.20298. [DOI] [PubMed] [Google Scholar]
- 39.Dupuis M, Schaerer E, Krause KH, Tschopp J. The calcium-binding protein calreticulin is a major constituent of lytic granules in cytolytic T lymphocytes. J Exp Med. 1993;177:1–7. doi: 10.1084/jem.177.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Habicht GS, Beck G, Ghebrehiwet B. C1q inhibits the expression of B lymphoblastoid cell line interleukin 1 (IL-1) J Immunol. 1987;138:2593–7. [PubMed] [Google Scholar]
- 41.Zhu J, Newkirk MM. Viral induction of the human autoantigen calreticulin. Clin Invest Med. 1994;17:196–205. [PubMed] [Google Scholar]
- 42.White TK, Zhu Q, Tanzer ML. Cell surface calreticulin is a putative mannoside lectin which triggers mouse melanoma cell spreading. J Biol Chem. 1995;270:15926–9. doi: 10.1074/jbc.270.27.15926. [DOI] [PubMed] [Google Scholar]
- 43.Kishore U, Sontheimer RD, Sastry KN, et al. Release of calreticulin from neutrophils may alter C1q-mediated immune functions. Biochem J. 1997;322:543–50. doi: 10.1042/bj3220543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stuart GR, Lynch NJ, Lu J, et al. Localisation of the C1q binding site within C1q receptor/calreticulin. FEBS Letters. 1996;397:245–9. doi: 10.1016/s0014-5793(96)01156-8. [DOI] [PubMed] [Google Scholar]